

Abstract
Pancreatitis is inflammation of pancreas and caused by a number of factors including pancreatic duct obstruction, alcoholism, and mutation in the cationic trypsinogen gene. Pancreatitis is represented as acute pancreatitis with acute inflammatory responses and; chronic pancreatitis characterized by marked stroma formation with a high number of infiltrating granulocytes (such as neutrophils, eosinophils), monocytes, macrophages and pancreatic stellate cells (PSCs). These inflammatory cells are known to play a central role in initiating and promoting inflammation including pancreatic fibrosis, i.e., a major risk factor for pancreatic cancer. A number of inflammatory cytokines are known to involve in promoting pancreatic pathogenesis that lead pancreatic fibrosis. Pancreatic fibrosis is a dynamic phenomenon that requires an intricate network of several autocrine and paracrine signaling pathways. In this review, we have provided the details of various cytokines and molecular mechanistic pathways (i.e., Transforming growth factor-β/SMAD, mitogen-activated protein kinases, Rho kinase, Janus kinase/signal transducers and activators, and phosphatidylinositol 3 kinase) that have a critical role in the activation of PSCs to promote chronic pancreatitis and trigger the phenomenon of pancreatic fibrogenesis. In this review of literature, we discuss the involvement of several pro-inflammatory and anti-inflammatory cytokines, such as in interleukin (IL)-1, IL-1β, IL-6, IL-8 IL-10, IL-18, IL-33 and tumor necrosis factor-α, in the pathogenesis of disease. Our review also highlights the significance of several experimental animal models that have an important role in dissecting the mechanistic pathways operating in the development of chronic pancreatitis, including pancreatic fibrosis. Additionally, we provided several intermediary molecules that are involved in major signaling pathways that might provide target molecules for future therapeutic treatment strategies for pancreatic pathogenesis.
Keywords: Pancreatitis, Pancreatic stellate cells, Transforming growth factor-β/SMAD, Janus kinase/signal transducers and activators, Mitogen-activated protein kinases
Core tip: Pancreatitis is an acute or chronic inflammatory disease of the pancreas and characterized by destruction of acinar cells, which lead activation of several inflammatory cells like macrophages and granulocytes which secrete number of pro-inflammatory cytokines. These pro-inflammatory cytokines activate pancreatic stellate cells, i.e., the key cells of pancreatic fibrosis. Various molecular signaling pathways (i.e., transforming growth factor-β/SMAD, mitogen-activated protein kinases, Rho kinase, Janus kinase/signal transducers and activators, and phosphatidylinositol 3 kinase) are known to have critical role in the activation of pancreatic stellate cells in chronic pancreatitis and development of pancreatic fibrosis that lead to the pancreatic carcinoma.
INTRODUCTION
Pancreatitis is a disease defined as acute or chronic inflammatory process of the pancreas characterized by premature activation of digestive enzymes within the pancreatic acinar cells and causing pancreatic auto-digestion[1]. In pancreatitis, a local inflammatory process initiated by release of pro- and anti-inflammatory cytokines and chemokines recruits granulocytes, monocytes and lymphocytes[2]. Annual incidence of acute pancreatitis varies from 13 to 45 per 100000 people in United States[3], whereas chronic pancreatitis ranges from 4.4 to 11.9 per 100000 per year, with a higher occurrence in Japan as compared to the United States[4-7]. Men are up to 1.5 times more likely to have chronic pancreatitis compared to women in the United States[7]. In 2009, there were 19724 admissions for chronic pancreatitis in the United States, with associated annual hospitalization costs of $172 million[5,8]. However, the pathogenesis of chronic pancreatitis is not fully understood, but it is believed that repeated episode of acute damage lead chronic pancreatitis. Recurrent pancreatic injury leads to scarring and remodeling that promotes fibrosis as well as calcification, and these calcifications develop into stones found within the tissue or pancreatic duct[5,9] (Figure 1). The main causes of pancreatitis are; obstruction in the main pancreatic duct, gallstones, alcohol misuse, smoking, hypercalcemia, hyperparathyroidism, drugs like valproate, thiazide toxicity, and genetic mutation[10-12]. During pancreatic injury, atrophic acinar cells activate several inflammatory key players like macrophages and granulocytes which release a number of pro-inflammatory cytokines [i.e., interleukin (IL)-1, IL-6, IL-8, IL-18, IL-33, and tumor necrosis factor (TNF)-α]. These pro-inflammatory cytokines further activate pancreatic stellate cells (PSCs) to promote chronic pancreatitis[13]. The detail of each cytokine involved in the pathogenesis of pancreatitis has been described independently.
Figure 1.
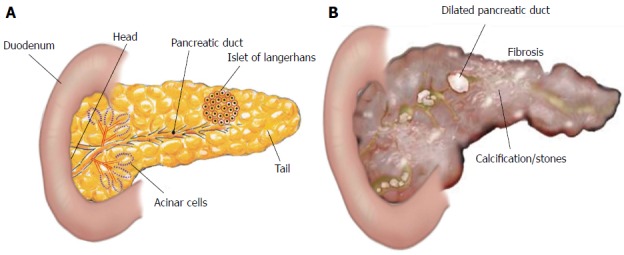
Structure of pancreas. A: The pancreas is a leaf-like structure and has two types of cells: Exocrine cells, that include acinar pancreatic duct cells, and endocrine cells, that include islets of Langerhans; B: The inflammatory process in the pancreas promotes fibrosis (scarring of tissue), calcifications or stones, and dilated pancreatic duct.
PRO-INFLAMMATORY CYTOKINES
IL-1
Induction of IL-1 has been reported in acute pancreatitis and numerous reports implicated the role of IL-1, and IL-1 receptor (IL-1R) in the pancreatic pathogenesis[14-18]. Interestingly, it has been shown that IL-1R gene-deficient mice or treatment with IL-1 receptor antagonist (rhIL-1Ra) attenuates cerulein-induced chronic pancreatitis in mice[16]. IL-1 converting enzyme (ICE) is responsible for the secretion of IL-1β from pro-IL-1β and experimental pancreatitis was significantly attenuated by pre-treatment with an ICE inactivator (VE-13045), resulting in reduced histological grading of pancreatitis and mortality. These findings were further supported by using ICE-knock out mice or intraperitoneal (i.p.) injection of ICE-inhibitor[19]. Additionally, IL-1β is also believed to play a role in the pathogenesis of pancreatitis. An elevated serum level of IL-1β has been associated with the development of acute pancreatitis[20]. Recently, Xu et al[20] have revealed that IL-1β can induce trypsin activation and decreases the cellular viability of pancreatic acinar cells. These effects depend on impaired autophagy via intracellular calcium changes. Ca2+ signaling may be a promising therapeutic target for the treatment of pancreatitis[20].
IL-6
IL-6 is a very important pro-inflammatory cytokine involved in inflammation and immune responses[21]. An important role of IL-6 has been shown in the development of acute and chronic pancreatitis as well as in pancreatic cancer. IL-6 mediates its action via gp130 protein and leads activation of Janus kinase/signal transducers and activators (JAK/STAT) signaling pathway[21]. Reported data have revealed that patients with pancreatitis indicated high serum levels of IL-6 as compare to healthy individuals[22,23]. In vitro studies have shown enhanced secretion of IL-6 from human pancreatic peri-acinar myofibroblast cells in the presence of several inflammatory mediators (i.e., TNF-α, IL-17, IL-1beta) and growth factors (i.e., fibroblast growth factor-2) and this data further supports the crucial role of IL-6 in the pathogenesis of acute pancreatitis[24,25]. Interestingly, neutralization of IL-6 by anti-IL-6 antibody therapy revealed suppression of STAT-3 activation in pancreatic acinar cells and consequently reduces the severity of acute pancreatitis[26]. The abnormal expression and deregulation of IL-6 in pancreatitis suggested that IL-6 serves as a valuable early marker for pancreatitis.
