

Abstract
BB-83698, a potent and selective inhibitor of peptide deformylase, was the first compound of this novel antibacterial class to progress to clinical trials. Single- and/or multiple-dose studies with doses ranging from 10 to 50 mg of BB-83698/kg of body weight were done with mice, rats, and dogs. Intravenous pharmacokinetics were characterized by low to moderate clearances and moderate volumes of distribution for all species. In dogs, but not in rodents, central nervous system (CNS) effects were dose limiting for intravenously administered BB-83698 and were suspected to be related to a high maximum concentration of the agent in plasma (Cmax) rather than to total systemic exposure. Controlled infusion studies with dogs demonstrated that CNS effects could be avoided without compromising systemic exposure by reducing the Cmax. A randomized, double-blind, placebo-controlled, five-way-crossover, single-dose-escalation, phase I study to explore the safety, tolerability, and pharmacokinetics of intravenous BB-83698 at doses ranging from 10 to 475 mg was performed with healthy male volunteers. Systemic exposures were generally in linear relationships with administered doses in animals and humans. Pharmacokinetics were consistent, predictable, and exhibited good allometric scaling among all species (r2 >0.98). Moreover, BB-83698 dosing in humans proceeded to a predicted efficacious exposure (the area under the concentration-time curve/MIC ratio, up to 184) without any clinically significant adverse effects.
Inhibition of bacterial peptide deformylase (PDF) is a novel antibacterial therapeutic approach (3, 4, 17). PDF is an essential bacterial metalloenzyme that deformylates the N-formylmethionine of newly synthesized bacterial polypeptides. The gene encoding PDF, def, is common to all sequenced pathogenic bacteria, and PDF inhibitors have been shown to be active against many pathogens (3, 4, 11, 16, 17). While PDF-like sequences have been identified in human mitochondria, studies indicate that human PDF has a low catalytic activity and is not essential for mammalian cell protein synthesis (15). PDF therefore represents an attractive target for antimicrobial chemotherapy because it is essential for bacterial growth, is highly conserved among bacteria, offers selectivity, and is novel.
Given the escalating problem of drug-resistant bacterial infections in both community and hospital settings (12, 14), PDF inhibition is attractive as a therapeutic alternative. PDF inhibitors have been shown to exhibit potent in vitro and in vivo activity against gram-positive organisms, including drug-resistant isolates (3, 4, 8, 9, 11; W. A. Craig and D. Andes, Abstr. 41st Intersci. Conf. Antimicrob. Agents Chemother., abstr. F-355, 2001). BB-83698 has demonstrated antibacterial activity against the most important community respiratory tract pathogens, including drug-resistant isolates, in particular, isolates of Streptococcus pneumoniae, group A streptococci (Streptococcus pyogenes), Moraxella catarrhalis, Mycoplasma pneumoniae, Chlamydia pneumoniae, and Legionella pneumophila (3, 6, 8, 13). The in vitro activities of BB-83698 and comparable drugs have been determined against a panel of organisms, including 281 streptococcal strains, 154 strains of Staphylococcus aureus, 110 strains of Haemophilus influenzae, and 50 Moraxella catarrhalis strains, all selected for their resistance phenotypes. The MICs at which 90% of the isolates tested were inhibited were between 0.25 and 0.5 mg/liter (depending on drug resistance phenotype) for S. pneumoniae strains, including penicillin-, erythromycin-, levofloxacin-, and multidrug-resistant strains (13). In vitro resistance development rates for S. pneumoniae strains are in line with those seen for the development of resistance to newer fluoroquinolones, such as moxifloxacin (A. Noel, K. E. Bowker, R. A. Howe, and A. P. MacGowan, Abstr. 42nd Intersci. Conf. Antimicrob. Agents Chemother., abstr. F-1680, 2002).
BB-83698 is a synthetic N-formyl hydroxylamine-based peptidomimetic (Fig. 1) and represents additional optimization from the early PDF inhibitor prototype BB-3497 (3, 5, 6). It is a potent and selective inhibitor of PDF (4) and is the first compound in this novel class to enter clinical trials. In preclinical studies, BB-83698 has demonstrated oral, subcutaneous, and intravenous (i.v.) efficacy against S. pneumoniae and S. aureus in peritoneal, lung, and thigh infections (1, 4; Craig and Andes, 41st ICAAC; data not shown). In addition, extensive pharmacokinetic (PK) and pharmacodynamic studies utilizing the murine thigh infection model have identified the area under the concentration-time curve (AUC)/MIC ratio as the parameter which best correlates with efficacy (Craig and Andes, 41st ICAAC). Such preclinical studies validate the therapeutic concept for consideration of BB-83698 as a novel antipneumococcal agent and provide guidance for initial clinical testing.
FIG. 1.

Chemical structure of first-in-class PDF inhibitor BB-83698. Mw, molecular weight.
The primary aim of this study was to describe the PK of BB-83698 in animals and humans. Clinical results from healthy human volunteers following i.v. administration at single, escalating dose levels are presented. Moreover, allometric projections from mouse, rat, and dog PK data are described. In preclinical safety testing, central nervous system (CNS) effects were dose limiting for BB-83698 in dogs but not in rodents and may have been related to the maximum concentration of the agent in plasma (Cmax). Accordingly, a secondary goal related to toxicokinetics and toxicodynamics involved controlled infusion studies in dogs to test the hypothesis that the CNS effects observed in dogs were due to high Cmaxs rather than total systemic exposure.
