

Abstract
Dorsolateral prefrontal cortex (dlPFC) mediates high-order cognitive functions that are impaired early in the aging process in monkeys and humans. Here, we report pronounced changes in mitochondrial morphology in dendrites of dlPFC neurons from aged rhesus macaques. Electron microscopy paired with 3D reconstruction from serial sections revealed an age-related increase in mitochondria with thin segments that intermingled with enlarged ones, the “Mitochondria-On-A-String” (MOAS) phenotype, similar to those recently reported in patients with Alzheimer's Disease. The thin mitochondrial segments were associated with endoplasmic reticulum cisterns, and the mitochondrial proteins Fis1 and Drp1, all of which initiate mitochondrial fission. These data suggest that the MOAS phenotype may reflect malfunction in mitochondrial dynamics, whereby fission is initiated, but the process is incomplete due to malfunction of subsequent step(s). Thus, aged rhesus monkeys may be particularly helpful in exploring the age-related changes that render higher cortical circuits so vulnerable to degeneration.
Keywords: mitochondrial dynamics, Alzheimer's disease, primate brain aging, cognition, electron microscopy, 3D reconstruction
1. Introduction
The newly evolved dorsolateral prefrontal cortex (dlPFC) mediates high-order cognitive functions (Fuster, 2001) that are impaired early in the aging process in both monkeys and humans (Herndon et al., 1997; Moore et al., 2006; Morrison and Baxter, 2012). A prototypic cognitive function executed by the dlPFC involves working memory, the ability to use representational knowledge to guide behavior, thought and emotion. Neurons in layer III of the dlPFC excite each other to maintain persistent firing across the delay period in a working memory task (Goldman-Rakic, 1995). Pyramidal cells in dlPFC deep layer III have extensive recurrent connections (Kritzer and Goldman-Rakic, 1995) on long, thin spines with N-Methyl-D-aspartate receptor (NMDAR)-NR2B synapses (M. Wang et al., 2013). These dlPFC layer III connections are weakened by increased calcium-cyclic adenosine monophosphate (cAMP)-cAMP-dependent protein kinase A (PKA) signaling, which opens nearby K+ channels to gate network connections (Arnsten, 2015; Arnsten et al., 2012). This process is regulated by the phosphodiesterase, PDE4A, which declines with advancing age (Carlyle et al., 2014), leading to increased PKA phosphorylation of tau (Jicha et al., 1999), and increased cAMP-PKA opening of K+ channels, which reduces neuronal firing during working memory (Wang et al., 2011). There is also a marked loss of thin dendritic spines from monkey layer III dlPFC with advancing age (Du et al., 2010; Dumitriu et al., 2010; Luebke et al., 2010), the spines that mediate recurrent excitatory connections and are the focus of cAMP-calcium-K+ channel gating. Thus, these circuits are particularly vulnerable to advancing age (Morrison and Baxter, 2012).
The persistent neuronal firing generated by dlPFC recurrent circuits during working memory is highly energy demanding. Thus, it is of interest that the dlPFC has higher mRNA expression of mitochondrial proteins compared to other cortical regions (Chandrasekaran et al., 1992). Importantly, oxidative stress indicative of mitochondrial pathology has been identified in the dlPFC as part of the progression in Alzheimer's disease (AD) (Ansari and Scheff, 2010). Ultrastructural studies of the aging rhesus monkey dlPFC may help to illuminate this process, as these analyses provide clarity rarely possible in human post-mortem tissue. For example, one recent study of axon terminals in layer III of the monkey dlPFC identified mitochondrial morphological abnormalities (i.e., “donut” morphology) with increasing age (Hara et al., 2014), indicating that mitochondrial changes may play a role in age-related cognitive decline. Given the great expansion of layer III dlPFC dendrites in primate evolution (Elston, 2003; Elston et al., 2006), the current study tested the hypothesis that mitochondrial morphology in dlPFC dendrites may also be altered with aging, and that changes in mitochondria might reveal laminar differences.
Mitochondria are extremely dynamic, multifunctional organelles that play important roles in maintaining cellular health. Proper cellular function demands a balance between opposing mitochondrial dynamics - fusion and fission. Fusion may allow mitochondria to compensate for defects by sharing components; while fission segregates the damaged segments of mitochondria that then undergo mitophagy in the autophagosome, preserving the integrity of the mitochondrial network (Cho et al., 2010; Friedman and Nunnari, 2014; Palmer et al., 2011; Youle and van der Bliek, 2012). An equilibrium between fusion and fission may also balance energy vs. toxic reactive oxygen species (ROS) production, where mitochondrial fission may provide a respite from high ROS production (Nasrallah and Horvath, 2014). Therefore, alterations in mitochondrial dynamics may lead to cell pathology.
Mitochondrial fission is initiated by interactions with the calcium-containing smooth endoplasmic reticulum (SER), where SER cisterns accumulate in the vicinity of the mitochondria, and determine the sites of constriction and subsequent division (Friedman et al., 2011; Friedman and Nunnari, 2014). Fission is initiated by the dynamin-like GTPase, dynamin-related protein 1 (Drp1; also known as DLP1; (Smirnova et al., 2001)), which translocates from the cytosol into the outer mitochondrial membrane (OMM), where it interacts with its primary receptor - mitochondrial fission protein 1 (Fis1). Drp1 oligomers assemble into rings/spirals around the OMM, leading to the final membrane constriction and scission (Ingerman et al., 2005; James et al., 2003; Yoon et al., 2003). The efficacy of Drp1 in fission is determined by its GTPase activity, which is inhibited by PKA signaling (Cereghetti et al., 2010; Cereghetti et al., 2008). As PKA activity is increased in the aged dlPFC (Carlyle et al., 2014), mitochondrial dynamics might be disrupted in these vulnerable circuits.
Here, we used electron microscopy (EM) paired with immunocytochemistry and three-dimensional (3D) reconstruction from serial sections to examine mitochondrial morphology and molecular expression patterns in the aging rhesus macaque dlPFC. We detected age-related morphogenetic abnormalities resulting in accumulation of mitochondria with intermingled thin (“pinched”) and enlarged segments, indicative of mitochondrial dysfunction. This phenotype has just been detected in AD brains, where it has been termed “mitochondria-on-a-string” (MOAS) (Zhang et al., 2016). Thus, the discovery of this phenotype in the aged rhesus monkey may help reveal the etiology of mitochondrial deficits in human AD.
