

Abstract
Fibrous dysplasia (FD) is a rare bone disease caused by postzygotic somatic activating mutations in the GNAS gene, which lead to constitutive activation of adenylyl cyclase, and elevated levels of cyclic AMP, which act on downstream signaling pathways, and cause normal bone to be replaced with fibrous tissue and abnormal (woven) bone. The bone disease may occur in one bone (monostotic), multiple bones (polyostotic), or in combination with hyperfunctioning endocrinopathies and hyperpigmented skin lesions (in the setting of McCune-Albright Syndrome). FD is common in the craniofacial skeleton, causing significant dysmorphic features, bone pain, and dental anomalies. This review summarizes the pathophysiology, clinical findings and treatment of FD, with an emphasis on the craniofacial and oral manifestations of the disease.
Keywords: Fibrous dysplasia, McCune-Albright syndrome, craniofacial bone disease, dental anomalies, GNAS gene
Introduction
Fibrous dysplasia (FD) (OMIM#1174800) is an uncommon skeletal disorder resulting in deformity, fractures, pain, and functional impairment (Boyce and Collins, 1993, Lichtenstein, 1938). The disease is mosaic and presents along a broad clinical spectrum, affecting one bone (monostotic) or multiple bones (polyostotic). FD may occur in isolation or in association with skin and endocrine features, termed the McCune-Albright syndrome (MAS) (Boyce and Collins, 1993). Patients may develop one or many combinations of features, leading to a complex and highly variable phenotype.
FD/MAS results from post-zygotic activating mutations in GNAS, which encodes the α-subunit of the Gs stimulatory protein (Figure 1) (Weinstein et al., 2004). Mutations occur at one of two positions: Arg201 (>95% of reported cases) (Lumbroso et al., 2004) or Gln227 (<5%) (Idowu et al., 2007). These mutations disrupt the intrinsic GTPase activity of Gsα, resulting in constitutive receptor activation and inappropriate cyclic AMP-mediated signaling. Because FD/MAS affects tissues derived from the endoderm, mesoderm, and ectoderm (i.e. the skin, bone, and endocrine systems), the mutation must occur early in embryogenesis, prior to derivation of the three germ layers (Happle, 1986). Therefore, the phenotype of an individual with FD/MAS results from both the distribution of the tissues that carry the GNAS mutation and the role that Gsα plays in those specific tissues.
Figure 1. Signaling pathway.
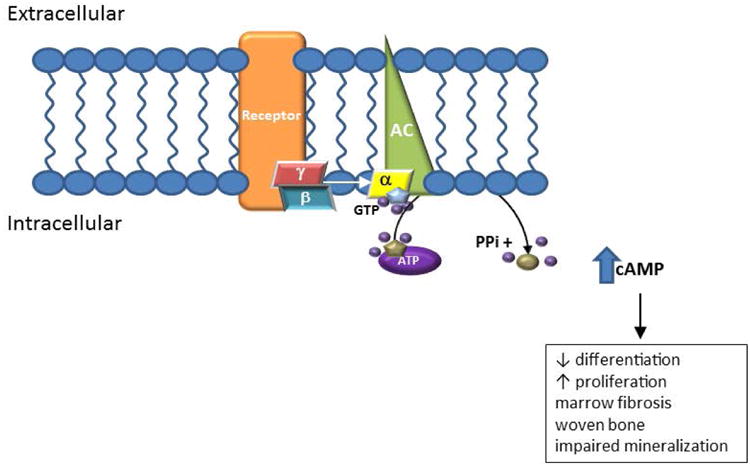
Fibrous dysplasia (FD) results from post-zygotic, activating mutations in the gene GNAS, which encodes for the Gsα subunit of the G protein complex. In FD, a mutated Gsα results in ligand-independent, continuous activation of adenylyl cyclase, resulting in excess production of intracellular cAMP. In bone, this causes proliferation of bone marrow stromal cells, leading to fibrosis, loss of hematopoiesis/marrow adipocytes, and a structurally abnormal matrix.
FD is a disease of the skeletal stem cell in which both bone formation and resorption are affected. Constitutive Gsα activation results in the formation of bone marrow stromal cells (BMSCs) that are unable to differentiate into normal marrow components, including osteoblasts, adipocytes, and hematopoiesis-supporting cells (Riminucci et al., 2006). Bone and marrow are replaced by proliferating bone marrow stromal cells, resulting in the formation of discrete fibro-osseous lesions (Riminucci et al., 1999). Overexpression of Receptor Activator of Nuclear Factor Kappa-B Ligand (RANKL) and Interleukin-6 (IL-6) has been associated with increased bone resorption, via activation of osteoclasts, and fibrous tissue deposition in FD (Riminucci et al., 2003b, Piersanti et al., 2010).
Clinical Presentation: Fibrous Dysplasia
Anatomically, FD lesions are often broadly distributed in a patchy pattern, reflecting the mosaic nature of the disease. FD exists along a broad clinical spectrum, ranging from an isolated, trivial lesion to severe disease involving nearly the entire skeleton. The clinical presentation is largely influenced by the location and distribution of the skeletal lesions (Figure 2A). In the appendicular skeleton, FD typically presents with fractures, deformity, limp, or pain. Repeated fractures and expansile deformities may lead to functional impairment and difficulty with ambulation, particularly of the proximal femorae, which may develop the classical “shepherd's crook” (coxa vara) deformity (Figure 2B, C) (Ippolito et al., 2014). Axial FD is common, and in rare cases may result in severe and progressive scoliosis (Figure 2D) (Leet et al., 2004b).
Figure 2. Axial and appendicular skeletal findings.
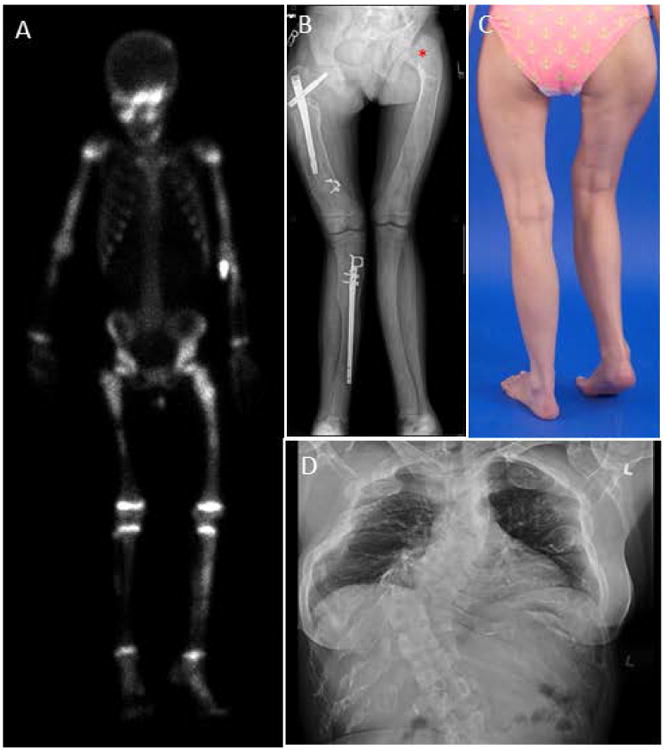
(A) Typical Technetium-99m (99mTc)bone scan, showing uptake in areas of fibrous dysplasia (FD) – including the skull, bilateral arms, legs and pelvis.
(B) Expansion and bowing of the lower extremities in a patient with polyostotic disease. Rods have been placed to help stabilize the right femur and tibia. “Shepherd's crook” deformity can be visualized at the left femoral neck angle (asterisk).
(C) Photograph of a patient with lower extremity deformity and leg-length discrepancy caused by FD.
(D) AP Chest radiograph with severe scoliosis caused by FD of the vertebral column.
Since FD is a rare disease, data regarding prevalence in the craniofacial skeleton is inconsistent, with reports ranging from 10-25% in monostotic disease and as high as 90% in polyostotic disease (Ricalde et al., 2012). In many reports, the sample size is small, and it is unclear if the subjects had thorough skeletal/endocrine screening to determine the extent of skeletal and extraskeletal disease. Many cases of monostotic disease are discovered incidentally on dental radiographic exam in the second decade of life. The most common location of monostotic disease is the zygomatico-maxillary complex (ZMC) (Valentini et al., 2009). The most typical monostotic presentation is a painless, slow-growing, unilateral swelling in the affected area (Figure 3A, B). When growth is very rapid, or presents in conjunction with pain or paresthesia, other diagnoses should be considered and a biopsy is recommended. FD lesions will often cross suture lines and involve adjacent bones, such as the sphenoid, temporal, frontal, occipital and parietal bones.
Figure 3. Craniofacial monostotic fibrous dysplasia.
