

Abstract
The airway epithelium is a semi-impermeable barrier whose disruption by growth factor reprogramming is associated with chronic airway diseases of humans. Transforming growth factor beta (TGFβ)-induced epithelial mesenchymal transition (EMT) plays important roles in airway remodeling characteristic of idiopathic lung fibrosis, asthma and chronic obstructive pulmonary disease (COPD). Inflammation of the airways leads to airway injury and tumor necrosis factor alpha (TNFα) plays an important pro-inflammatory role. Little systematic information about the effects of EMT on TNFα signaling is available. Using an in vitro model of TGFβ-induced EMT in primary human small airway epithelial cells (hSAECs), we applied quantitative proteomics and phosphoprotein profiling to understand the molecular mechanism of EMT and the impact of EMT on innate inflammatory responses. We quantified 7,925 proteins and 1,348 phosphorylation sites by stable isotope labeling with iTRAQ technology. We found that cellular response to TNFα is cell state dependent and the relative TNFα response in mesenchymal state is highly compressed. Combined bioinformatics analyses of proteome and phosphoproteome indicate that the EMT state is associated with reprogramming of kinome, signaling cascade of upstream transcription regulators, phosphor-networks, and NF-κB dependent cell signaling.
Keywords: epithelial mesenchymal transition, Proteomics, Phosphoproteomics, Mass spectrometry, Innate immune response
Graphical Abstract
1. Introduction
The human airways are lined with a highly differentiated cellular epithelium that forms a semi-impermeable barrier to insensible water losses and aeroallergens while promoting gas exchange [1, 2]. Chronic obstructive pulmonary disease and asthma represent common diseases associated with epithelial injury and repair, stimulating remodeling. Airway remodeling is a structural alteration of the airways produced by subepithelial fibrosis, myofibroblast hyperplasia and smooth muscle hypertrophy, linked to morbidity and a progressive decline in lung function [3]. Exposure to respiratory pathogens, allergens, environmental pollutants and cigarette smoke stimulates the release of growth factors [1, 4]. Chief among these is transforming growth factor (TGF)β whose upregulation has been observed in severe asthma, chronic obstructive pulmonary disease (COPD) and cystic fibrosis [5–8], and whose actions are to induce re-epithelialization, mucosal repair, and stem cell renewal [9, 10].
The actions of TGFβ are to induce a global transcriptional re-programming response known as epithelial-to-mesenchymal transition (EMT) [11]. During EMT, epithelial cells lose their adherens junctions, apical-basal polarity, reorganize their cytoskeleton, secrete extracellular matrix (ECM) proteins, and thereby transdifferentiate into motile, spindle-shaped mesenchymal cells. EMT plays a central role in normal tissue response to injury and tissue remodeling and repair [12]. Dysregulated EMT will cause disruption of the epithelial barrier, collagen overproduction, expansion of the myofibroblast population, and airway remodeling characteristic of idiopathic lung fibrosis [13], asthma [14] and COPD [15].
TGFβ signals in epithelial cells by binding a plasma membrane localized serine/threonine receptor complex, TGFβR, that coordinately activates two separate intracellular signaling pathways, known as the canonical TGFβR-SMAD-dependent signaling pathway and SMAD-independent pathways [16, 17]. The TGFβ-Smad pathway activates downstream transcriptional regulators Zinc finger protein SNAIL (SNAI1), Zinc finger E-box-binding homebox 1 (ZEB1), and Twist-related protein 1 (TWIST1). These factors drive EMT through downregulation of epithelial E-cadherin (CDH1) to destabilize adherens junctions, induction of mesenchymal intermediate filament proteins such as vimentin, activation of small GTPases to induce motility, and expression of matrix metalloproteinases (MMPs) and collagen to induce fibrogenesis. The non-canonical pathways include PI3 kinase (PI3K)-AKT, ERK/MAPK, p38 MAPK, WNT, NOTCH, Hedgehog, and NF-κB signaling pathways [16]. Both canonical and noncanonical arms are required to coordinate the complex genetic changes underlying EMT [16].
Airway mucosal injury/repair is a multicellular process involving activation of resident alveolar macrophages. In the respiratory tract, alveolar macrophages encountering invading microorganisms inducibly secrete tumor necrosis factor alpha (TNFα), a pleiotropic cytokine that mediates inflammatory signaling [18]. TNF binds to ubiquitously expressed single-pass type I transmembrane receptors of 55 and 75 kDa (TNFR-I and -II, respectively) to induces expression of inflammatory epithelial chemokine networks[19, 20] that amplify the inflammatory response. Intracellular signaling by TNFα is mediated primarily by the IκB kinase, responsible for release of NF-κB transcription factor from inactive stores [21], and phosphorylation by ribosomal S6 kinases required for formation of transcriptionally active complexes [22–24]. In this way, TNFα-induced inflammation is responsible for leukocyte and lymphocyte recruitment leading to oxidative tissue injury [25].
The TGFβ and TNFα pathways share common regulators, and participate in extensive cross-talk. Recent findings suggest that TNFα signaling can stabilize SNAI1 through the activation of the NF-κB pathway [26], and TGFβ can induce the activation of canonical and non-canonical NF-κB pathways [27]. Here, to obtain system-level insight into the mechanisms involved in the TGFβ-induced EMT in primary human small airway epithelial cells and to elucidate how EMT impacts on the TNFα signaling pathways, we used stable isotope labeling by iTRAQ technology analyzed by high-resolution mass spectrometry to investigate the changes in the abundance and phosphorylation state of proteins in hSAECs treated with TNFα in the absence or presence of TGFβ-induced EMT. Our proteomics and phosphoproteomics analysis identified many signaling pathways involved in TGFβ-induced EMT. We also found that EMT has profound reprogramming effects on NF-κB dependent cell signaling transduction mediating the innate immune response.
2. Materials and methods
2.1 Cell culture of hSAECs
An immortalized primary human small airway epithelial cell line was established by infecting primary hSAECs with human telomerase and cyclin dependent kinase (CDK)-4 retrovirus constructs [28]. The immortalized hSAECs were grown in SAGM small airway epithelial cell growth medium (Lonza, Walkersville, MD) in a humidified atmosphere of 5% CO2. hSAECs were stimulated with TGFβ (10 ng/mL, PeproTech, Rocky Hill, NJ) for 15 days to transformed into mesenchymal hSAECs [29]. The proteomics study had four experimental groups including normal hSAECs, TGFβ induced mesenchymal hSAECs, normal hSAECs treated with TNFα for 1 h, and mesenchymal hSAECs treated with TNFα for 1 h. Each experimental group has two biological replicates.
2.2 Protein extraction and trypsin digestion
The cells were washed three times with phosphate buffered Saline (PBS) solution before the harvest. The cells were collected and transferred to a conical tube for centrifugation. Cells were pelleted at 150g for 4 min and resuspended in 1.0 mL of Trizol reagent (Invitrogen, Carlsbad, CA). The cells were lysed thoroughly by repetitive pipetting. The proteins which are free of nucleic acids were extracted from this cell lysate using Trizol reagent according to the manufacturer’s instruction. The protein pellet was resuspended in 50 μL of 8 M Guanidine HCl. The protein concentration was measured using Bradford assay. One milligram of proteins from each sample was processed for digestion. The proteins were first reduced with 10 mM DTT at room temperature for 30 min, followed by alkylation with 30 mM iodoacetamine at room temperature for 2 h. Then, the sample was diluted with 50 μL of 100 mM triethylamonium bicarbonate (pH 8.0). An aliquot of Lys-C/Trypsin solution (Promega, Madison, WI) was added into each sample at the 100:1 protein: enzyme ratio. The samples were incubated at 37 ºC for overnight, and the solutions were further diluted with 400 μL of 100 μM of triethylammionium. An aliquot of Trypsin solution (Promega, Madison, WI) was added into each sample at the 50:1 protein: enzyme ratio. The samples were incubated at 37 ºC for 16 h. 10 μL of 10% trifluoroacetic acid was added into each sample to stop the trypsin digestion. Tryptic peptides were desalted on reversed phase tC18 SepPak columns (Waters, Milford, MA) and evaporated to dryness in a vacuum concentrator.
2.3 iTRAQ labeling of peptides
Desalted peptides were dissolved in 600 μl of 0.5 M triethylammionium pH 8.5 solution and were labeled with 8-plex iTRAQ reagents according to the manufacturer's instructions (AB Sciex, Foster City, CA). The iTRAQ regents used for each sample were as follows: 113 - hSAECs/Control, 114–hSAECs/Control, 115–hSAECs/TNFα, 116–hSAECs/TNFα, 117–EMT-hSAECs/Control, 118–EMT-hSAECs/Control, 119–EMT-hSAECs/TNFα, 121–EMT-hSAECs/TNFα. After 1 h incubation the reaction was stopped with 5% hydroxylamine. The Differentially labeled peptides were mixed and subsequently desalted on 500 mg SepPak columns.
2.4 Strong cation exchange chromatography of iTRAQ labeled peptides
The strong cation exchange chromatography of iTRAQ labeled peptides was performed as described previously with some modifications[30, 31]. Peptides were reconstituted in 50 μl strong cation exchange buffer A (7 mM KH2PO4, pH 2.65, 30% ACN) and separated on a Polysulfoethyl A strong cation exchange column (200 × 4.6 mm, 5 μm particle size, 300 Å pore size) from PolyLC using an Agilent 1260 HPLC system (Agilent, Santa Clara, CA). The column is initially equilibrated with buffer A (7 mM KH2PO4, pH 2.65, 350 mM KCl, 30% ACN) for 30 min. The gradient was 10 min of buffer A, followed by a 30-min linear gradient to 35% Buffer B (7 mM KH2PO4, pH 2.65, 350 mM KCl, 30% ACN), and 5-min linear gradient to 60% Buffer B, 60% Buffer B for 15 min, and a final equilibration with Buffer A for 20 min. The flow rate was 1.0 ml/min and the sample was injected after the initial 20 min equilibration phase. Upon injection, 60 fractions were collected throughout the run. The first 15 fractions were combined. Each SCX fraction was desalted with reversed phase tC18 SepPak columns (Waters, Milford, MA) and evaporated to dryness in a vacuum concentrator. For proteome analysis, 10% of each SCX fraction was aliquotted and analyzed by LC-MS/MS. The remaining 90% of each SCX fraction was subjected to immobilized metal affinity chromatography (IMAC) for enrichment of phosphopeptides.