2. Materials and methods
2.1. Animals
All animal protocols were approved by the Yale University Institutional Animal Care and Use Committee (IACUC) and comply with the NIH guidelines for animal care and use. The study utilized brains from young adult [female 7 year-old (yo), and 11 yo], and aged [female 26 yo, 27 yo, 31 yo; and male 33 yo] rhesus macaques from the Yale colony.
2.2. Tissue processing for immunolabeling
Animals were deeply anaesthetized with pentobarbital (0.03 ml/10g of body weight) and perfused transcardially by the fixative containing 4% paraformaldehyde (wt/vol), 0.2% picric acid, and 0.05% glutaraldehyde in 0.1M phosphate buffer (Carlyle et al., 2014). Coronal 60-μm-thick sections of the dlPFC were cut with a vibratome, cryoprotected with 30% sucrose and stored at -80°C. All sections of the dlPFC went through freeze-thaw cycles in liquid nitrogen to augment penetration of immunoreagents and were processed free-floating for immunocytochemistry and EM as described below.
After defrosting and extensive washes, the sections were blocked in 5% bovine serum albumin, and incubated with mouse anti-DLP1 monoclonal (Drp1; clone 8; dilution 1:100; BD Biosciences, San Jose, CA, USA), or rabbit anti-TTC11 polyclonal antisera (Fis1; 1:100; Novus Biologicals USA, Littleton, CO, USA) overnight at room temperature. The sections were then immersed in respective solutions of biotinylated goat anti-mouse or anti-rabbit IgGs (1:300) and developed by Elite ABC kit (all from Vector Laboratories, Burlingame, CA, USA) with Ni-intensified 3,3′-diaminobenzidine-4HCl (Ni-DAB) as a chromogen. Some of the sections incubated with anti-DLP1 serum were labeled with made-in-goat anti-mouse secondary serum conjugated with 1-nm gold particles (Aurion, Wageningen, The Netherlands; 1: 80). Silver-intensification of gold was performed with R-Gent SE-LM kit (Aurion) according to the manufacturer's instructions. Specificity of the methods was tested by omitting the primary antibody from the staining procedure. No staining was observed in negative control sections. The sections were post-fixed with 1% OsO4, (or 0.5% for gold/silver labeling) dehydrated and embedded in Durcupan (ACM; Fluka, Buchs, Switzerland) on microscope slides and coverslipped.
2.3. EM and 3D reconstruction
For EM investigations, cortical layers were identified at low magnification using a light microscope, then selected areas from distinct layers were dissected and re-embedded into Durcupan blocks. Serial 70-nm-thick ultrathin sections were obtained, contrasted and evaluated as previously described (Morozov et al., 2006; Morozov et al., 2016). All images were acquired using a JEM1010 (JEOL, Tokyo, Japan) transmission electron microscope at 80 kV and Multiscan 792 digital camera (Gatan, Pleasanton, CA, USA). For EM tomography of mitochondria, 250-nm-thick sections from Durcupan-embedded dlPFC layer III slices from 31 and 33 yo animals were prepared as above and submitted to Department of Cell Biology (Yale University, New Haven, CT, USA).
For 3D reconstruction of mitochondria, 30-35 serial images were made with 15,000× magnification. Neuropil fragments were chosen for the 3D reconstruction in a random manner (but, avoiding as possible, cell bodies, blood vessels and groups of the myelinated axons) by an investigator blinded to the animal's age and to laminar locations. The micrographs were aligned using the computer program Reconstruct (Fiala, 2005), publicly available at http://www.bu.edu/neural/Reconstruct.html. We concentrated on mitochondria within dendritic shafts that were identified in serial micrographs by straight shape (in contrast to curved axons and astroglial processes). Mitochondrial profiles were traced, and the 3D images of reconstructed mitochondria were obtained and analyzed using Reconstruct software.
2.4. Data Analysis
Neuropil segments (n=4-5/animal) from cortical layers II-VI of each animal were used for 3D reconstruction and quantitative analyses of mitochondria. The neuropil segments for each cortical layer was identified and dissected based on cytoarchitectural features of dlPFC. All identifiable dendritic mitochondria were used for quantitative EM with the exception of truncated mitochondria, which were defined as mitochondria with detectable segments shorter than 1 micron. Identity of the analyzed neurons (e.g., pyramidal cell vs. interneuron) and laminar location of the cell body was not possible in this study as it would require labeling with cell-type unique markers.
A mitochondrion was counted as “MOAS” if it's 3D reconstructed image had at least one segment with the diameter 100 nm or thinner, and the difference between thin and enlarged segments was at least 3 times. Among the MOAS mitochondria, we identified a subset with an exaggerated phenotype, where there was at least a five-fold difference in the diameter between the thin and enlarged segments; such mitochondria were defined as severe MOAS (sMOAS). The following morphological parameters were measured in 3D images for each analyzed neuropil fragment: (1), the percentages of MOAS and sMOAS among all dendritic mitochondria; (2), the total number of thin segments in the reconstructed mitochondria; (3), the total length of thin segments; and (4), the total length of the reconstructed mitochondria. The number and the length of thin segments were normalized per 100-micron length of reconstructed mitochondria. The ratio of maximal vs. minimal diameter was calculated for each reconstructed mitochondria. The measurements were combined in two groups - young (7 and 11 yo) and aged animals (26, 27, 31 and 33 yo) and the averages ± SEM were calculated. For analyses of laminar distributions of MOAS, we calculated mean ± SEM normalized number of thin segments in distinct cortical layers combined in two groups - young and aged animals. The relationship between quantified mitochondrial parameters and the age of the monkey was assessed by Pearson's correlation using the Excel software (Microsoft). All p values were two-tailed, with P<0.05 used as the threshold for significance.
3. Results
3.1. Qualitative analysis of MOAS
EM analysis and 3D reconstruction revealed age-dependent differences in mitochondrial morphology within dlPFC dendrites. In young adult animals (7 and 11 year-old [yo]), the majority of mitochondria (n=365 mitochondria analyzed in 3D across all cortical layers) were elongated along dendritic shafts, revealing uniform diameters of 200-300 nm throughout the entire length of the mitochondria (Fig. 1). In contrast, analysis of mitochondria (n=774 mitochondria analyzed in 3D across all cortical layers) in aged rhesus macaques (26, 27, 31 and 33 yo) revealed alterations in mitochondrial morphology with numerous mitochondria with differing diameters: thin segments had a diameter of ∼100 nm or less (“pinched segments”), and enlarged segments had diameters ranging 300-500 nm, i.e. a MOAS phenotype. These abnormally shaped mitochondria with intermingled thin and enlarged segments were most clearly seen in 3D, as shown in Fig. 2. Mitochondrial matrix and cristae were sometimes visible in the relatively thicker (∼100 nm) “pinches”, but were absent in the thinnest segments where only the double membrane connected the two enlarged segments. Ultrastructure of the enlarged segments was normal in most cases, i.e., fine structure of mitochondrial membranes and matrix, thickness of the intermembrane space, number and shape of crista were similar to those seen in young animals. Thus, abnormalities such as swelling of the mitochondria, characterized by reduced electron density of the matrix (Morozov et al., 2016), were not encountered in these enlarged segments.