IL-8
IL-8, known as chemokine (C-X-C motif) ligand 8 or CXCL8, acts as a potent chemo-attractor of neutrophils and affects neutrophil function during onset of inflammatory responses by regulating the trafficking of various types of leukocytes through interaction with transmembrane receptors. IL-8 is produced by several types of cells such as monocytes/macrophages and epithelial cells[27,28]. Systemic complications of acute pancreatitis are associated with higher levels of IL-8[29-31]. Induction of IL-8 was also reported in a patient with aggravation of pancreatitis which suggests that IL-8 takes part in the pathogenesis of pancreatitis[32]. Severity of acute pancreatitis is associated with polymorphisms of the IL-8 gene[33]. However, the mechanism of IL-8 mediated severity of acute pancreatitis is not yet well understood and requires further study in this area.
IL-18
Induction of IL-18 is now identified in a number of disorders, such as autoimmunity[34], cutaneous[35] and allergen-induced allergic responses[36]. IL-18 is a member of IL-1 family cytokine and implicated in numerous aspects of the innate and adaptive immune system, with some analogy to IL-1β[37]. Evidences indicate that IL-18 is induced in the blood of acute[38] and chronic pancreatitis patients[39,40]. Furthermore, higher serum level of IL-18 was also reported during mild and severe forms of acute pancreatitis compared to healthy controls[41]. Additionally, the induced IL-18 level was also reported in taurocholic acid and endotoxin-induced acute pancreatitis in rat[42]. Interestingly, IL-18 along with IL-12 induces severe acute pancreatitis in obese mice[43]. Notably, it is also reported that the IL-18 has an important role in the progression of disease from acute to chronic stages[40]. Overall, IL-18 seems to be released early during the course of acute pancreatitis and may act as a key immunomodulator of the inflammatory response in severe pancreatitis and associated fibrosis. However, the mechanistic pathway of IL-18-induced chronic pancreatic pathogenesis is yet not understood.
IL-33
IL-33, a new member of the IL-1 superfamily of cytokine, binds to a complex of the ST2L/IL1 receptor accessory protein (IL1RAcP), which mediates its function[44]. Several investigations suggest a crucial role of IL-33 in the pathogenesis of chronic pancreatitis and possibly pancreatic cancer[45,46]. IL-33 was found to activate acinar cell pro-inflammatory pathways and to exacerbate acute pancreatic inflammation in mice[47]. However, the activated PSCs express IL-33 in the nucleus and regulate the platelet-derived growth factor (PDGF)-induced proliferation in PSCs[48]. IL-33 also acts as a pro-inflammatory cytokine and modulates its receptor gene expression in Colo357 cells, i.e., human pancreatic carcinoma cells[45]. IL-33 and its receptor complex (ST2L and IL1RAcP) constitute a novel signaling system; therefore, this pathway may be important in promoting acute and chronic pancreatitis. Additionally, a role for IL-33 in the stimulation, proliferation and migration of pancreatic myofibroblasts is also reported[46].
TNF-α
TNF-α is a pleiotropic cytokine and acts as a central regulator of inflammation[49,50]. It is mainly secreted by monocytes and macrophages but is also released by pancreatic acinar cells after an inflammatory trigger[51-54]. A number of studies have revealed TNF-α plays an essential role in the pathogenesis of pancreatitis and contributes inflammatory responses to disease pathogenesis[51-53,55]. An in vitro-based study indicates cultured pancreatic acinar cells are able to produce, release, and respond to TNF-α[56], leading to the activation of nuclear factor-kappa B (NFκB); interestingly, inhibition of NFκB activity decreases the inflammatory response during experimental pancreatitis[57,58]. Serum levels of TNF-α have not been considered to be a good indicator of disease severity because the liver is able to rapidly clear TNF-α before it reaches the general circulation; therefore, it is often difficult to detect TNF-α in the serum of acute pancreatitis patients[59]. One study indicates that TNF-α levels were higher in acute pancreatitis as compared to the chronic form of the disease, but its concentration did not correlate with the severity of disease[60]. In contrast to this, a recent study has shown levels of TNF-α are also increased in patients with chronic pancreatitis and the concentration of TNF-α coordinately increases in advanced chronic pancreatitis[61]. Furthermore, TNF-α mediates its effect by two surface receptors, TNF-α receptor 1 (TNFR1), or p55, and TNFR2, or p75, and both receptors are expressed in the pancreas[54,62]. Interestingly, genetic deletion of TNFR1 prevents the activity of TNF-α and revealed beneficial effects on symptom severity and mortality in cerulein-induced pancreatitis[63].
ANTI-INFLAMMATORY CYTOKINES
IL-10
IL-10 is produced by a number of activated immune cells like monocytes/macrophages, Treg, and Th1 cells[64,65]. IL-10 gene deficient mice showed more inflammatory responses and lung injury during acute pancreatitis and chronic pancreatitis[66,67]. Pre-treatment of IL-10 agonist (i.e., IT 9302) was found to reduce lung injury and mortality in a rabbit pancreatitis model[68]. Plasma IL-10 level was found to correlate with the severity of pancreatitis and could be used as a marker for severity prediction[22,69]. Initial studies based on several rodent models of acute pancreatitis revealed a protective role of IL-10 by reducing the production of inflammatory cytokines from macrophages and also diminished the level of serum amylase, serum lipase, edema, necrosis and hemorrhage[70-72]. However, recombinant IL-10 treatment in human pancreatitis has given mix responses[73]. In summary, IL-10 holds the promise of a global attenuation of the cytokine response, and more work is needed to establish its beneficial use in pancreatitis.
GRANULOCYTES INFILTRATION IS CRITICAL IN THE PATHOGENESIS OF CHRONIC PANCREATITIS
Granulocytes infiltration in the pancreas is implicated in the initiation and progression of pancreatic inflammation. The major granulocytes identified in acute and chronic pancreatitis patients are neutrophils and eosinophils. Neutrophils play a crucial role in acute inflammatory pancreatitis, are attracted to the site of injury by the help of chemokines such as CXCL8 in humans as well as CXCL1 in mouse, and further regulate the immune responses. Neutrophils remain in the blood circulation and have a very short life of approximately 24 h[74]. However, in an inflammatory condition they became activated and their lifespan is prolonged for several days, during which they control inflammatory responses and activate several pro-inflammatory mediators[75]. Trypsinogen activation is the key step for progression of pancreatitis; and a report suggested that initial trypsinogen activation is not regulated by neutrophils, whereas later activation of trypsinogen during pancreatitis is dependent on neutrophils[76]. In addition, several cases have been reported in the literature indicating the presence of increased number of eosinophils in patients with pancreatitis and termed this condition as “Eosinophilic Pancreatitis”[77,78]. Eosinophilic pancreatitis is a rarely occurring disorder and reports indicate that eosinophilic pancreatitis is frequently diagnosed only after “false positive” pancreatic resection for suspected pancreatic tumor and mimic pancreatic neoplasm[78,79]. The first report of peripheral blood eosinophilia in a patient with chronic relapsing pancreatitis with pleural effusion was published by Juniper[80] in 1955 and thereafter, several evidences came in the literature[81-84]. Tokoo et al[81] performed a study of 122 patients with chronic pancreatitis and found marked eosinophilia in approximately 21 cases (17.2%). All of the affected patients were males; no females were found affected. Endocrine pancreatic function was normal in the chronic pancreatitis patients with eosinophilia, whereas marked exocrine pancreatic dysfunction was observed in these patients. The eosinophilia of chronic pancreatitis has been frequently developed in association with severe damage to adjacent organs (pleural effusion, pericarditis, and ascites), as well as an association with pancreatic pseudocyst. This finding suggests that there may be a close correlation between marked eosinophilia and severe tissue injury during acute exacerbations of chronic pancreatitis[81]. Another study revealed 28 cases (15.6%) of chronic pancreatitis with eosinophilia among 180 chronic pancreatitis patients and the ratio of male to female patients was 8.3:1. The occurrence of eosinophilia during the course of chronic pancreatitis might be responsible for the progression of pancreatic inflammation and fibrosis[82]. Additionally, reports indicate that peripheral eosinophilia, allergic disorders, and pancreatic eosinophil infiltration have been associated with autoimmune pancreatitis[83,84]. Diagnosis and treatment of eosinophilic pancreatitis is important as it promotes pancreatic fibrosis and neoplasm.
EXPERIMENTAL TOOLS TO DISSECT THE MECHANISM THAT PROMOTES PANCREATITIS
Pathogenesis of pancreatitis is essentially understood by using experimental animal models, because of the anatomical location of the pancreas and the difficulty in procuring human tissue at different stages of the inflammatory process. Several animal models are reported to understand the pathogenesis of pancreatitis, which enable us to develop more effective treatment therapies to improve the quality of life of patients suffering from pancreatitis-associated complications. In brief, we summarize some experimental models used for understanding the disease initiation and progression.