2.5 Immobilized metal affinity chromatography for enrichment of phosphopeptides
A Fe-NTA phosphopeptide enrichment kit (Thermo Scientific-Pierce) was used for enrichment of phosphopeptides according to manufacturer’s instruction. Briefly, peptides were reconstituted in 200 μl IMAC binding buffer and incubated for 1 h with IMAC beads. After incubation, the beads were washed twice with 100 μl IMAC washing buffer. Phosphorylated peptides were eluted from the beads with IMAC elution buffer. 5 μl of 10% trifluoroacetic acid (TFA) was added into each eluate, and the eluate was desalted with reversed phase tC18 SepPak columns (Waters, Milford, MA) and evaporated to dryness in a vacuum concentrator.
2.6 NanoLC-MS/MS Analysis
The desalted peptides or phosphopeptides were reconstituted in 20 μl 4% ACN/0.1% formic acid. All peptide samples were separated on an online nanoflow Easy nLC1000 UHPLC system (Thermo Scientific) and analyzed on a Q Exactive Orbitrap mass spectrometer (Thermo Scientific, San Jose, CA). 10 μl of sample was injected onto a capillary peptide trap column (Acclaim® Pepmap 100, 75 μm x 2 cm, C18, 3 μm, 100 Å, Thermo Scientific). After sample injection, the peptides were separated on a 25-cm UHPLC reversed phase column (Acclaim® Pepmap 100, 75 μm x 25 cm, C18, 2 μm, 100 Å, Thermo Scientific) at a flowrate of 300 nL/min. A 4-h linear gradient from 2% solvent A (0.1% formic acid in water) to 35% solvent B (0.1% formic acid in acetonitrile) was used for each liquid chromatography (LC)-MS/MS run. Data-dependent acquisition was performed using the Xcalibur 2.3 software in positive ion mode at a spray voltage of 2.1 kV. Survey spectra were acquired in the Orbitrap with a resolution of 70,000, the maximum injection time of 80 ms, an automatic gain control (AGC) of 1e6, and a mass range from 400 to 1400 m/z. The top 15 ions in each survey scan were selected for higher-energy collisional dissociation scans with a resolution of 17,500. For all higher-energy collisional dissociation scans, collision energy was set to 30%, the maximum inject time was 60 ms and the AGC was 1e5. Ions selected for MS/MS were dynamically excluded for 30 s after fragmentation.
2.7 Quantification and Identification of Peptides and Proteins
All mass spectra were processed using the Proteome Discoverer 1.4 (Thermo Scientific, San Jose, CA) which includes modules for iTRAQ-based quantification and probability of phosphosite localization. For peptide identification MS/MS spectra were searched against an SwissProt human protein sequence database (downloaded on February 2013, 20,247 protein entries) with full trypsin digestion; a mass tolerance of ± 20 ppm for precursor and product ions; maximum of two missed cleavage sites permitted; the static mass modifications included carbamidometheylation on cysteinyl residues, 8-plex iTRAQ labeling of lysines and peptide n-termini; Variable modifications were oxidation of methionine, phosphorylation of serine, threonine or tyrosine residues. The false discovery rate (FDR) cutoff for peptide and protein identification is 0.01 based on target-decoy-based approach. The probability of phosphosite assignment is calculated using PhosphoRS 3.1 Phospho-site localization algorithm. The phosphorylation sites with higher than 75% possibility of site assignment were considered as unambiguous site assignment. For iTRAQ quantification, iTRAQ ratios of peptides were calculated as the intensities of each reporter ion verse the intensity of reporter ion of 113 (I114/113, I115/I113, I116/ 113…etc). The iTRAQ protein ratios were obtained by calculating the median of all distinct peptides assigned to a protein group. The iTRAQ ratios of proteins and phosphopeptides quantified in each sample were log2 transformed. Based on the assumption of a dominating population of no-changing proteins, the log2 protein ratios in each iTRAQ channel were normalized to the most frequent value (log2 ratio of no-changing proteins) in that channel. The P-values were corrected from multiple hypothesis testing using Permutation-based false discovery rate.
2.8 Stable Isotope Dilution-Selected Reaction Monitoring-MS
The SID-SRM-MS assays of selected proteins were developed as described previously [32]. For each targeted protein, two or three peptides were initially selected and then the sensitivity and selectivity of these peptides were experimentally evaluated as described previously [33]. The peptide with best sensitivity and selectivity was selected as the surrogate for that protein. For each peptide, 3–5 SRM transitions were monitored. The signature peptides and SRM parameters are listed in Supplemental Table S1. The peptides were chemically synthesized incorporating isotopically labeled [13C615N4] arginine or [13C615N2] lysine to a 99% isotopic enrichment (Thermo Scientific, San Jose, CA). The amount of stable isotope labeled standard (SIS) peptides was determined by amino acid analysis. The proteins were trypsin digested on the beads as described above. The tryptic digests were then reconstituted in 30 μl of 5% formic acid-0.01% TFA. An aliquot of 10 μl of 50 fmol/μL diluted SIS peptides was added to each tryptic digest. These samples were desalted with a ZipTip C18 cartridge. The peptides were eluted with 80% ACN and dried. The peptides were reconstituted in 30 μl of 5% formic acid-0.01% TFA and were directly analyzed by LC-SRM-MS. LC-SRM-MS analysis was performed with a TSQ Vantage triple quadrupole mass spectrometer equipped with nanospray source (Thermo Scientific, San Jose, CA). 8–10 targeted proteins were analyzed in a single LC-SRM run. The online chromatography was performed using an Eksigent NanoLC-2D HPLC system (AB SCIEX, Dublin, CA). An aliquot of 10 μL of each of the tryptic digests was injected on a C18 reverse-phase nano-HPLC column (PicoFrit , 75 μm x 10 cm; tip ID 15 μm) at a flow rate of 500 nL/min with a 20-min 98% A, followed by a 15-min linear gradient from 2–30% mobile phase B (0.1 % formic acid-90% acetonitrile) in mobile phase A (0.1 % formic acid). The TSQ Vantage was operated in high-resolution SRM mode with Q1 and Q3 set to 0.2 and 0.7-Da Full Width Half Maximum (FWHM). All acquisition methods used the following parameters: 2100 V ion spray voltage, a 275°C ion transferring tube temperature, a collision-activated dissociation pressure at 1.5 mTorr, and the S-lens voltage used the values in S-lens table generated during MS calibration.
All SRM data were manually inspected to ensure peak detection and accurate integration. The chromatographic retention time and the relative product ion intensities of the analyte peptides were compared to those of the SIS peptides. The variation of the retention time between the analyte peptides and their SIS counterparts should be within 0.05 min, and the difference in the relative product ion intensities of the analyte peptides and SIS peptides were below 20%. The peak areas in the extract ion chromatograms of the native and SIS version of each signature peptide were integrated using Xcalibur® 2.1. The default values for noise percentage and base-line subtraction window were used. The ratio between the peak area of native and SIS version of each peptide was calculated.
2.9 Immunostaining and Confocal Immunofluorescence Microscopy
For confocal fluorescence microscopy, 25-mm round microscope cover glasses (Fisher Scientific, Pittsburgh, PA) were first pre-coated with sterilized collagen solution (Roche Applied Science) in a 6-well culture plate. hSAECs or EMT-hSAECs were plated over cover glass 2 days prior to the experiment. The cells were then fixed with 4% paraformaldehyde in PBS and incubated with 0.1 M ammonium chloride for 10 min. Afterwards, cells were permeabilized with 0.5% Triton-100 in PBS, followed by incubation in blocking buffer (5% goat serum, 0.1% IGEPAL CA-630, 0.05% NaN3, and 1% BSA) for 1 h and incubated with phalloidin-FITC in incubation buffer (0.1% IGEPAL CA-630, 0.05% NaN3, and 2% BSA) overnight at 4 °C. After washing, cells were imaged at the wavelength of 488 nm. Nuclei were counter stained with DAPI as previously [27, 34].
2.10 Quantitative real-time reverse transcription-PCR (RT-qPCR)
Total RNA was extracted using acid guanidium phenol extraction (Tri reagent; Sigma). For gene expression analyses, 1 μg of RNA was reverse transcribed using SuperScript III in a 2-μl reaction mixture. 1 μl of cDNA product was amplified in a 2 μl reaction mixture containing 10 μl of SYBR green Supermix (Bio-Rad) and the final concentration of 0.4 μM (each) forward and reverse gene-specific primers (Supplemental Table S2). The reaction mixtures were aliquoted into a Bio-Rad 96-well clear PCR plate, and the plate was sealed with Bio-Rad Microseal B film. The plates were denatured for 90 s at 95°C and then subjected to 40 cycles of 15 s at 94°C, 60 s at 60°C, and 1 min at 72°C in a CFX96TM Real-Time PCR Detection System (Bio-Rad). PCR products were subjected to melting curve analysis to ensure that a single amplification product was produced. Quantification of relative changes in gene expression was done using the threshold cycle (ΔΔCT) method. In brief, the ΔCT value was calculated [normalized to cyclophilin (PPIA)] for each sample by using the equation ΔCT = CT (target gene) CT (PPIA). Next, the ΔΔCT was calculated by using the equation ΔΔCT = ΔCT (experimental sample) ΔCT (control sample). Finally, the fold differences between the experimental and control samples were calculated using the formula 2 ΔΔCT.
