

Abstract
Sulfur dioxide (SO2) is being recognized as a possible endogenous gasotransmitter with importance on par with that of NO, CO, and H2S. Herein we describe a series of SO2 prodrugs that are activated for SO2 release via a bioorthogonal click reaction. The release rate can be tuned by adjusting the substituents on the prodrug.
SO2 gas is well known as an air pollutant and may cause respiratory and cardiovascular diseases upon chronic exposure.1–3 SO2 has also long been recognized as an antimicrobial agent, and widely used in food industry, especially in the brewery industry. Recent years have seen increasing evidence that SO2 can be produced endogenously by oxidation of sulfur-containing amino acids (cysteine, homocysteine).4–6 SO2 has also been shown to have possible physiological functions. There has been an increasing level of understanding of SO2’s mechanism of actions at the pathway level,7–9 and fluorescent probes have been reported that can detect endogenous production of SO2.10, 11 SO2 has been reported to exacerbate ischemia-reperfusion injury, and suppress hypertension, pulmonary hypertension, vascular remodeling, and inflammation.12 Thus, interest in exploring SO2’s functions as a gasotransmitter has increased in recent years. Up until now, knowledge of SO2’s physiological and pathological roles is still very limited, especially in comparison with the other three gasotransmistters NO, CO, and H2S.
There are three main methods currently used to provide SO2 for research purposes: gaseous SO2, HSO3−/SO32− pair, and SO2 donors. Despite the broad application in research activities, gaseous SO2 has obvious disadvantages including difficulties in preparing solutions of precise concentrations. Besides, the required equipment can be cumbersome with potential hazard for lab personnel. The HSO3−/SO32− ion pair method relies on the equilibrium established between hydrated SO2 (SO2·H2O) and HSO3−/SO32− ion pair in aqueous solution to release SO2.13, 14 However, using the HSO3−/SO32− ion pair for SO2 release does not allow easy control of its quantity and relies on conversion kinetics, which can be very complex. Another drawback of this method is that it is hard to de-convolute the biological effects of HSO3−/SO32− and molecular SO2, which have been demonstrated to be different.15 Thus, there is a need to develop prodrugs with controlled release of SO2 to imitate the process of endogenous production of SO2. Several groups have developed SO2 donor molecules with various triggering mechanisms. Chakrapani and colleagues have reported clever designs that utilize biological thiol (cysteine) and UV light to trigger the release of SO2 gas from donors.16, 17 They also developed elegant work on using benzosultines to generate SO2 under physiological conditions.18 Recently, Xian’s group reported benzothiazole sulfinate (BTS) as a pH dependent water soluble SO2 donor.19 We are interested in developing SO2 prodrug systems with controllable release rates under physiological conditions. Such prodrugs would complement what is already available in providing research tools to help advance this area of research.
Our group has a long-standing interest in studying gasotransmitters.20–26 Previously, we have demonstrated the feasibility of using an extrusion reaction as a way to deliver carbon monoxide (CO).21 Herein we describe a strategy to cage SO2 in thiophene dioxide and use a strained alkyne to trigger the release of SO2 in a controllable fashion (Scheme 1). By varying the substituents on the thiophene dioxide scaffold, the release rates can also be tuned.
Scheme 1.
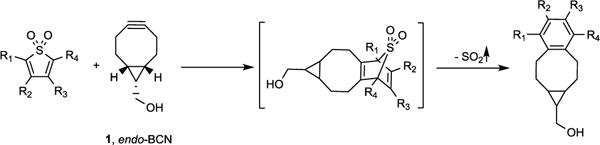
Proposed SO2 donor system
As early as 1976, Stille already examined the reaction of thiophene dioxide and a terminal alkyne under reflux condition in toluene. Though conditions were “harsh”, the cheletropic reaction yielded a cyclized product with the release of SO2.27 We planned to take advantage of this reaction to construct caged SO2 for release under near physiological conditions (room temperature to 37 °C, pH 7.4, and aqueous solution). A key issue in this project was how to lower the reaction temperature to ambient temperature for applications under near physiological conditions. 2,3,4,5-Tetrachlorothiophene dioxide has been shown to possess high reactivity in cycloaddition reactions with a variety of olefines. Specifically, it was shown to react steadily with ethylene and to give a cyclohexyldiene product and SO2 at 28 °C.28 We reasoned that a strained alkyne with increased HOMO energy would promote this cycloaddition reaction. The formation of a stable phenyl ring should also help to drive the subsequent cheletropic reaction with the concomitant release of SO2. Therefore, we synthesized 2,3,4,5-tetrachlorothiophene dioxide 3 from commercially available perchlorothiophene 2. Then we first examined the reaction of 3 (20 mM in MeOH) with endo-BCN (1, 4 equiv.) at room temperature as a proof-of-concept test. Indeed, after 5 min, we saw complete conversion of 3 into a new product, which was spectroscopically characterized as 4 (Scheme 2).