Fig. 1.
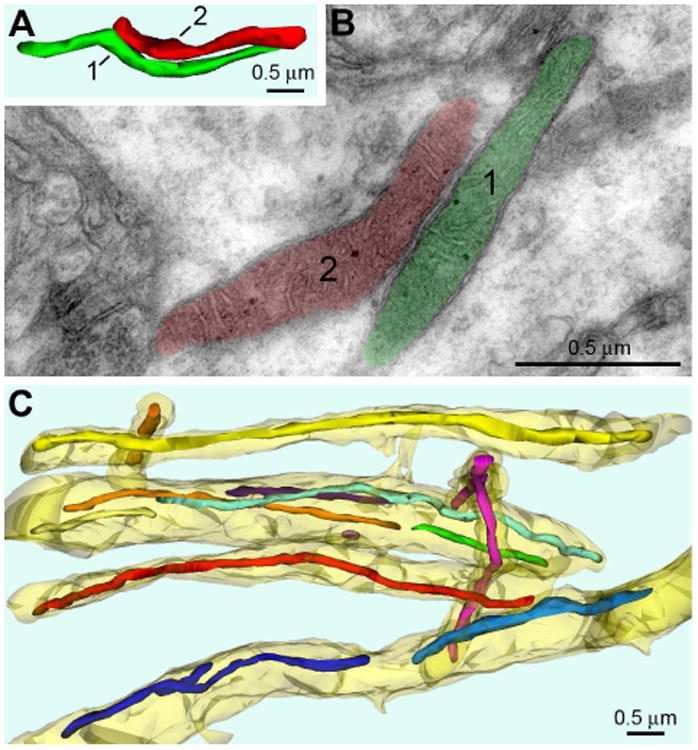
Shape and ultrastructure of mitochondria in dlPFC layer III dendritic shafts from 11 yo rhesus macaque. (A, B) 3D image and electron micrograph of two representative mitochondria (correspondingly labeled “1” and “2” and depicted green and red) located in the same dendrite. Shape of the mitochondria and ultrastructure of membranes and matrix are characteristic for normally functioning neurons. (C) 3D reconstruction from 33 serial sections from a randomly chosen segment of neuropil reveals numerous mitochondria elongated along dendritic shafts (shown semitransparent yellow). Notice similar diameters of all the mitochondria throughout entire length. Each mitochondrion is depicted with different color facilitating identification in 3D image.
Fig. 2.
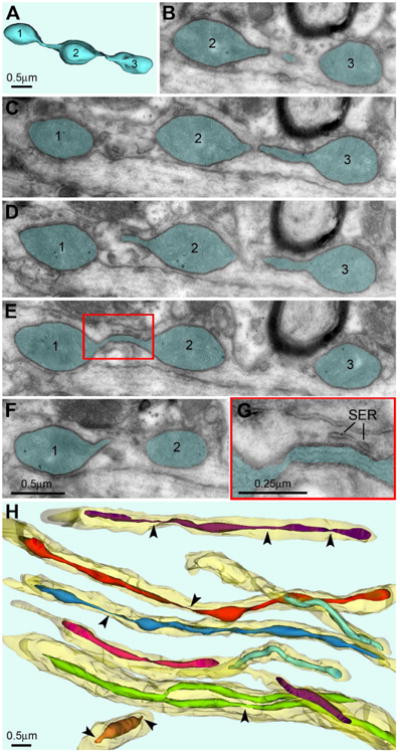
Shape and ultrastructure of mitochondria from dendritic shafts in dlPFC layer III of 31 yo monkey. (A-G) 3D and serial micrographs of an arbitrarily chosen MOAS. Three enlarged segments of the mitochondrion are equally numerated in the 3D and electron micrographs. Continuum of the mitochondrion (highlighted semitransparent blue) is seen in consecutive sections. Ultrastructure of crista and mitochondrial matrix are normal. Framed area in E is enlarged in G. Notice cisterns of smooth endoplasmic reticulum (SER) near thin segment of the mitochondrion. (H) 3D reconstruction from 30 serial sections of a randomly chosen dendrites (shown semitransparent yellow) reveals numerous thin segment (arrowheads) in many mitochondria.
3.2. Molecular characterization of MOAS as a fission-related phenotype
We hypothesized that the interleaved pinched and enlarged mitochondrial segments observed in aged monkey dlPFC may reflect malfunction in mitochondrial dynamics, whereby fission is initiated with appropriate constriction of the mitochondria, but the process of division is incomplete due to malfunction of subsequent step(s) in the fission process. To further evaluate this hypothesis, we examined the presence and localization of the smooth endoplasmic reticulum (SER), which is thought to be involved in the initiation of constriction, and two mitochondrial proteins that constitute the core proteins involved in the mitochondrial fission machinery, Fis1 and Drp1.
The SER has been shown to encircle mitochondria to initiate constriction and is thought to play an active role in defining the positions of mitochondrial division sites (Friedman et al., 2011; Friedman and Nunnari, 2014). Thus, we analyzed mitochondrial appositions with SER cisterns. We repeatedly found examples of SER-like cisterns accumulating near the “pinch” segments of mitochondria in serial ultrathin sections (Fig. 2G; 3B). This finding was confirmed using EM tomography as an alternative method that provides higher resolution (see movie in the Supplementary Materials). However, we also observed many examples where the SER was not evident near thin mitochondrial segments, consistent with previous data showing that apposition of the SER with the mitochondria is a transient phenomenon for executing the initial steps of fission (Friedman et al., 2011; Friedman and Nunnari, 2014). Quantitative analysis of SER frequency near constricted MOAS segments deserves further investigation, but is beyond the scope of the present study.
Fig. 3.