2.11 Bioinformatics analyses
For proteomics data analysis including normalization, statistics, principal component analysis, and Fisher's exact tests, we used the Perseus platform (http://141.61.102.17/perseus_doku/doku.php?id=start) [35, 36]. The Hierarchical clustering and heatmap based on the protein expressions were generated using Perseus software. The rows of heatmap indicate the proteins and the columns indicate the samples. The log2 ratios of each protein were z-score normalized for each row. Hierarchical clustering of the z-normalized log2 ratio was performed by using Euclidean distances between means. The number of clusters was set as 300. To predict the upstream transcriptional regulators and growth factors based on the observed protein abundance ratios, we used a Upstream Regulator Analysis tool [37], implemented in the Ingenuity® Pathway Analysis platform (IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity). Upstream regulator analysis was used to compare the known effect (transcriptional activation or repression) of a transcriptional regulator on its target genes to the observed changes in protein abundance to determine an overlap P-value measuring enrichment of network-regulated genes in the proteomic dataset and an activation Z-score which can be used to find likely regulating molecules based on a statistically significant pattern match of up- and downregulation, and also to predict the activation state (either activated or inhibited) of a putative regulator. In our analysis, we restricted the “Upstream Regulator Analysis” to the categories “transcriptional regulator” in the IPA filter settings. We considered proteins with an overlap P-value of < 0.01 that had an activation Z-score > 2.0 as activated and those with an activation Z-score of < 2.0 as inhibited.
Kinase family analysis was performed using kinome using Kinome Render [38] (http://bcb.med.usherbrooke.ca/kinomerender.php). Sequence logo analysis of the phosphopeptides was conducted using the Sequence Logo generator tool at (http://www.phosphosite.org/sequenceLogoAction.do). STRING v10 (http://string-db.org/) was used to generate protein interaction networks for phosphoproteins [39]. All interactions are predicted with high confidence threshold of 0.700, and all active predictive methods were allowed. KMEANS algorithm was used to cluster the phosphoproteins that are displayed in the networks. KMEANS use STRING global scores as input for clustering. Each cluster of phosphoproteins was assigned a color. The inter-cluster edges are represented by dashed-lines.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium [40] via the PRIDE partner repository with the dataset identifiers PXD003293, PXD003294, PXD003295, PXD003323, PXD003296, PXD003297, and PDX003974.
3. Results
3.1 Experimental design
The cellular model employed in this study is a telomerase-immortalized human small airway epithelial cell. hSAECs show a stable epithelial morphology and differentiated cytokeratin isoforms after over 100 population doublings, express the stem cell marker p63, the tumor suppressor protein p16INK4a, and have an intact p53 checkpoint pathway [27, 28]. To induce EMT, hSAECs were incubated in the presence of TGFβ (5 ng/mL) for 15 d. Cells were fixed, stained with FITC-conjugated phalloidin (for distribution of F actin) and DAPI (a nuclear DNA stain), and examined by confocal microscopy. In the absence of TGFβ stimulation, hSAECs assumed a normal cuboidal morphology with perinuclear variable F-actin cytoplasmic staining (Figure 1A). By contrast, TGFβ-treated hSAECs assumed an elongated shape with ruffled cytoplasmic extensions and markedly induced F-actin staining (Figure 1A). This morphological change of enhanced front-rear polarity, cytoskeletal actin rearrangement and the formation of ruffled leading edges are characteristic morphological changes of EMT and acquisition of enhanced motility [27, 28]. In addition, we used RT-qPCR to measure the expression of the EMT transcription regulators SNAI1, ZEB1, and TWIST1. We found that these genes were all upregulated in TGFβ-treated hSAECs, with SNAI1 mRNA being most highly upregulated by a 62-fold induction relative to control hSAECs (Figure 1B). Together, these data indicate that the hSAECs were in EMT state after the 15 d TGFβ1 treatment.
Figure 1. TGFβ induce ETM in hSAECs. (A) Imaging to show the cell phenotype change after EMT.

The fixed hSAECs (upper panel) and EMT-hSAECs (low-panel) were stained with phalloidin-FITC for F-actin and DAPI for nuclei (magnify x122). (B) Gene expression level of EMT transcription factors (SNAI1, ZEB1 and TWIST1) by RT-qPCR.
To identify the mechanism underlying the events of TGFβ-induced EMT and the effect of EMT on TNFα signaling at the systems level, we performed quantitative proteomics and phosphoproteomics analysis of cell lysates from normal hSAECs, hSAECs in TGFβ-induced EMT state, and TNFα treated hSAECs in the absence or presence of EMT. Figure 2 shows the schematic illustration of the experimental setup. A total of 8,268 unique protein groups were identified with 1% false discovery rate (FDR). Among them, 7,925 protein groups (95.8% of identified proteins groups) were quantified across all the eight samples (Supplemental Table S3). We calculated the fold change for the abundance of the 7,925 in each sample relative to unstimulated, hSAECs. The scatterplots in Supplemental Figure S1 illustrate the reproducibility of protein ratios of 7,925 quantified proteins in each biological replicates. Systematic pairwise analysis of all 7,925 quantified proteins between biological replicates resulted in Pearson correlation coefficients of at least 0.88 (0.88–0.93) indicating excellent reproducibility of quantification. Analysis of variance (ANOVA) identified 4,398 proteins which showed significant differences in at least one experimental group (1% Permutation-based FDR) (Supplemental Table S4). Systematic pairwise analysis of all 4,398 differentially expressed proteins between biological replicates shows excellent reproducibility of quantification with Pearson correlation coefficients being higher than 0.97 (Supplemental Figure S1D–E). This indicates that iTRAQ-based stable isotope labeling reveals highly reproducible quantitative data about thousands of proteins. In the phosphoproteomics study, 1,348 phosphorylation sites were identified with 1% FDR (Supplemental Table S5). Among these, 1,077 sites were unambiguously located. 781 phosphorylation sites were identified that show significant differences in at least one experimental group (ANOVA, 5% Permutation-based FDR are shown in Supplemental Table S6). The quantification of phosphorylation events also shows the same degree of high reproducibility as proteomics study. Pearson correlation coefficients of iTRAQ ratios of all 1,348 phosphorylation sites between biological replicates were at least 0.88 (0.88–0.94, Supplemental Figure S2A–C). The Pearson correlation coefficient between biological replicates was higher than 0.97 for these phosphorylation sites that were significantly different in at least one experimental group (Supplemental Figure S2D–E). Collectively, these data indicate that we have generated a robust and quantitative data set for systems-level analysis of the effects of EMT and understanding how the EMT state affects the TNFα signaling network. In each of the following sections, comparison of the EMT state will be followed by a comparison of the effects of TNFα.
Figure 2. Experimental workflow for systems-level proteomic profiling of TGF-β and TNFα signaling.
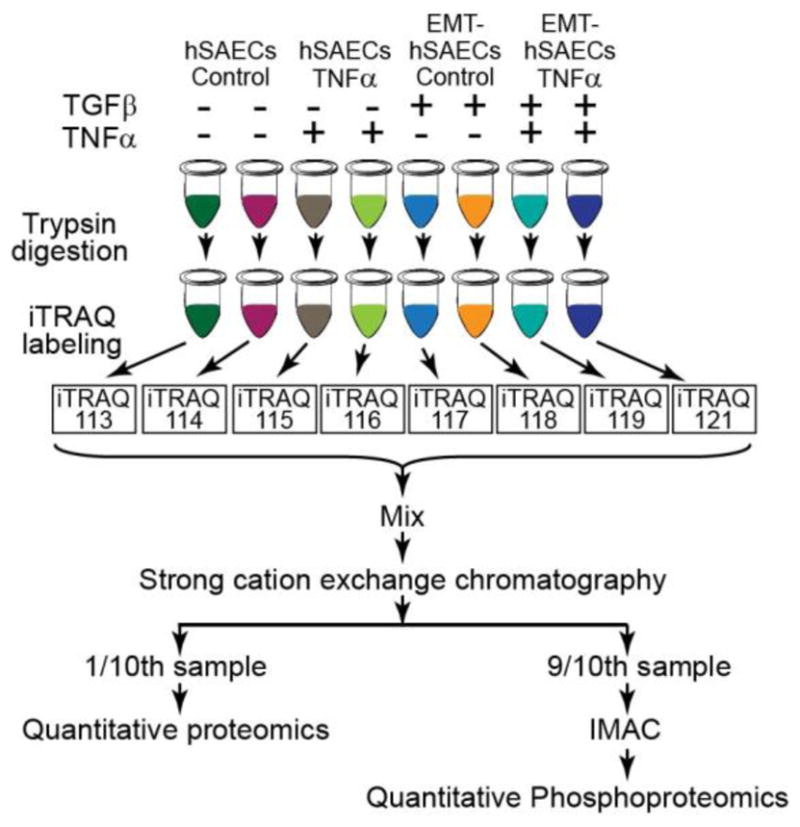
hSAECs cells were induced to EMT with TGFβ stimulation for 15 days. The hSAECs and EMT-hSAECs cells were stimulated with TNFα. After stimulation, cells were lysed, digested with Lys-C/trypsin and labeled with 8-plex iTRAQ reagents. Samples were mixed and separated via SCX. 90% of total peptides in each SCX fraction were further enriched for phosphopeptides with IMAC, whereas 10% of total peptides were used for quantitative proteomic profiling. All samples were analyzed by Q Exactive Orbitrap mass spectrometer. The proteins and phosphoproteins were identified and quantified with Proteome Discoverer.
