

Abstract
Numerous studies have demonstrated that administration of antigen (Ag)-pulsed dendritic cells (DCs) is an effective strategy for enhancing immunity to tumors and infectious disease organisms. However, the generation and/or isolation of DCs can require substantial time and expense. Therefore, using inactivated F. tularensis (iFt) Ag as a model immunogen, we first sought to determine if DCs could be replaced with peripheral blood mononuclear cells (PBMCs) during the ex-vivo pulse phase and still provide protection against Ft infection. Follow up studies were then conducted using the S. pneumoniae (Sp) vaccine Prevnar ®13 as the Ag in the pulse phase followed by immunization and Sp challenge. In both cases, we demonstrate that PBMCs can be used in place of DCs when pulsing with iFt and/or Prevnar ®13 ex vivo and re-administering the Ag-pulsed PBMCs as a vaccine. In addition, utilization of the i.n. route for Ag-pulsed PBMC administration is superior to use of the i.v. route in the case of Sp immunization, as well as when compared to direct injection of Prevnar ®13 vaccine i.m. or i.n. Furthermore, this PBMC-based vaccine strategy provides a more marked and enduring protective immune response and is also capable of serving as a multi-organism vaccine platform. The potential for this ex-vivo vaccine strategy to provide a simpler, less time consuming, and less expensive approach to DC-based vaccines and vaccination in general is also discussed.
Keywords: Vaccine, PBMCs, Dendritic cells, intranasal vaccine, mucosal vaccine platform, Francisella tularensis, Streptococcus pneumoniae
Graphical abstract
1. Introduction
Dendritic cells (DCs) are the most efficient antigen (Ag) presenting cells (APCs) at taking up, processing, and presenting Ags to naïve T cells [1]. This property of DCs has been harnessed to develop DC-based immunotherapeutics and vaccines against cancer and a number of infectious diseases, including HIV-1 and influenza [2–6]. Typically, use of DC-based immunotherapeutics and vaccines involve a number of ex vivo manipulations, including isolation of DCs or DC precursors followed by in vitro differentiation into DCs and, in some cases, induction of DC maturation. The DCs are then mixed ex vivo with vaccine Ags in the presence or absence of DC maturation factors for 3–7 days and subsequently administered back into the vaccine recipient [2, 4, 7–12]. Importantly, the process of DC isolation/differentiation is cumbersome, requires a high level of skill, time, and infrastructure, and is expensive. Thus, such a treatment can be difficult to utilize in the clinic, in particular in underdeveloped countries.
In contrast, peripheral blood mononuclear cells (PBMCs) contain numerous APC populations including: monocytes/macrophages, DCs, and B cells, all of which are fully capable of processing and presenting vaccine Ags to T cells and thereby stimulating an immune response. In addition, PBMCs are more easily and rapidly isolated, requiring minimal infrastructure and expense. However, successful studies utilizing PBMCs in place of DCs in an ex-vivo vaccine platform are rare. Specifically, in one case PBMCs were successfully used to deliver vaccine Ags, which was shown to ameliorate prostate cancer [13]. As a result of these studies, this vaccine has now been approved by the FDA as an immunotherapeutic for castration-resistant prostate cancer [14].
In this investigation, we replace DCs with PBMCs and pulse them ex vivo with inactivated F. tularensis LVS (iFt) and/or the S. pneumoniae (Sp) vaccine Prevnar ®13. Ft LVS is an attenuated form of the human virulent Ft, which is a category A biothreat agent [15]. Importantly, antibody (Ab) is the primary mediator of protection against Sp and can also play a role in the protection of mice against Ft LVS challenge [16, 17]. Thus, in these studies, we chose to focus on humoral immunity and the ability of Ag-pulsed PBMCs to generate a protective humoral immune response to Ft LVS and Sp. We demonstrate that PBMCs pulsed with iFt and/or Prevnar ®13 ex vivo generate both Ag-specific Ab responses and subsequent protection against both Ft and Sp either as individual vaccines or when combined as a multi-organism vaccine. In addition, the intranasal (i.n.) route of Ag-pulsed PBMC vaccine delivery is superior to the intravenous (i.v.) route in the case of Sp immunization and challenge, as well as when compared to the direct injection of Prevnar ®13 intramuscularly (i.m.) or i.n. Furthermore, this PBMC-based vaccine strategy provides a more marked and enduring protective immune response.
2. Materials and Methods
2.1. Mice
6–8 week old inbred female C57BL/6 mice were purchased from Taconic Laboratories (Hudson, NY) and housed in the Animal Resource Facility of Albany Medical College. The animal studies were reviewed and approved by the Institutional Animal Care and Use Committee at Albany Medical College utilizing NIH standards.