Serial electron micrographs of MOAS (highlighted semitransparent yellow) from dendritic shafts in dlPFC layer III of 26- (A, B) and 31 yo (C) rhesus macaque. Notice that both anti-Fis1 (A) and anti-Drp1 (B, C) immunolabeling (arrows) accumulates around thin segments of the mitochondria in accord with known function of Fis1 and Drp1 in mitochondrial fission (Friedman and Nunnari, 2014). Location of Drp1 on the OMM is confirmed with consistent DAB-Ni (B) and nano-gold/silver labeling (C) in serial sections. Numerous SER-like vesicles (highlighted semitransparent red in B) are seen in close proximity to some, but not all, MOAS. Framed areas in C1 and C2 are enlarged in C3 and C4, respectively.
Using immunolabeling and EM, we analyzed the location of Drp1, the GTPase that is recruited to constriction sites and subsequently cuts the mitochondrial membrane, and Fis1, its primary receptor on the OMM. As predicted, Fis1 labeling was observed on the “pinch” segments in mitochondria from aged animals (Fig. 3A). Similarly, both immunoperoxidase and immunogold labeling demonstrated Drp1 in contact with the outer surface of the OMM, in close proximity to the thin mitochondrial segments (Fig. 3B, C). Thus, both Fis1 and Drp1 could be localized to “pinch” segments, consistent with what has been seen in vitro during mitochondrial fission (Mozdy et al., 2000; Otsuga et al., 1998; Smirnova et al., 2001).
3.3 Quantitative and laminar analysis of MOAS in dlPFC
We quantified 3D-reconstructed mitochondria across the cortical layers in the young vs. aged dlPFC. Please note that laminar position refers to the dendritic location, and not the cell of origin, which is unknown. For example, for layer III, dendrites could include basilar dendrites from superficial layer III, basilar or apical dendrites from deep layer III, or apical dendrites of deeper neurons (layers V-VI). In our morphometric analysis, a mitochondrion was counted as MOAS if it had at least one segment with the diameter 100 nm or thinner, and at least a 3-fold difference in the diameter between thin vs. enlarged segments. We also identified a subset of mitochondria with the more pronounced MOAS phenotype if there was at least a 5-fold difference in the diameter between thin vs. enlarged segments; we defined such mitochondria as severe MOAS (sMOAS; Fig. 4).
Fig. 4.
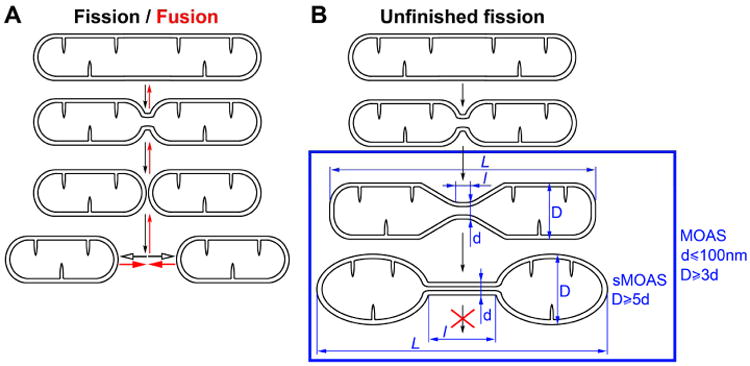
(A) In a healthy state, fission and fusion are opposing processes that occur in a dynamic fashion, resulting in very rapid changes in mitochondrial morphology (B) In case of pathological “unfinished fission”, numerous mitochondria with characteristic intermingling of thin and enlarged segments are accumulated in the cell. We counted a mitochondrion as “MOAS” if diameter of its thin segment (d) was at least 3 times less than diameter of the enlarged segments (D). Within the category of MOAS, the subgroup that demonstrated the most pronounced morphological deviation (D is more or equal to 5d) were identified as “sMOAS”. L is total length of reconstructed mitochondria; l is length of thin segment.
We detected pronounced age-related differences in the frequency of MOAS. In young adult macaques (7 and 11 yo), out of a total of 365 mitochondria analyzed across all laminar locations, only 8 mitochondria were identified as MOAS and only 1 of those 8 was identified as sMOAS. Two of these mitochondria were located in the layer V, and 6 mitochondria (including one sMOAS) were located in layer VI of the 11 yo animal; whereas there were no MOAS in layers II, III and IV. There was no instance of MOAS in the 7 yo monkey. In contrast, in the aged macaques (26, 27, 31 and 33 yo), the average frequency of MOAS was 26.9% (208 out of totally 774 mitochondria analyzed). Among these altered mitochondria, 35.6% (74 out of totally 208) belonged to the more severe sMOAS category (see also Table 1 and Fig. 5a).
Table 1. Percentage of MOAS in dendritic shafts of the rhesus macaque dlPFC.
| 7 y.-o. | 11 y.-o. | 26 y.-o. | 27 y.-o. | 31 y.-o. | 33 y.-o. | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | MOAS % |
sMOAS % |
n | MOAS % |
sMOAS % |
n | MOAS % |
sMOAS % |
n | MOAS % |
sMOAS % |
n | MOAS % |
sMOAS % |
n | MOAS % |
sMOAS % |
|
| Layer II | 33 | 0 | 0 | 38 | 0 | 0 | 78 | 55.1 | 25.6 | 58 | 8.6 | 1.7 | 46 | 43.5 | 6.5 | 38 | 2.6 | 0 |
| Layer III | 43 | 0 | 0 | 28 | 0 | 0 | 52 | 30.8 | 13.5 | 88 | 13.6 | 2.3 | 41 | 48.8 | 19.3 | 42 | 52.4 | 11.9 |
| Layer IV | 64 | 0 | 0 | 54 | 0 | 0 | 28 | 67.9 | 32.1 | 54 | 5.6 | 1.9 | 53 | 30.2 | 11.3 | 38 | 5.3 | 2.6 |
| Layers V-VI | 26 | 0 | 0 | 79 | 10.1 | 1.3 | 49 | 18.4 | 12.2 | 49 | 10.2 | 0 | 31 | 38.7 | 16.1 | 29 | 10.3 | 0 |
| All layers combined | 166 | 0 | 0 | 199 | 4.0 | 0.5 | 207 | 42.0 | 20.3 | 249 | 10.0 | 1.6 | 171 | 39.8 | 12.9 | 147 | 19.7 | 4.8 |
n – total number of mitochondria analyzed in 3D.
MOAS and sMOAS are determined according to geometric characteristic described in Fig. 4.
All brain tissue segments were chosen for 3D reconstruction in a random manner by investigator blinded for the animal age and laminar locations.
Fig. 5.