3.2 Protein expression of mesenchymal markers
It is well established that E-cadherin (CDH1) is expressed in epithelial cells, and suppressed by the core EMT transcription factors, SNAI1, ZEB1 and TWIST1 in response to TGFβ1 treatment [41]. Downregulation of CDH1 is a hallmark of EMT and is compensated by the increased expression of mesenchymal neural N-cadherin (CDH2). This EN-switch is known to cause the cell-state transition from an epithelial phenotype and acquisition of a mesenchymal phenotype character by homotypic N-cadherin interactions [16, 42]. We examined the expression of the EMT program in our proteomic datasets. We found that the simultaneous downregulation of CDH1 and upregulation of CDH2 in TGFβ1-treated cells (Figure 3A). Other well-known mesenchymal protein markers including ECM proteins [fibronectin (FN1) and collagens (COL8A1, COL4A2, and COL4A1)], mesenchymal intermediate filament protein (vimentin, VIM), matrix metalloproteinases (MMP2, MMP15, MMP17), and mesenchymal glycoproteins THBS1, SERPINE1 and SPARC were also strongly elevated in mesenchymal cells (Figure 3A). The expression of EMT markers was confirmed with stable isotope dilution (SID)-selected reaction monitoring (SRM)-mass spectrometry (MS) analysis (Figure 3B). Together, these data further confirmed that hSAECs cells were in mesenchymal state after 15 d of TGFβ1 stimulation.
Figure 3. Protein expression of EMT markers and proteins related to cytoskeletal architecture and cell motility.

(A) Expression level of EMT markers, quantified by iTRAQ based stable isotope tagging displayed as a heatmap of log2 expression values relative to untreated hSAECs. Abbreviations: Col, collagen; Ctrl, control; VIM, vimentin. (B) SID-SRM-MS validation of iTRAQ quantification of protein and phosphoprotein profiles. Shown are the relative changes of keratin (KRT)19, CDH1 , S100A9, MMP2, SPARC, and FN1 in response to EMT and or TNFα treatment. Data are expressed as the mean ± SD of the ratio of measured protein relative to stable isotope standard (native/SIS). Data represent biological replicates n=2 assayed in duplicate.
We observed that the expression of these mesenchymal markers in response to TNFα stimulation varies dependent on the cell state. In normal hSAECs, TNFα reduced the expression of CDH-1, 3 and -2, whereas TNFα treatment upregulated the expression of CDH2 in mesenchymal cells (Figure 3A). Similarly, we noted that TNFα repressed the expression of collagen (COL)-8A, 4A1/2 isoforms proteins in normal hSAECs, whereas in mesenchymal cells, the majority of identified collagen proteins were upregulated regardless of the absence or presence of TNFα stimulation.
3.3 Global analysis of the effect of mesenchymal transition on protein regulation
To gain systematic understanding of the biological pathways involved in TGFβ-induced EMT and how EMT perturbs the TNFα signaling pathways, we performed global bioinformatics analysis on the 4,398 proteins with abundance that are significantly different in at least one experimental group. Principle component analysis (PCA) of the expression level of these 4,398 proteins readily discriminated between the experimental groups (Figure 4A). In this analysis, the control hSAECs were tightly grouped together and clearly segregated from the TNFα-treated hSAECs and mesenchymal cells; whereas TNFα-treated mesenchymal cells were associated with mesenchymal hSAECs without TNFα stimulation. We noted that TNFα treatment induces no changes in the first principal component of mesenchymal cells in the PCA (Figure 4A) and only a minor shift in the second principle component. We interpret these results to indicate that TGFβ-induced EMT has altered the cell response to TNFα stimulation in hSAECs, and the relative TNFα response of the EMT-hSAECs is highly compressed. This finding is consistent with our earlier transcriptome study [29]. Next, we used unsupervised hierarchical clustering to group proteins based on their expression levels (Figure 4B). We found that TNFα and EMT caused dramatic and distinct changes in protein expression profiles of hSAECs, and TNFα-induced divergent cellular response in the absence or presence of EMT. This clustering identified five clusters (Figure 4B). To identify biological functions of these expression programs, we analyzed KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways, GO (Gene Ontology) biological functions (GOBF), molecular functions (GOMF) and cellular components (GOCC) in each cluster identifying significant differences by a Fisher exact test (P<0.001, Benjamini-Hochberg FDR 2%, Supplemental Table S7). Cluster 2 contains proteins upregulated in mesenchymal cells and is enriched for the annotations that are related to TGFβ-induced EMT, such as focal adhesion, extracellular matrix organization, angiogenesis and protein processing in the ER. In cluster 3 are the proteins that were downregulated in TNFα stimulated hSAECs. This cluster is enriched for the annotations that are related to chromatin modification, spliceosome, and cell adhesion molecules. In cluster 4 and 5 are proteins that were upregulated in TNFα stimulated hSAECs and silenced in mesenchymal cells. These proteins are enriched with the annotations of translational initiation/elongation/termination, glycolysis/gluconeogenesis and numerous annotations related to cellular metabolism.
Figure 4. Protein expression profiles in response to TNFα in the absence or presence of TGFβ-induced EMT.

(A) Principle component analysis of the 4,398 proteins with abundance that are significantly different in at least one experiment group; from two biological replicates of hSAECs control cells (black open circle), hSAECs treated with TNFα (blue square), EMT-hSAECs control cells (red closed circle), and EMT-hSAECs treated with TNFa (purple diamond) are shown. (B) Heatmap of z score- and averaged log2-transformed ratios of the abundance of the 4,398 proteins with abundance that is significantly different in at least one experiment group (ANOVA, FDR 1%). Proteins were grouped using unsupervised hierarchical clustering. Examples of significantly enriched functional annotations for each cluster are shown (Fisher’s exact test, P<0.05, FDR <2%).
3.4 Analysis of upstream transcription regulators induced by TGFβ and TNFα
To identify upstream transcription regulators that are either activated or inhibited in EMT and in response to TNFα in the absence or presence of EMT, we used Ingenuity Pathway IPA® Upstream Regulator analysis to analyze the proteomic profiling data. Upstream regulator analysis uses the observed protein abundance change in our datasets and prior knowledge of expected effects between transcriptional regulators and their target genes to identify the cascades of upstream regulators that can explain the observed protein expression changes in our study [37]. We identified 45 upstream transcription regulators that show significant activation or inhibition effect in at least one experimental group (activation z score > or <2, P value of overlap < 0.01) (Supplemental Table S8). Using unsupervised hierarchical clustering on the basis of the IPA activation score of these transcription regulators in each experiment, the 45 upstream transcription regulators grouped into three major clusters (Figure 5). Cluster 3 is composed of transcription regulators that have sustained activation profiles in mesenchymal cells regardless of the absence or presence of TNFα stimulation, but inhibition profiles in normal hSAECs with TNFα stimulation. Some of the proteins are key transcriptional regulators of EMT, such as SMAD4, TP53, NOTCH1, SRF and NFKBIA, indicating that the upstream analysis correctly predicted the key regulators of TGFβ signaling. We also identified several proteins in this cluster that are less well-characterized transcriptional regulators of TGFβ signaling such as X-box binding protein 1 (XBP1) which has the highest activation score in EMT-hSAECs (Z score 5.434, P value of overlap 8.54E-26) and Protein C-ets-1 (ETS1). Proteins in Cluster 1 have substantial activation profiles in TNFα treated hSAECs, but inhibitory effect in mesenchymal cells. Among them, SMAD7, MYC, and MYCN are key negative regulators of TGFβ signaling [43]. In this regard, MYC is known to stimulate the reverse process of mesenchymal-epithelial transition [44].
Figure 5. Upstream regulator analysis of transcription regulators of TGFβ-induced EMT and TNFα signaling in the absence or presence of EMT.

Heatmap of the activation Z scores for unstream transcription regulators predicted to be activated (red) or inhibited (blue) by stimulation of TNFα in absence or presence of TGFβ-induced EMT.
The heatmap (Figure 5) shows that the regulatory effects of these upstream transcription regulators on downstream target proteins in response to TNFα stimulation are dependent on the cell state. In normal hSAECs, TNFα induced dramatic changes in the activation profiles of these transcription regulators, with activation profiles in cluster 1 and inhibitory profiles in cluster 2. By contrast, in TNFα-treated mesenchymal cells, both the cluster 1 activation profiles and the inhibition profiles in cluster 2 are similar to that of the unstimulated mesenchymal cells. These data indicate to us that EMT alters the signaling cascade of upstream transcription regulators of theTNFα pathway.
3.5 Global bioinformatics analysis of TGFβ- and TNFα regulated kinases
Cell signaling networks often rely on protein phosphorylation of their components to transmit information. Protein phosphorylation is catalyzed by specific kinases and phosphatases. In our iTRAQ-based proteomics study, a total of 375 kinases and 142 phosphatases were quantified, and 169 kinases showed significant differences in at least one experimental group (ANOVA, 1% FDR). To visualize the protein profiles across the entire human kinase family, we placed the 375 kinases quantified in this proteomics study into the dendrogram of the human kinome using Kinome Render [38]. Kinome Render produced one separate image for each experimental group (Figure 6A-C). As shown in the Figure 6, kinases from all seven major kinome groups as well as atypical kinases were quantified in our proteomics study. Every kinase group exhibits upregulated and downregulated members in response to mesenchymal transition (Figure 6B). Further, we observed distinct, cell state-dependent expression profiles in response to TNFα. TNFα induced dramatic changes in hSAEC kinome profiles (Figure 6A), but less so in mesenchymal cells (Figure 6C). For example, Ephrin (Eph) A7, -A2, HER4 in tyrosine kinase group, Misshapen-Like Kinase (MINK/ZC33), mitogen and erk activating kinase (MEKK1/MAP3K1) in STE group, protein kinase N (PKN1/PRK1) in AGC group are downregulated in TNFα-stimulated hSAECs, but are upregulated or unchanged in mesenchymal cells with TNFα-stimulation (c.f. Figures 6A, C).
Figure 6. Expression profiles of protein kinases.

Protein kinases were mapped in the dendrogram of the human kinome. Kinome dendrogram of hSAECs with TNFα treatment (A), EMT-hSAECs control cells (B), and EMT-hSAECs with TNFα treatment (C) are shown. Kinases quantified in the proteomic study are marked with circles, where red circles (upregulated) and green circles (downregulated) are the kinases which abundance are significantly differences in at least one experiment group, while the grey circles are kinases which expression levels remain unchanged cross the four experimental groups; and large circles indicate higher degree of change in expression level as indicated in the figure legend.
