

Abstract
A novel nonnucleoside inhibitor of hepatitis C virus (HCV) RNA-dependent RNA polymerase (RdRp), [(1R)-5-cyano-8-methyl-1-propyl-1,3,4,9-tetrahydropyano[3,4-b]indol-1-yl] acetic acid (HCV-371), was discovered through high-throughput screening followed by chemical optimization. HCV-371 displayed broad inhibitory activities against the NS5B RdRp enzyme, with 50% inhibitory concentrations ranging from 0.3 to 1.8 μM for 90% of the isolates derived from HCV genotypes 1a, 1b, and 3a. HCV-371 showed no inhibitory activity against a panel of human polymerases, including mitochondrial DNA polymerase gamma, and other unrelated viral polymerases, demonstrating its specificity for the HCV polymerase. A single administration of HCV-371 to cells containing the HCV subgenomic replicon for 3 days resulted in a dose-dependent reduction of the steady-state levels of viral RNA and protein. Multiple treatments with HCV-371 for 16 days led to a >3-log10 reduction in the HCV RNA level. In comparison, multiple treatments with a similar inhibitory dose of alpha interferon resulted in a 2-log10 reduction of the viral RNA level. In addition, treatment of cells with a combination of HCV-371 and pegylated alpha interferon resulted in an additive antiviral activity. Within the effective antiviral concentrations of HCV-371, there was no effect on cell viability and metabolism. The intracellular antiviral specificity of HCV-371 was demonstrated by its lack of activity in cells infected with several DNA or RNA viruses. Fluorescence binding studies show that HCV-371 binds the NS5B with an apparent dissociation constant of 150 nM, leading to high selectivity and lack of cytotoxicity in the antiviral assays.
Hepatitis C virus (HCV) infection is the cause of significant long-term morbidity and mortality. While often asymptomatic, the majority of HCV infections result in chronic hepatitis that can progress to cirrhosis, end-stage liver disease, and hepatocellular carcinoma. It is estimated that about 170 million people worldwide, approximately 3% of the world's population, are infected with HCV (45). Presently, there is no specific antiviral agent directed against HCV and no vaccine for prevention of hepatitis C infection. The current approved treatments, interferon monotherapy or interferon in combination with ribavirin, have limited benefits. Interferon alone achieves a sustained viral response in only 10 to 20% of patients (32), while the recommended therapy of pegylated interferon in combination with ribavirin results in a sustained viral response in 54% of patients (23). The response rate is lower in patients who are infected with HCV genotype 1b (∼34%) (23, 47). Adverse side effects, such as severe flu-like symptoms, depression, psychoses, and anemia, are associated with these treatments, causing approximately 20% of patients to discontinue therapy (13, 17, 33). Consequently, there is an urgent need for the development of an HCV-specific antiviral that is more effective, less toxic, and easier to administer than the present therapy.
HCV, a member of the Flaviviridae family, is a positive-sense, single-stranded RNA virus with a genome size of ∼9.4 kb (26, 39). The genome RNA encodes a polyprotein of 3,010 to 3,011 amino acid residues in the order NH2-C-E1-E2-p7-NS2-NS3-NS4A-NS4B-NS5A-NS5B-COOH. This polyprotein is processed by host and viral proteases (18, 34). The nonstructural protein 5B (NS5B) is a virus-encoded RNA-dependent RNA polymerase (RdRp) that is responsible for replication of the viral RNA genome. A functional counterpart of NS5B does not exist in mammalian cells. For this reason, an inhibitor of NS5B could serve as an effective and selective agent for treating HCV infection.
The enzymatic activity of the NS5B enzyme in vitro has been extensively characterized (9, 19, 21, 40). Both the full-length and carboxyl-terminally truncated forms of NS5B have been shown to be functionally active in the presence of HCV or exogenous RNA templates. Although HCV replicates inefficiently in cell culture, viral replication, including the activity of the NS5B polymerase, can be studied in a cell culture system containing the HCV replicon (3, 20). The HCV replicon is a subgenomic RNA that consists of the HCV 5′ N-terminal repeat (NTR) upstream of a neomycin phosphotransferase gene, followed by the internal ribosome entry site of the encephalomyocarditis virus, the gene segment encoding the HCV NS3 to NS5B proteins, and the HCV 3′ NTR. Human hepatoma cells that harbor this replicon support high levels of autonomous HCV RNA replication and nonstructural protein production. Using this system, the effect of a compound on HCV replication can be measured by quantifying the amounts of viral RNA or protein in these cells. In addition, potential cytotoxic effects introduced by the compound can be measured by monitoring the levels of housekeeping genes in the same cells. The availability of these in vitro assays makes it possible to screen for compounds that might inhibit HCV replication.
The present report describes the discovery of a novel inhibitor, [(1R)-5-cyano-8-methyl-1-propyl-1,3,4,9-tetrahydropyano[3,4-b]indol-1-yl] acetic acid (HCV-371), that inhibits the HCV NS5B RdRp. The enzymatic selectivity against NS5B was demonstrated in several biochemical assays using a panel of human polymerases. The antiviral activity of this compound was evaluated and compared to that of alpha interferon (IFN-α) in the HCV replicon-cell culture model system. In addition, antiviral specificity and the lack of cytotoxicity were assessed. The mechanism of inhibition will be discussed.
MATERIALS AND METHODS
Materials.
Synthesis and purification of HCV-371 will be described elsewhere (9a). The plasmid pRC/CMV-HCV-BK, which contains all of the viral protein coding sequences of genotype 1b (BK), was derived from the cDNA clones provided by H. Okayama (39). The pOF RNA that was used for the RdRp assay was prepared as described previously (1). The expression plasmids carrying the NS5B gene of genotypes 1a (9606, A5, 207-248, 207-221, and 207-247), 1b (4a, FC 1-20, 320-10-3, 320-3-1, and 320-2-1), 3a (167-A1), and 4 were derived from sera from infected patients. Calf thymus DNA polymerase α enzyme was purchased from Chimerx Corp (Madison, Wis.). Human DNA polymerase α was purified from HeLa cells by using the method described by Wang et al. (44), with the exception that the monoclonal antibody SJK-132-20 was covalently conjugated to Affiprep Herz (Bio-Rad). Human DNA polymerase β was purchased from Trevigen. The α and β subunits of the human mitochondrial DNA polymerase γ were obtained from Laurie S. Kaguni (Michigan State University). To generate the holoenzyme form of DNA polymerase γ, the α and β subunits were combined at a 1:5 molar ratio and allowed to complex for 20 min at 4°C. Human RNA polymerase II was purified by conventional chromatography by using the method described by Maldonado et al. (22). Purified human immunodeficiency virus (HIV) reverse transcriptase was purchased from Worthington Biochemicals. All nucleotide triphosphates (NTPs) and RNasin inhibitor were purchased from Promega. [α-33P]NTP (3,000 Ci/mmol) was purchased from NEN. DEAE Multiscreen filter plates were purchased from Millipore. All other molecular biology reagents were obtained from suppliers as indicated.
Purification of NS5B.
The expression plasmid encoding the His-tagged C-terminal 21-amino-acid-deleted NS5B (NS5BΔCT21-His) was transformed in Escherichia coli cells. The bacterial cells were grown at 16°C, and the expression of NS5B was initiated by the addition of 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Following 4 to 6 h of incubation, the cells were harvested by centrifugation, and the cell pellet was either used immediately or stored at −80°C. Bacterial cells were lysed by at least three passages of the cells through the microfluidizer while the temperature was maintained at 4°C. The crude extract was batch loaded onto a nickel affinity resin (Nickel-nitrilotriacetic acid; QIAGEN) and washed successively with a buffer containing first 5 mM and then 25 mM imidazole. The protein was eluted with the application of 200 to 300 mM imidazole buffer. The eluted material was added to a cation exchange column (Poros HS; Perseptive Biosystem), followed by a wash using 20 mM HEPES (pH 7.4, 2.5% glycerol, 200 mM NaCl, 10 mM dithiothreitol [DTT]). The protein was then eluted with a NaCl gradient from 200 mM NaCl to 2 M NaCl in a mixture containing 20 mM HEPES, 2.5% glycerol, and 10 mM DTT. In the case of genotype 1b (BK) NS5BΔCT21-His, the purification was finished with size exclusion chromatography (Superdex 200; Amersham Pharmacia Biotech). For the preparation of enzymes from genotype 1b (isolate 320), 1a (207), and 3a (167), the last step involved purification on a heparin Sepharose column (Amersham Pharmacia Biotech). Upon loading of the sample, the column was washed with a mixture containing 20 mM Tris-HCl, pH 7.6, 350 mM NaCl, 2.5% glycerol, and 10 mM DTT. The bound proteins were eluted from the heparin column by using a linear gradient starting with the wash buffer and ending with a mixture containing 20 mM Tris-HCl, pH 7.6, 1 M NaCl, 2.5% glycerol, and 10 mM DTT. The proteins were concentrated up to 5 mg/ml, exchanged into buffer containing 50% glycerol, 25 mM HEPES, pH 7.5, 10 mM DTT, and 600 mM NaCl, and stored at −20°C.