2.2. PBMC isolation
PBMCs were isolated from freshly obtained whole blood from genetically identical C57Bl6 mice using density gradient separation [Histopaque 1083 (Sigma-Aldrich, St. Louis, MO)], following the manufacturer’s instructions. Typically, 200–300 μl of blood were obtained from each mouse, with the blood yielding approximately 3–5 X 106 PBMCs/ml, which is sufficient for the immunization of one mouse. Thus, pooled blood (5–6 ml) from 20–30 mice was generally required to conduct an experiment. Blood was mixed with an equal volume of 2% fetal bovine serum (FBS) in phosphate buffered saline (PBS) at room temperature. Subsequently, blood was mixed with the anticoagulant sodium citrate at a 1:9 (sodium citrate: blood) ratio and maintained at room temperature to avoid cell clumping. Next, 15 ml of Histopaque was poured into a 50 ml Sepmate tube (Vancouver, BC, Canada) and the pooled blood + FBS mixture gently layered onto the Histopaque. The tube was then centrifuged at 1200g for 10 min at room temperature. The upper layer then was quickly transferred to a fresh 50 ml tube and centrifuged at 500g for 5 minutes. Cell pellets were washed three times with PBS and finally re-suspended in 2 ml RPMI medium containing 10% FBS. Cell numbers were counted using a haemocytometer and adjusted as required. Importantly, the response of pooled cells would not be expected to differ if compared to the response of cells from a single animal, since inbred mice are being used as the source of blood. Specifically, even when mixed, the PBMCs from individual mice are genetically identical to and histocompatible with each other, as well as the mice into which they are being administered.
2.3. PBMC pulsing
PBMCs (4 X 106) were mixed with varying amounts of iFt and/or Prevnar ®13 Ag in 1 ml of RPMI 1640 medium supplemented with 10% FBS and then incubated for 3 h at 37°C (5% CO2). Subsequently, PBMCs were washed three times with 5 ml PBS. Finally, PBMCs were re-suspended in a sufficient volume of PBS to achieve the desired number of PBMCs/ml. The pulsed PBMCs were then used for immunization within 1 h after completion of Ag pulsing.
2.4. Immunization and challenge
Groups of 6– 8 female C57BL/6 mice were immunized on day 0 and 14 via the i.v., i.n., or i.m. route. Prior to administration, mice were anesthetized by intraperitoneal (i.p.) injection of 20% ketamine plus 5% xylazine before administering 28 μl (i.n.) or 50 μl (i.v.) of PBS (vehicle), Ag alone, PBMCs alone, Ag-pulsed PBMCs or challenge pathogens. Mice were then infected/challenged as described in each figure legend with Ft LVS or Sp serotype 3 (strain A66.1) via the i.n. route in 40 μl PBS and subsequently monitored for survival for at least 21 days.
2.5. Measuring Ab production
Ab responses to immunization were measured by ELISA for anti-Ft Ab as previously reported [18] and for anti-Sp Ab as follows. ELISA plates (Corning, Corning NY) were coated with 50 μl of live Sp (5 X107 CFU/ml) or live Ft LVS in carbonate buffer [4.3 g/L sodium bicarbonate (Sigma-Aldrich)] and 5.3 g/L sodium carbonate (Sigma-Aldrich) (at pH 9.4) for 16h at 4°C. Plates were then washed with washing buffer [PBS (Sigma) containing 0.5% BSA (Sigma)] and blocked for 2 hours with 200 μl of PBS containing 5% BSA. Serial 3-fold dilutions of sera (starting with 1:50) were added to the plates (50 ml /well) and incubated for 2h at 4°C. After three washes with washing buffer, Alkaline-phosphatase-conjugated anti-mouse Abs specific for IgG or IgM (Sigma) were added to each well (50 ml /well). ELISA plates were then incubated for 1 hour at 4°C and washed three more times with washing buffer. Next, 100 ml of BCIP/NBT (Alkaline-phosphatase substrate) (Sigma) was added and the plates were incubated for 1–3 h and optical density (OD) was read intermittently at 405 nm using a micro plate reader (Molecular Devices, Sunnyvale, CA). Using the Graph-Pad (Prizm) program, Ab titers were calculated as the EC50 (half maximal) value obtained by a 4-parameter non-linear regression curve between log reciprocal-dilution versus response (OD 405 nm).
2.6. Statistical analysis
The log-rank (Mantel–Cox) test was used for survival curves. For ELISA titers, 2-tailed Mann-Whitney U test was used to compare groups. Data analyses were performed using GraphPad Prism 5 (San Diego, CA).
3. Results
3.1. iFt-pulsed PBMCs generate protection against lethal Ft LVS challenge
Intravenous administration of PBMCs pulsed ex vivo with iFt conferred protection against Ft challenge (Fig. 1A), which correlated with the increased production Ft-specific Ab. (Fig 1B and C). This is similar to our previous studies in which i.n. administration of iFt-pulsed DCs also protected against Ft challenge [19]. However, while the number of iFt organisms/PBMC in this study was similar to that of iFt organisms/DC utilized in our previous study, one-third the number of PBMCs were needed to immunize as compared to DCs [19]. It should however also be noted that the immunization route for this experiment (i.v.) [Fig. 1 (PBMCs)] differed from the immunization route of our previous study (i.n.) utilizing DCs [19]. In regard to our focus on examining humoral immunity (Ab production), it should be noted that we have published data, as have others, demonstrating that Ab can play a role in protection against Ft LVS infection, even at the lower titers observed in these studies [20]. Lastly, it is also important to note that when immunizing with iFt-pulsed PBMCs administered i.v. and challenging with Ft LVS, bacteria were cleared in surviving mice (data not shown).