MATERIALS AND METHODS
Mouse PK.
ICR female mice, three animals per time point, were given i.v. 10- and/or 50-mg doses of a BB-83698 solution (pH 4.1) formulated in 50 mM citrate-140 mM NaCl per kg of body weight. Following a single dose, 0.5- to 0.7-ml blood samples were obtained by cardiac puncture. Sampling time points were 5 and 20 min and 1, 2, and 5 h. Animal care and use guidelines required that blood samples not be obtained from the same animal at all time points, due to the relatively small blood volumes in mice. Hence, animals were bled once, and blood samples were collected in microcentrifuge tubes coated with lithium heparin and placed on ice until they could be processed for plasma by centrifugation. BB-83698 was extracted from plasma samples by a protein precipitation method. A non-glycolipoprotein-validated liquid chromatography-tandem mass spectrometry (LC/MS/MS) method with appropriate quality controls (QCs) was used for quantitation of BB-83698 in plasma samples at Genesoft Pharmaceuticals, South San Francisco, Calif., essentially as previously described (8). Analysis of plasma drug concentrations yielded results wherein standard deviations at each time point were generally <25% of the means for the groups. All animal studies were done in accordance with federal guidelines in the Guide for the Care and Use of Laboratory Animals (14a) and/or the guidelines established by internal institutional animal care and use committees at GeneSoft Pharmaceuticals, Northview Labs (Hercules, Calif.), Central Toxicology Laboratory (CTL; Cheshire, United Kingdom), or RCC Ltd.
Twenty-eight-day rat toxicokinetics.
Sprague-Dawley rats (three of each sex per time point per treatment group) were given i.v. 10, 22, or 50 mg of BB-83698/kg/day for a total of 28 days. Serial plasma samples, from ∼0.2 ml of blood, were collected on days 1 and 14 or 28 for BB-83698 quantitation at the following time points: predose and 0.083, 0.33, 1, 2, 3, 8, and 24 h. One set of three animals was utilized for serial sampling at predose and 0.33, 2, 8, and 24 h, while another set of three was utilized for sampling at the other time points. A validated LC/MS/MS method with appropriate QCs was used for quantitation of BB-83698 in rat plasma samples at CTL.
Twenty-eight-day dog toxicokinetics.
Beagle dogs (three of each sex per treatment group) were given i.v. 10, 22, or 50 mg/kg/day for 14 to 28 days, and serial plasma samples from ∼1-ml bleeds were collected after the first and last administrations of the drug during a 28-day study at the following time points: predose and 0.083, 0.25, 0.5, 1, 2, 3, 4, 8, 12, and 24 h. For dogs, serial plasma samples were collected from the same animal. The in vivo dog studies were conducted at RCC Ltd., and a validated LC/MS/MS method with appropriate QCs was used for quantitation of BB-83698 in dog plasma samples at CTL.
Dog infusion range-finding study.
An i.v. infusion range-finding study was conducted with beagle dogs in two phases. In the first phase, pairs of animals (one male and one female) received single rising doses of BB-83698 (50, 75, 100, 150, and 175 mg/kg) as i.v. infusions over 1, 2, and/or 6 h to establish the maximum tolerated dose. This phase was followed by a 5-day repeat-dose phase at the maximum tolerated dose.
For the single-dose phase, a complete PK profiling with observation of clinical signs was done at each dose level. For the 5-day repeat-dose phase, plasma drug concentration analyses were done on days 1 and 5 of dosing. Serial blood (plasma) samples were collected up to the 24-h time point from individual animals.
Phase I clinical study of healthy volunteers.
A phase I, randomized, double-blind, placebo-controlled, five-way-crossover, single-dose-escalation study to explore the tolerability and PK of i.v. administration of 10 to 475 mg of BB-83698 was performed with healthy male volunteers at the PPD Development Clinic (Leicester, United Kingdom). The study was approved by the British Biotech Safety Review Board and the PPD Development Clinic Independent Ethics Committee. Healthy individuals, determined by physical examination to have no significant history of respiratory, cardiovascular, gastrointestinal, hepatic, renal, genitourinary, hematological, endocrine, metabolic, musculoskeletal, or neurologic disease, were included in the trial. Only males between 18 and 50 years of age with a body mass index between 19 and 29 kg/m2 were allowed to participate in this study. The mean body weight for each cohort ranged from 74 to 80 kg, the mean age ranged from 36 to 38 years, and the race distribution ranged from 66 to 100% white, 0 to 22% Asian, and 0 to 11% “other.” Exclusion criteria were as follows: (i) clinically significant findings upon physical examination, including systolic blood pressure of >140 mm Hg or diastolic blood pressure of >90 mm Hg, abnormal 24-h Holter monitoring, or abnormal electrocardiogram results as defined in the protocol; (ii) concomitant medical conditions or regular prescribed drug therapy, including over-the-counter and herbal remedies; (iii) a history of drug or alcohol abuse; and/or (iv) an immediate family history of prolonged QT intervals.