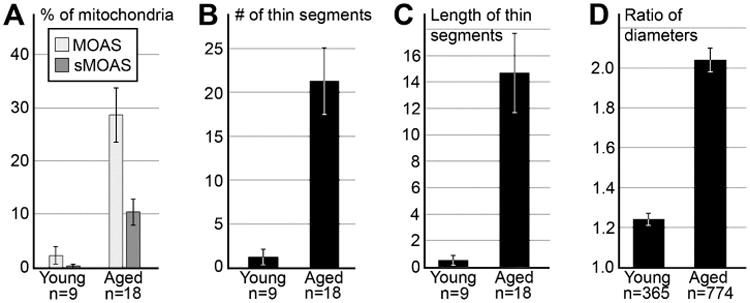
Mitochondrial morphological parameters in dendrites from dlPFC of young adult (7 and 11 yo) and aged (26, 27, 31 and 33 yo) rhesus macaques. (A) Percentages of MOAS and sMOAS dramatically differ between young and aged animals. (B, C) Number and length of thin segments in MOAS normalized per 100 micron length of reconstructed mitochondria. (D) Ratio of maximal vs. minimal diameters of mitochondria. Error bars show SEM. n is number of neuropils segments (A-C) or number of mitochondria analyzed (D).
As the reconstructed mitochondria differed in length, and many of the long mitochondria were incompletely reconstructed because of truncation on the edges of the analyzed volume, we calculated the number and length of the pinched segments normalized by the total length of the reconstructed mitochondria. This analysis revealed large increases in mitochondrial abnormalities in the aged monkey dlPFC (Table 2; Fig. 5b, c). There was limited evidence of MOAS in young monkey dlPFC, where abnormalities were only seen in layers V and VI of the 11yo monkey; and showed in average normalized number of pinch segments 5.8 and length of pinch segments 2.2μm per 100μm. In contrast, there were large normalized numbers and length of pinch segments in aged monkeys (Table 2; Fig. 5b, c). For example, in layer IV of the 26yo monkey, the normalized number of pinch segments was 48.9, and the length of pinch segments was 44.4μm per 100μm length of the reconstructed mitochondria.
Table 2. Length and number of thin segments in MOAS in dendritic shafts of the rhesus macaque dlPFC.
| 7 y.-o. | 11 y.-o. | 26 y.-o. | 27 y.-o. | 31 y.-o. | 33 y.-o. | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| L, μm |
lnorm, μm |
#norm | L, μm |
lnorm, μm |
#norm | L, μm |
lnorm, μm |
#norm | L, μm |
lnorm, μm |
#norm | L, μm |
lnorm, μm |
#norm | L, μm |
lnorm, μm |
#norm | |
| Layer II | 74.7 | 0 | 0 | 80.7 | 0 | 0 | 206.9 | 14.1 | 37.7 | 156.4 | 1.9 | 3.8 | 77.5 | 16.1 | 34.8 | 134.0 | 0.8 | 1.5 |
| Layer III | 90.8 | 0 | 0 | 97.0 | 0 | 0 | 63.5 | 24.9 | 31.5 | 235.7 | 4.8 | 6.8 | 128.3 | 19.4 | 28.8 | 131.4 | 27.6 | 33.5 |
| Layer IV | 112.4 | 0 | 0 | 115.1 | 0 | 0 | 89.9 | 44.4 | 48.9 | 120.9 | 3.7 | 3.3 | 78.3 | 19.8 | 25.5 | 78.0 | 2.2 | 3.8 |
| Layers V-VI | 25.0 | 0 | 0 | 155.7 | 2.2 | 5.8 | 43.8 | 17.1 | 27.4 | 108.4 | 3.0 | 6.5 | 47.7 | 17.6 | 35.6 | 66.8 | 2.8 | 7.5 |
| All layers combined | 302.9 | 0 | 0 | 447.9 | 0.8 | 2.0 | 404.1 | 22.9 | 38.1 | 621.4 | 3.5 | 5.3 | 331.8 | 18.5 | 30.4 | 410.2 | 10.0 | 13.2 |
L, total length of reconstructed mitochondria. Although many of the reconstructed mitochondria have different length, their summarized length correlates with the number of mitochondria analyzed (see Table 1).
lnorm, length of thin segments normalized by 100 μm-length of reconstructed mitochondria.
#norm, number of thin segments normalized by 100 μm-length of reconstructed mitochondria.
All brain tissue segments were chosen for 3D reconstruction in a random manner by investigator blinded for the animal age and laminar locations.
When we calculated the ratio of maximal vs. minimal diameters of the reconstructed mitochondria, we confirmed that the diameter of the mitochondria in young adult macaques is mostly uniform (ratio of maximum to minimum is close to 1); whereas the average range in diameter differences was 2.04±0.06 in the aged animals, suggesting that the diameter differences are significantly exaggerated in aged animals (Fig. 5d). Thus, our quantitative analysis demonstrated dramatic differences between young and aged monkeys in the number of MOAS, the normalized number and length of pinches, and the ratio of maximal to minimal diameters. These findings reinforce the idea that there are robust age-related alterations in mitochondrial morphometric parameters which might significantly contribute towards cognitive decline during aging.
Finally, we performed a laminar analysis of MOAS distribution (Tables 1, 2; Fig. 6). MOAS could be found in all layers in the aged monkeys (Tables 1, 2), and impaired mitochondrial dynamics were consistently observed across all layers in aged monkey dlPFC (Fig. 6A). However, when the laminar data were correlated with increasing age, there was a statistically significant correlation between the normalized number of pinches with increasing age in layer III (r=0.842, p=0.035), but not in other layers (p>0.2) (Fig. 6B). Thus, the clearest relationship between mitochondrial abnormalities and advancing age was observed in layer III, but the altered morphological phenotype was present in all layers of aged monkeys.
Fig. 6.