NS5B RdRp assay.
The RdRp assay was performed in a final volume of 50 μl per reaction. Twenty microliters of the NS5B enzyme mix containing 24 nM NS5B, 20 mM HEPES (pH 7.5), 5 mM MgCl2, 1 mM DTT, 0.05 mg of bovine serum albumin (BSA)/ml, 0.5 μM UTP, 1 μM ATP, 0.08 μM CTP, and 0.025 μM GTP was incubated in the presence of 10 μl of compounds for 15 min at room temperature. Concentrations of RNA and NTPs were kept at apparent Km levels. The final concentration of dimethyl sulfoxide (DMSO) in the reaction was 3%. The reaction was initiated by adding 3 nM pOF-transcribed RNA substrate, 0.4 U of RNasin/μl, and 0.125 μCi of [α-33P]GTP and incubated at room temperature for 2 h. The reaction was terminated by the addition of 50 μl of 150 mM EDTA. Product RNA containing incorporated radioactive nucleotides was collected by filtration through Millipore Multiscreen plates and washing three times with 200 μl of 0.5 M sodium phosphate buffer (pH 7.0) by using a Millipore Manifold. The filters containing the reaction products were allowed to dry at room temperature, and radioactivity was quantified by using a Wallac MicroBeta after the addition of 50 μl of Optiphase scintillant. The same product-harvesting procedure was applied to the other polymerase assays (see below).
HCV NS3 helicase expression, purification, and assay.
E. coli BL21 (DE3) (Stratagene) was transformed with an expression vector (pET 28b; Novagen Corp.) containing a genotype 1a, subtype H77 DNA sequence that encodes the C-terminal helicase domain of NS3 (amino acids 1193 to 1657 of the polyprotein; amino acids 167 to 631 of NS3). The nucleotide sequences encoding four amino acids were mutated to convert the sequence into one with a consensus type 1a-1b sequence (14). The expressed protein contains 20 additional amino acids at its N terminus derived from the vector, including six N-terminal His residues. After induction by overnight growth at room temperature in the presence of 0.5 mM IPTG, bacteria were lysed by sonication, and the enzyme was purified from the soluble fraction to >95% purity by chromatography on Ni2+-nitrilotriacetic acid resin (QIAGEN) and poly(A)-Sepharose (Pharmacia). Each helicase reaction (50 μl) contained buffer A (25 mM MOPS [morpholinepropanesulfonic acid], pH 7.0, 1.5 mM MgCl2, 2.5 mM DTT, 0.1 mg of BSA/ml, 1 mM ATP), 2 nM HCV helicase enzyme, and 0.5 nM partial double-stranded DNA substrate with a 3′ overhang (IGEN). For compound evaluation, 5 μl of the compound was dissolved in buffer A and incubated with enzyme diluted in buffer A for 10 min at room temperature. The reaction was initiated by the addition of ATP and DNA substrate. The reaction was carried out for 20 min at room temperature, terminated, and harvested by using the stop-capture oligonucleotide mix provided by IGEN. The amount of unwinding activity was quantified with the IGEN M8 instrument according to the manufacturer's instructions.
Human polymerase α, β, and γ and RNA polymerase II assays.
DNA polymerase α was assayed in a reaction containing 50 mM Tris-HCl (pH 8.0), 5 mM magnesium acetate, 0.05 mg of BSA/ml, 3% DMSO, 1 mM DTT, 1 μM dATP, 1 μM dCTP, 1 μM dGTP, 0.5 μM TTP, 0.06 mg of DNase 1-activated calf thymus DNA (CT DNA)/ml, 0.1 pmol of [α-33P]TTP (3,000 Ci/mmol; 3.3 μM), and 0.004 U of enzyme. The enzyme reaction was initiated by the addition of 5 μl of substrate solution composed of appropriate template primer, nucleotides, and RNase-free water. Enzyme assays were performed for 1 h at 37°C. For human DNA polymerase α, the Km values for deoxynucleoside triphosphates and DNase 1-activated CT DNA were determined empirically as ∼0.5 to 1 μM and 0.04 mg/ml, respectively. Human polymerase β was assayed in a reaction containing 50 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 100 mM KCl, 0.4 mg of BSA/ml, 3% DMSO, 1 mM DTT, and 0.04 U of enzyme, with substrate concentrations adjusted to the respective Km levels. The Km values for DNase 1-activated CT DNA, dATP, dCTP, dTTP, and dGTP were 0.015 mg/ml and 5, 10, 16, and 4 μM, respectively. The reaction was carried out for 2 h at room temperature. Human DNA polymerase γ holoenzyme was assayed using singly primed M13 DNA. Each reaction (25 μl) contained 50 mM Tris-HCl, pH 8.0, 2 mM MgCl2, 1 mM DTT, 50 μg of BSA/ml, 0.2 M potassium acetate (pH 8.0), 2.3 ng of the α subunit, and 3 ng of the β subunit. The final concentrations of substrates in the reaction were 1 μM (each) dATP, dCTP, and dGTP, 0.5 μM TTP, 1 pmol of [α-33P]TTP (3.3 μM; 3,000 Ci/mmol), and 6 nM (as nucleotide) singly primed M13 DNA. The enzyme reaction was initiated by the addition of 5 μl of substrate solution composed of appropriate template primer, nucleotides, and RNase-free water. The reaction was carried out for 2 h at room temperature. The Km values for each substrate were taken from the literature (5, 11, 16). Human RNA polymerase II activity was assayed at 37°C for 1.5 h in a final volume of 25 μl of a buffer containing 50 mM Tris-HCl (pH 7.9), 5% glycerol, 70 mM (NH4)2SO4, 2 mM MnCl2, 1 mM DTT, and 50 μg of BSA/ml. Each assay contained 1 to 2 μl of enzyme (0.060 mg/ml, ∼50% pure). The enzyme in reaction buffer was allowed to incubate in the presence or absence of compound delivered in 50 mM HEPES and 15% DMSO (5 μl) for 15 min. The enzyme reaction was initiated by the addition of 5 μl of substrate solution composed of 10 μM UTP and 0.05 μM poly(dA)-oligo(dT)12 (as nucleotide; Pharmacia).
Calf thymus DNA polymerase α assay.
The calf thymus DNA polymerase α activity was determined by using the conditions described by Chimerx, with the exception that the concentrations of NTPs were kept at Km levels. The Km values for dATP, dCTP, dGTP, and dTTP were determined empirically to be 12, 1.2, 1.5, and 2.7 μM, respectively. The reaction was incubated at room temperature for 2 h.
HIV reverse transcriptase assay.
Each reaction (25 μl) for the HIV reverse transcriptase assay contained 50 mM Tris-HCl (pH 8.5), 6 mM MgCl2, 80 mM KCl, 0.05% Triton X-100, 50 μg of BSA/ml, 1 mM DTT, 3% DMSO, and 10 fmol of enzyme. Enzyme and compound were incubated for 15 min at room temperature in a final volume of 15 μl. Reactions were initiated by the addition of substrate mix consisting of 16.25 μM TTP, 0.825 μCi of [α-33P]TTP, and 15 nM poly(rA-dT)12-18 and incubated for 2 h at room temperature. The Km values for TTP and poly(rA)-oligo(dT)12-18 were determined to be 7 and 0.05 μM, respectively.
Generation of HCV replicon cells.