3.6 TGFβ- and TNFα regulated phospho-networks
The iTRAQ quantification of some significantly differentially expressed phosphoproteins [Insulin receptor substrate 2 (IRS2) pSer 1100, Heat shock protein beta-1 (HSPB1) pSer 82 and pSer 15)] were further confirmed with quantitative SID-SRM-MS (Figure 7A). The SRM analysis shows very good agreement with iTRAQ quantification. We analyzed the phosphorylation profiles of 781 phosphorylation sites that showed significant differences in at least one experimental group (ANOVA, 5%FDR). A principle components analysis of the profiles of these phosphorylation sites was conducted. Each experimental point clusters closely with its replicate (Figure 7B). Principle component 2 separated experimental groups by the EMT state, whereas component 1 separated cell treatments. The greatest change in component 1 was induced by TNFα in normal hSAECs, whereas TNFα treatment produced a minor shift in this dimension in the mesenchymal cells. These data further support the conclusion that TNFα response is compressed in the mesenchymal state. Next, we used unsupervised hierarchical clustering to group these significantly changed phosphorylation sites into 3 clusters (Figure 7C). These data clearly show that EMT causes the rewiring of TNFα-regulated phosphorylation events. We performed analysis for enrichment of KEGG pathways, GOBF, GOMF, GOCC andPfam protein domains in each cluster (Figure 7C). In cluster 1, where the phosphorylation events are upregulated in mesenchymal cells, we found enrichment of phosphoproteins involved in positive regulation of MAPKKK cascade, actin-filament-based movement, focal adhesion and phosphoproteins with I-set domain and filament-head domain. In cluster 2 where phosphorylation events were downregulated in TNFα-treated hSAECs, but not in TNFα-treated mesenchymal cells, we found significant enrichment of phosphoproteins involved in regulating mRNA splicing and the mRNA surveillance pathway. Cluster 3 is enriched with phosphoproteins that regulate toll-like receptor signaling pathway, innate immune response and stress-activated MAPK cascade. The phosphorylation sites of these proteins were significantly upregulated in TNFα-treated hSAECs and suppressed in mesenchymal cells.
Figure 7. Expression profiles of phosphorylation evets in response to TNFα in the absence or presence of TGFβ-induced EMT.

(A) SID-SRM-MS validation of iTRAQ quantification of protein and phosphoprotein profiles. Shown are the relative changes of IRS2-phospho-Ser (pS) 1100, HSPB1 pS82 and pS15 in response to EMT and or TNFα treatment. Data are expressed as the mean ± SD of the ratio of measured protein relative to stable isotope standard (native/SIS). Data represent biological replicates n=2 assayed in duplicate. (B) Principle component analysis of the 781 phosphorylation sites with abundance that was significantly different in at least one experiment group; from two biological replicates of hSAECs control cells (black circle), hSAECs treated with TNFα (blue square), EMT-hSAECs control cells (red circle), and EMT-hSAECs treated with TNFa (purple diamond) are shown. (C) Heatmap of z score- and log2-transformed ratios of the average abundance of the 781phosphorylation sites with abundance that is significantly different in at least one experiment group. Phosphorylation sites were grouped using unsupervised hierarchical clustering. Examples of significantly enriched functional annotations for each cluster are shown. (Fisher’s exact test, P<0.05, FDR <2%). (D) Box-plots of all phosphorylation events within kinase substrate motifs identified as significantly altered TNFα or TNFα-induced EMT. (E) Sequence logo graphs of examples of significantly enriched kinase substrate motif for individual clusters of phosphorylation sites shown in (D).
To identify the potential difference in kinase substrate motifs in each cluster, we examined enrichment of kinase-substrate motifs using annotation from the PhosphositePlus® data base [45]. We found enrichment of three kinase substrate motifs in cluster 2 that included the MAPKAPK1 kinase substrate motif, GSK3, ERK1, ERK2 and CDK5 kinase substrate motif, and PKC kinase substrate motif (Fisher’s exact test, P<0.05, FDR <2%) (Figure 7D). Details of enrichment scores are given in Supplemental Table S9. The substrate proteins of these kinases are mainly involved in DNA-damage response, cell cycle, and PKA signaling based on IPA analysis. The enriched phospho-acceptor motifs are shown in the Sequence Logo graphs (Figure 7E).
To understand how EMT affects the TNFα induced phosphor-networks, we performed the STRING (Search Tool for Analysis of Interacting Genes/Proteins) analysis to establish the interaction networks for kinases and phosphoproteins that were significantly changed [39]. The STRING algorithms links genes or proteins into networks based on published functional or informatics-predicted interacts. Figure 8A and B are the STRING networks for kinases and phosphoproteins that are up-regulated upon TNFα stimulation, and Figure 8C and D are String networks for down-regulated kinases and phosphoproteins in response to TNFα stimulation.
Figure 8. String analysis of differentially expression protein kinases and phosphoproteins.

STRING networks for kinases and phosphoproteins that are up-regulated upon TNFα stimulation (A, B), and STRING networks for down-regulated kinases and phosphoproteins in response to TNFα stimulation (C,D). The confidence level of the predicted interactions was set as high (confidence score > 0.7). The nodes that did not interact with any other protein evaluated or networks that have less than three connections are not shown. (E) Quantification of phosphorylation of RelA S536, RSP6KA1 Thr 359/Ser 363, and Ser 221. RelA S536 and RSP6KA1 Thr 359/Ser 363 were quantified with ELISA; RSP6KA1 Ser 221 was quantified with mass spectrometry and iTRAQ stable isotope labeling.
Upon gross inspection, it was immediately evident that TNFα-induced phospho-networks in the absence or presence of EMT are topologically unique. Several phosphorylation events (GSK3B Ser215/Tyr216, RPS6KA1 Ser 221, CSNK1A1 Thr 321, and CDK1 Thr 14) that were significantly elevated in normal hSAECs by TNFα were lost in mesenchymal cells, whereas phospho-networks in mesenchymal cells are more related to EMT, for example, the elevated phosphorylation of proteins related to cytoskeleton reorganization in EMT and TGF-β signaling regulators - DNA binding coactivators AP1 (JUN) Ser 73 and activating transcription factor 2 (ATF2) Ser 112. Various gene responses to TGF-β require the presence of JUN and ATF2 transcriptional activity in the cell [43, 46]. In hSAECs, the most prominent TNFα phospho-network of down-regulated kinases and phosphoproteins is related to spliceosome. We found that the phosphorylation of the spliceosome complex including many serine/arginine-rich splicing factors and ribonucleoproteins was significantly inhibited in hSAECs, but not in mesenchymal cells. Together, these data indicate that EMT cause rewiring of TNFα phospho-networks.
3.7 EMT uncouples the ERK-RPS6K-RelA activation pathway
Here we note that the 90 kDa ribosomal S6 kinase (RPS6K) is a prominent component of TNFα induced phosphoprotein signaling in hSAECs, but is absent from the mesenchymal cells (Figure 8A, B). RPS6K is a MAPK-activated serine/threonine kinase sequentially activated by ERK1/2. Phosphorylation of the linker region (Thr 359 and Serine 363) and N-terminal kinase (Ser 221) are essential for enzymatic activation and its dissociation from ERK, resulting in downstream RelA Ser 536 phosphorylation [47–49]. We observed that TNFα induced a strong 3.6-fold activation of RPS6KA phosphorylation at Ser 221 and close to 2-fold activation at Thr 359/Ser 363 in hSAECs (Figure 8E), but this activation was lost in the mesenchymal state. To confirm the functional down-regulation of ERK-RPS6KA phosphorylation, we measured RelA Ser 536 phosphorylation in TNFα-stimulated hSAECs vs mesenchymal cells. In hSAECs, TNFa induced a robust activation of RelA Ser 536, however, RelA phosphorylation was lost in the mesenchymal state (Figure 8E). These data provide a mechanistic explanation for the compressed phospho-proteomic response to TNFα in the EMT state.
4. Discussion
The EMT is a dynamic cell state change that underlies normal development, organ injury/repair and cancer invasion. Triggered by the TGFβ growth factor, canonical and noncanonical signaling pathways coordinate the activation of the core transcriptional regulators including SNAIL, TWIST and ZEB1/2 that induce transcriptional and epigenetic reprogramming [12, 16, 50, 51]. Although individual contributions of the signaling pathways and their role in EMT have been studied extensively, the systematic characterization of molecular mechanisms of EMT in primary human cells and the effect of EMT on signaling transduction is still lacking. Recently, we used RNA-Seq to understand the gene program of EMT in primary human airway epithelial cells at the transcriptional level, and found that NF-κB/RelA controls a SMAD-independent gene network whose regulation is required for initiation of EMT[29]. In this study, we extend this previous RNA-Seq study to systematically quantify proteome-wide changes in protein abundance and phosphorylation of primary hSAECs in response to TGFβ-induced EMT and in a response to TNFα stimulation in the absence or presence of EMT. iTRAQ technology was selected because this stable isotope labeling approach provides increased reproducibility and accuracy for quantification of peptides and proteins from multiple biological states. By combining 2D-HPLC separation and improved data acquisition rate of an Orbitrap mass spectrometer, we were able to conduct a comprehensive proteome-wide analysis of the changes in the abundance of proteins or posttranslational modifications (PTMs). We identified 4,398 proteins and 781 phosphorylation sites that were significantly differentially expressed and independently validated critical findings with SID-SRM-MS analysis. This study is the first to our knowledge that uses a systems biology approach to study the molecular mechanism of the mesenchymal transition in primary human epithelial cells at the protein and posttranslational level and to provide a more understanding of reprogramming and compression of the TNFα signaling response..