Scheme 2.
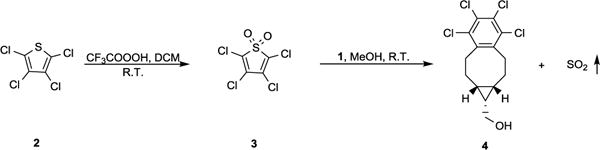
Proof-of-concept test of SO2 donor system
We then examined the reaction kinetics. The UV absorption of the product is significantly lower than that of the reactants at 328 nm. Therefore, we used UV absorbance decrease to monitor the reaction progress. The second order rate constant was determined by first using a large excess of 1 to examine the pseudo-first order reaction followed by plotting the pseudo-first order reaction rate constants against different 1 concentrations. The second order rate constant was determined to be 1.50 M−1 s−1 in MeOH at room temperature, allowing the reaction to be finished in about 3 hours at mid-μM levels (Figure S1B).
To further confirm the formation of SO2, we performed the DTNB test, which has been applied in SO2 measurement in atmosphere and food samples.29–31 DTNB would react with SO32− ion to give an reduced product, which can be quantitatively measured by UV absorbance at 412 nm (Figure 1A).29 To allow hydration of SO2, we performed this test in 5% DMSO/PBS. A reaction mixture was incubated at room temperature or 37 °C for 45 min (about 1 t1/2) and then the DTNB probe was added. Afterwards, the test solution was incubated at room temperature for another 15 min and subjected to UV absorbance reading at 412 nm. The experimental group showed higher absorbance at both room temperature and 37 °C than the negative control groups, confirming the formation of SO32−. Both experimental groups and positive control groups showed lower SO32− formation at 37 °C than at room temperature, presumably due to increased SO2 escape at higher temperature (Figure 1B).
Figure 1.
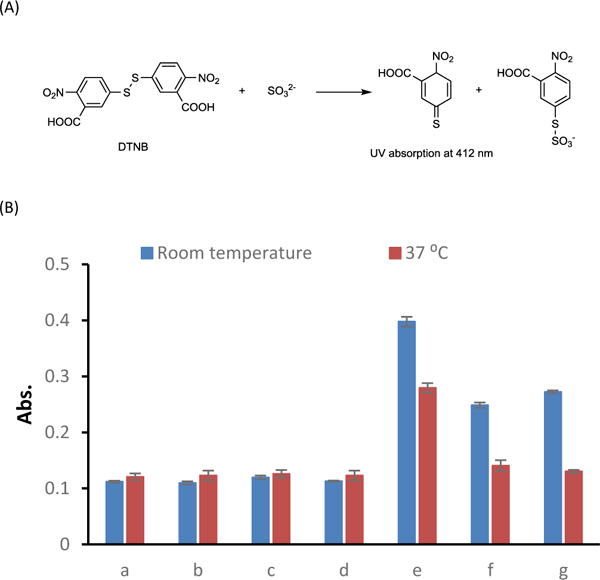
(A) DTNB test mechanism. (B) UV absorption at 412 nm of DTNB test after 45 min incubation at room temperature or 37 °C of (a) blank (5% DMSO/PBS), (b) 250 μM of 3 only, (c) 2.5 mM of 1 only, (d) 250 μM of 4 only, (e) 250 μM of 3 +2.5 mM of 1, (f) 50 μM of Na2SO3, (g) 50 μM of NaHSO3.