Clone A cells that contain an HCV genotype 1b replicon (derived from Huh-7 cells, a human hepatoma cell line) were licensed from Apath, LLC. A genotype 1a (H77 isolate; accession no. AF009606) replicon-containing cell line was established as follows. The genotype 1a replicon plasmid was prepared by substituting the 5′ end up to and including the sequences encoding the first 11 amino acids of NS3 (i.e., 5′ NTR, genes for the structural proteins and NS2, and sequences encoding the first 11 amino acids of NS3) of the genotype 1a infectious DNA clone (pCV-H77C; isolate H77) with the 5′ NTR sequences, the neo gene, the encephalomyocarditis virus internal ribosome entry site, and HCV sequences encoding the first 11 amino acids of NS3 from pBB7 (genotype 1b sequence BB7 plasmid licensed from Apath, LLC). The resulting genotype 1a replicon plasmid (pCV-H77CSR) encodes an NS3 protein with a single amino acid change relative to isolate H77 (alanine to serine at amino acid position 7 of NS3) and no changes in the 5′ NTR. The pCV-H77CSR DNA was linearized with XbaI or ScaI, and in vitro transcription was performed by using Ambion's Megascript T7 high-yield transcription kit. Purified RNA transcripts were electroporated into RNA transfection-susceptible clonal cell lines (CV-H77SCR) with a BTX ECM 830 Electro Square Porator (HV setting, high-voltage mode, 99-μs pulse length/3.0 kV, voltage of 0.96 kV, five pulses). A stably transfected genotype 1a cell line was selected with 0.3 mg of Geneticin/ml (G418 sulfate; Gibco/BRL). Sequence analysis of the genotype 1a replicon in cell line 2-5 after several passages revealed nucleotide changes that resulted in three single amino acid changes, one in the neo gene (methionine to valine at position 178), a second one in NS3 (arginine to lysine at position 110), and a third one in NS4B (cysteine to arginine at position 257).
Optimization of replicon cell-based screening assay.
HCV replicates inefficiently in cell culture, precluding detailed studies of viral replication by traditional tissue culture techniques. A replicon-containing cell system (Clone A cells) (3, 20) can be used to evaluate the antiviral effects of small-molecule inhibitors on HCV replication (28). HCV replication in this cell culture model has been demonstrated to be tightly coupled to the growth conditions of host cells (28). Accordingly, the Clone A replicon assay was optimized by using different cell seeding densities, culture media, and serum concentrations. The dynamics of HCV replication was monitored by measuring the levels of replicon RNA and protein under these conditions. When cells were seeded at 15 to 20% confluence, the level of the HCV RNA increased with the cell count during the first 3 days. As the cell density approached confluence from day 4 and on, a steady decrease of viral RNA levels was observed (data not shown). Thus, HCV replication appears to be stimulated when cell growth is occurring and diminished when the culture has reached confluence. In general, there was no significant difference in the overall cell viabilities and the levels of HCV RNA replication and protein production when the replicon-containing cells were maintained at 2 to 10% fetal calf serum in a 3-day period. Fetal calf serum obtained from different suppliers, however, resulted in different growth rates of the cells. The replicon-containing cells appeared to tolerate up to 1% DMSO; higher concentrations of DMSO led to suppression of cell growth and HCV replication (data not shown). Similar results were obtained with the genotype 1a replicon-containing cells. Based on these observations, the conditions used for compound evaluation were selected to ensure that cells were cultured in an exponential growth phase and that HCV replication remained linear for the duration of treatment.
HCV replicon assays.
Clone A and genotype 1a replicon-containing cells were propagated in Dulbecco's minimal essential medium (DMEM) containing 10% fetal calf serum (HyClone) supplemented with penicillin-streptomycin, 1× nonessential amino acids, and 1 mg of Geneticin/ml. For compound or interferon α2 (Accurate Chemical & Scientific) testing, G418 was eliminated and the fetal bovine serum (FBS) concentration was reduced to 2%. Cells were seeded at a density of 7 × 103 cells/well in a 96-well plate and incubated with compounds at various concentrations for 3 days at 37°C and 5% CO2. The final concentration of DMSO in the medium was 0.5%. Under these conditions, cells were approximately 25% confluent at the time of seeding and reached 80 to 90% confluence on day 3. After incubation, cells were either fixed with 0.05% glutaraldehyde for the detection of HCV protein with an enzyme-linked immunosorbent assay (ELISA) or processed for total RNA (RNeasy 96 kit; QIAGEN), from which HCV replicon RNA was quantified with Taqman reverse transcriptase PCR (RT-PCR). HCV-specific ELISA was carried out by using mouse anti-NS5A monoclonal antibody (Virostat) at a 1:400 dilution and goat anti-mouse-horseradish peroxidase-conjugated monoclonal antibody at a 1:500 dilution. The levels of HCV replicon RNA and the internal control (rRNA Pre-Development reagent; Applied Biosystems) were quantified in a single-step duplexed RT-PCR using the protocol recommended by the TaqMan One-Step RT-PCR Master Mix reagents kit (Applied Biosystems). The HCV primers (5′-CGTTGGCTACCCGTGATATTG-3′ and 5′-AATCGGGAGCGGCGAT-3′) and probe (5′-[6-car-boxyfluorescein]-TGACCGCTTCCTCGTGCTTTACGG-[6-carboxytetrameth-ylrhodamine]-3′), which encompass the neomycin region of the replicon, were designed with the Primer Express software provided by Applied Biosystems. The level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) RNA in the replicon-containing cells was quantified by using the GAPDH Pre-Development reagent mix (Applied Biosystems), with the same conditions for the RT-PCR amplification as those used for the HCV-rRNA duplexed RT-PCR.
The amounts of the HCV, 18S ribosomal, and GAPDH RNAs in each sample were estimated by comparing the threshold cycles with those in the corresponding standard curves. HCV RNA standards used for the construction of the standard curve were prepared by extracting the total RNA from the Clone A cells. The RNA sample was sent to the National Genetics Institute for quantification of the HCV RNA. Total RNA extracted from Clone A cells was quantified by measurement of the optical density at 260 nm and used for the construction of the standard curves of rRNA and GAPDH. Standard curves were plotted, and 50% inhibitory concentrations (IC50s) were determined using the MDL LSW data analysis software in Microsoft Excel. The amount of HCV RNA or GAPDH in the samples was expressed as the number of HCV RNA copies or GAPDH (nanograms) per microgram of total RNA (using rRNA as a marker for total RNA measurement).
Combination study.
Clone A cells were seeded in duplicate 96-well cell culture plates at subconfluent density (7 × 103 cells/well) in medium containing 2% FBS. A matrix of stock solutions containing a combination of HCV-371 and pegylated IFN-α (PEGASYS; Roche Pharmaceuticals) in various concentrations was prepared in a 96-well plate. Dose-response curves for the individual drugs were run in duplicate. HCV-371 was added to wells at final concentrations of 1, 0.3, 0.1, 0.03, 0.01, 0.003, and 0 μM. PEGASYS was added to the wells at concentrations of 1,000, 240, 120, 60, 15, 7.5, 3.75, and 0 pg/ml. All wells were adjusted to a final concentration of 1% DMSO. Plates were incubated for 72 h at 37°C and 5% CO2. At the end of the incubation period, total RNA was extracted and analyzed for HCV RNA, 18S rRNA, and GAPDH by using TaqMan RT-PCR as described above. Drug combination effects between HCV-371 and PEGASYS were calculated by MacSynergy II using the Bliss independence model (29-31). Results were displayed with DeltaGraph software.
Antiviral specificity.