Cerulein-induced pancreatitis model
The most widely used acute and chronic pancreatitis model, the cerulein-induced model is a highly reproducible and economical model in rats and mice[85-87]. Acute pancreatitis can be induced by intraperitoneal (i.p.) injection of cerulein (5 μg/kg per hour in rats and 50 μg/kg in mice) several times at hourly intervals, and repeated doses of cerulein can induce chronic pancreatitis[88,89]. Cerulein is an analog of cholecystokinin[90] and induces the secretion of digestive pancreatic enzymes from pancreatic acinar cells like amylase and lipase. Cerulein treatment further causes infiltration of inflammatory cells within the pancreas, pancreatic edema, and acinar cells vacuolization that are comparable to acute pancreatitis in humans. Cerulein-induced pancreatitis model has been considered as a representative model of mild acute pancreatitis of human.
L-arginine-induced pancreatitis model
Another experimental and reproducible pancreatitis model is L-arginine-induced model. This model is also widely used to study the pathophysiology of acute necrotizing pancreatitis to produce acinar cells necrosis. Initially, Mizunuma et al[91] and Tani et al[92] have demonstrated i.p. administration of excessive doses of L-arginine (500 mg/100 g body weight) in rat caused damage of pancreatic acinar cells. A single i.p. dose of 500 mg/100 g revealed necrosis in 70%-80% of acinar cells within 3 d[91,92]. Since, these observations, the L-arginine-induced acute pancreatitis rat model has been used by several investigators[93,94].
Bile salt-induced pancreatitis model
The first experimental biliary acute pancreatitis model was established by Bernard in 1856 via retrograde injection of bile and olive oil into the pancreas of a canine[95]. Since then, various bile salts such as sodium chenodeoxycholate[96], sodium taurocholate, sodium glycodeoxycholic acid[97], sodium-taurodeoxycholate and taurolithocholic acid 3-sulphate have been reported to induce acute pancreatitis in different animal models. Among these bile salts, the taurine-conjugated bile salt sodium taurocholate was the most widely used and best characterized chemical for the induction of acute pancreatitis[98]. Furthermore, a choline-deficient, ethionine-supplemented diet model is another established model to study the pathogenesis of acute and chronic pancreatitis[99,100].
Pancreatic duct ligation model
In the rat model of pancreatitis, bile reflux was first implicated in the disease pathogenesis and termed as biliary pancreatitis[101,102]. Biliary pancreatitis develops from obstruction by gallstone or bile reflux into the pancreatic duct, which causes induction of acute pancreatitis. The rat model shows that due to high pancreatic duct pressure, pancreatic juice refluxes into the bile duct in the presence of ampullary orifice obstruction, resulting in pancreatic edema, inflammatory cell infiltration, increased amylase production[103]. Chronic pancreatitis develops in these mice with time that includes atrophy, loss of acinar cells, and fibrosis[103,104].
Alcohol-induced pancreatitis model
Alcohol is another accountable factor for the pathogenesis of pancreatitis, and it has been used to trigger chronic pancreatitis in animal models[85,105,106]. Lieber and DeCarli have investigated the effects of ethanol on several organs by giving repeated feedings of ethanol as a part of the diet to rats and baboons[107] and the animals developed fatty liver disease, alcoholic hepatitis, and later on cirrhosis. Undesirably, alcohol ingestion alone did not induce chronic pancreatitis despite long experimental durations. However, the combination of alcohol with various agents such as cerulein or lipopolysaccharide exacerbated pancreatitis and resulted in fibrosis[108]. Activation of pancreatic stellate cells and fibrosis has been observed in the rat given isocaloric Lieber-DeCarli liquid diets along with alcohol for up to 10 wk and challenged with 1 or 3 repeated doses of lipopolysaccharide[109]. Alcohol-induced pancreatic damage is thought to be mediated by its metabolites, which activates ROS to cause acinar cells injury and activate pancreatic stellate cells, leading to fibrosis[110].
SNARE proteins mediating basolateral exocytosis in alcohol-induced pancreatitis
The important role of SNAREs [soluble NSF (N-ethylmaleimide-sensitive fusion proteins) attachment proteins receptors] mediating basolateral exocytosis in alcohol-induced pancreatic injury has been reported[111]. SNARE proteins are of two types (1) t-SNAREs present on the target membrane, and (2) v-SNAREs, positioned on the membrane of vesicles. The t-SNAREs, syntaxin and synaptosome-associated proteins, together make a SNARE complex which binds to v-SNAREs and triggers the fusion of vesicle and target membranes. In the pancreas, this leads to release of zymogen granules into the ducts for transport to the duodenum for their activation[112]. Additionally, a study indicates ethanol/cholecystokinin-evoked pancreatic acinar basolateral exocytosis has been mediated via protein kinase C alpha phosphorylation of Munc18c, which enables Syntaxin-4 to become receptive in forming a SNARE complex in the basolateral plasma membrane. The authors also considered this phenomenon as an operating mechanism contributing to alcoholic pancreatitis[111]. Importantly, displacement of Munc18c from the pancreatic acinar basal membrane surface has been observed in tissue samples from a patient suffering from alcohol-induced chronic pancreatitis[113].
CHRONIC PANCREATITIS LEADS FIBROSIS AND PANCREATIC CANCER
Chronic pancreatitis develops fibrosis and it is the common pathological characteristic feature and major risk factor for pancreatic cancer[114]. Recent data has shown 48960 new cases of pancreatic cancer arise and 40560 deaths occur annually in the United States because of pancreatic cancer[115]. Chronic pancreatitis is a long-standing inflammation of the pancreas that often leads to permanent damage of pancreas and serious complications, including pancreatic cancer. Chronic pancreatitis is characterized by marked stroma formation with an increased number of infiltrating macrophages and stellate cells, which are believed to play a central role in triggering inflammation and disease progression. The treatment of chronic pancreatitis and pancreatic cancer remains problematic as tissue becomes fibrotic due to injury that triggers several inflammatory, cellular as well as molecular signaling cascades that lead to formation and deposition of extra cellular matrix (ECM) at the site of injury. Several key cells are known to be involved in the process of fibrogenesis, such as inflammatory cells (e.g., macrophages and T cells), epithelial cells, fibrogenic effector cells, and endothelial cells. There are different types of effector cells in different organs, such as fibroblasts, myofibroblasts, and fibrocytes[116]. Among these cells, fibroblasts and myofibroblasts are the key cells in fibrosis and are responsible for secretion of ECM[117]. However, the function of fibrocytes is similar to the fibroblasts but to a lesser extent. Apart from this, macrophages have a more indirect contribution to fibrosis through their roles in chronic inflammation by producing a wide range of cytokines such as, transforming growth factor-β (TGF-β), PDGF, fibroblast growth factor 2 (FGF2) and insulin-like growth factor 1, all of them have pro-fibrotic effects on fibroblasts[118,119]. If fibrogenic processes persist for long time, parenchymal scarring, cellular dysfunction and organ failure take place[116]. Fibrosis is becoming a global problem and it can be of various types depending upon the tissue where it happened, such as cardiac, hepatic, renal, pulmonary, skin, liver and pancreatic fibrosis, etc. Fibrosis is an irreversible process and most of the drugs are not effective to treat fibrosis. Restriction of the progression of fibrogenesis might be a promising approach for the treatment of several fibrotic diseases.
Herein, our focus is on pancreatic fibrosis that happens during repeated injury to the pancreas. The normal pancreas has two major functions: (1) exocrine; and (2) endocrine. Exocrine pancreas comprises more than 95% of the pancreatic mass and consists of two types of pancreatic cells: (1) acinar cells, which produce digestive enzymes; and (2) ductal cells lining pancreatic ducts, which secrete a watery fluid to transport the digestive enzymes into the intestine. Endocrine pancreas mainly consists of the islets of Langerhans, which secrete insulin and other hormones[5,120] (Figure 1).