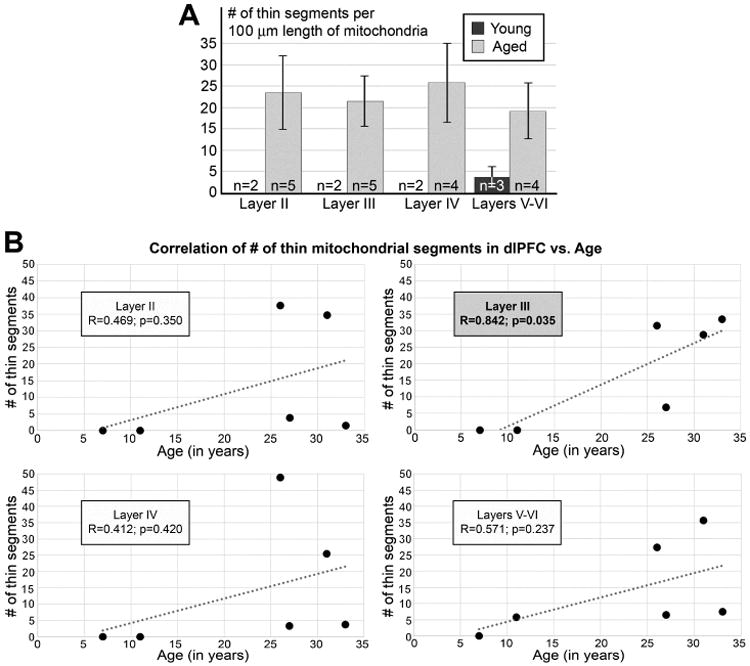
Laminar distribution of the number of thin segments normalized per 100 micron length of reconstructed mitochondria. (A) Average number of thin segments from four aged (26, 27, 31 and 33 yo; grey columns) and two young animals (7 and 11 yo; black column). In young animals, MOAS were identified only in layers V-VI, and were absent in the layers II-IV. Notice that the average numbers of thin segments are similar in all analyzed layers from aged animals. Error bars show SEM. n is number of neuropil segments analyzed. (B) Correlation of normalized number of thin segments in distinct layers during aging in the monkey dlPFC. Statistically significant correlation between MOAS and advancing age is seen only in layer III.
4. Discussion
Here, we report marked changes in mitochondrial morphology in dlPFC neurons from aged rhesus macaques. Namely, EM paired with 3D reconstruction from serial sections revealed numerous mitochondria with different size profiles characterized by thin segments that intermingle with enlarged ones, i.e., a MOAS phenotype. In contrast, altered mitochondria were extremely rare in the young adult macaques, suggesting pronounced age-related changes in mitochondrial morphology. Similar MOAS mitochondrial phenotypes have just been demonstrated in postmortem hippocampus from human subjects with AD, and in mice with human Alzheimer-related genetic mutations (Zhang et al., 2016). Although our studies were completely independent, our findings converge on the same conclusion, that alterations in mitochondrial morphology are a prominent feature of aged neurons in human and nonhuman primate cortical circuits that are vulnerable to atrophy.
4.1. Possible relationship of MOAS to altered mitochondrial fission dynamics
The changes in mitochondrial shape, and the pattern of immunolabeling, indicate that MOAS may arise from impairments in mitochondrial fission due to dysregulation of the mitochondrial fission machinery. Our findings suggest that mitochondrial division is initiated, producing constricted segments in the mitochondrial body, but that the process of fission is unable to proceed to completion, i.e., it is “unfinished fission”. This hypothesis is consistent with the findings that “pinched” segments were associated with SER cisterns, and Fis1 and Drp1 labeling. Although this idea needs to be confirmed in cell cultures where mitochondrial dynamics can be captured over time using time-lapse imaging, the current EM images from monkey brain are consistent with this interpretation.
Mitochondrial fission is part of a quality-control mechanism whereby damaged mitochondrial components are segregated from healthy components, followed by mitochondrial division and mitophagy of the damaged daughter mitochondrion (Youle and van der Bliek, 2012). A balance between fusion vs. fission is also necessary for limiting the production of toxic reactive oxygen species (ROS) and for normal cellular metabolism, whereas disruptions in these processes affect the cell and may be implicated in neurodegenerative diseases (Santos et al., 2015; Wang et al., 2009; D.B. Wang et al., 2013). In vitro studies have also shown that mitochondrial fission may be a part of a pathological process that impairs mitochondrial membrane permeability (i.e., opens mitochondrial permeability transition pores), resulting in release of cytochrome c in cytoplasm and activation of caspases that, in turn, initiate apoptotic or necrotic cell death pathways (Galluzzi et al., 2009; Green et al., 2011; Lin and Beal, 2006). Such pathological processes are characterized by fragmentation of long mitochondria into spherical ones that exhibit disordered ultrastructure, e.g., swelling of the mitochondrial matrix and disintegrated crista organization (Galluzzi et al., 2009; Green et al., 2011; Morozov et al., 2016). Our results demonstrate that MOAS is not associated with swollen mitochondrial matrix or disorganization of crista; mitochondrial profiles exhibit normal membrane integrity and electron density of the matrix. These findings are consistent with the fact that neuronal cell death in dlPFC is not associated with normal aging in monkeys (Morrison and Baxter, 2012), or early stages of Alzheimer's disease (Braak and Braak, 1991; Selkoe, 2002). The MOAS phenotype likely reflects deficits in the selective elimination of damaged mitochondria that may aggravate metabolic deficits and production of ROS, creating a vicious pathological cascade of events. Although Zhang et al. propose that the MOAS phenotype may be a “compensatory adaptation to bioenergetic stress providing protection against mitophagy” (Zhang et al., 2016), it may reflect a dysfunctional state of mitochondrial physiology that impacts cellular functioning. Further research will be needed to determine how these morphological changes affect neuronal physiology.
4.2. Possible molecular mechanisms of MOAS in aging dlPFC
Animal models may be helpful in determining the molecular mechanisms underlying changes in mitochondrial morphology with advancing age. The increased number of mitochondrial constrictions suggests that the initial steps of mitochondrial fission are not perturbed, but rather the final step - scission of the mitochondrial membranes - may be impaired. The final scission of the mitochondrial membranes is caused by Drp1 oligomerization around the OMM. Although Drp1 molecules appear to be capable of translocation to the mitochondrial surface undergoing fission (present study), Drp1 function may be compromised. Drp1 exerts its mechanical pressure through its GTPase activity, which can be inhibited by PKA signaling or other signaling cascades (Chang and Blackstone, 2010). Interestingly, cAMP-PKA activity is increased in the aged dlPFC, at least in part due to loss of phosphodiesterase 4A regulation of cAMP signaling (Carlyle et al., 2014; Wang et al., 2011). Thus, molecular changes in the aging brain may contribute to the MOAS phenotype by inhibiting Drp1 function. As inhibition of Drp1 GTPase activity contributes to MOAS in cultured neurons (Zhang et al., 2016), PKA inhibition of GTPase activity of Drp1 may similarly induce MOAS in vivo in aged dlPFC. Other pathways affecting Drp1 phosphorylation (e.g., CAMKIα, Cdk1-cyclin B, etc.) also deserve investigation (Westermann, 2010), especially as internal calcium release may also be increased in aged dlPFC (Carlyle et al., 2014).