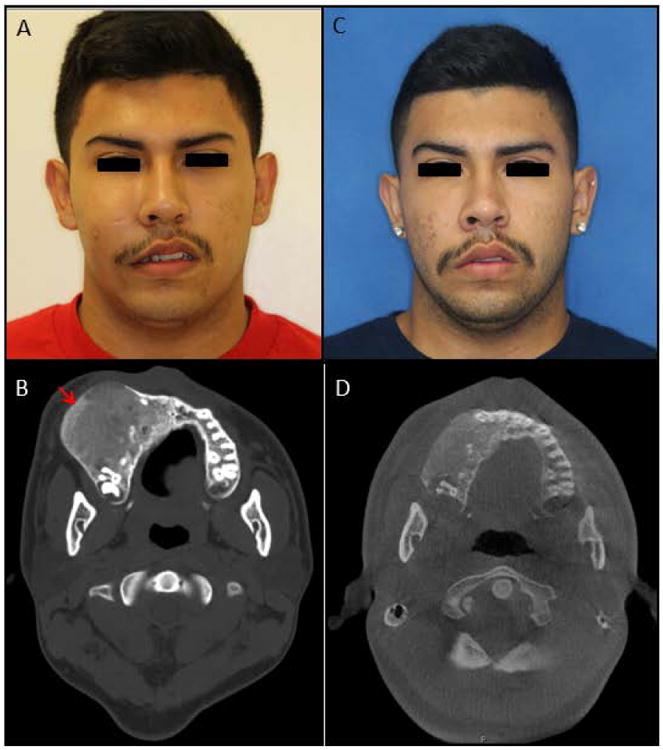
Patient with craniofacial monostotic fibrous dysplasia of the right maxilla. (A & B) Pre-operative photograph and axial computed tomography (CT) scan showing expansion of the right maxilla. The CT is remarkable for characteristic ground-glass appearance of lesion, extending from the alveolar bone (red arrow). (C & D) Post-operative photograph and axial CT scan after recontouring and debulking of the right maxillary lesion. Note the improved facial symmetry and contour of the right maxillary alveolus.
FD lesions typically manifest during the first few years of life, and expand during childhood and adolescence. Clinically significant bone lesions are usually apparent by age 5 years, with almost no significant lesions appearing after age 15 (Hart et al., 2007). FD lesions may become less active in adulthood, which may be related to apoptosis of mutation-bearing BMSCs (Kuznetsov et al., 2008). Craniofacial FD lesions are some of the earliest to be detected, but can remain “silent” until deformity or growth occurs. In a study reporting degree of skeletal burden, 90% of craniofacial FD lesions were detected by age 3.4 years (Hart et al., 2007). The involvement of the jaw bones and dental structures in FD has been previously described (Akintoye et al., 2013, Akintoye et al., 2003). Akintoye reported that 27 out of 36 patients (84%) had craniofacial FD involving the jaws, and 31% had disease in both the maxilla and mandible (Akintoye et al., 2003). The alveolar ridge cortices may expand, and there may be involvement of the hard palate. Teeth may become displaced as the lesion grows, while the arch form typically maintains its characteristic shape (Figure 3B).
Radiographically, the characteristic “ground glass” appearance, of mixed radiolucency/opacity, may be seen – this is a result of woven or abnormal bone superimposed on a fibrous tissue matrix. Typical features include cortical thinning and endosteal scalloping (Figure 4B). The radiographic appearance of FD varies by both age and location. Prior studies have shown that the radiographic appearance of FD will change over time: the homogeneous “ground-glass” appearance is most common during childhood and adolescence, whereas the lesions become less radiolucent, more mixed and heterogeneous with age; older subjects and those treated with bisphosphonates have been noted to have radiographically sclerotic lesions. (Figure 4 C,D) (Lee et al., 2012).
Figure 4.
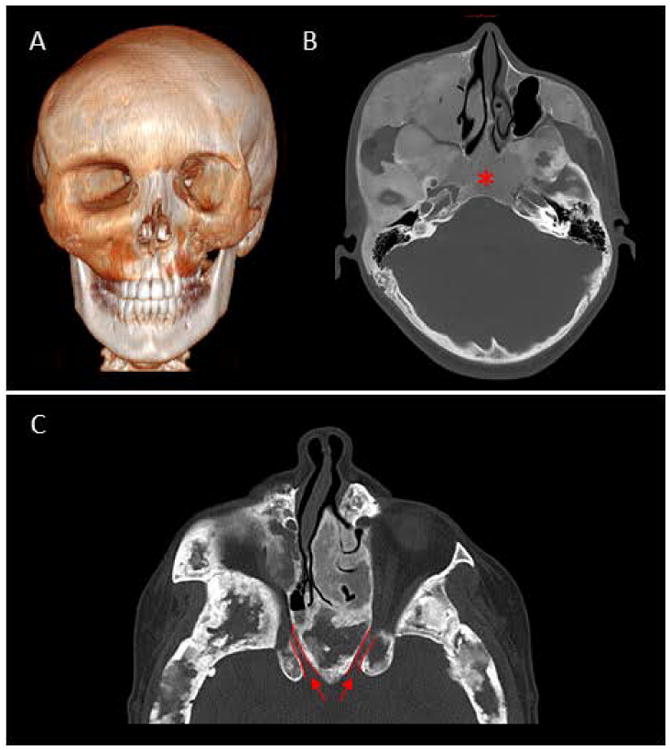
Craniofacial polyostotic fibrous dysplasia.
Imaging from two individuals with polyostotic craniofacial fibrous dysplasia (FD) in the setting of McCune-Albright Syndrome.
(A) 3D CT of a 17 year-old male, showing extensive disease of the midface and orbital asymmetry.
(B) Axial CT scan showing typical ground-glass appearance of the left midface and skull base, obliterating the region of the sphenoid sinuses (asterisk).
(C) Axial CT from a 33 year-old female showing the narrowing of the bilateral optic canals (outlined in red).
On imaging studies, craniofacial FD lesions are not well-demarcated, often crossing bony sutures, but not breaching the bony cortices. A CT scan can help to delineate the specific nature of the fibrous dysplasia lesion and surrounding structures. One of the most characteristic radiographic features of FD is the involvement of the sphenoid bones and the skull base. In cases of sphenoid involvement, the orbital canals should be evaluated for narrowing (Figure 4D).
The phosphaturic hormone fibroblast growth factor-23 (FGF23) is overproduced by FD cells, resulting in increased urinary phosphate excretion (Riminucci et al., 2003a). In patients with high skeletal disease burden, this may result in chronic hypophosphatemia and rickets/osteomalacia (Boyce et al., 2013a). Uncontrolled hypophosphatemia has been associated with increased risk of fracture, pain, and deformity in patients with FD (Leet et al., 2004a).
Clinical Presentation: Extraskeletal Features
Similar to FD lesions, the extraskeletal manifestations of MAS also reflect its underlying mosaicism. An individual may present with any combination of features, which may occur in isolation or in association with skeletal disease. Café-au-lait skin macules are generally the first manifestation of FD/MAS, presenting at or shortly after birth (Figure 5A). These lesions have characteristic jagged borders and tend to distribute along the midline of the body, reflecting patterns of embryonic cell migration. Endocrinopathies typically become apparent during childhood and persist into adulthood. Precocious puberty is the most common presenting symptom in girls, affecting nearly 80% of patients with MAS (Collins et al., 2012). Autonomously functioning ovarian cysts result in episodic estradiol production, which leads to recurrent vaginal bleeding episodes, skeletal maturation, and ultimately reduction in final adult height due to premature closure of the growth plates. Testicular involvement occurs in the majority of boys with FD/MAS, but precocious puberty occurs infrequently (Boyce et al., 2012a). Thyroid lesions are a common manifestation affecting 2/3 of patients, approximately half of whom develop hyperthyroidism (Figure 5B) (Boyce et al., 2012a). Pituitary involvement results in growth hormone excess in approximately 10-20% of patients, and appears to occur exclusively in individuals with comorbid skull base FD (Salenave et al., 2014). Hypercortisolism is a rare endocrinopathy that occurs exclusively in the neonatal period, resulting from cortisol overproduction by the fetal adrenal gland (Brown et al., 2010).
Figure 5. Extraskeletal findings.