4.1 Insights into control and consequences of EMT
Mesenchymal transition is a process that underlies organ injury and fibrotic response [50]. In normal cells, this program is distinct from that of the more intensively studied Type III EMT, arising in cells with prior oncogenic transformation produced by activating mutations of RAS GTPases upstream of noncanonical TGFβ activating pathways [reviewed in[50]]. Our earlier RNA-Seq analyses identified gene expression patterns in normal cells controlling integrin-extracellular matrix interaction that are distinct from those observed in KRAS-transformed airway epithelial cells [29]. Our study here extends this prior work demonstrating the activation of SMAD pathways, integrin signaling, PI3K-AKT, MAPK, NF-κB, WNT, HH, and Notch pathways at the protein level. The activation of SMAD pathway down regulates epithelial proteins CDH1, S100A8, S100A9, and keratin 19, and up-regulates mesenchymal fibronectin, collagens, MMPs, and VIM (Figure 3A). Bioinformatic analysis reveals that the proteins associated with focal adhesion, EMC-receptor interaction and cell adhesion molecules are upregulated in the mesenchymal state. In normal hSAECs treated with TNFα, the level of E-cadherin was significantly reduced. But unlike TGFβ induced mesenchymal cells in which the repression of E-cadherin is regulated by transcription regulators such as SNAIL, the mechanism of reduction of E-cadherin in TNFα treated normal hSAECs is likely due to the destabilization and degradation of E-cadherin on the cell membrane. In our proteomics study, we found that the level of p120 catenin was decreased in TNFα treated normal hSAECs. p120 catenin is a master regulator of cadherin retention and stability at the cell surface. E-cadherin is rapid internalized and degraded in the absence of p120 catenin [52, 53]. In addition, TNFα induced strong activation of NF-κB pathway which also suppresses the expression of E-cadherin [52, 54, 55].
Activation of the EMT is due to a coordinated interaction of transcription factors and repressors. Although SNAI1, TWIST and ZEB represent core transcription factors, integration of ChIP-Seq and RNA-Seq analysis into a global topographical map identified the participation of over 47 distinct transcription factors whose actions clustered in three distinct groups[29]. Our upstream transcription regulatory analysis of proteomics data suggested SMAD4, TP53, NOTCH1, SRF, and NFκBIA are major regulatory pathways activated while the actions of SMAD7, MYC and MYCN were inhibited in mesenchymal cells. Some less well-characterized transcription regulators of TGFβ were also identified, for example, XBP1 is a transcription factor known to regulate the expression immune response genes and plays a role in the endoplasmic reticulum (ER) stress response –known as the unfolded protein response (UPR) [56]. It was recently found that XBP1 acts as a novel regulator of EMT and can promote EMT and cell invasion in breast cancer cells by upregulating SNAI1 expression [57]. Protein ETS1 is another less known transcription regulator predicted to be activated in mesenchymal cells. ETS1 is known to play a key regulatory role in transcription control induced by TGFβ-Smad pathway [58] as an upstream transcription regulator of ZEB1 [59]. Further study will be required to understand the complex interactions produced by these clusters of transcription factors upstream of SNAI1.
Posttranslational modifications including phosphorylation play a predominant role in regulating EMT. Some of identified phosphorylation sites are known to regulate TGFβ-signaling. For example, two phosphorylation sites of HMGA2 (Ser 44 and Ser 102) are upregulated in EMT-hSEACs. HMGA2 is induced by the Smad pathway during EMT. HMGA2 provides transcriptional input for the expression control of EMT regulators, SNAI1/2 and TWIST. HMGA2 Ser 44 is within the CDK2 kinase substrate motif[60] and Ser 102 is within the ATM kinase substrate motif. Recent studies indicate that HMGA2 can be phosphorylated by ATM, and TGFβ1-induced transcription requires the interplay between HMGA2, ATM and H2AX phosphorylation [61, 62], implying that ATM kinase may play a role in regulating EMT through HMGA2. We also observed the upregulation of phosphorylation of Ser 73 of transcription factor AP1/c-JUN in EMT-hSAECs. Phosphorylation of Ser 73 within the c-JUN transactivation domain by JUN N-terminal kinase (JNK) is critical for its transactivation ability and upregulation of DNA binding domain. In response to TGFβ1 stimulation, JUN N-terminal kinase (JNK) phosphorylates AP1/c-Jun which mediates TGFβ induced transcription in cooperation with SMAD3–SMAD4[63] .
Alternative mRNA splicing is a post-transcriptional mechanism that contributes to the changes in cell adhesion, mobility and signal transduction in EMT [64]. We observed the downregulation of epithelial splicing regulatory protein 1 (ESRP1) and ESRP2. In cancer cells, the downregulation of ESRP1 and ESRP2 generates diverse protein isoforms including p120 catenin, fibroblast growth factor receptor (FGFR)2, and enabled homolog (ENAH) in EMT that affect motility and cellular adhesion in the epithelial or mesenchymal state [64, 65]. In our previous transcriptome study [29], we observed widespread alternative splicing in EMT-hSAECs (not shown). The role of alternative splicing, if any, in EMT will require further investigation. Phosphorylation is required for activity in splicing [66, 67], and there is clear evidence for phosphorylated serine/arginine-rich splicing factors engaging in protein/protein interactions and spliceosome assembly. Inhibition of protein phosphorylation will prevent pre-spliceosome early complex formation and stable binding of U2 and U4/U6. U5 snRNPs to the pre-RNA [67]. We note that there is global dephosphorylation of spliceosome proteins in normal hSAECs in response to TNFα stimulation (Figure 8C). In mesenchymal cells, phosphorylation of most of spliceosome protein remains unchanged upon TNF stimulation (Figure 8D). This result may be a consequence of global reorganization of kinases and phosphatases caused by this cell state change.
4.2 EMT globally reprograms signal transduction
Our study quantified a large number of kinases which account for ~ 72% of human kinome. We discovered that EMT caused profound changes in the expression profiles of kinases. For example, the kinases involved in the activation of MAPK pathway MAP2K5, MAP3K2, MAP4K5, MAPK3 and ALK1 were upregulated in EMT-hSAECs. ALK can induce tyrosine phosphorylation of MAP kinase MAPK1/ERK2 and MAPK3/ERK1. MAP2K5 acts as a scaffold for the formation of a ternary MAP3K2/MAP3K3-MAP3K5-MAPK7 signaling complex. MAP4K5 is an upstream kinase of c-JUN N-terminal pathway and may play a role in the response to environmental stress. MAPK3/ERK1 is an essential component of MAP kinase signal transduction pathway. The MAPK/ERK cascade mediates diverse biological process such as transcription, translation, cytoskeletal rearrangements. TGFβ activates ERK/MAPK, p38 and c-JUN N-terminal kinase MAPK pathways [16, 68]. Activation of ERK/MAPK increases TGFβ-induced transcription, while inhibition of ERK or p38 kinase activity represses TGFβ-induced EMT. In our recent study, we found that TNFα signaling pathway through the MAPKs is rewired as a result of EMT [69]. MAP3K2 regulates the JNK and ERK5 pathways by phosphorylating and activating MAP2K5 and MAP2K7. TGFβ-induced activation of ERK5/MAPK stabilizes SNAI1 by inhibiting GSK3β, which also contributes to EMT. The broad range changes of expression profiles of kinases will lead to the rewiring of signaling transduction. The full consequences of this reprogramming will require further investigation.
4.3 EMT reprograms ERK-RPS6KA-RelA signaling
Of particular interest for this study, we observe that the TNFα program is re-routed by mesenchymal transition to other pathways involved in maintaining EMT. Bioinformatics analysis shows that TNFα induces the activation of inflammation response and also the activation of metabolic processes such as the TCA cycle, respiratory electron transport chain, and fatty acid metabolism in normal hSAECs. Activation of these pathways is lost in mesenchymal cells. This rewiring appears to be mediated by differential coupling of phosphoprotein signaling networks.
TNFα induced signaling involves differential RelA phosphorylation. In particular, RelA phosphorylation of at Ser 536 is mediated by IKKβ and/or IKKα and is required for inducible RelA DNA binding and transcription activity [70]. It was shown earlier that insufficient ERK reduces the phosphorylation of RelA at S536 by reduced IKKβ activation mediated by the ERK downstream kinase RPS6K [48]. The activation of RPS6K occurs in several steps. First, ERK phosphorylates the activation loop of its C-terminal kinase (Thr 573) and the linker (Thr 359 and Ser 363) domains. In activation step two, the C-terminal kinase and/or a putative 3-phosphoinositide-dependent kinase (PDK2) phosphorylates Ser 380 and thereby increases the docking of 3-phosphoinositide-dependent kinase 1 (PDK1) which, in turn, phosphorylates Ser 221 in the activation loop of the N-terminal kinase domain of RPS6K [47, 71]. The phosphorylation of Ser 221 is essential for RPS6K activity [47]. Mutation of Ser 221 to Ala abolishes the ability of RPS6K to phosphorylate its substrates in vitro [72, 73]. In normal hSAECs, we found that the RPS6K was fully activated and the phosphorylation of Thr 359, Ser 363, and Ser 221 were significantly elevated in response to TNF stimulation. Consequently to the activation of RPS6K, RelA Ser 536 phosphorylation was up-regulated. However, in mesenchymal cells, the phosphorylation of Thr 359, Ser 363, and Ser 221 of RPS6K did not change upon TNFα stimulation, and RelA Ser 536 phosphorylation was absent. These findings indicate a functional defect in RPS6K activity, leading to the lower level of RelA Ser 536 phosphorylation. It is clearly evident that EMT causes the rewiring of phosphor-networks and this rewiring will redirect cell responses to TNFα differently depending on its underlying cell state.
5. Conclusion
In conclusion, our integrated proteomics-phosphoproteomics study provides a systems-level understanding of the mechanism of signaling pathways involved in EMT and the effect of EMT on TNFα signaling. Our findings suggest that the mesenchymal state reprograms signal transduction pathways producing distinct patterns of cellular responses dependent on the initial cell state.