Figure 1. iFt-pulsed PBMCs generate protection against lethal Ft LVS challenge and Ft LVS specific Abs.
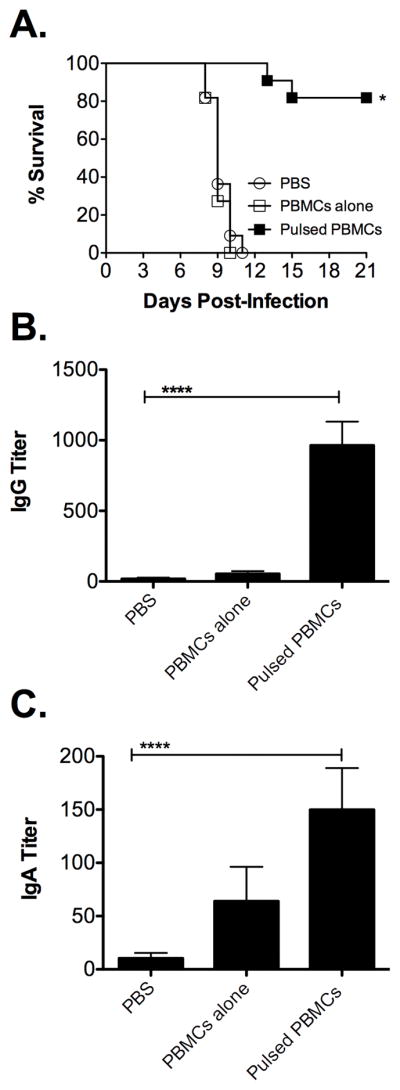
Freshly isolated PBMCs were pulsed with iFt at a ratio of 100 organisms/cell. Mice were administered (i.v.) two doses (two weeks apart) of 50 μl of PBS, 5 X 105 PBMCs alone, or 5 X 105 iFt-pulsed PBMCs. (A) Two weeks post-boost mice were infected with 1 X 104 CFU of Ft (i.n.) and their survival was monitored for 21 days. Values represent two independent experiments (n=12). (B and C) Two weeks post-boost, serum was collected and analyzed for Ft-specific IgG (B) and IgA (C) via ELISA. Values represent the mean ± SEM (n=12) from two independent experiments. Data were analyzed by Mann-Whitney U test. Significantly different groups (compared to PBS) are indicated with * (p<0.05) or ****(p<0.0001).
3.2. Prevnar ®13-pulsed PBMCs generate protection against lethal Sp challenge
In order to further demonstrate the potential usefulness of this PBMC-based immunization strategy, a more well-defined and commonly used vaccine candidate was also utilized. In this case, we selected the pneumococcal vaccine Prevnar ®13 and an established mouse model for evaluating vaccine efficacy against Sp [21]. As demonstrated in Figure 2A, 100% protection was observed when immunizing mice i.v. with Prevnar ®13-pulsed PBMCs versus direct injection of Prevnar ®13 i.v. or i.m. (50% and 63% protection, respectively). Furthermore, Prevnar ®13 pulsed PBMCs induced a slight increase in Sp-specific Ab titers compared to Prevnar ®13 administered alone (i.v.), while Prevnar ®13 administered i.m. alone induced titers of Sp-specific Ab similar to that of Prevnar ®13-pulsed PBMCs given i.m. (Fig. 2B). However, overall the Sp-specific Ab titers obtained following immunization were relatively low and the difference in survival between mice immunized with Prevnar ®13 i.m. (63%) and Prevnar ®13-pulsed PBMCs (100%) was not reflected in the Sp-specific Ab titers, suggesting that in this case and, as may also be the case in Figure 1, the Ab titers examined may not be the sole determinant of survival.
Figure 2. Prevnar ®13-pulsed PBMCs generate protection against lethal Sp challenge and Sp specific Ab.

PBMCs were pulsed with immunogen as described in Fig. 1 with slight modifications. Specifically, in this experiment, PBMCs were pulsed with 9.34 μg/ml Prevnar ®13. Mice were then administered i.v. two doses (two weeks apart) of 50 μl PBS, 5 X 105 PBMCs alone, or 5 X 105 Prevnar ®13-pulsed PBMCs. Positive controls included mice directly injected i.v. or i.m. with 1.7 μg of Prevnar ®13 alone. (A) Four weeks post-boost, mice were infected (i.n.) with 2 X 106 CFU of type 3 Sp and their survival was subsequently monitored for 21 days. Values represent one experiment (n= 6). (B) Twenty-five days post-boost, serum was collected and analyzed for total Sp-specific Ab via ELISA. Values represent the mean ± SEM (n=6). Data were analyzed by Mann-Whitney U test. Significantly different groups (compared to PBS) are indicated with *** (p<0.001) or *(p<0.05).