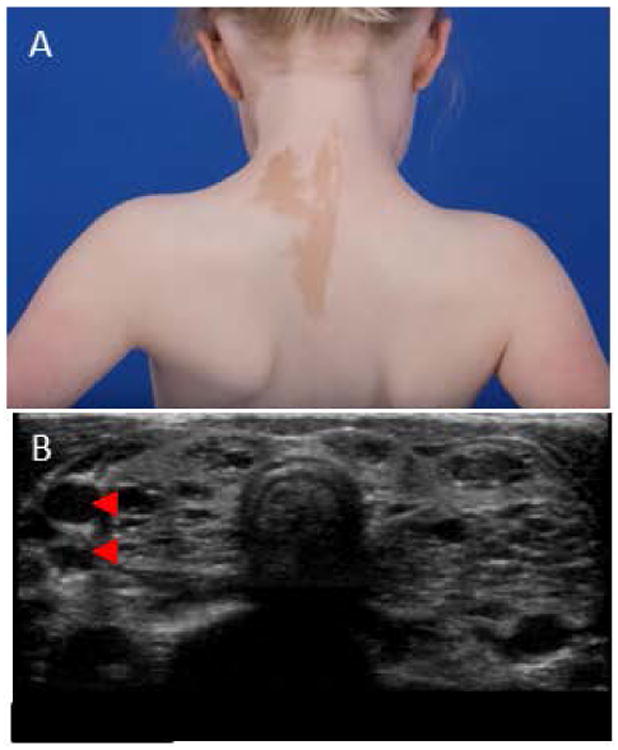
(A) Characteristic café-au-lait macule seen in McCune-Albright Syndrome. The borders are commonly referred to as resembling the “coast of Maine,” and the lesion typically respects the midline, following the lines of cellular migration (lines of Blaschko).
(B) Thyroid ultrasound (transverse view) showing multilocular hypoechoic lesions (arrowheads), common in McCune-Albright Syndrome
Rarely (in less than 1% of cases), the activating GNAS mutations (also referred to as “gsp” mutations) are thought to be responsible for a small increased risk of malignant transformation. Certain cancers have been reported in association with FD/MAS, including osteosarcoma, thyroid cancer, breast and testicular cancers (Ruggieri et al., 1994, Collins et al., 2003, Tanabeu et al., 1998, Huston and Simmons, 2004, Boyce et al., 2012a). In addition, the GNAS mutation has been found to play a role in the mutations responsible for precancerous pancreatic intraductal papillary mucinous neoplasms (Wu et al., 2011, Gaujoux et al., 2014, Parvanescu et al., 2014).
Diagnosis
The diagnosis of FD is based on a combination of clinical, radiographic, pathologic, and molecular information. The diagnosis of MAS is based on the presence of either polyostotic bone disease or the presence two or more clinical features of the syndrome (Boyce and Collins, 1993). In individuals whose only clinical feature is monostotic FD, biopsy and identification of a somatic GNAS mutation may be required to confirm the diagnosis. However, many experienced clinicians and radiologists will often note that a lesion appears “characteristic of FD” on radiographic exam, based on the ground-glass appearance, thus forming a diagnosis without the need for biopsy. The presence of FD or any features of MAS should prompt a comprehensive screening evaluation to identify any additional manifestations. This should include, at minimum, bone scintigraphy to identify polyostotic disease, biochemistries, and thyroid and gonadal ultrasonography to identify endocrine involvement. Detailed screening guidelines are included in reference (Boyce and Collins, 1993). Having more than one bony lesion, a café-au-lait spot, or endocrine involvement supports the diagnosis of FD (and MAS), thus eliminating the need to perform a bone biopsy.
Diagnosis can be made by histologic examination of the biopsied affected tissue; however, the diagnosis can be confounded by sampling error. The typical microscopic features of FD include a background of loosely arranged fibrous stroma, with irregularly-shaped bony trabeculae (woven bone) (Figure 6) (Riminucci et al., 1997). The trabeculae are often not connected to each other and have historically been described as “Chinese characters.” Osteoblastic rimming may or may not be seen along the trabeculae, with growing collagen (Sharpey's fibers) forming perpendicular to the sites of bone formation (Riminucci et al., 1999). Lamellar bone is not often seen in the areas of de novo bone formation within the lesion (Neville, 2009). No demarcation is noted where the FD and “normal” bone intersect – a distinction between FD and ossifying fibroma. Previous reports have suggested that FD in the skull and jaw bones tends to be more mature and ossified compared to the axial/appendicular skeleton – however, many of these studies have small sample sizes and have not been reliably reproduced (Neville, 2009, Riminucci et al., 1999).
Figure 6. Histology craniofacial fibrous dysplasia.
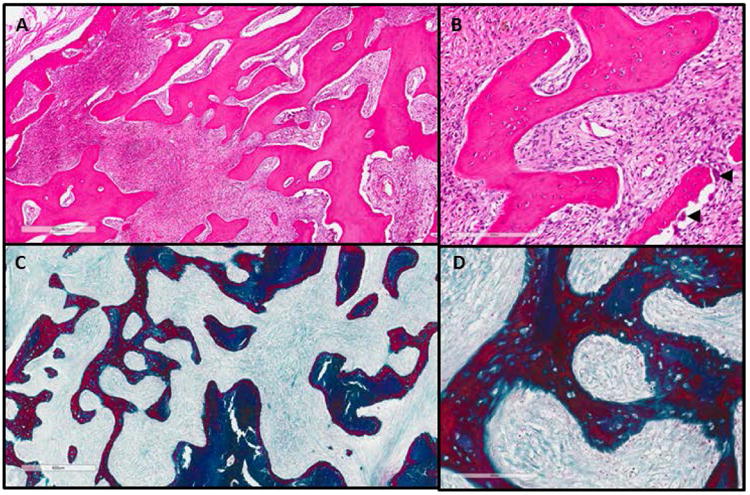
(A) H&E stain at 4× and (B) 20× of craniofacial fibrous dysplasia lesion. The boney matrix has been replaced by proliferating soft tissue. Osteoclasts are present in the lower panel of (B), arrowheads. (C) Masson trichrome stain from the same lesion at 4× and (D) 20×. (Dark blue = bone; Red = osteoid/woven bone; Light blue = fibrous tissue)
While GNAS mutation testing is considered the “gold standard” for FD diagnosis, testing for GNAS mutations may be confounded by the sensitivity of technique used and the level of mosaicism in the affected tissue (Boyce and Collins, 1993). Because of the somatic mosaic nature of the disease, standard polymerase chain reaction (PCR) techniques have limited utility for testing mutations in the affected blood and skin (Lumbroso et al., 2004). Alternate techniques such as modified primers (peptide nucleic acid) (Bianco et al., 2000) and next generation sequencing (Narumi et al., 2013) may have higher sensitivity. Further complicating matters is the fact that mutation levels can be variable and may change over time – with older individuals having fewer mutation-bearing cells (Riminucci et al., 1999).
Differential Diagnosis of Craniofacial Fibrous Dysplasia
FD falls under the umbrella of “benign fibro-osseous lesions” (BFOL) of the jaws, whose other members include ossifying fibromas, cemento-ossifying fibromas, cemento-osseous dysplasias, giant cell tumors, aneurysmal bone cysts, and simple bone cysts (Table 1). These lesions are grouped together because of their common histologic findings that include hyperproliferative fibrous material admixed with bony structures, and some elements of woven (irregular) bone (Neville, 2009). The clinical and radiographic appearances of these lesions also have substantial overlap. The correct diagnosis is important because it will help dictate treatment, and the current treatment paradigms vary depending on the diagnosis: for instance, most texts state that ossifying fibomas require complete excision and curettage, but FD and cement-ossifying fibroma may be observed and monitored in certain cases (Neville, 2009, Fletcher et al., 2013).
Table 1. Differential Diagnosis of Craniofacial Fibrous Dysplasia.
| Bone Diseases | Ossifying Fibroma |
| (Cemento-) Osseous Dysplasia | |
| Giant Cell Tumor/ Cherubism | |
| Aneurysmal Bone Cyst | |
| Simple (Idiopathic) Bone Cyst | |
| Osteosarcoma | |
| Osteoma/Cementoma | |
| Pagetoid Disease | |
| Langerhan's Cell Histiocytosis | |
| Odontogenic Diseases | Ameloblastoma |
| Ameloblastic Fibro-odontoma | |
| Adenomatoid odontogenic tumor | |
| Calcifying epithelial odontogenic tumor | |
| Inflammatory Process | Sclerosing osteomyelitis |
| Metabolic Bone Disease | Hyperparathyroidism (Brown's Tumor) |
Other pathologic lesions, such as aneurysmal bone cysts, mucoceles, or malignant degeneration should be considered when patients present with rapid growth, cortical expansion, severe pain, paresthesia, or displacement of adjacent structures (Lee et al., 2012). In the case of an aneurysmal bone cyst, these lesions can grow within the FD, and require surgical intervention.
FD lesions carry a very small risk (∼1%) of sarcomatous transformation (Qu et al., 2015, Ruggieri et al., 1994, Yabut et al., 1988, Sadeghi and Hosseini, 2011). As a result, the differential diagnosis of FD must include malignant neoplastic lesions, such as osteosarcomas, osteoblastomas, or metastatic bone disease. In the jaws, where there is overlap between dental and bony structures, the differential diagnosis can become even more extensive, including dental-specific pathologies, such as ameloblastoma or calcifying epithelial odontogenic tumor (Neville, 2009). Hyperparathyroidism, though rare, may also be mistaken and present as a fibro-osseous lesion (Brown tumor). Lesions of infectious origin, such as chronic sclerosing osteomyelitis must also be considered (though these pathoses tend to be a diagnosis of exclusion).