The study design involved an initial two cohorts of 9 or 10 healthy male subjects who received four ascending doses of BB-83698 and one randomized placebo dose. Clinical doses were selected based upon preclinical safety studies and simulations and predictions of initial human PK data. Nine subjects were recruited and dosed in cohort 1, each receiving 10, 25, 50, and 100 mg of BB-83698 along with the placebo. All nine subjects completed all dosing periods. Ten subjects were recruited and dosed in cohort 2. All subjects in cohort 2 completed three dosing periods and received 100 and 200 mg of BB-83698 along with the placebo. Following the dosing of the initial cohorts, an additional 10 subjects were recruited to cohort 3 and received doses of 200, 325, 400, and 475 mg of BB-83698. BB-83698 was administered via a 15-min i.v. infusion of a 50-ml citrate solution. A 2.7-ml sample of venous blood from a forearm vein was collected into a lithium heparin Monovette tube, and plasma was isolated. Samples for BB-83698 quantitation were obtained at the following time points: predose and 5, 10, and 15 min and 3.5, 4, 6, 8, 10, 12, 16, and 24 h after the start of the infusion. All plasma samples were analyzed at CTL by use of a validated LC/MS/MS method as described below.
Bioanalysis of rat, dog, and human plasma samples.
A protein precipitation method was used for extraction of BB-83698 from rat and dog plasma. Three hundred microliters of acetonitrile was added to 30 μl of plasma (for standard and unknown agents) in a Captiva 96-well protein precipitation plate. Vacuum was applied to draw the samples and solvent through, and the filtrates were collected in a 96-well collection plate. Samples were evaporated to dryness with a GeneVac DD4 centrifugal evaporator. Samples were reconstituted in 200 μl of 0.5% formic acid in methanol-water (40/60, vol/vol) and analyzed by LC/MS/MS using a Sciex API365 detector. Standards (0.010 to 50 μg/ml) were prepared in dog and rat control plasma samples. For each analysis, QC samples (low-, middle-, and high-QC samples) were prepared and analyzed in duplicate alongside test samples. The lower limit of quantitation was 0.01 μg/ml, and at least four out of the six individual QC samples were within ±15% of the nominal concentration in analytical runs. Plasma samples were diluted appropriately such that the final results were within the standard curve range. Appropriately diluted QC samples were included in the assay validation.
For human plasma samples (250 μl), following the addition of 50 μl of an internal standard (BB-85642), the plasma samples were acidified using 500 μl of orthophosphoric acid (0.2%) and extracted using a 96-well strata X (30-mg) solid-phase extraction plate. The extracts were evaporated to dryness and reconstituted in 200 μl of the mobile phase (0.5% formic acid in methanol-water [40/60, vol/vol]) prior to LC/MS/MS analysis using a Sciex API365 MS-MS detector. Standards (0.5 to 5,000 ng/ml) were prepared with control human plasma.
For each analysis, QC samples (low-, middle-, and high-QC samples) were prepared and analyzed in duplicate alongside test samples. The lower limit of quantitation was 0.5 ng/ml, and at least four out of the six individual QC samples were within ±15% of the nominal concentration in analytical runs. Plasma samples were diluted appropriately such that the final results were within the standard curve range. Appropriately diluted QC samples were included in the assay validation.
PK analysis.
Noncompartmental PK parameters were calculated from plasma drug concentration-time profiles by use of WinNonlin 4.0 Pro software (Pharsight, Mountain View, Calif.). Parameters were calculated for individual concentration-time profiles where possible. Otherwise, the mean values for the plasma drug concentration-time profiles were used (for mice and rats). The following relevant parameters were determined where possible: AUC from 0 to 24 h (AUC0-24), AUC from 0 to the last time point (AUC0-last) and/or extrapolated to infinity (AUC∞), Cmax, concentration in plasma extrapolated to time zero (Cp0), terminal elimination half-life (t1/2), volume of distribution at steady state (Vss), and clearance (CL). A regression analysis was performed to examine dose proportionality.
Allometric scaling.
CL (in milliliters per minute) and Vss (in milliliters) obtained after a single 10-mg/kg i.v. dose of BB-83698 (roughly equivalent to the highest dose tested in humans) was scaled across preclinical-study species (mouse, rat, and dog) as a function of body weight (in kilograms). The following standard allometric equation (2) was used:
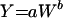 |
(1) |
where Y is the parameter of interest, coefficient a is the value of parameter at 1 unit of body weight, W is body weight, and b is the allometric exponent. For convenience, this equation was linearized to:
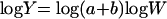 |
(2) |
Representative linearized plots for CL and Vss as functions of body weight were plotted along with predicted and observed CL and Vss values for humans. PK parameters for humans were predicted using equation 2. The parameter estimates for humans were predicted with 70 kg as the body weight.
RESULTS
Preclinical studies.
Relevant PK parameters after single and/or multiple dosing in mice, rats, and dogs are summarized in Tables 1 to 3, respectively. Mean plasma drug concentration-time profiles for mice, rats, and dogs after single administrations of 10-mg doses of BB-83698/kg are shown in Fig. 2. In separate studies not presented here, plasma protein binding was determined via an ultracentrifugation method and ranged from 78 to 82% for rats, mice, dogs, and humans (Report C11/NPK/006).
TABLE 1.