Vero cells (African green monkey kidney epithelial cells; American Type Culture Collection [ATCC]) were maintained in M199 medium (Mediatech) containing 10% heat-inactivated FBS (U.S. Biotechnologies) and 1% penicillin-streptomycin. HEp-2 cells (human carcinoma of the larynx epithelial cells; ATCC) were cultured in minimal essential medium (MEM; Mediatech) containing 10% heat-inactivated FBS and 1% penicillin-streptomycin. MRC-5 cells (human normal lung fibroblasts; ATCC) were cultured in MEM containing 10% heat-inactivated FBS, 1% penicillin-streptomycin, 1% l-glutamine, 1% nonessential amino acids, 1% sodium pyruvate, and 2% sodium bicarbonate. All cell lines were incubated at 37°C and 5% CO2. Respiratory syncytial virus (RSV; A isolate), lymphocytic choriomeningitis virus (LCMV; Armstrong E350 isolate), cytomegalovirus (CMV; AD-169 isolate), and herpes simplex virus type 1 (HSV-1; KOS isolate) were obtained from the ATCC. All experiments involving severe acute respiratory syndrome-associated coronavirus (SARS-CoV; clinical isolate) were conducted by collaborators at the U.S. Army Medical Research Institute for Infectious Diseases, Fort Detrick, Md. All of these assays were carried out in the appropriate medium containing 3% heat-inactivated FBS. Ninety-six-well cell culture plates were seeded 24 h before use with 1.5 × 104 (Vero), 2.2 × 104 (HEp-2), or 4.5 × 104 (MRC-5) cells per well. Cells were infected at the 90% tissue culture infective dose for SARS-CoV, RSV, and HSV-1 or at titers that generated an ELISA signal of 2.5 for CMV and LCMV at the end of the respective incubation periods. Compound was added to duplicate wells of cells at final concentrations of 100, 50, 25, 12.5, 6.3, 3.1, 1.6, 0.8, and 0 μΜ. The final concentration of DMSO in the assays was 0.5%. As controls, uninfected cells and cells receiving virus without compound were included on each assay plate. In addition, reference agents, when available, were included on each assay plate (ganciclovir for HSV-1 and CMV [Sigma] and ribavirin for LCMV and RSV [Sigma]. At the end of the incubation period (3 days for HSV-1 and LCMV and 4 days for SARS-CoV, RSV, and CMV), HSV-1-, SARS-CoV-, and RSV-infected cells were processed for crystal violet staining (cytopathic effect assay), while cells infected with CMV and LCMV were processed for ELISA analysis using standard techniques. LCMV- and CMV-specific ELISAs were carried out using a 1:20 dilution of mouse monoclonal antibody raised against LCMV nuclear protein (generous gift of Juan Carlos de la Torre, The Scripps Research Institute, La Jolla, Calif.) and a 1:200 dilution of CMV-specific cocktail of monoclonal antibodies generated from protein 52 and unique long gene 44 product (Dako), respectively. Goat anti-mouse horseradish peroxidase-conjugated monoclonal antibody (Bio-Rad) in a 1:4,000 dilution for LCMV and a 1:400 dilution for CMV was used as the secondary antibody for both ELISAs.
Cytotoxicity assays.
Huh-7, MT4 (transformed human lymphocyte; National Institutes of Health AIDS reagent program), HFF (primary human foreskin fibroblast), and Vero cells were seeded at 5 × 103, 10 × 103, 20 × 103, and 40 × 103 cells/well, respectively, in 96-well dishes to capture the actively dividing and stationary phases of cell growth. CHO-K12 (hamster ovary epithelial) and C6 (rat brain fibroblast) cells were seeded at a subconfluent density of 2 × 103 to 4 × 103 cells/well in 96-well dishes. Huh-7 cells were propagated in the same medium as that used to culture Clone A cells. Vero cells were propagated in DMEM containing 10% FBS supplemented with penicillin-streptomycin and 2 mM l-glutamine. MT4 cells were propagated in RPMI medium with 10% heat-shocked FBS, 1% penicillin-streptomycin, and 1% l-glutamine. HFF cells were isolated at Wyeth Research as previously described (12). Cells were grown in DMEM containing 10% FBS and 25 mM HEPES. CHO-K12 cells were cultured in 50% DMEM-50% F12 (Invitrogen) containing 10% FBS. C6 cells were cultured in MEM with Earle's salts containing 10% horse serum. Cells were incubated with increasing concentrations of the test compounds in their respective growth media at 37°C and 5% CO2 for 3 days. Cell metabolism and proliferation were measured by using either the CellTiter-Glo Luminescent cell viability assay (Promega) or the CellTiter 96 Aqueous One Solution cell proliferation assay (Promega). Cytotoxicity levels were expressed as the concentration that inhibited cell growth by 50% (CC50).
Fluorescence spectroscopy.
The fluorescence binding experiments with HCV-371 were done by using the changes in the intrinsic fluorescence of the enzyme to determine the binding affinity. To a quartz cuvette containing Tris buffer (pH 7.4), NS5B enzyme (NS5BΔCT21) was added to a final concentration of 1 μM (total reaction is 500 μl). The fluorescence emission spectrum of the enzyme was scanned from 310 to 400 nm with an excitation wavelength of 295 nm in a Jobin-Yvon Horiba fluorometer. The inhibitor does not fluoresce under these conditions. The fluorescence of the enzyme was acquired in the ratio mode (signal over reference) to correct for any variations in the output of the lamp. The excitation and emission wavelengths were set using the monochromator bandwidths of 1 and 3 nm, respectively. The instrument uses a 150-W xenon lamp as a continuous light source, with output in the wavelength range of 250 to 700 nm. The output of the lamp was checked periodically as a lamp spectrum, and the monochromators were calibrated with the water Raman spectrum. At the above-mentioned excitation and emission wavelengths, the tryptophan residues in the enzyme are selectively excited to emit at the maximum wavelength of ca. 335 nm. This initial fluorescence is designated F0. The inhibitor solutions were prepared by serially diluting the 10 mM inhibitor stock (prepared in 100% DMSO) in the Tris buffer (pH 7.4). Small aliquots of the diluted stock solution of the inhibitor, typically 1 to 2 μl, were successively combined with the enzyme solution. After each addition, the complex was incubated for 5 min prior to the recording of the fluorescence spectra as described above. The fluorescence intensity at 335 nm (designated F) was monitored as a function of the inhibitor concentration. The change in the fluorescence (F0 − F) that reflects the level of quenching was calculated from this data set after subtraction of the background due to the buffer, which contributes less than 5% under these conditions. The fluorescence change was fit to the following quadratic equation
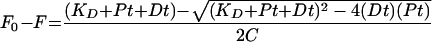 |
to obtain the dissociation constant (KD), where Pt and Dt are the concentrations of the protein and the drug, respectively. The factor C in the denominator corresponds to the ratio of Pt/(F0 − Fmax), where Fmax is the fluorescence intensity in the presence of saturating concentrations of the inhibitor (10).
RESULTS
Identification of [(1R)-5-cyano-8-methyl-1-propyl-1,3,4,9-tetrahydropyano[3,4-b]indol-1-yl] acetic acid (HCV-371).
High-throughput screening (HTS) of the Wyeth proprietary chemical library using a carboxyl-terminally truncated His-tagged NS5B fusion protein (NS5BΔCT21-His) and a hairpin-like single-stranded RNA substrate resulted in a series of structurally related compounds that inhibited HCV NS5B RdRp. The details of the structure-activity relationship optimization of this chemical series will be described elsewhere (9a). Among these compounds, [(1R)-5-cyano-8-methyl-1-propyl-1,3,4,9-tetrahydropyano[3,4-b]indol-1-yl] acetic acid (HCV-371) (Fig. 1) was selected for the following studies.
FIG. 1.

Chemical structure of HCV-371.
The activity of HCV-371 against four genotypes was evaluated by using purified enzymes, cloned and expressed in a format similar to that of the target enzyme from the HTS. This collection includes the following enzymes: genotypes 1a (five isolates), 1b (eight isolates), 3a (one isolate), and 4 (one isolate). The amino acid sequence divergence among the NS5B proteins derived from these naturally occurring HCV isolates ranges from 4 to 25% (Table 1). HCV-371 exhibited broad activity against the polymerase enzymes of HCV 1a, 1b, and 3a genotypes (IC50, 0.3 to 1.4 μM for 90% of genotype 1a and 1b enzymes and 1.8 μM for genotype 3a) (Table 1). The enzyme from the one genotype 4 isolate was less susceptible to HCV-371 (IC50, 17.8 μM).
TABLE 1.