3.3. Prevnar ®13-pulsed PBMCs administered i.n. also generate protection
The above studies focused on pulsed PBMCs given i.v., which is not a traditional route of human immunization. However, i.n. immunization offers a less invasive and potentially more desirable route of immunization. Thus, we sought to determine if the PBMC-based vaccine platform would be equally effective when administering Prevnar ®13-pulsed PBMCs i.n. Importantly, when evaluating the i.v. route we tested the administration of PBMCs pulsed with 9.34 μg/ml, as well as 18.68 μg/ml Prevnar ®13, with the latter giving a slightly higher response (data not shown). Thus, we chose to use the higher dose in subsequent i.n. immunization studies. As shown in Figure 3A, survival results obtained were similar to those using the i.v. administration route, in that Prevnar ®13-pulsed PBMCs administered i.n. induced 100% survival. Consistent with the latter, Prevnar ®13-pulsed PBMCs also induced Sp–specific Abs (Fig. 3B). Notably, in this case, Sp-specific Ab titers induced by i.n. immunization were substantially higher than those obtained when using the i.v. route (Fig. 3B versus Fig. 2B, respectively).
Figure 3. Prevnar ®13-pulsed PBMCs administered i.n. also generate protection and Sp specific Ab.

PBMCs were pulsed with 18.68 μg/ml Prevnar ®13. Mice were then administered i.n. 28 μl of PBS or in 28 μl PBS; 5 X 105 PBMCs alone, or 5 X 105 Prevnar ®13-pulsed PBMCs. Mice in the positive control group received 1.7 μg of Prevnar ®13 alone. (A) Four weeks post-boost, mice were infected with 2 X 106 CFU of type 3 Sp and their survival was subsequently monitored for 21 days. Values represent three independent experiments (n=18). Significantly different groups (compared to PBS) are indicated with *** (p<0.001). (B) Twenty-five days post-boost, serum was collected and analyzed for total Sp-specific Ab via ELISA. Values represent three independent experiments (n=18). Data were analyzed by Mann-Whitney U test. Significantly different groups (compared to PBS) are indicated with ** (p<0.05).
We also evaluated the impact of PBMC-based vaccination on bacterial burden in the lungs of i.n. immunized mice. As shown in Table S1, mice receiving PBS or PBMCs alone exhibited approximately a 2–3 log increase in bacterial burden on day 4 compared to day 2. In contrast, mice receiving the Prevnar ®13 vaccine alone or Prevnar ®13-pulsed PBMCs exhibited a 1-2 log reduction in bacterial burden on day 2 and a 3–5 log reduction in bacteria on day 4, when compared to mice immunized with PBS or PBMCs alone. On day 12 both the Prevnar®13 alone and Prevnar®13-pulsed PBMC groups had cleared the pathogen from their lungs, while those immunized with PBS or PBMCs alone had died.
3.4. Prevnar ®13-pulsed PBMCs administered i.n. induce a more marked and longer-lived Sp-specific Ab response than Prevnar ®13 administered alone and enhanced survival
We also sought to determine the strength and longevity of the response following i.n. administration of Prevnar ®13-pulsed PBMCs versus i.m. or i.n. administration of Prevnar ®13 vaccine alone. Again, increased Sp-specific Ab titers correlated with increased protection (Fig. 4). Furthermore, the Sp-specific Ab response following an i.n. boost with Prevnar ®13-pulsed PBMCs was stronger and longer-lived than that of i.n. administered Prevnar ®13 vaccine (Fig 4B). This difference was also greater compared to Prevnar ®13 administered alone via the clinically approved i.m. route. Another similar experiment also generated similar results (data not shown) following an Sp challenge 12 weeks post-boost.
Figure 4. Prevnar ®13-pulsed PBMCs administered i.n. induce a more marked and longer-lived Sp-specific Ab response, as well as enhanced survival.

PBMCs were pulsed with 18.68 μg/ml Prevnar ®13. Mice were immunized on days 0 and 14 (red font) i.n. or i.m. with PBS, PBMCs only, Prevnar ®13 alone, or Prevnar ®13-pulsed PBMCs (A) Mice were challenged on week 14 with 2X106 CFU of Sp (serotype 3, strain A66.1). The values presented represent those from one of two similar experiments with similar results (n=6). Significantly different groups (compared to PBS) are indicated with *** (p<0.001) or *(p<0.01). (B) Sp-specific serum IgG titers were measured at the time intervals indicated. A statistically significant difference was observed when comparing Ab titers from mice immunized with Prevnar ®13-pulsed PBMCs administered i.n. versus direct administration of Prevnar ®13 alone i.n. The values presented represent those from one of two similar experiments with similar results (n=6). Data were analyzed by Mann-Whitney U test. Significantly different groups (compared to PBS) are indicated with *(p<0.05).