Encouraged by the initial success, we went on to synthesize additional analogues to see whether we can tune the reaction rates for various applications. Thus, we synthesized compounds 5 and 6 with electron withdrawing groups at different positions (Figure 2A; for synthetic scheme see ESI). We reasoned that by varying the electron density on the thiophene ring, different LUMO energy will result in varied reaction rate with BCN. We also tested reaction with a trans-cyclooctene compound (8, equatorial isomer), which is known to have high strain energy and high HOMO, to see if the reaction rate can be further enhanced. In all these reactions at room temperature, we successfully isolated the cheletropic reaction products (see ESI). Interestingly, we had initial difficulties in analyzing the room temperature NMR spectra of the products containing the bicyclo[6.1.0]non-4-ene structure. This was due to peak overlap caused by line broadening in 1H NMR. Structure elucidation by 13C NMR is equally difficult because of missing peak(s) at high field. Given HRMS confirmation, we reasoned that this phenomenon is caused by the slow flipping of eight-member ring between the “chair” and “boat” conformations. We successfully observed two conformations by tracking the −CH2 group next to the −OH group at low temperature (270 K). As temperature increases, the exchange rate of the two populations increases, and the two peaks observed for the −CH2 group coalesced and then averaged to give a sharp peak. Unfortunately, within the temperature range tested, the exchange rate is not fast enough to average the −CH2 and −CH groups from the bicyclo[6.1.0]non-4-ene structure, which became broad above 278 K and hard to track (Figure 2C). However, this phenomenon has been observed exclusively with endo-product. As for product obtained from reaction between 6 and exo-BCN, sharp peaks at high field were observed at room temperature (see ESI). By contrast, with chlorine substitutions, we are able to track the carbons at room temperature, presumably because of its fast flipping rate. Edited HSQC experiment gave clear evidence of geminal protons splitting from the eight-membered ring, confirming the structures of cheletropic products (see ESI). We studied the reaction kinetics for these reactions and obtained k2 values ranging from 0.02 to 0.33 M−1s−1 (Table 1; Figure S1). However, compound 5 was found to be unstable in MeOH at room temperature, which was presumably ascribed to its strong electrophlicity. Therefore, compound 5 is not a suitable SO2 prodrug for biological applications due to this stability issue.
Figure 2.
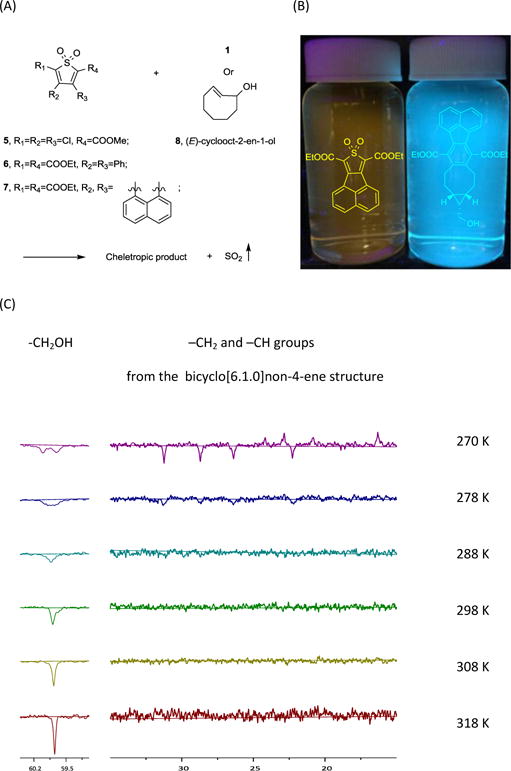
(A) SO2 donor pairs. (B) Fluorescent SO2 donor and product after SO2 release (100 μM in MeOH). (C) Coalescence of cheletropic product (6 + 1) observed in 13C NMR (DEPT-135) with increasing temperature.
Table 1.
Kinetics study of SO2 donor pairs
| SO2 donor pairs | 3 + 1 | 3 + 8 | 5 + 1 | 6 + 1 | 7 + 1 | 6 + exo-BCN |
| k2 (M−1s−1) | 1.50a | 0.02a | 0.33b | 0.05a | 0.01c | 0.04a |
Measured in MeOH under room temperature.
Measured in ACN under room temperature.
Measured in DMSO/PBS 4:1 at 37 °C.