Development of fibrosis is a dynamic phenomenon that requires an intricate network of several autocrine and paracrine signaling pathways[116]. In this process, ECM formation takes place in the interstitial spaces and in areas where the exocrine compartment, mainly acinar cells are damaged[121,122]. Pancreatic injury activates acinar cells, macrophages and neutrophils which induces pro-inflammatory cytokines (IL-1, IL-6, and IL-8), chemokines (monocyte chemoattractant protein-1, macrophage inflammatory protein-1) and growth factors, which further activate quiescent PSCs[2]. The available facts suggest that these activated PSCs are the main cells in the development of fibrosis during chronic pancreatitis via secretion of TGF-β, FGF and COX-2 which leads to synthesis of ECM[123,124]. A schematic mechanistic pathway involved in the progression of chronic pancreatitis is shown below in Figure 2.
Figure 2.
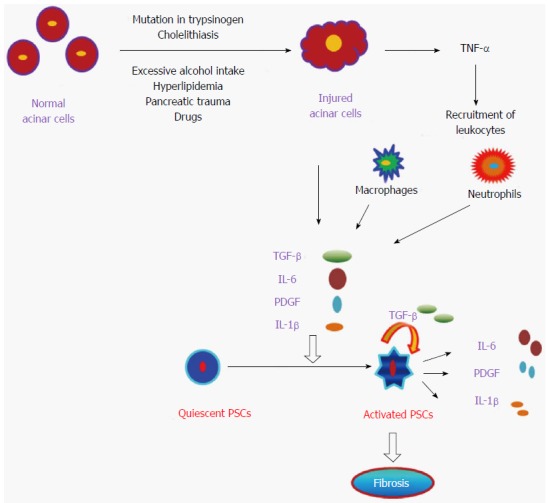
Pathogenesis of pancreatitis. Diagrammatic representation of the onset of pancreatitis by damaged pancreatic acinar cells which in turn activates quiescent pancreatic stellate cells (PSCs) to become activated PSCs and promote subsequent fibrosis of pancreas. TNF-α: Tumor necrosis factor-α; TGF-β: Transforming growth factor-β; PDGF: Platelet-derived growth factor; IL: Interleukin.
Furthermore, activated PSCs have the ability to synthesize and secrete several matrix proteins, matrix metalloproteinases (MMPs) and tissue inhibitors of matrix metalloproteinases, thus indicating that PSCs have dual functions to regulate the physiology of the exocrine pancreas, i.e., they can synthesize as well as degrade the extracellular matrix[125,126]. This indicates that PSCs have the ability to make a balance between fibrogenesis and matrix degradation to regulate the health of pancreatic tissue; that is, conservation of normal architecture or development of progressive fibrosis. Fibrosis is a complex process and the mechanism of pancreatic fibrosis is still not well understood. Due to pancreatic fibrosis, a number of therapies in pancreatic cancer have failed. In our standing, for proposing or designing any therapeutic strategy for chronic pancreatitis or pancreatic cancer, the mechanism of fibrosis development in the pancreas is important. Herein, we provide a summary of various molecular signaling pathways [i.e., TGF-β/SMAD, mitogen-activated protein kinase (MAPK), Rho kinase, JAK/STAT, and phosphatidylinositol 3 kinase (PI3K)] that have been shown to play a critical role in the activation of PSCs during chronic pancreatitis and trigger the phenomenon of fibrogenesis in pancreas (Figure 3).
Figure 3.
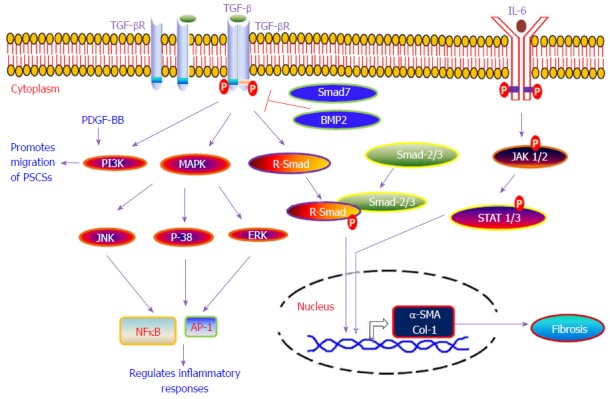
Various signaling pathways involved in the development of pancreatic fibrosis. Diagrammatic representation of various molecular signaling pathways which are involved in the development of pancreatic fibrosis. TGF-β: Transforming growth factor-β; PDGF: Platelet-derived growth factor; IL: Interleukin; MAPK: Mitogen-activated protein kinase; PI3K: Phosphatidylinositol 3 kinase; AP-1: Activator protein-1; NFκB: Nuclear factor kappa B.
TGF-β1/SMAD PATHWAY
TGF-β is a multipotent cytokine and exists in three isoforms (TGF-β1, TGF-β2 and TGF-β3) in mammals and plays an integral role in regulating immune responses, cell growth, cell differentiation and apoptosis[127,128]. TGF-β mediates its downstream signaling by binding to its specific receptors and triggers the activation of several SMAD proteins, which acts as chief transducers of the signal from the receptors to the nucleus. The receptor-regulated SMADs (R-SMADs), SMAD-2 and SMAD-3, are directly phosphorylated by the TGF-β1 receptor and make a complex with the common mediator SMAD (i.e., co-SMAD; SMAD-4) that translocate into the nucleus and activates the transcription of target genes[127,129,130]. Earlier studies have confirmed the involvement of TGF-β in the pathogenesis of acute pancreatitis, chronic pancreatitis and development of fibrosis[131-136]. PSCs play a key role in triggering pancreatic fibrosis and interestingly TGF-β was found to regulate activation and proliferation of PSCs in an autocrine manner via involvement of SMAD-2, SMAD-3 and ERK pathways[137,138]. Amelioration of pancreatic fibrosis in cerulein-treated mice was observed with defective TGF-β signaling by overexpressing a dominant-negative mutant form of TGF-β type 2 receptor (pS2-dnR II) only in the pancreas under control of pS2/TFF1 promoter[139]. Subsequent study has revealed suppression of TGF-β signaling halts cerulein-induced pancreatitis[140]. These studies indicate a functional TGF-β signaling pathway might be required for cerulein to induce acute pancreatitis in these mice[139,140]. In contrast, deactivation of TGF-β signaling induces autoimmune pancreatitis in mice, indicating the important role of TGF-β either in maintaining immune homeostasis and suppressing autoimmunity or in preserving the integrity of pancreatic acinar cells[141]. Transgenic mice with an S100A4/fibroblast-specific protein 1 Cre-mediated conditional knockout of TGF-β type 2 receptor spontaneously developed autoimmune pancreatitis in 6 wk. This indicates autoimmune pancreatitis resulted from loss of TGF-β signaling in S100A4-positive dendritic cells[142].
Plenty of evidence suggests the involvement of TGF-β in pancreatic fibrosis, however TGF-α was also found to increase the proliferation as well as migration of PSCs via up-regulation of MMP-1, which might contribute to the pathogenesis of chronic pancreatitis[143]. A recent report has shown loss of SMAD-4 synergizes with TGF-α over-expression in promoting pancreatic metaplasia, PanIN development, and fibrosis[144]. Furthermore, a higher level of TGF-β1 during pancreatic inflammation triggers the deregulation of the micro-RNA-217-SIRT1 pathway and then promotes EMT and subsequent fibrosis in the pancreas[145]. Although, TGF-β still remains elusive in terms of our understanding of its multifunctional modes of action and TGF-β also activates SMAD-independent signaling pathways including MAPK pathways and phosphoinositide (PI) 3-kinase[129,146,147] but the detailed mechanisms are not well understood.