Supplementary Material
Significance.
Epithelial mesenchymal transition and inflammation have important implications for clinical and physiologic manifestations of chronic airway diseases such as severe asthma, COPD, and lung fibrosis. Little systematic information on the interplay between EMT and innate inflammation is available. This study combined quantitative proteomics and phosphorproteomics approach to obtain system-level insight into the upstream transcription regulators involved in the TGFβ-induced EMT in primary human small airway epithelial cells and to elucidate how EMT impacts on the TNFα signaling pathways. The proteomics and phosphoproteomics analysis indicates that many signaling pathways involved in TGFβ-induced EMT and EMT has profound reprogramming effects on innate inflammation response.
Highlights.
Quantitative proteomics and phosphoproteomics were integrated to understand the impact of epithelial mesenchymal transition (EMT) on innate inflammatory responses.
7,925 proteins and 1,348 phosphorylation sites were quantified with iTRAQ technology.
Cellular response to TNFα is cell state dependent and the relative TNFα response in mesenchymal state is highly compressed.
45 upstream transcription regulators of EMT and/or TNFα signaling were identified.
Combined bioinformatics analyses of proteome and phosphoproteome indicate that the EMT state is associated with reprogramming of kinome, signaling cascade of upstream transcription regulators, phosphor-networks, and NF-κB cell signaling.
Acknowledgments
This work was supported by National Institutes of Health Grants NCATS UL1TR001439 (to ARB), DMS-1361411/DMS-1361318 (ARB, YZ), NIAID AI062885 (ARB), NIEHS P30 ES006676 (ARB) and pilot funding from the Sealy Center for Molecular Sciences.
Footnotes
Author contributions
YXZ and ARB conceived and initiated the study. YXZ and ARB designed the study and interpreted results. YXZ developed methods, performed the mass spectrometry experiments, and analyzed the data. BT and ARB performed cell culture and protein extraction. BT performed immunostaining, microscopy experiments, and RT-qPCR experiment. RGS performed the data analysis to quantify phosphorylation of RelA Ser 536 and RPS6K Thr 359, Ser 363, and Ser 221 . YXZ and YQZ performed SRM validation. YXZ and ARB wrote the manuscript. All authors read the manuscript and discussed the interpretation of results.
Additional information
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Knight DA, Holgate ST. The airway epithelium: structural and functional properties in health and disease. Respirology. 2003;8:432–46. doi: 10.1046/j.1440-1843.2003.00493.x. [DOI] [PubMed] [Google Scholar]
- 2.Lambrecht BN, Hammad H. The airway epithelium in asthma. Nature medicine. 2012;18:684–92. doi: 10.1038/nm.2737. [DOI] [PubMed] [Google Scholar]
- 3.Al-Muhsen S, Johnson JR, Hamid Q. Remodeling in asthma. The Journal of allergy and clinical immunology. 2011;128:451–62. doi: 10.1016/j.jaci.2011.04.047. quiz 63–4. [DOI] [PubMed] [Google Scholar]
- 4.Fedorov IA, Wilson SJ, Davies DE, Holgate ST. Epithelial stress and structural remodelling in childhood asthma. Thorax. 2005;60:389–94. doi: 10.1136/thx.2004.030262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakir J, Shannon J, Molet S, Fukakusa M, Elias J, Laviolette M, Boulet LP, Hamid Q. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. The Journal of allergy and clinical immunology. 2003;111:1293–8. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- 6.Hackett TL, Warner SM, Stefanowicz D, Shaheen F, Pechkovsky DV, Murray LA, Argentieri R, Kicic A, Stick SM, Bai TR, Knight DA. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. American journal of respiratory and critical care medicine. 2009;180:122–33. doi: 10.1164/rccm.200811-1730OC. [DOI] [PubMed] [Google Scholar]
- 7.Minshall EM, Leung DY, Martin RJ, Song YL, Cameron L, Ernst P, Hamid Q. Eosinophil-associated TGF-beta1 mRNA expression and airways fibrosis in bronchial asthma. American journal of respiratory cell and molecular biology. 1997;17:326–33. doi: 10.1165/ajrcmb.17.3.2733. [DOI] [PubMed] [Google Scholar]
- 8.Harris WT, Kelly DR, Zhou Y, Wang D, Macewen M, Hagood JS, Clancy JP, Ambalavanan N, Sorscher EJ. Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PloS one. 2013;8:e70196. doi: 10.1371/journal.pone.0070196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holgate ST, Holloway J, Wilson S, Bucchieri F, Puddicombe S, Davies DE. Epithelial-mesenchymal communication in the pathogenesis of chronic asthma. Proceedings of the American Thoracic Society. 2004;1:93–8. doi: 10.1513/pats.2306034. [DOI] [PubMed] [Google Scholar]
- 10.Bartram U, Speer CP. The role of transforming growth factor beta in lung development and disease. Chest. 2004;125:754–65. doi: 10.1378/chest.125.2.754. [DOI] [PubMed] [Google Scholar]
- 11.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell research. 2009;19:156–72. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. The Journal of clinical investigation. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. American journal of physiology. Lung cellular and molecular physiology. 2007;293:L525–34. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 14.Holgate ST, Davies DE, Puddicombe S, Richter A, Lackie P, Lordan J, Howarth P. Mechanisms of airway epithelial damage: epithelial-mesenchymal interactions in the pathogenesis of asthma. The European respiratory journal. Supplement. 2003;44:24s–9s. doi: 10.1183/09031936.03.00000803. [DOI] [PubMed] [Google Scholar]
- 15.Nowrin K, Sohal SS, Peterson G, Patel R, Walters EH. Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways: fibrosis, remodeling and cancer. Expert review of respiratory medicine. 2014;8:547–59. doi: 10.1586/17476348.2014.948853. [DOI] [PubMed] [Google Scholar]
- 16.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews. Molecular cell biology. 2014;15:178–96. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McDonald OG, Wu H, Timp W, Doi A, Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nature structural & molecular biology. 2011;18:867–74. doi: 10.1038/nsmb.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beutler B. TNF, immunity and inflammatory disease: Lessons of the past decade. J Invest Med. 1995;43:227–35. [PubMed] [Google Scholar]
- 19.Tian B, Nowak DE, Brasier AR. A TNF-induced gene expression program under oscillatory NF-kappaB control. BMC Genomics. 2005;6:137–54. doi: 10.1186/1471-2164-6-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian B, Nowak DE, Jamaluddin M, Wang S, Brasier AR. Identification of Direct Genomic Targets Downstream of the Nuclear Factor-{kappa}B Transcription Factor Mediating Tumor Necrosis Factor Signaling. Journal of Biological Chemistry. 2005;280:17435–48. doi: 10.1074/jbc.M500437200. [DOI] [PubMed] [Google Scholar]
- 21.Karin M. The beginning of the end: IkappaB kinase (IKK) and NF-kappaB activation. J Biol Chem. 1999;274:27339–42. doi: 10.1074/jbc.274.39.27339. [DOI] [PubMed] [Google Scholar]
- 22.Chen Lf, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, Greene WC. NF-{kappa}B RelA Phosphorylation Regulates RelA Acetylation. Molecular and Cellular Biology. 2005;25:7966–75. doi: 10.1128/MCB.25.18.7966-7975.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowak DE, Tian B, Jamaluddin M, Boldogh I, Vergara LA, Choudhary S, Brasier AR. RelA Ser276 Phosphorylation Is Required for Activation of a Subset of NF-{kappa}B-Dependent Genes by Recruiting Cyclin-Dependent Kinase 9/Cyclin T1 Complexes. Molecular and Cellular Biology. 2008;28:3623–38. doi: 10.1128/MCB.01152-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNFalpha in pulmonary pathophysiology. Respiratory research. 2006;7:125. doi: 10.1186/1465-9921-7-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lukacs NW, Strieter RM, Chensue SW, Widmer M, Kunkel SL. TNF-alpha mediates recruitment of neutrophils and eosinophils during airway inflammation. J Immunol. 1995;154:5411–7. [PubMed] [Google Scholar]
- 26.Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer cell. 2009;15:416–28. doi: 10.1016/j.ccr.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalita M, Tian B, Gao B, Choudhary S, Wood TG, Carmical JR, Boldogh I, Mitra S, Minna JD, Brasier AR. Systems approaches to modeling chronic mucosal inflammation. BioMed research international. 2013;2013:505864. doi: 10.1155/2013/505864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, Dimaio JM, Milchgrub S, Smith AL, Souza RF, Gilbey L, Zhang X, Gandia K, Vaughan MB, Wright WE, Gazdar AF, Shay JW, Minna JD. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer research. 2004;64:9027–34. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 29.Tian B, Li X, Kalita M, Widen SG, Yang J, Bhavnani SK, Dang B, Kudlicki A, Sinha M, Kong F, Wood TG, Luxon BA, Brasier AR. Analysis of the TGFbeta-induced program in primary airway epithelial cells shows essential role of NF-kappaB/RelA signaling network in type II epithelial mesenchymal transition. BMC genomics. 2015;16:529. doi: 10.1186/s12864-015-1707-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadygov RG, Zhao Y, Haidacher SJ, Starkey JM, Tilton RG, Denner L. Using power spectrum analysis to evaluate (18)O-water labeling data acquired from low resolution mass spectrometers. Journal of proteome research. 2010;9:4306–12. doi: 10.1021/pr100642q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, Denner L, Haidacher SJ, LeJeune WS, Tilton RG. Comprehensive analysis of the mouse renal cortex using two-dimensional HPLC - tandem mass spectrometry. Proteome science. 2008;6:15. doi: 10.1186/1477-5956-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Y, Brasier AR. Applications of selected reaction monitoring (SRM)-mass spectrometry (MS) for quantitative measurement of signaling pathways. Methods. 2013;61:313–22. doi: 10.1016/j.ymeth.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Y, Tian B, Edeh CB, Brasier AR. Quantitation of the dynamic profiles of the innate immune response using multiplex selected reaction monitoring-mass spectrometry. Molecular & cellular proteomics : MCP. 2013;12:1513–29. doi: 10.1074/mcp.M112.023465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalita MK, Sargsyan K, Tian B, Paulucci-Holthauzen A, Najm HN, Debusschere BJ, Brasier AR. Sources of cell-to-cell variability in canonical nuclear factor-kappaB (NF-kappaB) signaling pathway inferred from single cell dynamic images. The Journal of biological chemistry. 2011;286:37741–57. doi: 10.1074/jbc.M111.280925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cox J, Mann M. 1D and 2D annotation enrichment: a statistical method integrating quantitative proteomics with complementary high-throughput data. BMC bioinformatics. 2012;13(Suppl 16):S12. doi: 10.1186/1471-2105-13-S16-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tyanova S, Mann M, Cox J. MaxQuant for in-depth analysis of large SILAC datasets. Methods in molecular biology. 2014;1188:351–64. doi: 10.1007/978-1-4939-1142-4_24. [DOI] [PubMed] [Google Scholar]