PK parameters for mice following single-dose i.v. administrations of BB-83698
| PK parameter | Value at dose of:
|
|
|---|---|---|
| 10 mg/kg | 50 mg/kg | |
| AUC∞ (h · μg/ml) | 3.8 | 55.1 |
| Cp0 (μg/ml) | 8.8 | 54.6 |
| Cmax (μg/ml) | 6.3 | 44.6 |
| t1/2 (h) | 1.0 | 2.6 |
| CL (ml/min/kg) | 44.4 | 15.1 |
| Vss (ml) | 2.0 | 2.6 |
TABLE 3.
PK parameters for dogs following i.v. administration of BB-83698 over 28 days
| Time point (day) | Sexa |
Cmax (μg/ml) at indicated dose (mg/kg/day)b
|
AUC0-last (μg · h/ml) at indicated dose (mg/kg/day)b
|
||||
|---|---|---|---|---|---|---|---|
| 10 | 22 | 50 | 10 | 22 | 50 | ||
| 1 | M | 9.6 ± 1.5 | 19.3 ± 3.3 | 48.4 ± 1.9 | 15.4 ± 2.4 | 24.4 ± 2.8 | 59.6 ± 3.4 |
| F | 11.1 ± 7.2 | 21.0 ± 2.5 | 39.5 ± 4.1 | 15.3 ± 9.1 | 40.2 ± 12.3 | 64.9 ± 14.1 | |
| 28 | M | 11.2 ± 2.1 | 18.1 ± 3.3 | 39.0 ± 7.1 | 15.1 ± 3.4 | 21.4 ± 3.9 | 47.6 ± 4.4 |
| F | 11.5 ± 5.3 | 23.7 ± 2.7 | 44.3 ± 17.5 | 13.9 ± 9.2 | 24.3 ± 3.1 | 50.0 ± 11.1 | |
M, male; F, Female.
Means ± standard deviations.
FIG. 2.

Mean plasma drug concentration-time profiles following single 10-mg doses of BB-83698/kg administered i.v. to mice (three animals per time point) (open squares), rats (three animals per sex per time point) (open diamonds), and dogs (three animals of each sex) (open circles).
In mice, the plasma drug concentration-time profiles were biphasic, with t1/2s of 1 to 3 h after 10- and 50-mg/kg doses. BB-83698 exhibited low CL and a moderate Vss. The AUCs and Cmax increased in a more-than-dose-proportional manner as the doses increased. CL was almost threefold lower after a 50-mg/kg dose than after a 10-mg/kg dose. The Vss remained relatively unchanged with increasing dose.
For rats, as observed for mice, plasma drug concentration-time curves were biphasic, with an initial decline phase of up to 3 h followed by a slower decay phase. Cmax and AUC0-last increased with dose, although slightly more than dose proportionally at the high dose. On day 1, after 22- and 50-mg/kg doses, exposure in females appeared to be higher than in males, but a difference in results between the sexes was not apparent on day 14 or 28. Since the AUC0-last did not alter consistently between the first and last doses, it may be concluded that there was no enzyme induction or inhibition which would influence its PK. The overall mean terminal t1/2 in male and female rats was approximately 3.4 h, with values ranging from 1.2 to 4.6 h.
For dogs, Cmaxs and AUCs were dose proportional, indicating linear PK over the 10- to 50-mg/kg/day dose range. As for rats, there were no changes in Cmaxs and AUC0-lasts between the first and last doses. Many of the plasma drug concentration-time curves exhibit biphasic patterns similar to those seen for rats, with initial decreases in concentrations up to 4 to 8 h, followed by slower declines. The overall mean terminal t1/2 appears to be around 6 h. There was no evidence of differences in AUC0-lasts or Cmaxs between the sexes for the animals at any dose level.
Administration of BB-83698 to dogs in preclinical safety studies was often accompanied by emesis, which occurred across a wide range of dose levels, was attenuated with repeat dosing, and rarely interfered with systemic exposure. Dose-limiting toxicity for BB-83698 in dogs, observed initially at i.v. bolus doses of 75 to 100 mg/kg, appeared CNS related and included tremor, unsteady gait, and convulsions at the higher dose. Controlled i.v. infusions offered a means of better understanding toxicokinetic determinants of the acute CNS effects. The Cmaxs and corresponding AUCs with escalating doses of BB-83698 administered as an infusion over 1, 2, and 6 h are summarized in Table 4 along with major clinical findings. A Cmax of ∼30 μg/ml, obtained following an i.v. infusion of 50 mg of BB-83968/kg over 1 h to beagle dogs, was well tolerated. The corresponding AUC∞ value was ∼74 μg · h/ml. Higher doses (75 and 100 mg/kg) administered as a 1-h infusion resulted in more profound CNS effects. The 75-mg/kg dose resulted in unsteadiness, tremor, and some buckling of the hind limbs. However, the same dose administered as a 2-h infusion, wherein the AUC was maintained but the Cmax was reduced by 40%, was well tolerated. It is obvious from these comparisons that an increase in Cmax, particularly at concentrations of >50 μg/ml, correlated with increased CNS toxicity, since a 100-mg/kg dose administered over 1 h with a Cmax of approximately 60 μg/ml resulted in convulsions. However, longer (6-h) infusions at higher doses (150 and 175 mg/kg) resulted in higher AUCs but lower Cmaxs (30 to 35 μg/ml) (Fig. 3) and were not associated with CNS effects.