Activities of HCV-371 against HCV NS5B polymerases from different isolates of genotypes 1a, 1b, 3a, and 4
| Genotype and enzyme isolate | HCV-371 IC50 ± SDa (μM) | Amino acid sequence divergence from genotype 1b BK
|
|
|---|---|---|---|
| % | No. of changes | ||
| Genotype 1b | |||
| BK | 0.3 ± 0.1 (n = 51) | ||
| 4a | 0.4 ± 0.3 (n = 14) | 3.4 | 19 |
| 320-10-3 | 0.5 ± 0.3 (n = 8) | 4.4 | 25 |
| 320-3-1 | 0.4 ± 0.2 (n = 12) | 4.6 | 27 |
| BB7 | 0.4 ± 0.2 (n = 15) | 3.7 | 22 |
| 320-2-1 | 0.5 ± 0.2 (n = 14) | 4.2 | 25 |
| J4 | 0.4 ± 0.1 (n = 6) | 4.7 | 28 |
| FC 1-20 | 0.5 ± 0.6 (n = 17) | 4.7 | 27 |
| Genotype 1a | |||
| 9606 | 4.8 ± 2.7 (n = 14) | 11.5 | 67 |
| A5 | 1.4 ± 0.8 (n = 15) | 11.5 | 68 |
| 207-248 | 0.6 ± 0.2 (n = 16) | 11.3 | 66 |
| 207-221 | 0.7 ± 0.4 (n = 15) | 11.4 | 64 |
| 207-247 | 1.0 ± 0.5 (n = 16) | 11.8 | 69 |
| Genotype 3a | |||
| 167-A1 | 1.8 ± 1.3 (n = 22) | 24.7 | 144 |
| Genotype 4 | 17.8 ± 8.0 (n = 3) | 21.5 | 126 |
Inhibitory activities are expressed as the mean IC50 ± standard deviation. n, number of determinations.
Biochemical selectivity of HCV-371.
The effect of HCV-371 on other nucleic acid-metabolizing enzymes was evaluated in standard biochemical assays optimized for the respective enzymes. The enzymatic selectivity of HCV-371 was determined by calculating the ratio of the IC50 for the HCV NS5B polymerase (1b, BK) to that for the other purified enzymes (selectivity index). HCV-371 did not inhibit purified DNA-dependent DNA polymerases, including human polymerases α, β, and γ and calf thymus polymerase α; a human DNA-dependent RNA polymerase II; or a viral RNA-dependent DNA polymerase, such as HIV reverse transcriptase and HCV helicase. The IC50s for these enzymes were all >80 μM, compared to an IC50 of 0.3 μM for HCV NS5B (genotype 1b, BK). This results in a selectivity index of >267 for all of the enzymes tested.
Antiviral effect of HCV-371 in HCV replicon cells.
Antiviral activity of HCV-371 was assessed by growing genotype 1b (Clone A) and genotype 1a replicon-containing cells in medium containing increasing concentrations of HCV-371. After 3 days of treatment, the medium was removed, and the inhibitory activity of HCV-371 in the treated cells was evaluated by using two independent assays. The first assay quantified the level of viral protein produced from the replicon by using an NS5A-specific ELISA. The second assay was a fluorescent quantitative TaqMan RT-PCR that determined the steady-state levels of the HCV RNA and the internal control (18S rRNA) simultaneously. HCV-371 is active in the replicon system. A single application of HCV-371 to Clone A cells led to a dose-dependent reduction of the HCV RNA level (50% effective concentration [EC50], 4.8 ± 0.5 μM and 6.1 ± 1.9 μM for genotype 1b and 1a replicons, respectively) (Table 2 and Fig. 2), with a concomitant decrease in viral protein level (EC50, 13 ± 3 μM in 1b replicon). About a 2-log10 decrease in the level of HCV RNA was observed with a single maximum dose of HCV-371 at 40 μM. While there was a dose-dependent reduction of the HCV RNA level, the levels of GAPDH and rRNA mRNAs remained relatively constant within the effective antiviral concentrations (Fig. 2). This result indicated that the antiviral activity of HCV-371 was specific to HCV, since there was no decrease in the levels of the cellular housekeeping gene.
TABLE 2.
Antiviral specificity of HCV-371
| Virus | Family | Classification | HCV-371 EC50 ± SDa (μM) |
|---|---|---|---|
| HCV | Flaviviridae | Positive single-stranded RNA | |
| 1b replicon | 4.8 ± 0.5b | ||
| 1a replicon | 6.1 ± 1.9b | ||
| SARS-CoV | Coronaviridae | Positive single-stranded RNA | >100c |
| RSV | Paramyxoviridae | Negative single-stranded RNA | >80c |
| LCMV | Arenaviridae | Ambisense RNA | >100d |
| CMV | Herpesviridae | Double-stranded DNA | >100d |
| HSV-1 | Herpesviridae | Double-stranded DNA | >100d |
Each value represents the mean from at least two determinations.
Values are based on the measurement of the HCV RNA.
EC50s were assessed by cytopathic effect protection in infected cells.
EC50s were determined by virus-specific ELISA.
FIG. 2.

TaqMan analysis of HCV replicon and GAPDH RNA levels in Clone A cells treated with HCV-371. Clone A cells were grown in medium containing increasing concentrations of HCV-371. After 3 days, the medium was removed and RNA was extracted for HCV-18S rRNA and GAPDH-18S rRNA quantitative RT-PCR analyses. Each point represents an average from eight independent determinations with the associated standard error bars. The x axis represents compound concentrations from 0.3 to 160 μM. The y axis represents the amount of HCV or GAPDH RNA as a percentage of that from the untreated controls.
Multiple treatments of replicon-containing cells.
Antiviral activity of HCV-371 was evaluated in another study in which Clone A cells were treated with HCV-371 every 3 to 4 days (a total of six times) over a 20-day period. In this experiment, cells were grown in medium containing 20 μM (about five times the EC50 in the single-dose 3-day assay) HCV-371. When cells reached 80% confluence (after about 3 to 4 days), they were passaged with a 1:4 dilution, and the medium was replaced with fresh medium containing HCV-371 at the same concentration. As a control, Clone A cells were passaged in parallel under the same conditions, with the tissue culture medium containing either no compound or 20 U of IFN-α (about five times the EC50 in a single-dose 3-day assay) (data not shown). At the end of the fifth treatment (day 16), cells that were treated with HCV-371 had a >3-log10 reduction (from 2,000 to 2 copies/cell) of the HCV RNA level. About a 2-log10 decrease of the HCV RNA level was observed in the cells treated with IFN-α (Fig. 3A). The level of HCV RNA in the DMSO control cells remained relatively constant throughout the treatment (Fig. 3A). Prolonged treatment with HCV-371 or IFN-α did not introduce any notable fluctuation in the level of the GAPDH mRNA, suggesting that both compounds are well tolerated by the replicon-containing cells (Fig. 3B).
FIG. 3.

Multiple treatments of Clone A cells with HCV-371. Clone A cells were treated with 20 μM HCV-371, 20 U of IFN-α, or DMSO every 3 to 4 days over a 20-day period. When cells reached about 80% confluence, they were passaged with a 1:4 dilution, and the medium was replaced with fresh medium containing the respective compounds at the same concentration. Total RNA from the cells was prepared for HCV-18S rRNA and GAPDH-18S rRNA duplexed RT-PCR analyses. The amounts of HCV RNA or GAPDH RNA were expressed as the number of copies per microgram of total RNA and nanograms of GAPDH per microgram of total RNA for the HCV RNA and GAPDH mRNA, respectively. Each point represents an average from four replicate samples with the associated standard error bars. (A) Effect on HCV replicon RNA. (B) Effect on GAPDH mRNA.
Combination treatment of HCV-371 with pegylated interferon.
HCV-371 in combination with other HCV therapeutics may improve cure rates and reduce or prevent the emergence of variants with reduced susceptibility. To characterize the drug combination effects between HCV-371 and pegylated IFN-α (PEGASYS), a three-dimensional analytical method was used (29, 31). This method examines drug combinations by using the Bliss independence null model that is based on statistical probability and assumes that two drugs act independently to inhibit replication. Using this method, the theoretical additive interactions are calculated from the dose-response curves of the individual drugs acting alone. The predicted additive effects are then subtracted from the experimentally determined effects to reveal a difference in dose-response surface. The resulting surface appears as a horizontal plane at 0% difference if the interactions are additive. Any peaks above the plane are indicative of a greater-than-expected effect (synergy). Conversely, peaks appearing below the plane are indicative of a less-than-expected effect (antagonism). The confidence intervals around the experimental dose-response surface are used to evaluate the data statistically, and the volume of the peaks is calculated to quantify the synergy or antagonism produced.