Currently, several elegant fluorescent probes for SO2 have been developed based on the nucleophilicity of SO32−/HSO3−. However, few of them are suitable for real-time monitoring of SO2 generation due to delayed response.32 One alternative strategy for real time monitoring of SO2 release is to devise such SO2 prodrugs that would become fluorescent after SO2 release. Therefore, we were interested in designing SO2 prodrugs that would lead to the formation of a fluorescent reporter, which allows for real-time monitoring of SO2 release. We reasoned that attaching an aromatic substituent to the thiophene ring would allow formation of an expanded conjugation system after the cheletropic reaction and thus result in pronounced changes in the spectroscopic behavior of the aromatic system after the reaction. Therefore, we synthesized thiophene dioxide with naphthalene conjugated at the 3,4 positions (7, Figure 2A). The compound itself has weak yellow fluorescence with an excitation wavelength of 299 nm, emission wavelength of 556 nm, and a quantum yield of 0.006. After reaction with BCN and SO2 release, the resulting product shows strong cyan fluorescence with an emission wavelength of 470 nm, excitation wavelength of 296 nm, and quantum yield of 0.138 (Figure 2B). We also established the kinetics profile of this reaction by monitoring the fluorescence increase at 470 nm. A k2 of 0.01 M−1s−1 was obtained (Table 1; Figure 3; Figure S2).
Figure 3.
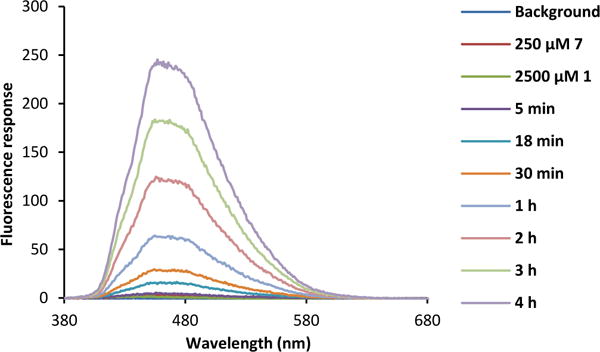
Fluorescence response increases as cycloaddition reaction between 7 and 1 proceeds (λex=350 nm). Condition: 250 μM 7 + 2500 μM 1 in 4:1 DMSO/PBS at 37 °C.
In summary, we have developed a SO2 donor system, which releases SO2 through a Diels-Alder reaction with a strained alkyne/alkene as a trigger. We explored the initial tunability of the releasing rate by varying the electron density of thiophene dioxide ring. With a small set of compounds, we were able to tune the reaction rate in the range of 0.01–1.50 M−1s−1, giving a 150-fold range in reactivity.
Additional work is underway in further extending the tunability of reaction rate and in testing the donor system in triggering various biological responses.
Supplementary Material
Acknowledgments
Partial financial support from the National Institutes of Health (CA180519) is gratefully acknowledged.
Footnotes
Electronic Supplementary Information (ESI) available: Additional experimental data. See DOI: 10.1039/x0xx00000x
References
- 1.Li R, Meng Z, Xie J. Toxicol Lett. 2007;175:71–81. doi: 10.1016/j.toxlet.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Min J, Min K, Cho S, Paek D. Int J Cardiol. 2009;133:119–121. doi: 10.1016/j.ijcard.2007.08.139. [DOI] [PubMed] [Google Scholar]
- 3.Liao D, Duan Y, Whitsel E, Zheng Z, Heiss G, Chinchilli V, Lin H. Am J Epidemiol. 2004;159:768–777. doi: 10.1093/aje/kwh109. [DOI] [PubMed] [Google Scholar]
- 4.Ubuka T, Yuasa S, Ohta J, Masuoka N, Yao K, Kinuta M. Acta Med Okayama. 1990;44:55–64. doi: 10.18926/AMO/30442. [DOI] [PubMed] [Google Scholar]
- 5.Du S, Jin H, Bu D, Zhao X, Geng B, Tang C, Du J. Acta Pharmacol Sin. 2008;29:923–930. doi: 10.1111/j.1745-7254.2008.00845.x. [DOI] [PubMed] [Google Scholar]
- 6.Stipanuk M. Annu Rev Nutr. 2004;24:539–577. doi: 10.1146/annurev.nutr.24.012003.132418. [DOI] [PubMed] [Google Scholar]