TABLE 4.
PK parameters and clinical observations for dogs following i.v. infusion of BB-83698
| Dose (mg/kg/day) | Sexa | No. of dogs | Infusion time (h) | Clinical observation(s) | AUC∞ (μg · hr/ml) | Cmax (μg/ml) |
|---|---|---|---|---|---|---|
| 50 | M and F | 2 | 1 | None | 74 | 29 |
| 75 | M and F | 3 | 1 | Vomiting, unsteadiness, tremor, buckling of hind limbs | 127-157 | 52-55 |
| 75 | F | 1 | 2 | None | 118 | 34 |
| 100 | M | 1 | 1 | Vomiting, restlessness, convulsion at end | 178 | 60 |
| 150 | M and F | 2 | 6 | Lip licking | 213, 169b | 30, 23 |
| 175 | M and F | 2 | 6c | |||
| Day 1 | Occasional vomiting, decreased activity | 228, 214 | 35, 34 | |||
| Day 5 | Occasional vomiting, decreased activity | 159, 147 | 25, 23 |
M, male; F, Female.
Values separated by commas are for male and female dogs, respectively.
For the 175 mg/kg/day dose, the infusion was done for 6 h/day for 5 days.
FIG. 3.

Mean plasma drug concentration-time profiles on day 1 (first dose) and day 5 (last dose) following 6-h i.v. infusions of 175 mg of BB-83698/kg for a total of 5 days. Following 6-h infusions of 175 mg of BB-83698/kg, two serial plasma samples were obtained at times up to 24 h on day 1 after the first dose (open circles), and two more were obtained on day 5 after the last dose (open squares). Plasma samples were analyzed by a validated LC/MS/MS method with appropriate QCs.
Clinical study.
In the phase I study, eight dose levels, ranging from 10 to 475 mg, were tested. There were no significant adverse events at any dose level or clear trends in treatment-related effects. As the safety findings are outside of the scope and capacity of this study, they will be presented in detail elsewhere.
Following the administration of escalating doses of BB-83698 to healthy volunteers, maximum plasma drug concentrations were observed after termination of infusion at 15 min. Thereafter, concentrations declined rapidly over the first 4 to 6 h, followed by more gradual declines, including relatively prolonged terminal elimination phases. Mean plasma drug concentration-time profiles determined following 10- to 475-mg doses of BB-83698 are shown in Fig. 4. Relevant PK parameters following i.v. administration of BB-83698 are summarized in Table 5. The overall mean CL was approximately 238 ± 130 ml/min, with mean CL values of 189 ml/min at a dose of 475 mg to 521 ml/min after the lowest dose. In general, AUCs and Cmaxs increased linearly with dose, and there was no evidence of dose dependence (Fig. 5 and 6). While the degree of linearity of the AUC across all dose levels appears to deviate somewhat at the lowest and highest doses, additional analytical procedures (e.g., various dilution parameters) and additional time point sampling would need to be performed for final resolution of the nature of this relationship.
FIG. 4.

Mean plasma drug concentration-time profiles following i.v. infusions of BB-83698 (10 to 475 mg) to healthy male volunteers. The following dose levels, represented in the key, were tested (n = number of subjects): 10 mg (n = 9), 25 mg (n = 9), 50 mg (n = 9), 100 mg (n = 19), 200 mg (n = 20), 325 mg (n = 10), 400 mg (n = 10), and 475 mg (n = 10). Plasma samples were analyzed by a validated LC/MS/MS method.
TABLE 5.
PK parameters following i.v. infusion of BB-83698 to healthy male volunteersa
| Dose (mg) | Cmax (μg/ml) | AUC0-24 (μg · h/ml) | AUC∞ (μg · h/ml) | t1/2 (h) | CL (ml/min) | Vss (liters) |
|---|---|---|---|---|---|---|
| 10 | 0.5 ± 0.1 | 0.3 ± 0.1 | 0.4 ± 0.1 | 4.8 ± 0.7 | 520.6 ± 139.4 | 93.2 ± 20.7 |
| 25 | 1.2 ± 0.3 | 1.3 ± 0.2 | 1.3 ± 0.2 | 5.0 ± 0.8 | 335.1 ± 70.4 | 68.0 ± 14.7 |
| 50 | 2.7 ± 0.6 | 3.6 ± 0.7 | 3.7 ± 0.7 | 6.0 ± 1.8 | 233.5 ± 43.4 | 56.7 ± 21.9 |
| 100 | 5.3 ± 1.4 | 9.5 ± 3.0 | 10.3 ± 3.0 | 10.1 ± 5.8 | 173.3 ± 46.6 | 69.8 ± 34.3 |
| 200 | 9.6 ± 2.1 | 21.8 ± 6.7 | 26.6 ± 7.9 | 15.4 ± 11.8 | 135.4 ± 38.1 | 106.2 ± 73.0 |
| 325 | 15.6 ± 3.4 | 32.8 ± 8.1 | 41.3 ± 10.1 | 16.7 ± 10.6 | 141.5 ± 46.8 | 106.1 ± 67.4 |
| 400 | 18.5 ± 3.6 | 36.0 ± 7.6 | 39.2 ± 7.5 | 8.7 ± 7.5 | 175.9 ± 33.3 | 70.1 ± 38.5 |
| 475 | 19.3 ± 4.0 | 38.9 ± 8.9 | 45.8 ± 12.0 | 8.5 ± 4.8 | 188.5 ± 68.3 | 87.8 ± 39.9 |
Values are given in means±standard deviations.