4.3. Relevance of MOAS to impaired working memory circuits
A recent study of aging rhesus monkeys indicates that changes in mitochondrial morphology are associated with cognitive deficits. Particularly, this study showed that impairments in working memory correlated with the accumulation of donut-shaped mitochondria in presynaptic boutons in layer III of dlPFC (Hara et al., 2014, 2016). Donut-shaped mitochondria are a predominant source of reactive oxygen species (Ahmad et al., 2013), suggesting this phenotype reflected metabolic abnormalities in the circuits that underlie working memory function. The aged monkeys in the current study had been tested on a working memory task for many years and for differing amounts of time, and thus their cognitive abilities cannot be accurately estimated for relationship to MOAS. However, our finding of an age-related increase in MOAS in layer III dendrites indicates that deregulated mitochondrial dynamics occurs in the postsynaptic compartment as well.
Layer III microcircuits are the source of the “Delay cells” that generate neural representations needed for working memory (Goldman-Rakic, 1995), and are a focus of age-related pathology (Morrison and Baxter, 2012). For example, there is significant loss of layer III spines during aging, which correlates with the degree of cognitive impairment (Dumitriu et al., 2010; Peters et al., 2008). Spine loss, as well as increased cAMP-PKA opening of K+ channels, likely contributes to a progressive reduction in persistent Delay cell firing with advancing age (Wang et al., 2011). The current finding of age-related increases in MOAS in layer III of dlPFC dendrites indicates that dysfunction of mitochondria may occur in parallel, or may even be an upstream etiological mechanism driving these age-related changes (Li et al., 2004). It should be emphasized that we do not know the cells of origin for the dendrites examined in the current study, and thus the link to layer III pyramidal cells remains speculative. However, persistent Delay cell firing is energy-intensive, and thus may falter with inadequate energy production. Furthermore, mitochondrial dysfunction can aggravate inflammatory cascades (West et al., 2011) that can contribute to neuronal atrophy and spine loss (Stephan et al., 2012). Thus, understanding the physiological consequences of the MOAS phenotype may illuminate how neurons become vulnerable with advancing age.
4.4. Relevance to Alzheimer's disease
The severe MOAS phenotype was recently observed within hippocampal neurons in patients with AD (Zhang et al., 2016). Thus, the aging rhesus monkey may provide an important animal model for understanding the mechanisms underlying the etiology of mitochondrial insults, and how these contribute to neuronal atrophy or degeneration. It should be noted that MOAS were not seen in the aged human control group (Zhang et al., 2016), i.e. it was not evident in the aged human hippocampus, whereas MOAS were observed in the aged rhesus monkey dlPFC (present study). This apparent discrepancy may arise from differences in regional sensitivity, as the hippocampus is less vulnerable to aging and stress than is the extremely susceptible dlPFC (Moore et al., 2006; Morrison and Baxter, 2012; Voytko, 1999).
Several lines of evidence suggest that MOAS contribute to the neurodegenerative process. For example, mitochondrial deficits appear early in mouse models of AD (Du et al., 2010; Yao et al., 2009). Consistent with the basic research, deficits in the balance of mitochondrial fusion/fission have been reported in neurodegenerative diseases such as AD and Parkinson's disease (Santos et al., 2015; Wang et al., 2009). Understanding this process and protecting mitochondrial integrity may provide a strategy for improving the health and function of the aging brain. Future studies involving a larger number of cognitively-assessed monkeys may help to elucidate how changes in mitochondrial morphology relate to the functional integrity of the aging brain.
Supplementary Material
Electron microscopy reveals changes in mitochondria in aged monkey prefrontal cortex
Changes resemble “mitochondria-on-a-string” (MOAS) as seen in Alzheimer's Disease
MOAS pinch points linked with SER cisterns, Fis1 and Drp1, suggesting altered fission
Increased calcium-cAMP-PKA inhibition of Drp1 may drive age-related changes
Studies of aged monkey cortex may help reveal etiology of Alzheimer's pathology
Acknowledgments
We are grateful to Dr. Xinran Liu (Department of Cell Biology, Yale University) for EM tomography. This research was funded by NIH Pioneer Award DP1AG047744-01 to AFTA.
Footnotes
Disclosure statement: The authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad T, Aggarwal K, Pattnaik B, Mukherjee S, Sethi T, Tiwari BK, Kumar M, Micheal A, Mabalirajan U, Ghosh B, Sinha Roy S, Agrawal A. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013;4:e461. doi: 10.1038/cddis.2012.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, Scheff SW. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol. 2010;69:155–167. doi: 10.1097/NEN.0b013e3181cb5af4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF. Stress weakens prefrontal networks: molecular insults to higher cognition. Nat Neurosci. 2015;18:1376–1385. doi: 10.1038/nn.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsten AF, Wang MJ, Paspalas CD. Neuromodulation of thought: flexibilities and vulnerabilities in prefrontal cortical network synapses. Neuron. 2012;76:223–239. doi: 10.1016/j.neuron.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Carlyle BC, Nairn AC, Wang M, Yang Y, Jin LE, Simen AA, Ramos BP, Bordner KA, Craft GE, Davies P, Pletikos M, Sestan N, Arnsten AF, Paspalas CD. cAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc Natl Acad Sci USA. 2014;111:5036–5041. doi: 10.1073/pnas.1322360111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereghetti GM, Costa V, Scorrano L. Inhibition of Drp1-dependent mitochondrial fragmentation and apoptosis by a polypeptide antagonist of calcineurin. Cell Death Differ. 2010;17:1785–1794. doi: 10.1038/cdd.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci USA. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekaran K, Stoll J, Giordano T, Atack JR, Matocha MF, Brady DR, Rapoport SI. Differential expression of cytochrome oxidase (COX) genes in different regions of monkey brain. J Neurosci Res. 1992;32:415–423. doi: 10.1002/jnr.490320313. [DOI] [PubMed] [Google Scholar]
- Chang CR, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann N Y Acad Sci. 2010;1201:34–39. doi: 10.1111/j.1749-6632.2010.05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Lipton SA. Mitochondrial dynamics in cell death and neurodegeneration. Cell Mol Life Sci. 2010;67:3435–3447. doi: 10.1007/s00018-010-0435-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer's disease mouse model. Proc Natl Acad Sci USA. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitriu D, Hao J, Hara Y, Kaufmann J, Janssen WG, Lou W, Rapp PR, Morrison JH. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging-related cognitive impairment. J Neurosci. 2010;30:7507–7515. doi: 10.1523/JNEUROSCI.6410-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston GN. Cortex, cognition and the cell: new insights into the pyramidal neuron and prefrontal function. Cereb Cortex. 2003;13:1124–1138. doi: 10.1093/cercor/bhg093. [DOI] [PubMed] [Google Scholar]