MAPK
MAPK are of three types, ERK, JNK, and p-38, and play an important role in a variety of cellular processes, including cell proliferation, cell survival, apoptosis, and cytokine production[148]. In alcohol-induced pancreatic injury, ethanol and its metabolite acetaldehyde were found to induce activator protein-1 (AP-1) and MAPK signaling in PSCs[149,150]. Furthermore, CX3CL1 is a chemokine that serves as an adhesion molecule as well as a migration factor, and was elevated in patients with alcoholic chronic pancreatitis[151]. A recent report indicates ethanol induces CX3CL1 release via ERK activation in PSCs[152]. However, H2O2 induces oxidative stress, AP-1, MAP-kinase pathway and expression of α (I) procollagen in PSCs[153]. Apart from this, PDGF induces rapid activation of Raf-1, ERK 1/2, and AP-1 protein and further indicates a correlation between ERK activity and PSC activation[154]. Furthermore, the involvement of protease-activated receptor-2 (PAR-2) was also found in the pathogenesis of pancreatitis and PAR-2 agonists increased collagen synthesis via activation of JNK and p-38 MAP kinase pathways in PSCs, suggesting the role of PAR-2 during induction of pancreatic fibrosis[155]. PD98059 is an inhibitor of MAP/ERK kinase-1 (MEK-1) pathway and was able to protect against cerulein-induced acute pancreatitis in mice[156]. Apart from this, angiotensin II-treated PSCs start proliferation and increase DNA synthesis via an epidermal growth factor receptor transactivation-ERK activation pathway, indicating the possible role of angiotensin II in development of pancreatic fibrosis[157,158]. Taken together, these studies have broadened our knowledge to understand the role of the MAP kinase signaling pathway in the development of pancreatitis-associated fibrosis, but still the existence of several molecular signaling pathways which may cross-talk to each other have an important role in the development of fibrosis and need to be explored further.
RHO KINASE PATHWAY
In chronic pancreatitis, activation of PSCs and induced stress fiber formation suggest the reorganization of cytoskeletal proteins is involved in this disease process[159]. The Rho family proteins RhoA, Rac and Cdc42 are considered the core molecules that induce stress fiber formation and regulate cellular adherence by remodeling of the cytoskeleton in response to external signals[160,161]. Further, the inhibition of Rho A signaling diminished the endothelial hyper-permeability that was induced by sera from severe acute pancreatitis patients with lung injury via inhibiting F-actin aggregates[162]. Inhibitors of Rho kinase such as Y-27632 and HA-1077 (fasudil) block activity of PSCs, via reducing α-SMA, proliferation, chemotaxis, and type I collagen production in culture-activated PSCs[163]. During cerulein-induced pancreatitis in mice, Y-27632 caused induction of serum amylase levels, higher interstitial edema and vacuolization at 12-18 h after the first injection of cerulein. Y-27632 in turn inhibited the recovery of protein expression of ROCK-II at 18 h after the first cerulein injection. These results indicate that RhoA and ROCK-II accumulate in normal CCK-stimulated pancreatic enzyme secretion and prevent cerulein-induced acute pancreatitis[164]. Rho-kinase signaling was found to regulate trypsinogen activation and its release from the pancreatic acinar cells during acute pancreatitis and subsequent CXC chemokine formation, neutrophil infiltration and tissue injury[165]. Thus, these results indicate that Rho-kinase may serve as a novel molecular target for future treatment of acute pancreatitis, but there is need for vast effort to understand the Rho-Kinase signaling in pancreatitis. This area opens up a new avenue for future research.
JAK/STAT SIGNALING PATHWAY
The JAK/STAT signaling pathway regulates several cellular functions such as cell proliferation, differentiation, and inflammatory responses[166-168]. IL-6 is a well-known pro-inflammatory cytokine and mediates its action via JAK/STAT signaling pathway[21] and plays a crucial role in the progression of pancreatitis. Various reports have indicated higher serum levels of IL-6 in patients with pancreatitis as compared to healthy individuals[22,23]. Furthermore, an in-vitro study also indicates induced secretion of IL-6 from the human pancreatic peri-acinar myofibroblast cells under the influence of several inflammatory mediators, such as TNF-α, IL-17, IL-1β and FGF-2; this data further indicates the crucial role of IL-6 in the pathogenesis of acute pancreatitis[24,25]. Interestingly, blockade of IL-6 using anti-IL-6 antibody suppresses STAT-3 activation in the pancreatic acinar cells and consequently diminishes the severity of acute pancreatitis by induction of pancreatic acinar cell apoptosis[26]. Apart from this, another report suggests that PDGF induces the proliferation of PSCs[169] by activating the JAK-2/STAT-3 pathway[155]. The inhibition of JAK-1/STAT-1 improves the severity of cerulein-stimulated pancreatic injury by inhibiting the activation of NFĸB, and this indicates that activation of JAK-1/STAT-1 is involved in the early events of pancreatic injury[170]. Still, a better understanding of the JAK/STAT signaling pathway is required to know its role in the proliferation of PSCs and progression of fibrosis in chronic pancreatitis.
PI3K-AKT PATHWAY
PI3K-Akt is a major intracellular signaling pathway that belongs to a family of lipid and protein kinases. When growth factors bind to membrane bound receptor tyrosine kinase, it activates PI3K and its downstream regulators Akt and mTOR and regulates several aspects such as cell growth, survival, apoptosis and inflammation[171-173]. Earlier, it has been shown the PI3K pathway inhibitor wortmannin reduces the intra pancreatic activation of trypsinogen in acinar cells[174] and decreases inflammatory cytokines in severe acute pancreatitis in rats[175]. These reports suggest involvement of the PI3K pathway in the pathogenesis of acute pancreatitis. PI3Kγ is an isoform of PI3K known to regulate pathologic responses of the pancreatic acinar cells during pancreatitis[176]. The role of PI3Kγ was studied in two different models of acute pancreatitis, cerulein and choline-deficient/ethionine-supplemented diet. Mice lacking the PI3Kγ gene are protected from acinar cell injury/necrosis and show reduced severity of acute pancreatitis, indicating PI3K inhibitors may provide a possible therapy for acute pancreatitis[172,177].
CLINICAL CHARACTERISTICS AND DIAGNOSIS OF PANCREATITIS
The major clinical characteristics of pancreatitis are abdominal pain localized to the upper-to-middle abdomen, abdominal distension, nausea, fever, flank pain, vomiting, back pain, jaundice, hematemesis, melena diarrhea with foul-smelling, oily bowel movements and weight loss[178]. Abdominal pain is the most common symptom found in 50% to 80% of cases, and it is the major cause for hospitalizations of patients related to pancreatitis. Although the pancreatic pain is low in the abdomen, following food intake it worsens and becomes localized to the epigastric area[179]. Ammann et al[180] have identified two types of pancreatic pain (type A and type B) on the basis of natural history of alcoholic chronic pancreatitis. In type A pain, there are short (< 10 d) episodes of acute pain with long pain-free periods, whereas type B pain persists for a longer period of time (1-2 mo) with intervals of intense pain. Type A pain is experienced more often and is typically easier to treat. Several serum-based biomarkers have been identified for the diagnosis of acute pancreatitis such as amylase, lipase and trypsinogen[181]. In acute pancreatitis, the level of amylase (glycoside hydrolase) is rapidly induced within 4 to 6 h of disease onset, remains high for 3 to 4 d and sensitivity decreases with time from onset[182-184]. Higher levels of lipase have been found during the onset of acute pancreatitis, and it is more specific and sensitive than amylase for detecting acute pancreatitis because serum level of lipase remain elevated for around 2 wk before it returns to the normal level[183,185]. The sensitivity and specificity of amylase is about 63.6% and 99.4%, whereas sensitivity and specificity of lipase were 95.5% and 99.2%, respectively[186,187]. Pancreatic lipase is four times more active than amylase and it is less affected by exocrine pancreatic deficiency occurring in patients with chronic pancreatitis[183,188]. Trypsinogen is the inactive form of the enzyme trypsin and is cleaved by duodenal enterokinase to produce the active enzyme trypsin and trypsinogen activated peptide[183,189]. Normally trypsinogen is secreted in very low levels from pancreatic acinar cells but during pancreatitis secreted trypsinogen enzyme moves into the systemic circulation due to increased vascular permeability, and consequently there is increased clearance in the urine. During the onset of disease, trypsinogen concentration is elevated in the serum as well as urine and declines to normal level within 3 to 5 d[183,185,190].