Craniofacial and Dental Specific Findings
About 90% of patients with MAS have craniofacial FD involvement, which may involve the maxilla and mandible (Ricalde et al., 2012). FD in the craniofacial skeleton can cause significant expansion of the bones, with varying degrees of facial asymmetry and disfigurement. In some cases, FD can cause displacement of structures such as the orbits, leading to dystopia and orbital canal involvement that can result in visual disturbances. The external or internal auditory canals, as well as the ossicles, may be involved, which can lead to hearing impairment (Frisch et al., 2015). Nasal congestion can also occur, from FD involvement of the turbinates and nasal obstruction.
Craniofacial FD can affect the alveolar bones of the maxilla and mandible, subsequently affecting occlusion and teeth (Figure 7A). Furthermore, since a significant proportion of craniofacial FD patients may experience bone pain, it is important to rule out odontogenic pathology. The maxillary sinuses may be obliterated or diminutive if the FD affects the maxillary bones, appearing as a radiopacity on panoramic radiograph (Figure 7B). Teeth may or may not be displaced or rotated; roots will often show splaying around a FD lesion (Figure 8). The most reported dental anomaly in FD patients has been malocclusion and dental crowding or spacing – this is secondary to the alveolar bone expanding at a rapid rate, causing improper positioning of the dentition.
Figure 7. Fibrous dysplasia of the jaws.
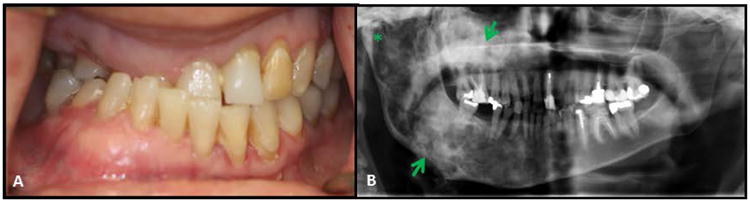
(A) Significant malocclusion in a patient with craniofacial FD. This patient has a right unilateral crossbite, dental crowding and rotation of teeth.
(B) Panoramic radiograph of the same patient showing the right maxilla and mandible (arrows) affected by fibrous dysplasia. The right mandibular condyle is also affected (asterisk). The left side is unaffected by the disease.
Figure 8. Dental anomalies in fibrous dysplasia.
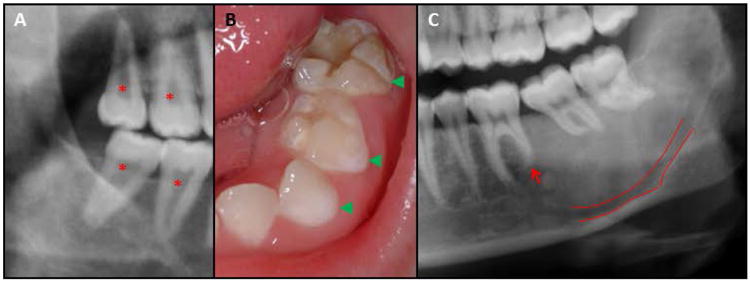
Dental anomalies can be seen when fibrous dysplasia (FD) affects the jaws. (A) Taurodontism and fused pulp chambers (asterisk) seen in the molars of a FD patient. (B) Hypomineralization of primary dentition (arrow heads). (C) Mandibular FD lesion causing splaying and dilaceration of the molar roots, and blunting of the distal root of the first molar (arrow). The inferior alveolar nerve is also displaced inferiorly (green outline).
Akintoye (Akintoye et al., 2003) performed a retrospective study analyzing 32 patients with craniofacial FD/MAS in the NIH natural history study. Dental attrition, enamel hypomineralization and hypoplasia were reported; however, these findings were not significantly correlated with the other clinical findings of FD/MAS. Other described dental anomalies include dentin dysplasia, taurodontic pulp chambers, odontomas, and high caries index (Figure 8) (Akintoye et al., 2003). This study also noted that there were no reported changes in the patients' craniofacial FD after dental procedures, including orthodontic treatment, exodontia, biopsy, and dental restorations (Akintoye et al., 2003).
For teeth within FD lesions, the periodontal ligament can be difficult to visualize on periapical radiographs, with an ill-defined lamina dura (Neville, 2009). The Inferior Alveolar Nerve (IAN) canal may also be challenging to identify, as the structure itself may be displaced (Figure 8C). A conebeam CT scan will assist in detecting vital structures in the event that surgery is needed.
Taurodontism can also be seen on dental radiographs, and may be associated with endocrine disorders, such as growth hormone excess (Figure 8A) (Akintoye et al., 2003). Thus, the metabolic dysfunction seen in MAS can have a potential effect on tooth development and eruption sequence. However, there is no current evidence that FD in the maxilla or mandible has an effect on tooth development and/or function (Akintoye et al., 2013).
Clinical Management and Treatment
Clinical management of FD/MAS is best performed by a team approach, depending on the needs of the individual patient. Identifying and treating extraskeletal disease is an essential component of management. Endocrinopathies can generally be managed medically. Precocious puberty in girls may be treated with the aromatase inhibitor, letrozole (Feuillan et al., 2007), while boys generally require the use of both an aromatase inhibitor and a testosterone receptor antagonist (Boyce et al., 2012a). Patients with growth hormone excess generally respond to somatostatin analogues and/or the growth hormone receptor antagonist, pegvisomant, which may be used alone in combination (Akintoye et al., 2006, Salenave et al., 2014). Similar to other disorders of FGF23 excess, hypophosphatemia is managed by supplementation with phosphorus and vitamin D analogues (Boyce et al., 2013a, Boyce and Collins, 1993).
Currently, there are no medical therapies of proven efficacy to treat FD. Approximately 40% of patients with craniofacial FD present with pain, which is often treated with bisphosphonates (Collins et al., 2005, Kelly et al., 2008). Bisphosphonates are medications that decrease bone turnover by inhibiting the activity of bone-resorbing osteoclasts, and are commonly used to treat osteoporosis and skeletal metastases. Bisphosphonates have been advocated as a potential treatment for FD based upon the high levels of osteoclastogenesis present in some FD lesions (Riminucci et al., 2003b). Early reports showed beneficial effects on bone pain, but inconsistent effects on FD radiographic appearance (Plotkin et al., 2003, Liens et al., 1994, Chapurlat et al., 1997). A placebo-controlled trial of the oral bisphosphonate, alendronate, did not show effects on pain, radiographic appearance, or skeletal disease burden (Boyce et al., 2014). Of concern, recent evidence establishes that patients with FD are at risk for osteonecrosis of the jaw (ONJ) as a consequence of bisphosphonate therapy (Metwally et al., 2016). Based on the current literature, intravenous bisphosphonates likely have utility in treatment of FD-related bone pain, however they should be used at the lowest dose and interval needed so as to minimize the risk of ONJ.
The orthopedic goals for managing skeletal FD lesions include optimizing function and decreasing morbidity with regard to bone pain, fractures and deformity. Conservative techniques are recommended, particularly in the pediatric population. It is important that any underlying endocrine abnormalities be treated prior to surgical intervention. Additionally, physical therapy is integral to maintain strength and range-of-motion. Orthopedic devices are used to correct limb-length discrepancies while promoting function (Paul et al., 2014). Bone grafting in the axial/appendicular skeleton has been shown to have high rates of graft resorption, particularly in patients less than 18 years (Leet et al., 2016, Leet and Collins, 2007). Intramedullary devices are preferred over plates and screws in the lower extremities. Scoliosis can become severe, and is often managed with standard fusion techniques (Leet and Collins, 2007, Mancini et al., 2009).
Craniofacial and Dental Management
The goals of management in craniofacial FD largely revolve around the improvement of function and aesthetics. Management is based primarily on the age of the patient, the extent of bony disease and the clinical behavior of the lesion. Lesions can be characterized as: quiescent (stable, no growth), non-aggressive (slow growth), or aggressive (rapid growth with or without pain, paresthesia) (Lee et al., 2012). Treatment includes observation with close monitoring, consisting of clinical examination, patient photographs, sensory nerve testing and periodic maxillofacial CT imaging. Routine audiologic testing and ophthalmologic exams should be performed in those individuals with evidence of external/internal auditory canal or orbital involvement. Biopsy is indicated if the diagnosis is questionable, or if there is concern for aggressive growth and/or atypical/unusual clinical behavior.