The combined antiviral effect of a wide range of concentrations of HCV-371 (0.3 to 30 μM) and PEGASYS (0.1 to 30 pg/ml) was assessed in the replicon-containing Clone A cells by TaqMan RT-PCR analysis. The results from three independent determinations indicated that HCV-371 and PEGASYS interact additively (Fig. 4). The volumes of synergy and antagonism at the 95% confidence level (5.01 and −3.27, respectively) are below those considered to be significant (+50 and −50 for synergy and antagonism, respectively [M. N. Prichard, K. R. Aseltine, and J. C. Shipman, personal communication]). In addition, evaluation of the levels of 18S rRNA and GAPDH by TaqMan RT-PCR analysis revealed no toxicity for any of the drug combinations tested (data not shown), confirming the absence of cytotoxic effects.
FIG. 4.

Synergy at the 95% confidence level for the HCV-371 and PEGASYS combination study. Clone A cells were seeded in 96-well cell culture plates at subconfluent density in medium containing 2% FBS. Combinations of HCV-371 (0.3 to 30 μM) and PEGASYS (0.1 to 30 pg/ml) were added to the plates, forming a matrix. Dose-response curves for the individual drugs were run in duplicate. Plates were incubated for 72 h at 37°C and 5% CO2. Total RNA was extracted from replicon-containing cells and analyzed for HCV RNA, 18S rRNA, and GAPDH RNA with a TaqMan RT-PCR. The resulting data were calculated at the 95% confidence level by MacSynergy II and displayed with DeltaGraph software.
Cytotoxicity.
To evaluate whether HCV-371 has any potential cytotoxic effect, six cell lines of multiple mammalian origins were evaluated under stationary and dividing conditions in two independent assays. The first assay measured the metabolic condition of the cells by determining the amount of intracellular ATP production. A 3-day treatment of HCV-371 produced no cytotoxic effect in stationary and dividing Vero (African green monkey kidney), MT4 (human lymphocytes), HFF (primary human foreskin fibroblast), and Huh-7 (human liver epithelial) cells. The concentration (CC50) of HCV-371 required to inhibit 50% of the intracellular ATP production in these four cell lines was >160 μM, the highest concentration tested (Table 3). The second assay measured the effect on cell proliferation and metabolism after the cells were treated with HCV-371 for 3 days. This assay is based on the bioreduction of a dye (MTT) by NADPH or NADH produced from dehydrogenase enzymes in metabolically active cells. HCV-371 was not cytotoxic to dividing CHO-K12 (hamster ovary epithelial), C6 (rat brain fibroblast), and Huh-7 cells. The concentration of HCV-371 required to inhibit by 50% the proliferation of these four cell lines ranged from 63 μM to >160 μM (Table 3). The results from the above studies suggested that the inhibitory effect of HCV-371 is specific to HCV and that HCV-371 is well tolerated by mammalian cells.
TABLE 3.
Effect of HCV-371 on mammalian cell metabolic functions
| Cell line | Growing condition | Species of origin | Cell type and source | CC50 ± SDc (μM) |
|---|---|---|---|---|
| Huh-7 | Dividing and stationarya,b | Human | Liver epithelial cells | >160 |
| MT4 | Dividing and stationarya | Human | Lymphocytes | >160 |
| HFF | Dividing and stationarya | Human | Foreskin fibroblasts | >160 |
| Vero | Dividing and stationarya | African green monkey | Kidney epithelial cells | >160 |
| CHO-K12 | Dividingb | Hamster | Ovary epithelial cells | 63 ± 6.4 |
| C6 | Dividingb | Rat | Brain fibroblasts | 80 ± 7.8 |
Measured by ATP production.
Measured by a standard MTT bioreduction assay.
Inhibitory activities are expressed as mean CC50. Each reported value represents the mean of results from at least two independent determinations.
Antiviral specificity.
HCV-371 was evaluated for intracellular antiviral activity against several RNA and DNA viruses. The susceptibility of these viruses to HCV-371 was assessed in cell culture assays that measured either the cytoprotection by the compound in infected cells or the viral load reduction in the presence of the compound. Table 2 shows that the EC50s of HCV-371 for these DNA or RNA viruses were all greater than 80 μM, compared to the EC50s for HCV replicons of 4.8 to 6.1 μM, demonstrating excellent antiviral specificity.
Binding of HCV-371 to NS5B.
Fluorescence spectroscopy methods were used to determine the binding potency of HCV-371 to the NS5B enzyme. Interactions of HCV-371 with HCV NS5B induce significant quenching (roughly 50%) of the fluorescence of tryptophan residues. The binding isotherm shown in Fig. 5 shows that the dissociation constant for this interaction corresponds to a value of about 150 nM. HCV-371 binds the enzyme saturably, and the data fit is consistent with a binding stoichiometry of one inhibitor molecule binding per enzyme monomer unit, as deduced from the fluorescence data. The stoichiometry is confirmed by isothermal titration calorimetry (data not shown).
FIG. 5.

Binding isotherm of HCV-371 with NS5BΔCT21-His. Enzyme was incubated with increasing concentrations of the compounds. The fluorescence changes in the protein were monitored at the emission maximum of 335 nm with excitation at 295 nm. The changes in the fluorescence were fitted to a quadratic equation to determine the binding affinity.
DISCUSSION
With an escalating number of patients who are infected with HCV for more than 15 years, many of whom are likely to develop advanced liver diseases by 2012, there remains an urgent unmet need for more-effective, less-toxic, and less-complex treatment regimens with shorter durations. The NS5B RNA-dependent RNA polymerase, which is strictly required for HCV genome replication, has been the target of intense antiviral research (6-8, 37, 38). Inhibitors of other viral polymerases, such as stavudine, adefovir, and tenofovir for HIV (4, 25, 36, 46), lamivudine and entecavir for hepatitis B virus (2, 27), and acyclovir and valacyclovir for HSV (35, 41), have proven clinical effectiveness in patients infected with the corresponding viruses. This report describes the discovery of a novel class of HCV polymerase inhibitors, the prototype of which has demonstrated potency, specificity, and safety in biochemical and cell-based assays. HCV-371, a representative of over 300 compounds synthesized in this series, displays potent inhibition against the NS5B enzyme and HCV replication. In the biochemical assay, HCV-371 inhibits the polymerases derived from genotypes 1a, 1b, and 3a that are found in the major HCV-infected patient population. In the persistent HCV replication cell culture model, HCV-371 reduces the viral load by 3 orders of magnitude. Furthermore, combination treatment of HCV-371 and PEGASYS results in an additive antiviral effect in the replicon-containing cells. The antiviral activity associated with HCV-371 is specific for HCV at both the biochemical and the cellular levels. No inhibition is observed in the biochemical assays containing purified human polymerases or other unrelated mammalian or viral nucleic acid-metabolizing enzymes, and no antiviral activity is found in cells infected with other DNA and RNA viruses. HCV-371 is noncytotoxic and has a large therapeutic window. Within the effective antiviral concentrations, there are no detectable toxicities in cellular metabolism and cell proliferation. In addition, long-term treatment of cells with HCV-371 does not result in adverse effects. The in vitro pharmacological results suggest that HCV-371 has all the hallmarks of a potent, specific, and safe antiviral agent that might have clinical utility.
The specific activity of HCV-371 can be attributed to its binding to the NS5B enzyme in an allosteric site in the thumb domain of NS5B near the interface with the C-terminal domain. This site has been previously reported to accommodate substituted phenylalanine inhibitors (43). Interactions of HCV-371 with HCV NS5B induce significant quenching (roughly 50%) of the fluorescence of tryptophan residues. The fluorescence quenching is due to changes in the solvent environment of the residue, to which it is very sensitive. Free tryptophan has a fluorescence emission maximum at 350 nm in aqueous systems due to complete exposure of the indole ring to a hydrophilic solvent environment. In fluorescence spectroscopy, tryptophan residues are not typically exposed, and the average environment of the residues is such that the maximum emission wavelength is blueshifted to about 335 to 340 nm, as in the case with HCV NS5B enzyme. Due to changes in the local environment of the protein or to direct electronic interactions of HCV-371 with the enzyme, the intrinsic fluorescence is quenched. Details of the crystal structure of the NS5B-HCV-371 complex and the mechanism of action will be described elsewhere. HCV-371 and the related analogues have been shown to bind at a region distant from the catalytic site. Noncompetitive allosteric inhibition of HCV RNA synthesis is believed to be one of the plausible mechanisms.