3.5 The Ag-pulsed PBMC vaccine strategy can also serve as a multi-organism vaccine platform
Given the positive results using the PBMC-based vaccine strategy and Ft and Sp immunization and challenge models, we investigated whether the PBMC-based vaccine platform could induce protection against multiple pathogens simultaneously using iFt plus Prevnar ®13-pulsed PBMCs administered i.n. Due to initial concerns that the vaccine Ags (iFt or Prevnar ®13), if combined during the pulse phase, may compromise the immunostimulatory capacity of the other, we first pulsed PBMCs individually with either iFt or Prevnar ®13, and subsequently combined the iFt and Prevnar 13-pulsed PBMCs just prior to immunization. Following Ft or Sp challenge of mice immunized with either the individual or combined PBMC-based vaccines, we observed that approximately 60–75% of mice immunized with either iFt or iFt plus Prevnar ®13-pulsed PBMCs survived Ft LVS challenge. Survival of Sp-challenged mice immunized with either Prevnar ®13 or iFt plus Prevnar ®13-pulsed PBMCs was 85–100%, with no survival being observed in mice immunized with PBS or PBMCs alone (Fig. 5A and B). Furthermore, no significant difference in protection was observed when comparing the effectiveness of the single versus combined vaccines. These studies were then repeated with iFt and Prevnar ®13 being combined during the pulse phase (Fig. 5C and D). Results were similar to those in Figures 5A and 5B re-affirming that this PBMC-based vaccine approach can serve as a multi-organism vaccine platform. Also as observed when using the single pathogen PBMC-based vaccination protocols for Ft or Sp (Figs. 1–4), both Ft-specific and Sp-specific Ab titers were increased substantially following immunization (Fig. 5E and F).
Figure 5. The Ag-pulsed PBMC vaccine strategy can also serve as a multi-organism vaccine platform.

Mice were immunized with PBS (A, B, C, D, E and F), PBMCs alone (A, B, C, D, E and F), PBMCs pulsed with iFt alone (12 μg/ml) (A, C, and E), PBMCs pulsed with Prevnar ®13 alone (18.68 μg/ml) (B, D, and F), PBMCs pulsed with iFt (12 μg/ml) and Prevnar ®13 (18.68 μg/ml) separately and mixed before immunization (A and B) or PBMCs pulsed simultaneously with iFt (12 μg/ml) and Prevnar ®13 (18.68 μg/ml) (C, D, E, and F). Four weeks post-boost mice were challenged with either 2100 CFU of Ft (A and C) or 2 X 106 CFU of Sp (B and D). Survival was monitored for 21 days. Values represent one experiment (n=6 mice/group for PBS and PBMC groups and 8 mice/group for Ag-pulsed PBMCs groups). Significantly different groups (compared to PBS) are indicated with *(p<0.05) **** (p < 0.0001), *** (p< 0.001) or * (p < 0.05). (E and F) Three weeks post-boost, sera was obtained from all the groups and analyzed for Ft-specific IgG (C) or Sp-specific IgG (D). Values represent the mean ± SEM (n=12) from two independent experiments. Data were analyzed by Mann-Whitney U test. Significantly different groups (compared to PBS) are indicated with * (p < 0.05) or **** (p < 0.01).
4. Discussion
In this study we demonstrate that PBMCs can efficiently deliver vaccine Ags and elicit immune protection against bacterial infection when pulsed ex vivo and re-administered as a vaccine either i.v. or i.n. Specifically, iFt or Prevnar ®13-pulsed PBMCs induced protection against lethal challenge with Ft LVS or Sp, respectively. The protection also correlated with increased Ft LVS or Sp-specific Abs. In regard to our central focus on the generation of humoral (Ab) responses, evidence indicates that Ab is key to protection against Sp [16]. In addition, Ab can also play a role in protection against Ft LVS [17, 22]. Nevertheless, cellular immunity plays a key role in the latter and thus is the focus of an in depth but separate manuscript on this topic.
The precise mechanism(s) by which Ag-pulsed PBMC’s induce protective immunity remains to be determined. However, it is worth noting that APCs within the PBMC population not only include DCs, but also monocytes/macrophages and B cells, all of which could synergize to produce a more diverse and potent immune response. Furthermore, analysis of PBMCs for activation markers pre and post-PBMC isolation indicate that cellular activation within the PBMC population following density gradient separation does occur (Fig. S1). Importantly, activated APCs within the PBMC population would be better equipped to stimulate an immune response in terms of processing and presenting Ags, secreting pro-inflammatory cytokines, and migrating to lymphoid tissues, once introduced into host tissues. During the Ag-pulsing of PBMCs, APCs may also be activated by adjuvants such as that contained in Prevnar ®13, through the engagement of Pathogen Associated Molecular Pattern molecules (PAMPs) [23, 24] found on iFt, and/or cellular debris resulting from the in vitro isolation/processing of PBMCs. Thus, we expect such activation likely does contribute to a positive impact on the downstream immune response generated by Ag-pulsed PBMCs. In addition, the experimental parameters utilized in this study, such as the PBMC isolation method, medium used during pulsing, use of APC activation factors, time of incubation, number of PBMCs, Ag dose, ratio of Ag to PBMC, and the method of administration of Ag-pulsed PBMCs may impact the potency and efficacy of this approach. Thus, further optimization will be required, in particular when applied to humans.