- Elston GN, Benavides-Piccione R, Elston A, Zietsch B, Defelipe J, Manger P, Casagrande V, Kaas JH. Specializations of the granular prefrontal cortex of primates: implications for cognitive processing. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:26–35. doi: 10.1002/ar.a.20278. [DOI] [PubMed] [Google Scholar]
- Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Nunnari J. Mitochondrial form and function. Nature. 2014;505(7483):335–43. doi: 10.1038/nature12985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JM. The prefrontal cortex--an update: time is of the essence. Neuron. 2001;30:319–333. doi: 10.1016/s0896-6273(01)00285-9. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10:481–494. doi: 10.1038/nrn2665. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–85. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Yuk F, Puri R, Janssen WG, Rapp PR, Morrison JH. Presynaptic mitochondrial morphology in monkey prefrontal cortex correlates with working memory and is improved with estrogen treatment. Proc Natl Acad Sci USA. 2014;111:486–491. doi: 10.1073/pnas.1311310110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Yuk F, Puri R, Janssen WG, Rapp PR, Morrison JH. Estrogen Restores Multisynaptic Boutons in the Dorsolateral Prefrontal Cortex while Promoting Working Memory in Aged Rhesus Monkeys. J Neurosci. 2016;36:901–910. doi: 10.1523/JNEUROSCI.3480-13.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndon JG, Moss MB, Rosene DL, Killiany RJ. Patterns of cognitive decline in aged rhesus monkeys. Behav Brain Res. 1997;87:25–34. doi: 10.1016/s0166-4328(96)02256-5. [DOI] [PubMed] [Google Scholar]
- Ingerman E, Perkins EM, Marino M, Mears JA, McCaffery JM, Hinshaw JE, Nunnari J. Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol. 2005;170:1021–1027. doi: 10.1083/jcb.200506078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–36379. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Weaver C, Lane E, Vianna C, Kress Y, Rockwood J, Davies P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer's disease. J Neurosci. 1999;19:7486–7494. doi: 10.1523/JNEUROSCI.19-17-07486.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritzer MF, Goldman-Rakic PS. Intrinsic circuit organization of the major layers and sublayers of the dorsolateral prefrontal cortex in the rhesus monkey. J Comp Neurol. 1995;359:131–143. doi: 10.1002/cne.903590109. [DOI] [PubMed] [Google Scholar]
- Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Luebke J, Barbas H, Peters A. Effects of normal aging on prefrontal area 46 in the rhesus monkey. Brain Res Rev. 2010;62:212–232. doi: 10.1016/j.brainresrev.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore TL, Killiany RJ, Herndon JG, Rosene DL, Moss MB. Executive system dysfunction occurs as early as middle-age in the rhesus monkey. Neurobiol Aging. 2006;27:1484–1493. doi: 10.1016/j.neurobiolaging.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Morozov YM, Ayoub AE, Rakic P. Translocation of synaptically connected interneurons across the dentate gyrus of the early postnatal rat hippocampus. J Neurosci. 2006;26:5017–5027. doi: 10.1523/JNEUROSCI.0272-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov YM, Sun YY, Kuan CY, Rakic P. Alteration of SLP2-like immunolabeling in mitochondria signifies early cellular damage in developing and adult mouse brain. Eur J Neurosci. 2016;43:245–257. doi: 10.1111/ejn.13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13:240–250. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozdy AD, McCaffery JM, Shaw JM. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol. 2000;151:367–380. doi: 10.1083/jcb.151.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasrallah CM, Horvath TL. Mitochondrial dynamics in the central regulation of metabolism. Nat Rev Endocrinol. 2014;10:650–658. doi: 10.1038/nrendo.2014.160. [DOI] [PubMed] [Google Scholar]
- Otsuga D, Keegan BR, Brisch E, Thatcher JW, Hermann GJ, Bleazard W, Shaw JM. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J Cell Biol. 1998;143:333–349. doi: 10.1083/jcb.143.2.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CS, Osellame LD, Stojanovski D, Ryan MT. The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cell Signal. 2011;23:1534–1545. doi: 10.1016/j.cellsig.2011.05.021. [DOI] [PubMed] [Google Scholar]
- Peters A, Sethares C, Luebke JI. Synapses are lost during aging in the primate prefrontal cortex. Neuroscience. 2008;152:970–981. doi: 10.1016/j.neuroscience.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos D, Esteves AR, Silva DF, Januario C, Cardoso SM. The Impact of Mitochondrial Fusion and Fission Modulation in Sporadic Parkinson's Disease. Mol Neurobiol. 2015;52:573–586. doi: 10.1007/s12035-014-8893-4. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–89. doi: 10.1146/annurev-neuro-061010-113810. [DOI] [PubMed] [Google Scholar]
- Voytko ML. Impairments in acquisition and reversals of two-choice discriminations by aged rhesus monkeys. Neurobiol Aging. 1999;20:617–627. doi: 10.1016/s0197-4580(99)00097-4. [DOI] [PubMed] [Google Scholar]
- Wang DB, Garden GA, Kinoshita C, Wyles C, Babazadeh N, Sopher B, Kinoshita Y, Morrison RS. Declines in Drp1 and parkin expression underlie DNA damage-induced changes in mitochondrial length and neuronal death. J Neurosci. 2013;33:1357–1365. doi: 10.1523/JNEUROSCI.3365-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Gamo NJ, Yang Y, Jin LE, Wang XJ, Laubach M, Mazer JA, Lee D, Arnsten AF. Neuronal basis of age-related working memory decline. Nature. 2011;476:210–213. doi: 10.1038/nature10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, Morrison JH, Wang XJ, Arnsten AF. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron. 2013;77:736–749. doi: 10.1016/j.neuron.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. doi: 10.1038/nrm3013. [DOI] [PubMed] [Google Scholar]
- Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y, Krueger EW, Oswald BJ, McNiven MA. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol. 2003;23:5409–5420. doi: 10.1128/MCB.23.15.5409-5420.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Trushin S, Christensen TA, Bachmeier BV, Gateno B, Schroeder A, Yao J, Itoh K, Sesaki H, Poon WW, Gylys KH, Patterson ER, Parisi JE, Diaz Brinton R, Salisbury JL, Trushina E. Altered brain energetics induces mitochondrial fission arrest in Alzheimer's Disease. Sci Rep. 2016;6:18725. doi: 10.1038/srep18725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.