HCV-371 displays differential inhibitory activity against the isolates derived from different genotypes. All eight isolates from genotype 1b are equally sensitive to HCV-371, while some isolates derived from genotypes 1a, 3a, and 4 are 5- to 59-fold less susceptible to the compound (Table 1). Although the present collection of isolates is too small to determine an accurate structure-activity relationship, the amino acid alignment within NS5B among these isolates suggests that there are differences in the amino acids near the HCV-371 binding pocket. The impact of these amino acid changes in each genotype has yet to be determined.
Although the present HTS utilized the C-terminal truncated NS5B as the target enzyme, HCV-371 and the related analogues were found to inhibit the full-length NS5B enzyme with similar potencies (data not shown). Notably, the biochemical activity can be translated into antiviral activity in cells. HCV replicon cells presumably contain the polymerase and other virus-host factors in the form of a replication complex. One possibility suggested by these results is that the HCV-371 binding site on the polymerase is accessible in cells and/or that the overall conformation of this truncated form of NS5B resembles that of the full-length enzyme in the replication complex.
The ability of an antimicrobial agent to maintain an effective concentration at the infected site in vivo is dependent on many physicochemical and biopharmaceutical properties which directly determine a drug's absorption, distribution, metabolism, elimination, and pharmacodynamics in vivo. The chimpanzee and the chimeric mouse transplanted with human liver models are the two infectious HCV animal models that might help to establish the in vitro-in vivo correlation (24, 42). In particular, the chimeric mouse model has recently linked the replicon activity of an HCV NS3 protease inhibitor to clinical efficacy (IC50s for BILN 2061 in genotype 1b NS3 protease and replicon assays are 0.7 and 3 nM, respectively [15]). Direct in vitro-in vivo correlation using HCV polymerase inhibitors in these animal models has not been reported. In addition to HCV-371, several classes of NS5B inhibitors, including diketo acids (38), benzothiadiazine (8), and modified nucleoside analogues (6, 37), have shown in vitro activities. With the discovery of these inhibitors and the availability of the in vitro and in vivo systems, it is hopeful that an effective HCV therapeutic will soon be developed.
Acknowledgments
We thank Wyeth's HTS group for screening the compound libraries. We are indebted to our chemistry colleagues at ViroPharma Incorporated for their helpful support and suggestions throughout the compound optimization. Special thanks are extended to Christopher J. Rizzo, Patricia A. McDuffie, Olga Palant, Stephen Plotch, and Anthony Amin for providing technical assistance and helpful discussion.
REFERENCES
- 1.Baginski, S. G., D. C. Pevear, M. Seipel, S. C. Sun, C. A. Benetatos, S. K. Chunduru, C. M. Rice, and M. S. Collett. 2000. Mechanism of action of a pestivirus antiviral compound. Proc. Natl. Acad. Sci. USA 97:7981-7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Billich, A. 2001. Entecavir (Bristol-Myers Squibb). Curr. Opin. Investig. Drugs 2:617-621. [PubMed] [Google Scholar]
- 3.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 290:1972-1974. [DOI] [PubMed] [Google Scholar]
- 4.Buti, M., and R. Esteban. 2003. Adefovir dipivoxil. Drugs Today 39:127-135. [DOI] [PubMed] [Google Scholar]
- 5.Campbell, J. L. 1986. Eukaryotic DNA replication. Annu. Rev. Biochem. 55:733-771. [DOI] [PubMed] [Google Scholar]
- 6.Carroll, S. S., J. E. Tomassini, M. Bosserman, K. Getty, M. W. Stahlhut, A. B. Eldrup, B. Bhat, D. Hall, A. L. Simcoe, R. LaFemina, C. A. Rutkowski, B. Wolanski, Z. C. Yang, G. Migliaccio, R. De Francesco, L. C. Kuo, M. MacCoss, and D. B. Olsen. 2003. Inhibition of hepatitis C virus RNA replication by 2′-modified nucleoside analogs. J. Biol. Chem. 278:11979-11984. [DOI] [PubMed] [Google Scholar]
- 7.Chan, L., T. J. Reddy, M. Proulx, S. K. Das, O. Pereira, M. Courchesne, C. Roy, C. Yannopoulos, C. Poisson, N. Nguyen-Ba, L. Halab, R. Bethell, M.-Q. Zhang, M. David, L. L'Heureux, J. Bedard, M. Hamel, and O. Nicolas. 2003. Discovery of a novel class of HCV NS5B RNA dependent RNA polymerase inhibitors: SAR studies and activity in replicon cells. Antivir. Res. 57:A76. [Google Scholar]
- 8.Dhanak, D., K. J. Duffy, V. K. Johnston, J. Lin-Goerke, M. Darcy, A. N. Shaw, B. Gu, C. Silverman, A. T. Gates, M. R. Nonnemacher, D. L. Earnshaw, D. J. Casper, A. Kaura, A. Baker, C. Greenwood, L. L. Gutshall, D. Maley, A. DelVecchio, R. Macarron, G. A. Hofmann, Z. Alnoah, H. Y. Cheng, G. Chan, S. Khandekar, R. M. Keenan, and R. T. Sarisky. 2002. Identification and biological characterization of heterocyclic inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. J. Biol. Chem. 277:38322-38327. [DOI] [PubMed] [Google Scholar]
- 9.Ferrari, E., J. Wright-Minogue, J. W. S. Fang, B. M. Baroudy, J. Y. N. Lau, and Z. Hong. 1999. Characterization of soluble hepatitis C virus RNA-dependent RNA polymerase expressed in Escherichia coli. J. Virol. 73:1649-1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jamieson, E. R., M. P. Jacobson, C. M. Barnes, C. S. Chow, and S. J. Lippard. 1999. Structural and kinetic studies of a cisplatin-modified DNA icosamer binding to HMG1 domain B. J. Biol. Chem. 274:12346-12354. [DOI] [PubMed] [Google Scholar]
- 11.Johnson, L. M., M. Snyder, L. M. Chang, R. W. Davis, and J. L. Campbell. 1985. Isolation of the gene encoding yeast DNA polymerase I. Cell 43:369-377. [DOI] [PubMed] [Google Scholar]
- 12.Jones, T. R., V. P. Muzithras, and Y. Gluzman. 1991. Replacement mutagenesis of the human cytomegalovirus genome: US10 and US11 gene products are nonessential. J. Virol. 65:5860-5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khakoo, S., P. Glue, L. Grellier, B. Wells, A. Bell, C. Dash, I. Murray-Lyon, D. Lypnyj, B. Flannery, K. Walters, and G. M. Dusheiko. 1998. Ribavirin and interferon alfa-2b in chronic hepatitis C: assessment of possible pharmacokinetic and pharmacodynamic interactions. Br. J. Clin. Pharmacol. 46:563-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim, J. L., K. A. Morgenstern, J. P. Griffith, M. D. Dwyer, J. A. Thomson, M. A. Murcko, C. Lin, and P. R. Caron. 1998. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure 6:89-100. [DOI] [PubMed] [Google Scholar]
- 15.Lamarre, D., P. C. Anderson, M. Bailey, P. Beaulieu, G. Bolger, P. Bonneau, M. Bos, D. R. Cameron, M. Cartier, M. G. Cordingley, A. M. Faucher, N. Goudreau, S. H. Kawai, G. Kukolj, L. Lagace, S. R. LaPlante, H. Narjes, M. A. Poupart, J. Rancourt, R. E. Sentjens, R. St George, B. Simoneau, G. Steinmann, D. Thibeault, Y. S. Tsantrizos, S. M. Weldon, C. L. Yong, and M. Llinas-Brunet. 2003. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 426:186-189. [DOI] [PubMed] [Google Scholar]
- 16.Lehman, I. R., and L. S. Kaguni. 1989. DNA polymerase alpha. J. Biol. Chem. 264:4265-4268. [PubMed] [Google Scholar]