While different routes of immunization may be preferable dependent on the pathogen and immune mechanisms(s) required for protection, we observed that the i.n. route induced better protection against Sp compared to the i.v route. Conversely, the i.v. route evoked a more favorable immune response in the case of Ft. Importantly, the i.n. route offers numerous advantages including ease of administration and the ability to induce strong mucosal and parenteral immune responses [25–27]. The differences based on vaccination route that we observed may be due to a number of factors such as inherent differences in vaccine Ags (Prevnar 13, which is a protein-carbohydrate conjugate vaccine versus iFt, a whole cell inactivated vaccine). Also, recent studies have noted the role of addressin in adhesion of naive lymphocytes to high endothelial venules at mucosal sites [28]. In the case of Ag-pulsed PBMCs, the pulsed immunocytes might more easily gain direct access to lymphoid tissues through such mechanisms, as compared to the i.v. route. Alternatively, differential immune requirements of the respective pathogens for immune protection could also explain this difference. In addition to the above, there are a multitude of additional factors that may also explain this observation. First, the polysaccharide Ags in Prevnar ®13 versus the whole cell Ags of iFt may have distinct pharmacokinetics. Second, Ft is known to induce distinct immune responses depending on the route of introduction. Specifically, when utilizing the i.n. route, an IL-17 dominant response is produced, whereas a response favoring IFN-γ production is produced via the dermal route [29]. Also, the i.v. route has been found to be less effective at inducing an immune response to Ag (peptide)-pulsed DC vaccines as compared to s.c., i.p. and i.d. routes [30, 31]. Thus, it is possible that polysaccharide conjugate vaccines such as Prevnar ®13 follow a similar pattern. If so, Prevnar 13-pulsed PBMCs may produce considerably lower immune responses by the i.v. route as compared to the i.n. route. Nevertheless, studies in this regard are very limited and further study will be needed to determine the optimal immunization route based on the specific pathogen for which the vaccine is being developed. In addition, the oral route also remains a future area of investigation, given that animal studies suggest that human infants very likely absorb white blood cells from colostral breast milk [32, 33].
The observation that protective immune responses to two different vaccine Ags can be achieved when pulsing PBMCs simultaneously with those Ags suggests that this approach might also provide a multi-organism vaccine platform. If confirmed in humans, this vaccine strategy could significantly reduce the costs and trauma of immunization by a) reducing vaccine dose magnitude; b) reducing vaccination clinic visits needed to achieve protection against multiple diseases; and c) reducing the number of injections required for currently recommended immunizations. Importantly, while the studies presented suggest this to be a very promising approach to multivalent vaccination, as well as vaccination in general, further research is needed to determine the maximal number of vaccine Ags, which can induce protective responses after simultaneous PBMC pulsing, the lowest doses of Ags and shortest pulse periods required, and the relationship of the findings in mice to those in human subjects. It is also important to note that the pooling of blood from multiple donors required to conduct mouse studies would not be necessary in the case of humans, since the pulsed PBMCs will be isogenic (either obtained from the recipients blood or cord blood) in which case MLR reactions would not be a concern. Also, we do not know the number of PBMCs that will be required for translation of this strategy to humans. This will require clinical studies. However, in one DC-based immunotherapy study approximately 50 X 106 Ag-pulsed DCs were delivered into a human donor [13, 14]. To obtain 50 X 106 PBMCs approximately 25–50 ml blood would be required, which is easily achieved. While this could prove problematic in the case of infants, future studies will also focus on reducing the number of PBMCs required.
Demonstration of polyvalent simultaneous immunization using the i.n. method also provides a basis for studies to determine whether this method/route can be made practical for field use by further simplification (elimination of wash steps, separation of PBMC’s by simple centrifugation, possible use of cord blood, etc.). Other applications could include: multiple simultaneous immunizations against bioterrorism agents, testing of adjuvants, reducing adjuvant toxicity, and evaluation of potential utility with DNA vaccines, as well as prophylactic or therapeutic immunization against diseases for which currently available vaccines are either suboptimal or nonexistent.
5. Conclusions
A new method of vaccination based on ex vivo Ag pulsing of PBMCs has been shown to protect mice from lethal challenge with Ft and Sp. In regard to the latter, administration of Ag-pulsed PBMC’s induced more marked and longer-lived Sp specific Ab responses and enhanced survival. Furthermore, potential for the use of this strategy as a multi-organism/multivalent vaccine platform was demonstrated. If adaptable for human use, this new method could potentially reduce the number of injections, clinic visits, and costs needed to provide protection against vaccine preventable or vaccine treatable diseases by making possible multiple simultaneous vaccinations in a single clinic visit (See Supplemental Text and Table S2).
Supplementary Material
Highlights.
Antigen-pulsed PBMCs effectively induce protection against cognate pathogens.
Antigen-pulsed PBMCs can be delivered as an effective vaccine intranasally.