Surgery remains the cornerstone of treatment for craniofacial FD. Conservative debulking and recontouring procedures are frequently performed, but can be complicated by post-operative regrowth of the lesion (Figure 3C, D). We have previously shown that 68% of operations exhibited regrowth, particularly in those patients with untreated growth hormone excess (Boyce et al., 2016). For this reason, we strongly suggest that all underlying endocrinopathies be screened for and treated prior to surgery To date, we still do not have a good understanding of why some FD lesions exhibit a pattern of postoperative regrowth, nor are we able to predict its recurrence. Studies of craniofacial FD surgery have shown that biopsies are not associated with regrowth and recurrence, as compared to more extensive surgical procedures (Boyce et al., 2016). In an effort to combat recurrence, many elective procedures are postponed until after skeletal maturity, when the FD lesions tend to become more quiescent. Furthermore, while resection/reconstruction procedures have increased morbidity, they appear to have less rate of recurrence than recontouring procedures, 45% versus 82%, respectively (Boyce et al., 2016).
Special consideration is given to optic neuropathy in the setting of FD, one of the most dreaded complications of the disease, albeit rare. Prophylactic optic nerve decompression is no longer advocated to treat FD of the optic canal, and is noted to increase the risk of vision loss (Lee et al., 2002). Optic nerve decompression surgery has high peri-operative morbidity. Therefore, we prefer close observation (routine ophthalmologic exams with ocular coherence tomography (OCT) and evoked potentials, medical treatment of growth hormone excess when present) as the favored approach to patients with FD-encasement of the optic canal (Burke et al., 2016). A meta-analysis from the NIH patient cohort revealed that surgery in asymptomatic patients is associated with worse prognosis, as compared to patients managed expectantly (Amit et al., 2011). Visual loss occurred in growth hormone excess patients who delayed treatment until adulthood; none of the growth hormone excess subjects that were treated in childhood had visual disturbances (Boyce et al., 2013b).
Hearing loss has also been associated with FD, and is thought to be a result from stenosis of the external auditory canal, secondary to overgrowth of temporal bones (Morrissey et al., 1997, Megerian et al., 1995). An abnormal tympanogram is the most common audiologic finding in FD. While conductive hearing loss is more common, sensorineural and mixed hearing loss have also been reported in FD patients (Megerian et al., 1995, Frisch et al., 2015). Using the Coll(2.3)+/Rs1+ mouse model, which resembles the FD bone phenotype, Akil et al. explored the etiology of FD-associated hearing loss: they noted that the cochlea was largely spared from the bone disease and cited that the severity of hearing loss correlated with the deregulation of bone remodeling factors, including RANKL, SOST, MMP-13 and TRAP (Akil et al., 2014). In addition to hearing loss, FD patients can develop ear canal cholesteatomas which can lead to bone destruction and hearing loss (Megerian et al., 1995).
Craniofacial surgeries should be planned utilizing a team approach, including oral and maxillofacial/craniofacial surgeons, plastic and reconstructive surgeons, otolaryngology, neurosurgery, pediatric dentists, orthodontists, speech and language pathologists, and nursing specialists. Minimizing operating time, particularly in pediatric populations, is important with regard to timing and morbidity. The use of 3-D treatment planning methods has helped to greatly decrease operative time in such complex patient populations.
There is a paucity of data regarding dental therapies in patients with FD/MAS. Due to the complex co-morbidities associated with FD/MAS, the dental needs can sometimes be overlooked. Variable presentations cause some dental practitioners to delay or avoid dental procedures. Dental procedures are safe to perform in patients with FD and do not adversely affect the course of the disease (Akintoye et al., 2003).
Patients with craniofacial FD have an observed predisposition to malocclusion (Akintoye et al., 2013), often requiring them to need orthodontic treatment. Previous reports have indicated that orthodontic treatment in FD bone takes more time, as compared to the typical patient population, and as a result should be delayed until skeletal maturity (Akintoye et al., 2013). Anecdotally, our experience has been that teeth move more quickly in FD-involved bone and that there is a tendency to have orthodontic relapse; however, data to support this are currently lacking. The use of bonded retainers can often help with the risk of relapse. Currently, we assure patients and families that there are no absolute contraindications to orthodontic treatment in FD.
Orthognathic surgery (jaw repositioning surgery), in combination with orthodontic therapy, can also be performed in FD patients (Yeow and Chen, 1999, Cheung et al., 1995). Many of these procedures also involve conservative recountouring and debulking of the FD bone. In a study performed by Cheung et al., 4 FD patients underwent recontouring and osteotomies of the jaws, with titanium plate/screw fixation. The titanium plates were sectioned and the screws were removed en bloc at re-operation (between 9 and 32 months following initial surgery). In comparison to normal controls, there was no difference in bone-titanium surface area contact, healing or inflammation (Cheung et al., 1995). Yeow and colleagues noted that osteotomies and maxillary downfracture should be performed with care, particularly in cases where the maxillary sinuses have been obliterated and there is extensive skull base disease (Yeow and Chen, 1999).
It has previously been recommended that FD patients have more frequent recall visits for dental hygiene, secondary to the higher caries index (Akintoye et al., 2003). Prescribers are advised to recommend topical fluoride application and electric toothbrushes to aid this patient population. It is important for patients to have routine dental evaluations to prevent the need for extensive dental procedures, and to minimize oral inflammation – two factors that have been implicated in osteonecrosis of the jaws. Regarding endodontic treatment, FD patients can safely undergo root canal therapy. Concern may be raised in those patients with MAS-related endocrinopathies, which can cause taurodontic pulp chambers and pulpal pathology (Akintoye et al., 2013).
Oral and maxillofacial surgeons will routinely evaluate FD patients. It is crucial that craniofacial FD patients be placed on long-term follow-up to monitor for lesion growth and progression. Maxillofacial CT scans should be performed periodically while the patient is growing or sooner if clinically indicated. Pain and paresthesia (secondary to nerve compression) are concerning symptoms which could indicate malignant transformation or another process, such as an aneurysmal bone cyst. Radiographic features with irregular borders or cortical perforation should also be suspicious for a secondary process. Such patients may require a repeat biopsy or more extensive procedure to rule-out malignant degeneration. The oral surgeon should also be cognizant when performing procedures on patients who have a history of bisphosphonates, anti-osteoclastic (denosumab), anti-neoplastic or other anti-angiogenic drugs. Patients with MAS-related endocrinopathies, such as hyperthyroidism, or growth-hormone excess, should be treated in conjunction with the patient's endocrinologist.
Though there is a lack of literature and long-term data, dental implants have been use to replace dentition in FD bone. Bajwa et al. (Bajwa et al., 2008) reported the case of a 32 year-old female with FD/MAS who underwent successful osseointegration and loading of dental implants in the maxilla and mandible. Her lesions were reportedly quiescent for 3 years prior to procedure, and the dental implants were at least 15 mm in length; the patient was noted to be functional at 5 years – the longest follow-up reported to date. Based upon the literature, there is an unclear risk of implant failure, with some reports stating that the bone healing and integration may occur more slowly secondary to the quality of the bone. We recommend that implants be placed after skeletal maturity and once the growth of the FD lesion has abated.
Future Directions
Currently, there are no definitive medical treatments for FD. One approach that holds promise for the future is skeletal stem cell/bone marrow stromal cell transplantation; however more translational research is needed before such applications can be implemented (Bianco et al., 2013). The report of denosumab treatment in FD patients (Boyce et al., 2012b, Benhamou et al., 2014) has questioned the role of RANK-L inhibition in the treatment of FD. Denosumab, a humanized monoclonal antibody to RANKL, is currently approved in the treatment of osteoporosis, giant cell tumors of the long bones, and prevention of skeletal-related events from bone metastases (Thomas et al., 2010). Denosumab inhibits RANKL binding to its receptor, RANK, thereby preventing the activation and function of osteoclasts (Xu et al., 2013). The overexpression of RANKL has been reported in FD-like bone cells (Piersanti et al., 2010) and FD tissue (Wang et al., 2014), which suggests that it may play a role in the pathophysiology of FD. In one patient, denosumab was associated with hypophosphatemia and secondary hyperparathyroidism while on treatment, and severe, life-threatening hypercalcemia on treatment discontinuation (Boyce et al., 2012b). The etiology of these reactions in unclear, however rebound hypercalcemia post-denosumab discontinuation has been reported in association with giant cell tumors (Gossai et al., 2015) and other conditions of high bone turnover (Grasemann et al., 2013). Additional studies are needed to determine the safety and efficacy of denosumab treatment in FD patients.