- 17.Liang, T. J., B. Rehermann, L. B. Seeff, and J. H. Hoofnagle. 2000. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann. Intern. Med. 132:296-305. [DOI] [PubMed] [Google Scholar]
- 18.Lohmann, V., J. O. Koch, and R. Bartenschlager. 1996. Processing pathways of the hepatitis C virus proteins. J. Hepatol. 24:11-19. [PubMed] [Google Scholar]
- 19.Lohmann, V., F. Korner, U. Herian, and R. Bartenschlager. 1997. Biochemical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol. 71:8416-8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110-113. [DOI] [PubMed] [Google Scholar]
- 21.Lohmann, V., A. Roos, F. Korner, J. O. Koch, and R. Bartenschlager. 1998. Biochemical and kinetic analyses of NS5B RNA-dependent RNA polymerase of the hepatitis C virus. Virology 249:108-118. [DOI] [PubMed] [Google Scholar]
- 22.Maldonado, E., R. Drapkin, and D. Reinberg. 1996. Purification of human RNA polymerase II and general transcription factors. Methods Enzymol. 274:72-100. [DOI] [PubMed] [Google Scholar]
- 23.Manns, M. P., J. G. McHutchison, S. C. Gordon, V. K. Rustgi, M. Shiffman, R. Reindollar, Z. D. Goodman, K. Koury, M. Ling, and J. K. Albrecht. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358:958-965. [DOI] [PubMed] [Google Scholar]
- 24.Mercer, D. F., D. E. Schiller, J. F. Elliott, D. N. Douglas, C. Hao, A. Rinfret, W. R. Addison, K. P. Fischer, T. A. Churchill, J. R. Lakey, D. L. Tyrrell, and N. M. Kneteman. 2001. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 7:927-933. [DOI] [PubMed] [Google Scholar]
- 25.Miller, M. D., N. A. Margot, P. D. Lamy, M. D. Fuller, K. E. Anton, A. S. Mulato, and J. M. Cherrington. 2001. Adefovir and tenofovir susceptibilities of HIV-1 after 24 to 48 weeks of adefovir dipivoxil therapy: genotypic and phenotypic analyses of study GS-96-408. J. Acquir. Immune Defic. Syndr. 27:450-458. [DOI] [PubMed] [Google Scholar]
- 26.Mizokami, M., and K. Ohba. 1993. Molecular classification of hepatitis C virus. Gastroenterol. Jpn. 28:42-44. [DOI] [PubMed] [Google Scholar]
- 27.Oh, J. M., J. Kyun, and S. W. Cho. 2002. Long-term lamivudine therapy for chronic hepatitis B in patients with and without cirrhosis. Pharmacotherapy 22:1226-1234. [DOI] [PubMed] [Google Scholar]
- 28.Pietschmann, T., V. Lohmann, G. Rutter, K. Kurpanek, and R. Bartenschlager. 2001. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 75:1252-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prichard, M. N., L. E. Prichard, and C. Shipman. 1993. Strategic design and three-dimensional analysis of antiviral drug combinations. Antimicrob. Agents Chemother. 37:540-545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prichard, M. N., and C. Shipman. 1996. Analysis of combinations of antiviral drugs and design of effective multidrug therapies. Antivir. Res. 1:9-20. [PubMed] [Google Scholar]
- 31.Prichard, M. N., and C. Shipman. 1990. A three-dimensional model to analyze drug-drug interactions. Antivir. Res. 14:181-205. [DOI] [PubMed] [Google Scholar]
- 32.Reichard, O., G. Norkrans, A. Fryden, J. H. Braconier, A. Sonnerborg, O. Weiland, and the Swedish Study Group. 1998. Randomised, double-blind, placebo-controlled trial of interferon alpha-2b with and without ribavirin for chronic hepatitis C. Lancet 351:83-87. [DOI] [PubMed] [Google Scholar]
- 33.Russo, M. W., and M. W. Fried. 2003. Side effects of therapy for chronic hepatitis C. Gastroenterology 124:1711-1719. [DOI] [PubMed] [Google Scholar]
- 34.Simmonds, P. 1996. Virology of hepatitis C virus. Clin. Ther. 18:9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spruance, S. L., S. K. Tyring, B. DeGregorio, C. Miller, K. Beutner, and the Valaciclovir HSV Study Group. 1996. A large-scale, placebo-controlled, dose-ranging trial of peroral valaciclovir for episodic treatment of recurrent herpes genitalis. Arch. Intern. Med. 156:1729-1735. [PubMed] [Google Scholar]
- 36.Squires, K., A. L. Pozniak, G. Pierone, Jr., C. R. Steinhart, D. Berger, N. C. Bellos, S. L. Becker, M. Wulfsohn, M. D. Miller, J. J. Toole, D. F. Coakley, A. Cheng, and T. Study. 2003. Tenofovir disoproxil fumarate in nucleoside-resistant HIV-1 infection: a randomized trial. Ann. Intern. Med. 139:313-320. [DOI] [PubMed] [Google Scholar]
- 37.Stuyver, L. J., T. Whitaker, T. R. McBrayer, B. I. Hernandez-Santiago, S. Lostia, P. M. Tharnish, M. Ramesh, C. K. Chu, R. Jordan, J. Shi, S. Rachakonda, K. A. Watanabe, M. J. Otto, and R. F. Schinazi. 2003. Ribonucleoside analogue that blocks replication of bovine viral diarrhea and hepatitis C viruses in culture. Antimicrob. Agents Chemother. 47:244-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Summa, V., A. Petrocchi, P. Pace, V. G. Matassa, R. De Francesco, S. Altamura, L. Tomei, U. Koch, and P. Neuner. 2004. Discovery of α,γ-diketo acids as potent selective and reversible inhibitors of hepatitis C virus NS5b RNA-dependent RNA polymerase. J. Med. Chem. 47:14-17. [DOI] [PubMed] [Google Scholar]
- 39.Takamizawa, A., C. Mori, I. Fuke, S. Manabe, S. Murakami, J. Fujita, E. Onishi, T. Andoh, I. Yoshida, and H. Okayama. 1991. Structure and organization of the hepatitis C virus genome isolated from human carriers. J. Virol. 65:1105-1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomei, L., R. L. Vitale, I. Incitti, S. Serafini, S. Altamura, A. Vitelli, and R. De Francesco. 2000. Biochemical characterization of a hepatitis C virus RNA-dependent RNA polymerase mutant lacking the C-terminal hydrophobic sequence. J. Gen. Virol. 81:759-767. [DOI] [PubMed] [Google Scholar]
- 41.Wald, A., J. Zeh, G. Barnum, L. G. Davis, and L. Corey. 1996. Suppression of subclinical shedding of herpes simplex virus type 2 with acyclovir. Ann. Intern. Med. 124:8-15. [DOI] [PubMed] [Google Scholar]
- 42.Walker, C. M. 1997. Comparative features of hepatitis C virus infection in humans and chimpanzees. Springer Semin. Immunopathol. 19:85-98. [DOI] [PubMed] [Google Scholar]
- 43.Wang, M. T., K. K. S. Ng, M. M. Cherney, L. Chan, C. G. Yannopoulos, J. Bedard, N. Morin, N. Nguyen-Ba, M. H. Alaoui-Ismaili, R. C. Bethell, and M. N. G. James. 2003. Non-nucleoside analogue inhibitors bind to an allosteric site on HCV NS5B polymerase. Crystal structures and mechanism of inhibition. J. Biol. Chem. 278:9489-9495. [DOI] [PubMed] [Google Scholar]
- 44.Wang, T. S., W. C. Copeland, L. Rogge, and Q. Dong. 1995. Purification of mammalian DNA polymerases: DNA polymerase alpha. Methods Enzymol. 262:77-84. [DOI] [PubMed] [Google Scholar]
- 45.World Health Organization. 1998. WHO concerns on hepatitis C. Lancet 351:1415. [Google Scholar]
- 46.Yogev, R., S. Lee, A. Wiznia, S. Nachman, K. Stanley, S. Pelton, L. Mofenson, S. Fiscus, E. Jimenez, M. H. Rathore, M. E. Smith, L. Y. Song, K. McIntosh, and the Pediatrics AIDS Clinical Trials Group 388 Study Team. 2002. Stavudine, nevirapine and ritonavir in stable antiretroviral therapy-experienced children with human immunodeficiency virus infection. Pediatr. Infect. Dis. J. 21:119-125. [DOI] [PubMed] [Google Scholar]
- 47.Zeuzem, S., S. V. Feinman, J. Rasenack, E. J. Heathcote, M. Y. Lai, E. Gane, J. O'Grady, J. Reichen, M. Diago, A. Lin, J. Hoffman, and M. J. Brunda. 2000. Peginterferon alfa-2a in patients with chronic hepatitis C. N. Engl. J. Med. 343:1666-1672. [DOI] [PubMed] [Google Scholar]