FIG. 5.

Relationship plot for AUC∞ versus dose following i.v. infusions of BB-83698 to healthy male volunteers. Data points represent, in order of increasing dose (10, 25, 50, 100, 200, 325, 400, and 475 mg), the mean AUC∞s (±standard deviations) obtained at those dose levels. The solid line is the linear regression line predicted by the equation y = 0.11x (r2 = 0.96).
FIG. 6.

Relationship plot for Cmax versus dose following i.v. infusions of 10 to 475 mg of BB-83698 to healthy male volunteers. Data points represent, in order of increasing dose (10, 25, 50, 100, 200, 325, 400, and 475 mg), mean Cmaxs (±standard deviations) obtained at those dose levels. The solid line is the linear regression line predicted by the equation y = 0.05x (r2 = 0.99).
Interspecies scaling.
Representative linearized plots for CL and Vss as functions of body weight are shown for BB-83698 across animal species (mouse, rat, and dog) along with predicted and observed CL and Vss in healthy volunteers in Fig. 7. The exponent b in equation 1 was 0.8 and 1.0 for CL and Vss, respectively. The exponents were within the acceptable ranges of 0.6 to 0.8 for CL and 0.8 to 1.0 for Vss. Predicted CL of BB-83698 in humans was 450 ml/min. Observed CL values in healthy volunteers following administration of 10 to 25 mg of BB-83698 were within 25% of the prediction, i.e., 335 to 521 ml/min. However, at higher dose levels, observed CL values were lower than predicted, with values ranging from 135 to 234 ml/min. The observed Vss over the dose range of 10 to 475 mg was approximately 93 to 88 liters, respectively, and was reasonably close to the predicted value of 140 liters. Overall, the linearity for allometric scaling of PK between animals and humans was strong.
FIG. 7.

Linearized plots for CL (milliliters per minute) and Vss (milliliters) as a function of body weight (BW; kilograms) for BB-83698 across three species (mouse, rat, and dog) along with predicted and observed CL values and Vsss for humans. Data points in the graph, shown as closed diamonds, represent, in order of increasing weight, average values of the pharmacokinetic parameters for mice, rats, and dogs, along with the predicted values for humans. Actual data values are included in the figure. The solid lines represent the lines of best fit to the linearized form of the allometric equation. The observed CL values and Vss for healthy male volunteers after the 10-, 25-, and 475-mg doses are shown as open squares.
DISCUSSION
This work is the first description of the single- and/or multidose PK in rodents, dogs, and humans of a PDF inhibitor, a novel class antibacterial. In single- and repeat-dose studies with broad dose ranges, i.v. administration of BB-83698 to mice, rats, and dogs resulted in biphasic plasma drug concentration-time profiles. There were dose-related increases in AUCs and Cmaxs with increasing dosages in all three preclinical-study species. However, while there was good dose proportionality for Cmax and AUC between the 10- and 50-mg/kg doses in dogs, the values were more than dose proportional in rats and mice at the 50-mg/kg dose level. The magnitude of nonproportionality in the rodents was not substantial, and the basis for it was unclear. In the clinical study, overall linear PK were observed over the 10- to 475-mg dose ranges tested, albeit some deviation was observed at the lowest and highest dose levels. The highest dose resulted in an AUC comparable to that observed at 50 mg/kg in the rodents. Moreover, the PK in healthy volunteers were well predicted by allometric scaling of data from animals. As seen in preclinical studies, BB-83698 exhibited slow systemic CL and moderate Vss in healthy volunteers. BB-83698 exhibited a mean t1/2 in humans of approximately 9 h that, combined with observed AUCs, supports once-daily dosing.
In terms of insights related to BB-83698 metabolism, preliminary studies indicated that the major route of metabolism in rats, based on analysis of bile and urine, and in humans (via analysis of urine) was glucuronidation (data not shown). As comparisons between day 1 and day 28 AUCs in the repeat-dosing rat and dog studies were similar, one might conclude that there was not significant induction or inhibition of BB-83698 metabolism. In contrast, we cannot rule out the possibility of induction in the 175-mg/kg/day 5-day controlled infusion study of beagle dogs, as day 5 AUCs and Cmaxs were 25 to 30% lower than day 1 values.
The design and implementation of controlled i.v. infusion studies in dogs provided valuable data of a complementary nature to infusion PK in humans and also pertained to the relationship between toxicokinetics and toxicodynamics. Emesis was of a similar magnitude across a broad range of Cmaxs and AUCs and often attenuated with repeat dosing. Dose-limiting toxicity in dogs, apparently related to CNS dysfunction, was of increasing severity with increasing plasma Cmaxs but not necessarily with increasing AUCs (Table 4). At concentrations exceeding 50 μg/ml, tremors, unsteady gait, and hind limb buckling were noted, and convulsions were observed at concentrations approaching 60 μg/ml. Interestingly, no CNS effects were observed in mice or rats following i.v. administration with Cmaxs of ≥50 μg/ml (Tables 1 and 2; also data not shown), and the mechanism for CNS effects in dogs was unclear, as BB-83698 brain penetration, at least in rodents (K. W. Johnson, unpublished data), was very low.