Multiple animal models have been used to study FD, but not met with success. Initial studies by Bianco et al. used the murine transplantation model to successfully reproduce the FD phenotype in dysplastic ossicles, noting that both wild-type and mutant cells were required to support FD lesions (Bianco et al., 1998). As referenced above, the Coll (2.3)+/Rs1+ mouse model, with constitutive Gs signaling, was created by Hsiao and colleagues, replicating the histologic and phenotypic features of FD (Hsiao et al., 2008, Hsiao et al., 2010). Although this model does not effectively reproduce FD's mosaic nature, and the phenotype could be reversed by suppression of Rs1, it does support the notion that Gs signaling is age-dependent on bone formation. In another model, Saggio et al. developed an inherited, histopathologic replica of human FD by generating mice using lentiviral constructs and constitutive promoters that express the mutation globally (Saggio et al., 2014). FD lesions developed postnatally, progressing temporally through three histopathologic stages: the first, a modeling phase of endosteal/medullary excessive bone formation; the second, with aberrant bone remodeling; and the third, developing into mature fibrous dysplasia in mice greater than 1 year of age. A second mouse model was subsequently developed by Remoli et al., using the 2.3kb Col1a1 promoter to express Gsα R201C in mature osteoblasts. Despite developing postnatally and exhibiting the high bone mass phenotype, this mouse phenotype (similar to the Hsiao mouse model) did not represent the other characteristic pathologic changes seen in FD, such as marrow fibrosis, loss of hematopoietic tissue, osteomalacia, and osteolytic changes (Remoli et al., 2015). This model supports the notion that the FD-related tissue changes result from cells other than mature osteoblasts. Finally, in ongoing studies that target Gsα activity, a small molecule library of 343,786 compounds was screened to identify modulators of Gsα activity (Bhattacharyya et al., 2014). Compounds were screened for inhibitory activity against the mutant Gsα protein and clustered for further testing. These molecules may have significant future therapeutic potential.
Conclusions
The FD/MAS patient population is complex and diverse, with unique needs that span multiple medical and dental specialties. It is crucial for FD/MAS patients to be treated by a team, including physicians, surgeons, and dental specialists. The initial findings of FD/MAS may be found incidentally by the dental practitioner on routine examination. It is important to be able to recognize the characteristic signs of FD/MAS, in addition to knowing how to refer the patient for an appropriate workup. It is also imperative to understand the implications of MAS-related endocrinopathies, such as growth hormone excess, and how these conditions can inform treatment decisions. For MAS/FD, in particular, the dental professional must learn to work as a team with his or her medical colleagues to develop appropriate treatment strategies.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NIDCR.
References
- Akil O, Hall-Glenn F, Chang J, Li A, Chang W, Lustig LR, Alliston T, Hsiao EC. Disrupted bone remodeling leads to cochlear overgrowth and hearing loss in a mouse model of fibrous dysplasia. PLoS One. 2014;9:e94989. doi: 10.1371/journal.pone.0094989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akintoye SO, Boyce AM, Collins MT. Dental perspectives in fibrous dysplasia and McCune-Albright syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116:e149–55. doi: 10.1016/j.oooo.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akintoye SO, Kelly MH, Brillante B, Cherman N, Turner S, Butman JA, Robey PG, Collins MT. Pegvisomant for the treatment of gsp-mediated growth hormone excess in patients with McCune-Albright syndrome. J Clin Endocrinol Metab. 2006;91:2960–6. doi: 10.1210/jc.2005-2661. [DOI] [PubMed] [Google Scholar]
- Akintoye SO, Lee JS, Feimster T, Booher S, Brahim J, Kingman A, Riminucci M, Robey PG, Collins MT. Dental characteristics of fibrous dysplasia and McCune-Albright syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96:275–82. doi: 10.1016/s1079-2104(03)00225-7. [DOI] [PubMed] [Google Scholar]
- Amit M, Collins MT, Fitzgibbon EJ, Butman JA, Fliss DM, Gil Z. Surgery versus watchful waiting in patients with craniofacial fibrous dysplasia--a meta-analysis. PLoS One. 2011;6:e25179. doi: 10.1371/journal.pone.0025179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajwa MS, Ethunandan M, Flood TR. Oral rehabilitation with endosseous implants in a patient with fibrous dysplasia (McCune-Albright syndrome): a case report. J Oral Maxillofac Surg. 2008;66:2605–8. doi: 10.1016/j.joms.2007.06.669. [DOI] [PubMed] [Google Scholar]
- Benhamou J, Gensburger D, Chapurlat R. Transient improvement of severe pain from fibrous dysplasia of bone with denosumab treatment. Joint Bone Spine. 2014;81:549–50. doi: 10.1016/j.jbspin.2014.04.013. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya N, Hu X, Chen CZ, Mathews Griner LA, Zheng W, Inglese J, Austin CP, Marugan JJ, Southall N, Neumann S, Northup JK, Ferrer M, Collins MT. A high throughput screening assay system for the identification of small molecule inhibitors of gsp. PLoS One. 2014;9:e90766. doi: 10.1371/journal.pone.0090766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco P, Barker R, Brustle O, Cattaneo E, Clevers H, Daley GQ, De Luca M, Goldstein L, Lindvall O, Mummery C, Robey PG, Sattler De Sousa EBC, Smith A. Regulation of stem cell therapies under attack in Europe: for whom the bell tolls. EMBO J. 2013;32:1489–95. doi: 10.1038/emboj.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco P, Kuznetsov SA, Riminucci M, Fisher LW, Spiegel AM, Robey PG. Reproduction of human fibrous dysplasia of bone in immunocompromised mice by transplanted mosaics of normal and Gsalpha-mutated skeletal progenitor cells. J Clin Invest. 1998;101:1737–44. doi: 10.1172/JCI2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco P, Riminucci M, Majolagbe A, Kuznetsov SA, Collins MT, Mankani MH, Corsi A, Bone HG, Wientroub S, Spiegel AM, Fisher LW, Robey PG. Mutations of the GNAS1 gene, stromal cell dysfunction, and osteomalacic changes in non-McCune-Albright fibrous dysplasia of bone. J Bone Miner Res. 2000;15:120–8. doi: 10.1359/jbmr.2000.15.1.120. [DOI] [PubMed] [Google Scholar]
- Boyce AM, Bhattacharyya N, Collins MT. Fibrous dysplasia and fibroblast growth factor-23 regulation. Curr Osteoporos Rep. 2013a;11:65–71. doi: 10.1007/s11914-013-0144-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce AM, Burke A, Cutler Peck C, Dufresne CR, Lee JS, Collins MT. Surgical Management of Polyostotic Craniofacial Fibrous Dysplasia: Long-Term Outcomes and Predictors for Postoperative Regrowth. Plast Reconstr Surg. 2016;137:1833–9. doi: 10.1097/PRS.0000000000002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce AM, Chong WH, Shawker TH, Pinto PA, Linehan WM, Bhattacharryya N, Merino MJ, Singer FR, Collins MT. Characterization and management of testicular pathology in McCune-Albright syndrome. J Clin Endocrinol Metab. 2012a;97:E1782–90. doi: 10.1210/jc.2012-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce AM, Chong WH, Yao J, Gafni RI, Kelly MH, Chamberlain CE, Bassim C, Cherman N, Ellsworth M, Kasa-Vubu JZ, Farley FA, Molinolo AA, Bhattacharyya N, Collins MT. Denosumab treatment for fibrous dysplasia. J Bone Miner Res. 2012b;27:1462–70. doi: 10.1002/jbmr.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce AM, Collins MT. Fibrous Dysplasia/McCune-Albright Syndrome. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editors. GeneReviews(R) Seattle WA: University of Washington, Seattle; 1993. [Google Scholar]