CURRENT TREATMENTS STRATEGY
The first-line of treatment involves fasting along with intravenous fluids if the pancreatitis is very painful and this help the pancreas to rest and recover. Depending on the underlying cause of pancreatitis, management may vary to address the specific cause. Currently, several medications and treatment options are available such as analgesics likes paracetamol or non-steroidal anti-inflammatory drugs or both followed by tramadol, perhaps coupled with a neuroleptic antidepressant. Another option is steroid therapy in which prednisolone is used for the treatment of autoimmune pancreatitis[191,192]. Furthermore, micronutrient therapy seems to be promising and it includes vitamin C, E, B6, B12, folic acid, methionine, and β-carotene. Braganza et al[10] have revealed that micronutrient therapy is designed to supply methyl and thiol moieties, which are helpful to restrict the generation of reactive oxygen species and deactivate pro-inflammatory oxidation products, reduce mast cell degranulation, decrease necrosis of pancreatic acinar cells and lessen pro-fibrotic induction. The outcome of these six clinical trial-based studies revealed that micronutrient therapy controls the pain and curbs attacks in patients suffering with chronic pancreatitis[193-199]. If pancreatitis is due to an obstructing gallstone, surgical intervention may be needed to remove the gallstone. Intervention may also be required to treat a pseudocyst or surgically remove the part of affected pancreas. Micronutrient treatment seems to substantially reduce the need for surgery.
CONCLUSION
The current review provides a comprehensive understanding of the development of chronic pancreatitis and the role of cells and cytokines involved in promoting pathogenesis. Briefly, we have discussed disease characteristics, molecular mechanisms involved in pancreatitis, the role of granulocytes such as neutrophils and eosinophils, the details of associated cytokines and chemokines implicated in the progression along with major signaling pathways such as TGF-β/SMAD, MAP kinase, PI3K, Rho kinase, and JAK/STAT that are crucial in the development of pancreatic fibrosis following pancreatic injury. This review will help to understand the intricate process of several autocrine and paracrine pathways involved in pancreatitis pathogenesis including remodeling. We provided details regarding the disease that might be useful for investigators to focus on, and cells and their associated mediators that might be helpful for future strategies for diagnostic and therapeutic interventions in the treatment of pancreatitis.
ACKNOWLEDGMENTS
Dr. Mishra is Endowed Schlieder Chair; therefore, we thank Edward G Schlieder Educational Foundation for their support in conducting our research activities.
Footnotes
Supported by National Institutes of Health, Nos. R01 DK067255 and R01 AI080581.
Conflict-of-interest statement: All authors declare no conflict of interests for this review.
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): D
Grade E (Poor): 0
Peer-review started: June 13, 2016
First decision: July 20, 2016
Article in press: August 16, 2016
P- Reviewer: Laura CB, Fujino Y, Zhang ZM S- Editor: Gong ZM L- Editor: A E- Editor: Wu HL
References
- 1.Shah AU, Sarwar A, Orabi AI, Gautam S, Grant WM, Park AJ, Shah AU, Liu J, Mistry PK, Jain D, et al. Protease activation during in vivo pancreatitis is dependent on calcineurin activation. Am J Physiol Gastrointest Liver Physiol. 2009;297:G967–G973. doi: 10.1152/ajpgi.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng L, Xue J, Jaffee EM, Habtezion A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology. 2013;144:1230–1240. doi: 10.1053/j.gastro.2012.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yadav D, Lowenfels AB. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1252–1261. doi: 10.1053/j.gastro.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang AL, Vadhavkar S, Singh G, Omary MB. Epidemiology of alcohol-related liver and pancreatic disease in the United States. Arch Intern Med. 2008;168:649–656. doi: 10.1001/archinte.168.6.649. [DOI] [PubMed] [Google Scholar]
- 5.Elham A. Introduction to Pancreatic Disease: Chronic Pancreatitis. Available from: https://www.pancreapedia.org/reviews/introduction-to-pancreatic-disease-chronic-pancreatitis.
- 6.Hirota M, Shimosegawa T, Masamune A, Kikuta K, Kume K, Hamada S, Kihara Y, Satoh A, Kimura K, Tsuji I, et al. The sixth nationwide epidemiological survey of chronic pancreatitis in Japan. Pancreatology. 1979;12:79–84. doi: 10.1016/j.pan.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Yang SJ, Chen HM, Hsieh CH, Hsu JT, Yeh CN, Yeh TS, Hwang TL, Jan YY, Chen MF. Akt pathway is required for oestrogen-mediated attenuation of lung injury in a rodent model of cerulein-induced acute pancreatitis. Injury. 2011;42:638–642. doi: 10.1016/j.injury.2010.07.242. [DOI] [PubMed] [Google Scholar]
- 8.Peery AF, Dellon ES, Lund J, Crockett SD, McGowan CE, Bulsiewicz WJ, Gangarosa LM, Thiny MT, Stizenberg K, Morgan DR, et al. Burden of gastrointestinal disease in the United States: 2012 update. Gastroenterology. 2012;143:1179–1187.e1-3. doi: 10.1053/j.gastro.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Löhr JM. What are the useful biological and functional markers of early-stage chronic pancreatitis? J Gastroenterol. 2007;42 Suppl 17:66–71. doi: 10.1007/s00535-006-1932-9. [DOI] [PubMed] [Google Scholar]
- 10.Braganza JM, Dormandy TL. Micronutrient therapy for chronic pancreatitis: rationale and impact. JOP. 2010;11:99–112. [PubMed] [Google Scholar]
- 11.Braganza JM, Lee SH, McCloy RF, McMahon MJ. Chronic pancreatitis. Lancet. 2011;377:1184–1197. doi: 10.1016/S0140-6736(10)61852-1. [DOI] [PubMed] [Google Scholar]
- 12.Lankisch PG, Apte M, Banks PA. Acute pancreatitis. Lancet. 2015;386:85–96. doi: 10.1016/S0140-6736(14)60649-8. [DOI] [PubMed] [Google Scholar]
- 13.Zhang H, Neuhöfer P, Song L, Rabe B, Lesina M, Kurkowski MU, Treiber M, Wartmann T, Regnér S, Thorlacius H, et al. IL-6 trans-signaling promotes pancreatitis-associated lung injury and lethality. J Clin Invest. 2013;123:1019–1031. doi: 10.1172/JCI64931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka N, Murata A, Uda K, Toda H, Kato T, Hayashida H, Matsuura N, Mori T. Interleukin-1 receptor antagonist modifies the changes in vital organs induced by acute necrotizing pancreatitis in a rat experimental model. Crit Care Med. 1995;23:901–908. doi: 10.1097/00003246-199505000-00019. [DOI] [PubMed] [Google Scholar]
- 15.Fink GW, Norman JG. Specific changes in the pancreatic expression of the interleukin 1 family of genes during experimental acute pancreatitis. Cytokine. 1997;9:1023–1027. doi: 10.1006/cyto.1997.0260. [DOI] [PubMed] [Google Scholar]
- 16.Norman JG, Fink GW, Sexton C, Carter G. Transgenic animals demonstrate a role for the IL-1 receptor in regulating IL-1beta gene expression at steady-state and during the systemic stress induced by acute pancreatitis. J Surg Res. 1996;63:231–236. doi: 10.1006/jsre.1996.0253. [DOI] [PubMed] [Google Scholar]
- 17.Norman JG, Fink GW, Denham W, Yang J, Carter G, Sexton C, Falkner J, Gower WR, Franz MG. Tissue-specific cytokine production during experimental acute pancreatitis. A probable mechanism for distant organ dysfunction. Dig Dis Sci. 1997;42:1783–1788. doi: 10.1023/a:1018886120711. [DOI] [PubMed] [Google Scholar]