TABLE 2.
PK parameters for rats following i.v. administration of BB-83698 over 28 days
| Time point (day) | Sexb |
Cmax (μg/ml) at indicated dose (mg/kg/day)
|
AUC0-last (μg · h/ml) at indicated dose (mg/kg/day)
|
||||
|---|---|---|---|---|---|---|---|
| 10 | 22 | 50 | 10 | 22 | 50 | ||
| 1 | M | 10.5 | 26.4 | 51.1 | 8.0 | 17.5 | 53.1 |
| F | 10.0 | 25.8 | 66.0 | 10.8 | 33.0 | 95.5 | |
| 28a | M | 12.4 | 42.6 | 41.0 | 6.9 | 23.7 | 99.7 |
| F | 15.3 | 40.1 | 64.9 | 8.5 | 23.3 | 80.3 | |
Day 14 for animals receiving dose of 50 mg/kg/day.
M, male; F, Female.
In the phase I study with dosing up to 475 mg, neither were significant observations made nor were clear trends for adverse effects observed, despite significant systemic exposures. In terms of realized AUC and efficacy projections, an AUC/MIC ratio for S. pneumoniae of approximately 184, calculated using AUC∞s and an MIC of 0.25 μg/ml (for penicillin-resistant S. pneumoniae) (13), was achieved at 475 mg. Such an exposure is within the predicted efficacious range (AUC/MIC ratio of ≥133) for once-daily dosing derived from dedicated PK-pharmacodynamic collaborative studies of Craig and Andes (41st ICAAC). Moreover, antipneumococcal murine thigh infection and lethal peritonitis studies with i.v. administration of BB-83698 affirmed significant efficacy at AUC/MIC ratios in the range of 100 to 200 (K. W. Johnson and M. Gross, unpublished data). If one were to target a predicted reliable antipneumococcal AUC/MIC ratio of ∼200 or higher in all treated subjects in further clinical trials, then a repeat dose level of ≥600 mg would be projected, assuming consistent linearity for AUC with increasing doses. Such projections suggest that a 600-mg dose may approach a Cmax of 30 μg/ml if administered as a 15-min infusion, which would be anticipated to be well tolerated based on preclinical results. Nonetheless, if one were inclined to limit the Cmax, higher dose administration in humans could utilize longer infusion durations to maintain AUCs but reduce Cmaxs based on the dog infusion study outcomes. Indeed, predictions from the PK data for healthy volunteers project that increasing the infusion length from 15 min to 1 or 2 h would result in a 50 or 60% decrease, respectively, in Cmax.
Hydroxamate-type metalloproteinase inhibitors, such as matrix metalloproteinase inhibitors (MMPI), have been pursued for several years as candidate therapeutics for multiple clinical indications. A challenge for many of the discovery programs has been inadequate PK, including poor oral bioavailability (7, 10). Although PDF inhibitors exhibit unique structural features compared to MMPI, even early hydroxamate-based PDF inhibitors suffered PK setbacks in vivo (9). BB-83698 and a recently described homolog, BB-81384, are unique in that they are N-formyl hydroxylamine (reverse hydroxamate)-type metalloproteinase inhibitors with favorable substitutions at P2′ and P3′ positions which enhance their in vivo PK and pharmacodynamic properties (8).
In conclusion, described herein are the PK and/or toxicokinetics of BB-83698 in animals and humans. These studies are particularly novel because they represent the first multispecies preclinical and clinical reports of a novel type of MMPI as well as the first antibacterial PDF inhibitor entry into humans. As a study of a first-in-class drug, the results were promising for further development of BB-83698 or related PDF inhibitors for a number of reasons. First, with the exception of some deviation at the lowest and highest dose levels in humans, systemic exposure was generally linear with administered doses in animals and humans. Second, the PK exhibited good allometric scaling among all species. Third, dose-limiting CNS effects, observed only in dogs, were apparently linked to high Cmaxs. Dose escalation in dogs could be easily continued by adjusting infusion duration to reduce the Cmax without compromising the AUC, the apparent primary pharmacodynamic determinant. Last, single-dose escalation in healthy human volunteers proceeded to a predicted efficacious level. No significant adverse events were noted, and the PK, including affirmation of the feasibility of once-daily dosing, were encouraging.
Acknowledgments
Financial support was provided by Genesoft Pharmaceuticals and British Biotech Pharmaceuticals.
We thank Matt Gross for pharmacology support, staff members from Bay Biolabs (Northview, Calif.), CTL, and RCC Ltd. for outsourced experimental support, and Peter Hoyle (Hoyle Inc.) for consulting services.