Antigen-pulsed PBMCs induce enhanced antigen-specific antibody production.
Antigen-pulsed PBMCs induce longer lasting immunity and protection.
This PBMC-based approach can serve as a multivalent vaccine platform.
Acknowledgments
This work was supported by the National Institutes of Health (R01 AI100138), a gift to AMC (DN), and a matching gift from the Merck Partnership For Giving. We thank the Animal Research Facility and DIMD Immunology Core for expert technical assistance during these studies.
Abbreviations
- DCs
Dendritic cells
- PBMCs
Peripheral Blood Mononuclear Cells
- Ab
Antibody
- Ft
F. tularensis
- iFt
Inactivated Ft
- Sp
Streptococcus pneumoniae
- Ag
Antigen
- APCs
Ag Presenting Cells
- FBS
Fetal Bovine Serum
- PBS
Phosphate Buffered Saline
- i.n
Intranasal
- i.v
Intravenous
- i.m
Intramuscular
- i.p
Intraperitoneal
Footnotes
Conflict of Interest
Dr. Nalin owns three patents on the PBMC vaccination method and a related device. None of the other authors have conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998 Mar 19;392(6673):245–52. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Apostolopoulos V, Thalhammer T, Tzakos AG, Stojanovska L. Targeting Antigens to Dendritic Cell Receptors for Vaccine Development. Journal of drug delivery. 2013;2013:869718. doi: 10.1155/2013/869718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia F, Plana M, Climent N, Leon A, Gatell JM, Gallart T. Dendritic cell based vaccines for HIV infection: The way ahead. Human vaccines & immunotherapeutics. 2013 Aug 2;9(11) doi: 10.4161/hv.25876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boonnak K, Vogel L, Orandle M, Zimmerman D, Talor E, Subbarao K. Antigen-activated dendritic cells ameliorate influenza A infections. J Clin Invest. 2013 Jul 1;123(7):2850–61. doi: 10.1172/JCI67550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nature reviews Immunology. 2007 Oct;7(10):790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 6.Gil M, Bieniasz M, Wierzbicki A, Bambach BJ, Rokita H, Kozbor D. Targeting a mimotope vaccine to activating Fcgamma receptors empowers dendritic cells to prime specific CD8+ T cell responses in tumor-bearing mice. J Immunol. 2009 Nov 15;183(10):6808–18. doi: 10.4049/jimmunol.0900364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Su H, Messer R, Whitmire W, Fischer E, Portis JC, Caldwell HD. Vaccination against chlamydial genital tract infection after immunization with dendritic cells pulsed ex vivo with nonviable Chlamydiae. J Exp Med. 1998 Sep 7;188(5):809–18. doi: 10.1084/jem.188.5.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garcia F, Climent N, Guardo AC, Gil C, Leon A, Autran B, et al. A dendritic cell-based vaccine elicits T cell responses associated with control of HIV-1 replication. Science translational medicine. 2013 Jan 2;5(166):166ra2. doi: 10.1126/scitranslmed.3004682. [DOI] [PubMed] [Google Scholar]
- 9.Ozawa Y, Suda T, Nagata T, Hashimoto D, Nakamura Y, Enomoto N, et al. Mucosal vaccine using CTL epitope-pulsed dendritic cell confers protection for intracellular pathogen. American journal of respiratory cell and molecular biology. 2009 Oct;41(4):440–8. doi: 10.1165/rcmb.2008-0446OC. [DOI] [PubMed] [Google Scholar]
- 10.Zhou Y, Zhang Y, Yao Z, Moorman JP, Jia Z. Dendritic cell-based immunity and vaccination against hepatitis C virus infection. Immunology. 2012 Aug;136(4):385–96. doi: 10.1111/j.1365-2567.2012.03590.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015 Mar 19;519(7543):366–9. doi: 10.1038/nature14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Constantino J, Gomes C, Falcao A, Cruz MT, Neves BM. Antitumor dendritic cell-based vaccines: lessons from 20 years of clinical trials and future perspectives. Translational research : the journal of laboratory and clinical medicine. 2016 Feb;168:74–95. doi: 10.1016/j.trsl.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 13.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. The New England journal of medicine. 2010 Jul 29;363(5):411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 14.Carballido E, Fishman M. Sipuleucel-T: Prototype for development of anti-tumor vaccines. Current oncology reports. 2011 Apr;13(2):112–9. doi: 10.1007/s11912-011-0152-5. [DOI] [PubMed] [Google Scholar]
- 15.Oyston PC, Sjostedt A, Titball RW. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nature reviews Microbiology. 2004 Dec;2(12):967–78. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 16.Wilson R, Cohen JM, Jose RJ, de Vogel C, Baxendale H, Brown JS. Protection against Streptococcus pneumoniae lung infection after nasopharyngeal colonization requires both humoral and cellular immune responses. Mucosal immunology. 2015 May;8(3):627–39. doi: 10.1038/mi.2014.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drabick JJ, Narayanan RB, Williams JC, Leduc JW, Nacy CA. Passive protection of mice against lethal Francisella tularensis (live tularemia vaccine strain) infection by the sera of human recipients of the live tularemia vaccine. The American journal of the medical sciences. 1994 Aug;308(2):83–7. doi: 10.1097/00000441-199408000-00003. [DOI] [PubMed] [Google Scholar]