- 18.Norman J, Yang J, Fink G, Carter G, Ku G, Denham W, Livingston D. Severity and mortality of experimental pancreatitis are dependent on interleukin-1 converting enzyme (ICE) J Interferon Cytokine Res. 1997;17:113–118. doi: 10.1089/jir.1997.17.113. [DOI] [PubMed] [Google Scholar]
- 19.Paszkowski AS, Rau B, Mayer JM, Möller P, Beger HG. Therapeutic application of caspase 1/interleukin-1beta-converting enzyme inhibitor decreases the death rate in severe acute experimental pancreatitis. Ann Surg. 2002;235:68–76. doi: 10.1097/00000658-200201000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu B, Bai B, Sha S, Yu P, An Y, Wang S, Kong X, Liu C, Wei N, Feng Q, et al. Interleukin-1β induces autophagy by affecting calcium homeostasis and trypsinogen activation in pancreatic acinar cells. Int J Clin Exp Pathol. 2014;7:3620–3631. [PMC free article] [PubMed] [Google Scholar]
- 21.Lesina M, Wörmann SM, Neuhöfer P, Song L, Algül H. Interleukin-6 in inflammatory and malignant diseases of the pancreas. Semin Immunol. 2014;26:80–87. doi: 10.1016/j.smim.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Berney T, Gasche Y, Robert J, Jenny A, Mensi N, Grau G, Vermeulen B, Morel P. Serum profiles of interleukin-6, interleukin-8, and interleukin-10 in patients with severe and mild acute pancreatitis. Pancreas. 1999;18:371–377. doi: 10.1097/00006676-199905000-00007. [DOI] [PubMed] [Google Scholar]
- 23.Hansen M, Nielsen AR, Vilsbøll T, Lund A, Krarup T, Knop FK, Vestergaard H. Increased levels of YKL-40 and interleukin 6 in patients with chronic pancreatitis and secondary diabetes. Pancreas. 2012;41:1316–1318. doi: 10.1097/MPA.0b013e31824d9b93. [DOI] [PubMed] [Google Scholar]
- 24.Shimada M, Andoh A, Hata K, Tasaki K, Araki Y, Fujiyama Y, Bamba T. IL-6 secretion by human pancreatic periacinar myofibroblasts in response to inflammatory mediators. J Immunol. 2002;168:861–868. doi: 10.4049/jimmunol.168.2.861. [DOI] [PubMed] [Google Scholar]
- 25.Andoh A, Bamba S, Fujino S, Inatomi O, Zhang Z, Kim S, Takayanagi A, Shimizu N, Fujiyama Y. Fibroblast growth factor-2 stimulates interleukin-6 secretion in human pancreatic periacinar myofibroblasts. Pancreas. 2004;29:278–283. doi: 10.1097/00006676-200411000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Chao KC, Chao KF, Chuang CC, Liu SH. Blockade of interleukin 6 accelerates acinar cell apoptosis and attenuates experimental acute pancreatitis in vivo. Br J Surg. 2006;93:332–338. doi: 10.1002/bjs.5251. [DOI] [PubMed] [Google Scholar]
- 27.Singh JK, Simões BM, Howell SJ, Farnie G, Clarke RB. Recent advances reveal IL-8 signaling as a potential key to targeting breast cancer stem cells. Breast Cancer Res. 2013;15:210. doi: 10.1186/bcr3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jundi K, Greene CM. Transcription of Interleukin-8: How Altered Regulation Can Affect Cystic Fibrosis Lung Disease. Biomolecules. 2015;5:1386–1398. doi: 10.3390/biom5031386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKay CJ, Gallagher G, Brooks B, Imrie CW, Baxter JN. Increased monocyte cytokine production in association with systemic complications in acute pancreatitis. Br J Surg. 1996;83:919–923. doi: 10.1002/bjs.1800830712. [DOI] [PubMed] [Google Scholar]
- 30.Rau B, Steinbach G, Gansauge F, Mayer JM, Grünert A, Beger HG. The potential role of procalcitonin and interleukin 8 in the prediction of infected necrosis in acute pancreatitis. Gut. 1997;41:832–840. doi: 10.1136/gut.41.6.832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daniel P, Leśniowski B, Mokrowiecka A, Jasińska A, Pietruczuk M, Małecka-Panas E. Circulating levels of visfatin, resistin and pro-inflammatory cytokine interleukin-8 in acute pancreatitis. Pancreatology. 2010;10:477–482. doi: 10.1159/000276986. [DOI] [PubMed] [Google Scholar]
- 32.Zhukova EN. Blood interleukin-8 in patients with different stages of chronic relapsing pancreatitis and its role in the pathogenesis of pancreatitis. Ross Gastroenterol Zh. 2001;(1):15–18. [PubMed] [Google Scholar]
- 33.Hofner P, Balog A, Gyulai Z, Farkas G, Rakonczay Z, Takács T, Mándi Y. Polymorphism in the IL-8 gene, but not in the TLR4 gene, increases the severity of acute pancreatitis. Pancreatology. 2006;6:542–548. doi: 10.1159/000097363. [DOI] [PubMed] [Google Scholar]
- 34.Sedimbi SK, Hägglöf T, Karlsson MC. IL-18 in inflammatory and autoimmune disease. Cell Mol Life Sci. 2013;70:4795–4808. doi: 10.1007/s00018-013-1425-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee JH, Cho DH, Park HJ. IL-18 and Cutaneous Inflammatory Diseases. Int J Mol Sci. 2015;16:29357–29369. doi: 10.3390/ijms161226172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niranjan R, Rajavelu P, Ventateshaiah SU, Shukla JS, Zaidi A, Mariswamy SJ, Mattner J, Fortgang I, Kowalczyk M, Balart L, et al. Involvement of interleukin-18 in the pathogenesis of human eosinophilic esophagitis. Clin Immunol. 2015;157:103–113. doi: 10.1016/j.clim.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 binding protein. Front Immunol. 2013;4:289. doi: 10.3389/fimmu.2013.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ueda T, Takeyama Y, Yasuda T, Matsumura N, Sawa H, Nakajima T, Ajiki T, Fujino Y, Suzuki Y, Kuroda Y. Significant elevation of serum interleukin-18 levels in patients with acute pancreatitis. J Gastroenterol. 2006;41:158–165. doi: 10.1007/s00535-005-1735-4. [DOI] [PubMed] [Google Scholar]
- 39.Yuan BS, Zhu RM, Braddock M, Zhang XH, Shi W, Zheng MH. Interleukin-18: a pro-inflammatory cytokine that plays an important role in acute pancreatitis. Expert Opin Ther Targets. 2007;11:1261–1271. doi: 10.1517/14728222.11.10.1261. [DOI] [PubMed] [Google Scholar]
- 40.Schneider A, Haas SL, Hildenbrand R, Siegmund S, Reinhard I, Nakovics H, Singer MV, Feick P. Enhanced expression of interleukin-18 in serum and pancreas of patients with chronic pancreatitis. World J Gastroenterol. 2006;12:6507–6514. doi: 10.3748/wjg.v12.i40.6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wereszczynska-Siemiatkowska U, Mroczko B, Siemiatkowski A. Serum profiles of interleukin-18 in different severity forms of human acute pancreatitis. Scand J Gastroenterol. 2002;37:1097–1102. doi: 10.1080/003655202320378310. [DOI] [PubMed] [Google Scholar]
- 42.Pastor CM, Morel DR, Vonlaufen A, Schiffer E, Lescuyer P, Frossard JL. Delayed production of IL-18 in lungs and pancreas of rats with acute pancreatitis. Pancreatology. 2010;10:752–757. doi: 10.1159/000317283. [DOI] [PubMed] [Google Scholar]
- 43.Sennello JA, Fayad R, Pini M, Gove ME, Ponemone V, Cabay RJ, Siegmund B, Dinarello CA, Fantuzzi G. Interleukin-18, together with interleukin-12, induces severe acute pancreatitis in obese but not in nonobese leptin-deficient mice. Proc Natl Acad Sci USA. 2008;105:8085–8090. doi: 10.1073/pnas.0804091105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 45.Schmieder A, Multhoff G, Radons J. Interleukin-33 acts as a pro-inflammatory cytokine and modulates its receptor gene expression in highly metastatic human pancreatic carcinoma cells. Cytokine. 2012;60:514–521. doi: 10.1016/j.cyto.2012.06.286. [DOI] [PubMed] [Google Scholar]
- 46.Nishida A, Andoh A, Imaeda H, Inatomi O, Shiomi H, Fujiyama Y. Expression of interleukin 1-like cytokine interleukin 33 and its receptor complex (ST2L and IL1RAcP) in human pancreatic myofibroblasts. Gut. 2010;59:531–541. doi: 10.1136/gut.2009.193599. [DOI] [PubMed] [Google Scholar]