REFERENCES
- 1.Azoulay-Dupuis, E., J. Mohler, and J. P. Bédos. 2004. Efficacy of BB-83698, a novel peptide deformylase inhibitor, in a mouse model of pneumococcal pneumonia. Antimicrob. Agents Chemother. 48:80-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boxenbaum, H. 1982. Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. J. Pharmacokinet. Biopharm. 10:201-227. [DOI] [PubMed] [Google Scholar]
- 3.Clements, J. M., R. P. Beckett, A. Brown, G. Catlin, M. Lobell, S. Palan, W. Thomas, M. Whittaker, S. Wood, S. Salama, P. J. Baker, H. F. Rodgers, V. Barynin, D. W. Rice, and M. G. Hunter. 2001. Antibiotic activity and characterization of BB-3497, a novel peptide deformylase inhibitor. Antimicrob. Agents Chemother. 45:563-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clements, J. M., A. P. Ayscough, K. Keavey, and S. P. East. 2002. Peptide deformylase inhibitors, potential for a new class of broad spectrum antibacterials. Curr. Med. Chem. Anti-Infect. Agents 1:239-249. [Google Scholar]
- 5.Davies, S. J., A. P. Ayscough, R. P. Beckett, J. M. Clements, S. Doel, L. M. Pratt, Z. M. Spavold, S. W. Thomas, and M. Whittaker. 2003. Structure-activity relationships of the peptide deformylase inhibitor BB-3497: modification of the P2′ and P3′ side chains. Bioorg. Med. Chem. Lett. 13:2715-2718. [DOI] [PubMed] [Google Scholar]
- 6.East, S., R. P. Beckett, D. C. Brookings, J. M. Clements, S. Doel, K. Keavey, G. Pain, H. K. Smith, W. Thomas, A. J. Thompson, R. S. Todd, and M. Whittaker. 2003. Peptide deformylase inhibitors with activity against respiratory tract pathogens. Bioorg. Med. Chem. Lett. 14:59-62. [DOI] [PubMed] [Google Scholar]
- 7.Eskens, F. A., N. C. Levitt, A. Sparreboom, L. Choi, R. Mather, J. Verweij, and A. L. Harris. 2000. Effect of food on the pharmacokinetics of oral MMI270B (CGS 27023A), a novel matrix metalloproteinase inhibitor. Clin. Cancer Res. 6:431-433. [PubMed] [Google Scholar]
- 8.Gross, M., J. Clements, R. P. Beckett, W. Thomas, S. Taylor, D. Lofland, S. Ramanathan-Girish, M. Garcia, S. Difuntorum, U. Hoch, H. Chen, and K. W. Johnson. 2004. Oral anti-pneumococcal activity and pharmacokinetic profiling of a novel peptide deformylase inhibitor. J. Antimicrob. Chemother. 53:487-493. [DOI] [PubMed] [Google Scholar]
- 9.Hackbarth, C. J., D. Z. Chen, J. G. Lewis, K. Clark, J. B. Mangold, J. A. Cramer, P. S. Margolis, W. Wang, J. Koehn, C. Wu, S. Lopez, G. Withers III, H. Gu, E. Dunn, R. Kulathila, S.-H. Pan, W. L. Porter, J. Jacobs, J. Trias, D. V. Patel, B. Weidmann, R. J. White, and Z. Yuan. 2002. N-Alkyl urea hydroxamic acids as a new class of peptide deformylase inhibitors with antibacterial activity. Antimicrob. Agents Chemother. 46:2752-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hadden, M., and D. Orton. 2004. Trip report: the Screentech World Summit Conference on “Protease Inhibitors,” San Diego, California, March 23-26, 2003. Albany Molecular Research, Inc., technical report, vol. 8. [Online.] www.albmolecular.com/features/tekreps/vol08/no14/.
- 11.Jones, R., and P. Rhomberg. 2003. Comparative spectrum and activity of NVP-PDF386 (VRC4887), a new peptide deformylase inhibitor. J. Antimicrob. Chemother. 51:157-161. [DOI] [PubMed] [Google Scholar]
- 12.Kollef, M. H., and V. J. Fraser. 2001. Antibiotic resistance in the intensive care unit. Ann. Intern. Med. 134:298-314. [DOI] [PubMed] [Google Scholar]
- 13.Lofland, D., S. Difuntorum, A. Waller, J. M. Clements, M. K. Weaver, J. A. Karlowsky, and K. W. Johnson. 2004. In vitro antibacterial activity of the peptide deformylase inhibitor BB-83698. J. Antimicrob. Chemother. 53:664-668. [DOI] [PubMed] [Google Scholar]
- 14.Low, D. E. 2001. Antimicrobial drug use and resistance among respiratory pathogens in the community. Clin. Infect. Dis. 33(Suppl. 3):S206-S213. [DOI] [PubMed] [Google Scholar]
- 14a.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academy Press, Washington, D.C.
- 15.Nguyen, K., C. Colton, R. Chakrabarti, M. Zhu, and D. Pei. 2003. Characterization of a human peptide deformylase: implications for antibacterial drug design. Biochemistry 42:9952-9958. [DOI] [PubMed] [Google Scholar]
- 16.Wise, R., J. M. Andrews, and J. Ashby. 2002. In vitro activities of peptide deformylase inhibitors against gram-positive pathogens. Antimicrob. Agents Chemother. 46:1117-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan, Z., J. Trias, and R. White. 2001. Deformylase as a novel antibacterial target. Drug Discov. Today 6:954-961. [DOI] [PubMed] [Google Scholar]