- 37.Kramer A, Green J, Pollard J, Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics. 2014;30:523–30. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chartier M, Chenard T, Barker J, Najmanovich R. Kinome Render: a stand-alone and web-accessible tool to annotate the human protein kinome tree. PeerJ. 2013;1:e126. doi: 10.7717/peerj.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, Kuhn M, Bork P, Jensen LJ, von Mering C. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vizcaino JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Rios D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz PA, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus HJ, Albar JP, Martinez-Bartolome S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nature biotechnology. 2014;32:223–6. doi: 10.1038/nbt.2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature reviews. Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 42.Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer metastasis reviews. 2009;28:15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- 43.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. The EMBO journal. 2000;19:1745–54. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li R, Liang J, Ni S, Zhou T, Qing X, Li H, He W, Chen J, Li F, Zhuang Q, Qin B, Xu J, Li W, Yang J, Gan Y, Qin D, Feng S, Song H, Yang D, Zhang B, Zeng L, Lai L, Esteban MA, Pei D. A Mesenchymal-to-Epithelial Transition Initiates and Is Required for the Nuclear Reprogramming of Mouse Fibroblasts. Cell Stem Cell. 2010;7:51–63. doi: 10.1016/j.stem.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 45.Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. PhosphoSite: A bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–61. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y, Derynck R. Regulation of Smad signalling by protein associations and signalling crosstalk. Trends in cell biology. 1999;9:274–9. doi: 10.1016/s0962-8924(99)01579-2. [DOI] [PubMed] [Google Scholar]
- 47.Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Molecular and cellular endocrinology. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 48.Hu J, Nakano H, Sakurai H, Colburn NH. Insufficient p65 phosphorylation at S536 specifically contributes to the lack of NF-kappaB activation and transformation in resistant JB6 cells. Carcinogenesis. 2004;25:1991–2003. doi: 10.1093/carcin/bgh198. [DOI] [PubMed] [Google Scholar]
- 49.Zhang L, Cheng J, Ma Y, Thomas W, Zhang J, Du J. Dual pathways for nuclear factor kappaB activation by angiotensin II in vascular smooth muscle: phosphorylation of p65 by IkappaB kinase and ribosomal kinase. Circulation research. 2005;97:975–82. doi: 10.1161/01.RES.0000190589.52286.41. [DOI] [PubMed] [Google Scholar]
- 50.Ijaz T, Pazdrak K, Kalita M, Konig R, Choudhary S, Tian B, Boldogh I, Brasier AR. Systems Biology Approaches To Understanding Epithelial Mesenchymal Transition (EMT) In Mucosal Remodeling And Signaling In Asthma. World Allergy Organization Journal. 2014;7:13. doi: 10.1186/1939-4551-7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katsuno Y, Lamouille S, Derynck R. TGF-beta signaling and epithelial-mesenchymal transition in cancer progression. Current opinion in oncology. 2013;25:76–84. doi: 10.1097/CCO.0b013e32835b6371. [DOI] [PubMed] [Google Scholar]
- 52.Davis MA, Ireton RC, Reynolds AB. A core function for p120-catenin in cadherin turnover. The Journal of cell biology. 2003;163:525–34. doi: 10.1083/jcb.200307111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishiyama N, Lee SH, Liu S, Li GY, Smith MJ, Reichardt LF, Ikura M. Dynamic and static interactions between p120 catenin and E-cadherin regulate the stability of cell-cell adhesion. Cell. 2010;141:117–28. doi: 10.1016/j.cell.2010.01.017. [DOI] [PubMed] [Google Scholar]
- 54.Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711–24. doi: 10.1038/sj.onc.1209808. [DOI] [PubMed] [Google Scholar]
- 55.Schmitz H, Fromm M, Bentzel CJ, Scholz P, Detjen K, Mankertz J, Bode H, Epple HJ, Riecken EO, Schulzke JD. Tumor necrosis factor-alpha (TNFalpha) regulates the epithelial barrier in the human intestinal cell line HT-29/B6. Journal of cell science. 1999;112(Pt 1):137–46. doi: 10.1242/jcs.112.1.137. [DOI] [PubMed] [Google Scholar]
- 56.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature reviews. Molecular cell biology. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 57.Li H, Chen X, Gao Y, Wu J, Zeng F, Song F. XBP1 induces snail expression to promote epithelial- to-mesenchymal transition and invasion of breast cancer cells. Cellular signalling. 2015;27:82–9. doi: 10.1016/j.cellsig.2014.09.018. [DOI] [PubMed] [Google Scholar]
- 58.Koinuma D, Tsutsumi S, Kamimura N, Taniguchi H, Miyazawa K, Sunamura M, Imamura T, Miyazono K, Aburatani H. Chromatin immunoprecipitation on microarray analysis of Smad2/3 binding sites reveals roles of ETS1 and TFAP2A in transforming growth factor beta signaling. Molecular and cellular biology. 2009;29:172–86. doi: 10.1128/MCB.01038-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shirakihara T, Saitoh M, Miyazono K. Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta. Molecular biology of the cell. 2007;18:3533–44. doi: 10.1091/mbc.E07-03-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schwanbeck R, Manfioletti G, Wisniewski JR. Architecture of high mobility group protein I-C.DNA complex and its perturbation upon phosphorylation by Cdc2 kinase. The Journal of biological chemistry. 2000;275:1793–801. doi: 10.1074/jbc.275.3.1793. [DOI] [PubMed] [Google Scholar]
- 61.Palmieri D, Valentino T, D'Angelo D, De Martino I, Postiglione I, Pacelli R, Croce CM, Fedele M, Fusco A. HMGA proteins promote ATM expression and enhance cancer cell resistance to genotoxic agents. Oncogene. 2011;30:3024–35. doi: 10.1038/onc.2011.21. [DOI] [PubMed] [Google Scholar]
- 62.Singh I, Ozturk N, Cordero J, Mehta A, Hasan D, Cosentino C, Sebastian C, Kruger M, Looso M, Carraro G, Bellusci S, Seeger W, Braun T, Mostoslavsky R, Barreto G. High mobility group protein-mediated transcription requires DNA damage marker gamma-H2AX. Cell research. 2015;25:837–50. doi: 10.1038/cr.2015.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Santibanez JF. JNK mediates TGF-beta1-induced epithelial mesenchymal transdifferentiation of mouse transformed keratinocytes. FEBS letters. 2006;580:5385–91. doi: 10.1016/j.febslet.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 64.Warzecha CC, Jiang P, Amirikian K, Dittmar KA, Lu H, Shen S, Guo W, Xing Y, Carstens RP. An ESRP-regulated splicing programme is abrogated during the epithelial-mesenchymal transition. The EMBO journal. 2010;29:3286–300. doi: 10.1038/emboj.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shapiro IM, Cheng AW, Flytzanis NC, Balsamo M, Condeelis JS, Oktay MH, Burge CB, Gertler FB. An EMT-driven alternative splicing program occurs in human breast cancer and modulates cellular phenotype. PLoS genetics. 2011;7:e1002218. doi: 10.1371/journal.pgen.1002218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanopka A, Muhlemann O, Petersen-Mahrt S, Estmer C, Ohrmalm C, Akusjarvi G. Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature. 1998;393:185–7. doi: 10.1038/30277. [DOI] [PubMed] [Google Scholar]
- 67.Mermoud JE, Cohen PT, Lamond AI. Regulation of mammalian spliceosome assembly by a protein phosphorylation mechanism. The EMBO journal. 1994;13:5679–88. doi: 10.1002/j.1460-2075.1994.tb06906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 69.Desai P, Yang J, Tian B, Sun H, Kalita M, Ju H, Paulucci-Holthauzen A, Zhao Y, Brasier AR, Sadygov RG. Mixed-effects model of epithelial-mesenchymal transition reveals rewiring of signaling networks. Cellular signalling. 2015;27:1413–25. doi: 10.1016/j.cellsig.2015.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. The Journal of biological chemistry. 1999;274:30353–6. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- 71.Ballif BA, Roux PP, Gerber SA, MacKeigan JP, Blenis J, Gygi SP. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:667–72. doi: 10.1073/pnas.0409143102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Banerjee P, Ahmad MF, Grove JR, Kozlosky C, Price DJ, Avruch J. Molecular structure of a major insulin/mitogen-activated 70-kDa S6 protein kinase. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:8550–4. doi: 10.1073/pnas.87.21.8550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dalby KN, Morrice N, Caudwell FB, Avruch J, Cohen P. Identification of regulatory phosphorylation sites in mitogen-activated protein kinase (MAPK)-activated protein kinase-1a/p90rsk that are inducible by MAPK. The Journal of biological chemistry. 1998;273:1496–505. doi: 10.1074/jbc.273.3.1496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.