- Boyce AM, Glover M, Kelly MH, Brillante BA, Butman JA, Fitzgibbon EJ, Brewer CC, Zalewski CK, Cutler Peck CM, Kim HJ, Collins MT. Optic neuropathy in McCune-Albright syndrome: effects of early diagnosis and treatment of growth hormone excess. J Clin Endocrinol Metab. 2013b;98:E126–34. doi: 10.1210/jc.2012-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce AM, Kelly MH, Brillante BA, Kushner H, Wientroub S, Riminucci M, Bianco P, Robey PG, Collins MT. A randomized, double blind, placebo-controlled trial of alendronate treatment for fibrous dysplasia of bone. J Clin Endocrinol Metab. 2014 doi: 10.1210/jc.2014-1371. jc20141371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RJ, Kelly MH, Collins MT. Cushing syndrome in the McCune-Albright syndrome. J Clin Endocrinol Metab. 2010;95:1508–15. doi: 10.1210/jc.2009-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke AB, Boyce AM, Collins MT. Fibrous Dysplasia: Management of the Optic Canal. Plast Reconstr Surg. 2016;137:1060e–1e. doi: 10.1097/PRS.0000000000002210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapurlat RD, Delmas PD, Liens D, Meunier PJ. Long-term effects of intravenous pamidronate in fibrous dysplasia of bone. J Bone Miner Res. 1997;12:1746–52. doi: 10.1359/jbmr.1997.12.10.1746. [DOI] [PubMed] [Google Scholar]
- Cheung LK, Samman N, Pang M, Tideman H. Titanium miniplate fixation for osteotomies in facial fibrous dysplasia--a histologic study of the screw/bone interface. Int J Oral Maxillofac Surg. 1995;24:401–5. doi: 10.1016/s0901-5027(05)80467-9. [DOI] [PubMed] [Google Scholar]
- Collins MT, Kushner H, Reynolds JC, Chebli C, Kelly MH, Gupta A, Brillante B, Leet AI, Riminucci M, Robey PG, Bianco P, Wientroub S, Chen CC. An instrument to measure skeletal burden and predict functional outcome in fibrous dysplasia of bone. J Bone Miner Res. 2005;20:219–26. doi: 10.1359/JBMR.041111. [DOI] [PubMed] [Google Scholar]
- Collins MT, Sarlis NJ, Merino MJ, Monroe J, Crawford SE, Krakoff JA, Guthrie LC, Bonat S, Robey PG, Shenker A. Thyroid carcinoma in the McCune-Albright syndrome: contributory role of activating Gs alpha mutations. J Clin Endocrinol Metab. 2003;88:4413–7. doi: 10.1210/jc.2002-021642. [DOI] [PubMed] [Google Scholar]
- Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis. 2012;7(Suppl 1):S4. doi: 10.1186/1750-1172-7-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuillan P, Calis K, Hill S, Shawker T, Robey PG, Collins MT. Letrozole treatment of precocious puberty in girls with the McCune-Albright syndrome: a pilot study. J Clin Endocrinol Metab. 2007;92:2100–6. doi: 10.1210/jc.2006-2350. [DOI] [PubMed] [Google Scholar]
- Fletcher CDM World Health Organization & International Agency for Research on Cancer. WHO classification of tumours of soft tissue and bone. Lyon: IARC Press; 2013. [Google Scholar]
- Frisch CD, Carlson ML, Kahue CN, Pelosi S, Haynes DS, Lane JI, Neff BA, Link MJ, Driscoll CL. Fibrous dysplasia of the temporal bone: a review of 66 cases. Laryngoscope. 2015;125:1438–43. doi: 10.1002/lary.25078. [DOI] [PubMed] [Google Scholar]
- Gaujoux S, Salenave S, Ronot M, Rangheard AS, Cros J, Belghiti J, Sauvanet A, Ruszniewski P, Chanson P. Hepatobiliary and Pancreatic neoplasms in patients with McCune-Albright syndrome. J Clin Endocrinol Metab. 2014;99:E97–101. doi: 10.1210/jc.2013-1823. [DOI] [PubMed] [Google Scholar]
- Gossai N, Hilgers MV, Polgreen LE, Greengard EG. Critical hypercalcemia following discontinuation of denosumab therapy for metastatic giant cell tumor of bone. Pediatr Blood Cancer. 2015;62:1078–80. doi: 10.1002/pbc.25393. [DOI] [PubMed] [Google Scholar]
- Grasemann C, Schundeln MM, Hovel M, Schweiger B, Bergmann C, Herrmann R, Wieczorek D, Zabel B, Wieland R, Hauffa BP. Effects of RANK-ligand antibody (denosumab) treatment on bone turnover markers in a girl with juvenile Paget's disease. J Clin Endocrinol Metab. 2013;98:3121–6. doi: 10.1210/jc.2013-1143. [DOI] [PubMed] [Google Scholar]
- Happle R. The McCune-Albright syndrome: a lethal gene surviving by mosaicism. Clin Genet. 1986;29:321–4. doi: 10.1111/j.1399-0004.1986.tb01261.x. [DOI] [PubMed] [Google Scholar]
- Hart ES, Kelly MH, Brillante B, Chen CC, Ziran N, Lee JS, Feuillan P, Leet AI, Kushner H, Robey PG, Collins MT. Onset, progression, and plateau of skeletal lesions in fibrous dysplasia and the relationship to functional outcome. J Bone Miner Res. 2007;22:1468–74. doi: 10.1359/jbmr.070511. [DOI] [PubMed] [Google Scholar]
- Hsiao EC, Boudignon BM, Chang WC, Bencsik M, Peng J, Nguyen TD, Manalac C, Halloran BP, Conklin BR, Nissenson RA. Osteoblast expression of an engineered Gs-coupled receptor dramatically increases bone mass. Proc Natl Acad Sci U S A. 2008;105:1209–14. doi: 10.1073/pnas.0707457105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EC, Boudignon BM, Halloran BP, Nissenson RA, Conklin BR. Gs G protein-coupled receptor signaling in osteoblasts elicits age-dependent effects on bone formation. J Bone Miner Res. 2010;25:584–93. doi: 10.1002/jbmr.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huston TL, SIMMONS RM. Ductal carcinoma in situ in a 27-year-old woman with McCune-Albright syndrome. Breast J. 2004;10:440–2. doi: 10.1111/j.1075-122X.2004.21490.x. [DOI] [PubMed] [Google Scholar]
- Idowu BD, Al-Adnani M, O'Donnell P, Yu L, Odell E, Diss T, Gale RE, Flanagan AM. A sensitive mutation-specific screening technique for GNAS1 mutations in cases of fibrous dysplasia: the first report of a codon 227 mutation in bone. Histopathology. 2007;50:691–704. doi: 10.1111/j.1365-2559.2007.02676.x. [DOI] [PubMed] [Google Scholar]
- Ippolito E, Farsetti P, Boyce AM, Corsi A, De Maio F, Collins MT. Radiographic classification of coronal plane femoral deformities in polyostotic fibrous dysplasia. Clin Orthop Relat Res. 2014;472:1558–67. doi: 10.1007/s11999-013-3380-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MH, Brillante B, Collins MT. Pain in fibrous dysplasia of bone: age-related changes and the anatomical distribution of skeletal lesions. Osteoporos Int. 2008;19:57–63. doi: 10.1007/s00198-007-0425-x. [DOI] [PubMed] [Google Scholar]
- Kuznetsov SA, Cherman N, Riminucci M, Collins MT, Robey PG, Bianco P. Age-dependent demise of GNAS-mutated skeletal stem cells and “normalization” of fibrous dysplasia of bone. J Bone Miner Res. 2008;23:1731–40. doi: 10.1359/jbmr.080609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Fitzgibbon E, Butman JA, Dufresne CR, Kushner H, Wientroub S, Robey PG, Collins MT. Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med. 2002;347:1670–6. doi: 10.1056/NEJMoa020742. [DOI] [PubMed] [Google Scholar]
- Lee JS, Fitzgibbon EJ, Chen YR, Kim HJ, Lustig LR, Akintoye SO, Collins MT, Kaban LB. Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis. 2012;7(Suppl 1):S2. doi: 10.1186/1750-1172-7-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leet AI, Boyce AM, Ibrahim KA, Wientroub S, Kushner H, Collins MT. Bone-Grafting in Polyostotic Fibrous Dysplasia. J Bone Joint Surg Am. 2016;98:211–9. doi: 10.2106/JBJS.O.00547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leet AI, Chebli C, Kushner H, Chen CC, Kelly MH, Brillante BA, Robey PG, Bianco P, Wientroub S, Collins MT. Fracture incidence in polyostotic fibrous dysplasia and the McCune-Albright syndrome. J Bone Miner Res. 2004a;19:571–7. doi: 10.1359/JBMR.0301262. [DOI] [PubMed] [Google Scholar]
- Leet AI, Collins MT. Current approach to fibrous dysplasia of bone and McCune-Albright syndrome. J Child Orthop. 2007;1:3–17. doi: 10.1007/s11832-007-0006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leet AI, Magur E, Lee JS, Wientroub S, Robey PG, Collins MT. Fibrous dysplasia in the spine: prevalence of lesions and association with scoliosis. J Bone Joint Surg Am. 2004b;86-A:531–7. [PubMed] [Google Scholar]
- Lichtenstein L. Polyostotic fibrous dysplasia. Archives of Surgery. 1938;36:874–898. [Google Scholar]
- Liens D, Delmas PD, Meunier PJ. Long-term effects of intravenous pamidronate in fibrous dysplasia of bone. Lancet. 1994;343:953–4. doi: 10.1016/s0140-6736(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Lumbroso S, Paris F, Sultan C European Collaborative, S. Activating Gsalpha mutations: analysis of 113 patients with signs of McCune-Albright syndrome--a European Collaborative Study. J Clin Endocrinol Metab. 2004;89:2107–13. doi: 10.1210/jc.2003-031225. [DOI] [PubMed] [Google Scholar]