- 18.Bitsaktsis C, Rawool DB, Li Y, Kurkure NV, Iglesias B, Gosselin EJ. Differential requirements for protection against mucosal challenge with Francisella tularensis in the presence versus absence of cholera toxin B and inactivated F. tularensis. J Immunol. 2009 Apr 15;182(8):4899–909. doi: 10.4049/jimmunol.0803242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pham GH, Iglesias BV, Gosselin EJ. Fc receptor-targeting of immunogen as a strategy for enhanced antigen loading, vaccination, and protection using intranasally administered antigen-pulsed dendritic cells. Vaccine. 2014 Sep 8;32(40):5212–20. doi: 10.1016/j.vaccine.2014.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rawool DB, Bitsaktsis C, Li Y, Gosselin DR, Lin Y, Kurkure NV, et al. Utilization of Fc receptors as a mucosal vaccine strategy against an intracellular bacterium, Francisella tularensis. J Immunol. 2008 Apr 15;180(8):5548–57. doi: 10.4049/jimmunol.180.8.5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bitsaktsis C, Iglesias BV, Li Y, Colino J, Snapper CM, Hollingshead SK, et al. Mucosal immunization with an unadjuvanted vaccine that targets Streptococcus pneumoniae PspA to human Fcgamma receptor type I protects against pneumococcal infection through complement- and lactoferrin-mediated bactericidal activity. Infection and immunity. 2012 Mar;80(3):1166–80. doi: 10.1128/IAI.05511-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubelkova K, Krocova Z, Balonova L, Pejchal J, Stulik J, Macela A. Specific antibodies protect gamma-irradiated mice against Francisella tularensis infection. Microbial pathogenesis. 2012 Nov-Dec;53(5–6):259–68. doi: 10.1016/j.micpath.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 23.Diebold SS. Activation of dendritic cells by toll-like receptors and C-type lectins. Handbook of experimental pharmacology. 2009;(188):3–30. doi: 10.1007/978-3-540-71029-5_1. [DOI] [PubMed] [Google Scholar]
- 24.Owen JL, Mohamadzadeh M. Microbial activation of gut dendritic cells and the control of mucosal immunity. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2013 Nov;33(11):619–31. doi: 10.1089/jir.2013.0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis SS. Nasal vaccines. Advanced drug delivery reviews. 2001 Sep 23;51(1–3):21–42. doi: 10.1016/s0169-409x(01)00162-4. [DOI] [PubMed] [Google Scholar]
- 26.Pabst R. Mucosal vaccination by the intranasal route. Nose-associated lymphoid tissue (NALT)-Structure, function and species differences. Vaccine. 2015 Aug 26;33(36):4406–13. doi: 10.1016/j.vaccine.2015.07.022. [DOI] [PubMed] [Google Scholar]
- 27.Riese P, Sakthivel P, Trittel S, Guzman CA. Intranasal formulations: promising strategy to deliver vaccines. Expert opinion on drug delivery. 2014 Oct;11(10):1619–34. doi: 10.1517/17425247.2014.931936. [DOI] [PubMed] [Google Scholar]
- 28.Csencsits KL, Jutila MA, Pascual DW. Nasal-associated lymphoid tissue: phenotypic and functional evidence for the primary role of peripheral node addressin in naive lymphocyte adhesion to high endothelial venules in a mucosal site. J Immunol. 1999 Aug 1;163(3):1382–9. [PubMed] [Google Scholar]
- 29.Woolard MD, Hensley LL, Kawula TH, Frelinger JA. Respiratory Francisella tularensis live vaccine strain infection induces Th17 cells and prostaglandin E2, which inhibits generation of gamma interferon-positive T cells. Infection and immunity. 2008 Jun;76(6):2651–9. doi: 10.1128/IAI.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okada N, Tsujino M, Hagiwara Y, Tada A, Tamura Y, Mori K, et al. Administration route-dependent vaccine efficiency of murine dendritic cells pulsed with antigens. British journal of cancer. 2001 Jun 1;84(11):1564–70. doi: 10.1054/bjoc.2001.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mullins DW, Sheasley SL, Ream RM, Bullock TN, Fu YX, Engelhard VH. Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. The Journal of experimental medicine. 2003 Oct 6;198(7):1023–34. doi: 10.1084/jem.20021348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tuboly S, Bernath S. Intestinal absorption of colostral lymphoid cells in newborn animals. Advances in experimental medicine and biology. 2002;503:107–14. doi: 10.1007/978-1-4615-0559-4_12. [DOI] [PubMed] [Google Scholar]
- 33.Goldman AS, Goldblum RM. Transfer of maternal leukocytes to the infant by human milk. Current topics in microbiology and immunology. 1997;222:205–13. doi: 10.1007/978-3-642-60614-4_10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.