- 7.Meng Z, Li Y, Li J. Arch Biochem Biophys. 2007;467:291–296. doi: 10.1016/j.abb.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Q, Meng Z. Eur J Pharmacol. 2009;602:117–123. doi: 10.1016/j.ejphar.2008.11.030. [DOI] [PubMed] [Google Scholar]
- 9.Wang Y, Ren A, Yang X, Wang L, Rong W, Tang C, Yuan W, Lin L. Physiol Res. 2009;58:521–527. doi: 10.33549/physiolres.931456. [DOI] [PubMed] [Google Scholar]
- 10.Li G, Chen Y, Wang J, Lin Q, Zhao J, Ji L, Chao H. Chem Sci. 2013;4:4426. [Google Scholar]
- 11.Yu F, Han X, Chen L. Chem Commun. 2014;50:12234–12249. doi: 10.1039/c4cc03312d. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Jin H, Tang C, Du J. Clin Exp Pharmacol Physiol. 2010;37:745–752. doi: 10.1111/j.1440-1681.2009.05249.x. [DOI] [PubMed] [Google Scholar]
- 13.Jones L, McLaren E. J Chem Phys. 1958;28:995–995. [Google Scholar]
- 14.Townsend T, Allanic A, Noonan C, Sodeau J. J Phys Chem A. 2012;116:4035–4046. doi: 10.1021/jp212120h. [DOI] [PubMed] [Google Scholar]
- 15.Li J, Meng Z. Nitric Oxide. 2009;20:166–174. doi: 10.1016/j.niox.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 16.Malwal S, Sriram D, Yogeeswari P, Konkimalla V, Chakrapani H. J Med Chem. 2012;55:553–557. doi: 10.1021/jm201023g. [DOI] [PubMed] [Google Scholar]
- 17.Malwal S, Chakrapani H. Org Biomol Chem. 2015;13:2399–2406. doi: 10.1039/c4ob02466d. [DOI] [PubMed] [Google Scholar]
- 18.Malwal S, Gudem M, Hazra A, Chakrapani H. Org Lett. 2013;15:1116–1119. doi: 10.1021/ol400190f. [DOI] [PubMed] [Google Scholar]
- 19.Day J, Yang Z, Chen W, Pacheco A, Xian M. ACS Chem Biol. 2016;11:1647–1651. doi: 10.1021/acschembio.6b00106. [DOI] [PubMed] [Google Scholar]
- 20.Wang K, Peng H, Wang B. J Cell Biochem. 2014;115:1007–1022. doi: 10.1002/jcb.24762. [DOI] [PubMed] [Google Scholar]
- 21.Wang D, Viennois E, Ji K, Damera K, Draganov A, Zheng Y, Dai C, Merlin D, Wang B. Chem Commun. 2014;50:15890–15893. doi: 10.1039/c4cc07748b. [DOI] [PubMed] [Google Scholar]
- 22.Wang K, Peng H, Ni N, Dai C, Wang B. J Fluoresc. 2014;24:1–5. doi: 10.1007/s10895-013-1296-5. [DOI] [PubMed] [Google Scholar]
- 23.Zheng Y, Ji X, Ji K, Wang B. Acta Pharm Sin B. 2015;5:367–377. doi: 10.1016/j.apsb.2015.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji X, Damera K, Zheng Y, Yu B, Otterbein L, Wang B. J Pharm Sci. 2016;105:406–416. doi: 10.1016/j.xphs.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng Y, Yu B, Ji K, Pan Z, Chittavong V, Wang B. Angew Chem, Int Ed. 2016;55:4514–4518. doi: 10.1002/anie.201511244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ji X, Zhou C, Ji K, Aghoghovbia R, Pan Z, Chittavong V, Ke B, Wang B. Angew Chem, Int Ed. 2016;55:15846–15851. doi: 10.1002/anie.201608732. [DOI] [PubMed] [Google Scholar]
- 27.Nelb R, Stille J. J Am Chem Soc. 1976;98:2834–2839. [Google Scholar]
- 28.Raasch M. J Org Chem. 1980;45:856–867. [Google Scholar]
- 29.Humphrey R, Ward M, Hinze W. Anal Chem. 1970;42:698–702. [Google Scholar]
- 30.Guo Z, Li Y, Zhang X, Chang W, Ci Y. Anal Bioanal Chem. 2002;374:1141–1146. doi: 10.1007/s00216-002-1567-5. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Zhao M. Food Control. 2006;17:975–980. [Google Scholar]
- 32.Lin V, Chen W, Xian M, Chang C. Chem Soc Rev. 2015;44:4596–4618. doi: 10.1039/c4cs00298a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.