- Mancini F, Corsi A, De Maio F, Riminucci M, Ippolito E. Scoliosis and spine involvement in fibrous dysplasia of bone. Eur Spine J. 2009;18:196–202. doi: 10.1007/s00586-008-0860-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megerian CA, Sofferman RA, Mckenna MJ, Eavey RD, Nadol JB., JR Fibrous dysplasia of the temporal bone: ten new cases demonstrating the spectrum of otologic sequelae. Am J Otol. 1995;16:408–19. [PubMed] [Google Scholar]
- Metwally T, Burke A, Tsai JY, Collins MT, Boyce AM. Fibrous Dysplasia and Medication-Related Osteonecrosis of the Jaw. J Oral Maxillofac Surg. 2016 doi: 10.1016/j.joms.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrissey DD, Talbot JM, Schleuning AJ., 2ND Fibrous dysplasia of the temporal bone: reversal of sensorineural hearing loss after decompression of the internal auditory canal. Laryngoscope. 1997;107:1336–40. doi: 10.1097/00005537-199710000-00008. [DOI] [PubMed] [Google Scholar]
- Narumi S, Matsuo K, Ishii T, Tanahashi Y, Hasegawa T. Quantitative and sensitive detection of GNAS mutations causing mccune-albright syndrome with next generation sequencing. PLoS One. 2013;8:e60525. doi: 10.1371/journal.pone.0060525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville BW. Oral and maxillofacial pathology. St Louis, Mo: Saunders/Elsevier; 2009. [Google Scholar]
- Parvanescu A, Cros J, Ronot M, Hentic O, Grybek V, Couvelard A, Levy P, Chanson P, Ruszniewski P, Sauvanet A, Gaujoux S. Lessons from McCune-Albright syndrome-associated intraductal papillary mucinous neoplasms: : GNAS-activating mutations in pancreatic carcinogenesis. JAMA Surg. 2014;149:858–62. doi: 10.1001/jamasurg.2014.535. [DOI] [PubMed] [Google Scholar]
- Paul SM, Gabor LR, Rudzinski S, Giovanni D, Boyce AM, Kelly MR, Collins MT. Disease severity and functional factors associated with walking performance in polyostotic fibrous dysplasia. Bone. 2014;60:41–7. doi: 10.1016/j.bone.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piersanti S, Remoli C, Saggio I, Funari A, Michienzi S, Sacchetti B, Robey PG, Riminucci M, Bianco P. Transfer, analysis, and reversion of the fibrous dysplasia cellular phenotype in human skeletal progenitors. J Bone Miner Res. 2010;25:1103–16. doi: 10.1359/jbmr.091036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin H, Rauch F, Zeitlin L, Munns C, Travers R, Glorieux FH. Effect of pamidronate treatment in children with polyostotic fibrous dysplasia of bone. J Clin Endocrinol Metab. 2003;88:4569–75. doi: 10.1210/jc.2003-030050. [DOI] [PubMed] [Google Scholar]
- Qu N, Yao W, Cui X, Zhang H. Malignant transformation in monostotic fibrous dysplasia: clinical features, imaging features, outcomes in 10 patients, and review. Medicine (Baltimore) 2015;94:e369. doi: 10.1097/MD.0000000000000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remoli C, Michienzi S, Sacchetti B, Consiglio AD, Cersosimo S, Spica E, Robey PG, Holmbeck K, Cumano A, Boyde A, Davis G, Saggio I, Riminucci M, Bianco P. Osteoblast-specific expression of the fibrous dysplasia (FD)-causing mutation Gsalpha(R201C) produces a high bone mass phenotype but does not reproduce FD in the mouse. J Bone Miner Res. 2015;30:1030–43. doi: 10.1002/jbmr.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricalde P, Magliocca KR, Lee JS. Craniofacial fibrous dysplasia. Oral Maxillofac Surg Clin North Am. 2012;24:427–41. doi: 10.1016/j.coms.2012.05.004. [DOI] [PubMed] [Google Scholar]
- Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, White KE, Waguespack S, Gupta A, Hannon T, Econs MJ, Bianco P, Gehron Robey P. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. 2003a;112:683–92. doi: 10.1172/JCI18399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riminucci M, Fisher LW, Shenker A, Spiegel AM, Bianco P, Gehron Robey P. Fibrous dysplasia of bone in the McCune-Albright syndrome: abnormalities in bone formation. Am J Pathol. 1997;151:1587–600. [PMC free article] [PubMed] [Google Scholar]
- Riminucci M, Kuznetsov SA, Cherman N, Corsi A, Bianco P, Gehron Robey P. Osteoclastogenesis in fibrous dysplasia of bone: in situ and in vitro analysis of IL-6 expression. Bone. 2003b;33:434–42. doi: 10.1016/s8756-3282(03)00064-4. [DOI] [PubMed] [Google Scholar]
- Riminucci M, Liu B, Corsi A, Shenker A, Spiegel AM, Robey PG, Bianco P. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site-specific patterns and recurrent histological hallmarks. J Pathol. 1999;187:249–58. doi: 10.1002/(SICI)1096-9896(199901)187:2<249::AID-PATH222>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Riminucci M, Saggio I, Robey PG, Bianco P. Fibrous dysplasia as a stem cell disease. J Bone Miner Res. 2006;21(Suppl 2):P125–31. doi: 10.1359/jbmr.06s224. [DOI] [PubMed] [Google Scholar]
- Ruggieri P, Sim FH, Bond JR, Unni KK. Malignancies in fibrous dysplasia. Cancer. 1994;73:1411–24. doi: 10.1002/1097-0142(19940301)73:5<1411::aid-cncr2820730516>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Sadeghi SM, Hosseini SN. Spontaneous conversion of fibrous dysplasia into osteosarcoma. J Craniofac Surg. 2011;22:959–61. doi: 10.1097/SCS.0b013e31820fe2bd. [DOI] [PubMed] [Google Scholar]
- Saggio I, Remoli C, Spica E, Cersosimo S, Sacchetti B, Robey PG, Holmbeck K, Cumano A, Boyde A, Bianco P, Riminucci M. Constitutive expression of Gsalpha(R201C) in mice produces a heritable, direct replica of human fibrous dysplasia bone pathology and demonstrates its natural history. J Bone Miner Res. 2014;29:2357–68. doi: 10.1002/jbmr.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salenave S, Boyce AM, Collins MT, Chanson P. Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab. 2014 doi: 10.1210/jc.2013-3826. jc20133826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabeu Y, Nakahara S, Mitsuyama S, Ono M, Toyoshima S. Breast Cancer in a Patient with McCune-Albright Syndrome. Breast Cancer. 1998;5:175–178. doi: 10.1007/BF02966691. [DOI] [PubMed] [Google Scholar]
- Thomas D, Henshaw R, Skubitz K, Chawla S, Staddon A, Blay JY, Roudier M, Smith J, Ye Z, Sohn W, Dansey R, Jun S. Denosumab in patients with giant-cell tumour of bone: an open-label, phase 2 study. Lancet Oncol. 2010;11:275–80. doi: 10.1016/S1470-2045(10)70010-3. [DOI] [PubMed] [Google Scholar]
- Valentini V, Cassoni A, Marianetti TM, Terenzi V, Fadda MT, Iannetti G. Craniomaxillofacial fibrous dysplasia: conservative treatment or radical surgery? A retrospective study on 68 patients. Plast Reconstr Surg. 2009;123:653–60. doi: 10.1097/PRS.0b013e318196bbbe. [DOI] [PubMed] [Google Scholar]
- Wang HD, Boyce AM, Tsai JY, Gafni RI, Farley FA, Kasa-Vubu JZ, Molinolo AA, Collins MT. Effects of denosumab treatment and discontinuation on human growth plates. J Clin Endocrinol Metab. 2014;99:891–7. doi: 10.1210/jc.2013-3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein LS, Liu J, Sakamoto A, Xie T, Chen M. Minireview: GNAS: normal and abnormal functions. Endocrinology. 2004;145:5459–64. doi: 10.1210/en.2004-0865. [DOI] [PubMed] [Google Scholar]
- Wu J, Matthaei H, Maitra A, Dal Molin M, Wood LD, Eshleman JR, Goggins M, Canto MI, Schulick RD, Edil BH, Wolfgang CL, Klein AP, Diaz LA, JR, Allen PJ, Schmidt CM, Kinzler KW, Papadopoulos N, Hruban RH, Vogelstein B. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3:92ra66. doi: 10.1126/scitranslmed.3002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu SF, Adams B, Yu XC, Xu M. Denosumab and giant cell tumour of bone-a review and future management considerations. Curr Oncol. 2013;20:e442–7. doi: 10.3747/co.20.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabut SM, JR, Kenan S, Sissons HA, Lewis MM. Malignant transformation of fibrous dysplasia. A case report and review of the literature. Clin Orthop Relat Res. 1988:281–9. [PubMed] [Google Scholar]
- Yeow VK, Chen YR. Orthognathic surgery in craniomaxillofacial fibrous dysplasia. J Craniofac Surg. 1999;10:155–9. doi: 10.1097/00001665-199903000-00012. [DOI] [PubMed] [Google Scholar]