- 47.Kempuraj D, Twait EC, Williard DE, Yuan Z, Meyerholz DK, Samuel I. The novel cytokine interleukin-33 activates acinar cell proinflammatory pathways and induces acute pancreatic inflammation in mice. PLoS One. 2013;8:e56866. doi: 10.1371/journal.pone.0056866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masamune A, Watanabe T, Kikuta K, Satoh K, Kanno A, Shimosegawa T. Nuclear expression of interleukin-33 in pancreatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2010;299:G821–G832. doi: 10.1152/ajpgi.00178.2010. [DOI] [PubMed] [Google Scholar]
- 49.Zhang JM, An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin. 2007;45:27–37. doi: 10.1097/AIA.0b013e318034194e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Esposito E, Cuzzocrea S. TNF-alpha as a therapeutic target in inflammatory diseases, ischemia-reperfusion injury and trauma. Curr Med Chem. 2009;16:3152–3167. doi: 10.2174/092986709788803024. [DOI] [PubMed] [Google Scholar]
- 51.Malleo G, Mazzon E, Siriwardena AK, Cuzzocrea S. Role of tumor necrosis factor-alpha in acute pancreatitis: from biological basis to clinical evidence. Shock. 2007;28:130–140. doi: 10.1097/shk.0b013e3180487ba1. [DOI] [PubMed] [Google Scholar]
- 52.Malleo G, Mazzon E, Siriwardena AK, Cuzzocrea S. TNF-alpha as a therapeutic target in acute pancreatitis--lessons from experimental models. ScientificWorldJournal. 2007;7:431–448. doi: 10.1100/tsw.2007.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Norman JG, Fink GW, Franz MG. Acute pancreatitis induces intrapancreatic tumor necrosis factor gene expression. Arch Surg. 1995;130:966–970. doi: 10.1001/archsurg.1995.01430090052018. [DOI] [PubMed] [Google Scholar]
- 54.Hohmann HP, Remy R, Brockhaus M, van Loon AP. Two different cell types have different major receptors for human tumor necrosis factor (TNF alpha) J Biol Chem. 1989;264:14927–14934. [PubMed] [Google Scholar]
- 55.Schäfer C, Tietz AB, Göke B. Pathophysiology of acute experimental pancreatitis: lessons from genetically engineered animal models and new molecular approaches. Digestion. 2005;71:162–172. doi: 10.1159/000086138. [DOI] [PubMed] [Google Scholar]
- 56.Gukovskaya AS, Gukovsky I, Zaninovic V, Song M, Sandoval D, Gukovsky S, Pandol SJ. Pancreatic acinar cells produce, release, and respond to tumor necrosis factor-alpha. Role in regulating cell death and pancreatitis. J Clin Invest. 1997;100:1853–1862. doi: 10.1172/JCI119714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Virlos I, Mazzon E, Serraino I, Di Paola R, Genovese T, Britti D, Thiemerman C, Siriwardena A, Cuzzocrea S. Pyrrolidine dithiocarbamate reduces the severity of cerulein-induced murine acute pancreatitis. Shock. 2003;20:544–550. doi: 10.1097/01.shk.0000093543.78705.aa. [DOI] [PubMed] [Google Scholar]
- 58.Virlos I, Mazzon E, Serraino I, Genovese T, Di Paola R, Thiemerman C, Siriwardena A, Cuzzocrea S. Calpain I inhibitor ameliorates the indices of disease severity in a murine model of cerulein-induced acute pancreatitis. Intensive Care Med. 2004;30:1645–1651. doi: 10.1007/s00134-004-2328-z. [DOI] [PubMed] [Google Scholar]
- 59.Grewal HP, Kotb M, el Din AM, Ohman M, Salem A, Gaber L, Gaber AO. Induction of tumor necrosis factor in severe acute pancreatitis and its subsequent reduction after hepatic passage. Surgery. 1994;115:213–221. [PubMed] [Google Scholar]
- 60.Kıyıcı A, İbiş M, Akbulut S, Köklü S, Uçar E, Ünlü A. Serum TNF-alpha levels in acute and chronic pancreatitis. Eur J Gen Med. 2009;6:103–107. [Google Scholar]
- 61.Gasiorowska A, Talar-Wojnarowska R, Kaczka A, Borkowska A, Czupryniak L, Małecka-Panas E. Subclinical Inflammation and Endothelial Dysfunction in Patients with Chronic Pancreatitis and Newly Diagnosed Pancreatic Cancer. Dig Dis Sci. 2016;61:1121–1129. doi: 10.1007/s10620-015-3972-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115:1–20. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Denham W, Yang J, Fink G, Denham D, Carter G, Ward K, Norman J. Gene targeting demonstrates additive detrimental effects of interleukin 1 and tumor necrosis factor during pancreatitis. Gastroenterology. 1997;113:1741–1746. doi: 10.1053/gast.1997.v113.pm9352880. [DOI] [PubMed] [Google Scholar]
- 64.Sabat R, Grütz G, Warszawska K, Kirsch S, Witte E, Wolk K, Geginat J. Biology of interleukin-10. Cytokine Growth Factor Rev. 2010;21:331–344. doi: 10.1016/j.cytogfr.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 65.Iyer SS, Cheng G. Role of interleukin 10 transcriptional regulation in inflammation and autoimmune disease. Crit Rev Immunol. 2012;32:23–63. doi: 10.1615/critrevimmunol.v32.i1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gloor B, Todd KE, Lane JS, Rigberg DA, Reber HA. Mechanism of increased lung injury after acute pancreatitis in IL-10 knockout mice. J Surg Res. 1998;80:110–114. doi: 10.1006/jsre.1997.5289. [DOI] [PubMed] [Google Scholar]
- 67.Demols A, Van Laethem JL, Quertinmont E, Degraef C, Delhaye M, Geerts A, Deviere J. Endogenous interleukin-10 modulates fibrosis and regeneration in experimental chronic pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2002;282:G1105–G1112. doi: 10.1152/ajpgi.00431.2001. [DOI] [PubMed] [Google Scholar]
- 68.Osman MO, Jacobsen NO, Kristensen JU, Deleuran B, Gesser B, Larsen CG, Jensen SL. IT 9302, a synthetic interleukin-10 agonist, diminishes acute lung injury in rabbits with acute necrotizing pancreatitis. Surgery. 1998;124:584–592. [PubMed] [Google Scholar]
- 69.Chen CC, Wang SS, Lu RH, Chang FY, Lee SD. Serum interleukin 10 and interleukin 11 in patients with acute pancreatitis. Gut. 1999;45:895–899. doi: 10.1136/gut.45.6.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kusske AM, Rongione AJ, Ashley SW, McFadden DW, Reber HA. Interleukin-10 prevents death in lethal necrotizing pancreatitis in mice. Surgery. 1996;120:284–288; discussion 289. doi: 10.1016/s0039-6060(96)80299-6. [DOI] [PubMed] [Google Scholar]
- 71.Van Laethem JL, Marchant A, Delvaux A, Goldman M, Robberecht P, Velu T, Devière J. Interleukin 10 prevents necrosis in murine experimental acute pancreatitis. Gastroenterology. 1995;108:1917–1922. doi: 10.1016/0016-5085(95)90158-2. [DOI] [PubMed] [Google Scholar]
- 72.Rongione AJ, Kusske AM, Reber HA, Ashley SW, McFadden DW. Interleukin-10 reduces circulating levels of serum cytokines in experimental pancreatitis. J Gastrointest Surg. 1997;1:159–165; discussion 165-166. doi: 10.1016/s1091-255x(97)80104-7. [DOI] [PubMed] [Google Scholar]
- 73.Dumot JA, Conwell DL, Zuccaro G, Vargo JJ, Shay SS, Easley KA, Ponsky JL. A randomized, double blind study of interleukin 10 for the prevention of ERCP-induced pancreatitis. Am J Gastroenterol. 2001;96:2098–2102. doi: 10.1111/j.1572-0241.2001.04092.x. [DOI] [PubMed] [Google Scholar]
- 74.Witko-Sarsat V, Pederzoli-Ribeil M, Hirsch E, Sozzani S, Cassatella MA. Regulating neutrophil apoptosis: new players enter the game. Trends Immunol. 2011;32:117–124. doi: 10.1016/j.it.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 75.Yang ZW, Meng XX, Xu P. Central role of neutrophil in the pathogenesis of severe acute pancreatitis. J Cell Mol Med. 2015;19:2513–2520. doi: 10.1111/jcmm.12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Abdulla A, Awla D, Thorlacius H, Regnér S. Role of neutrophils in the activation of trypsinogen in severe acute pancreatitis. J Leukoc Biol. 2011;90:975–982. doi: 10.1189/jlb.0411195. [DOI] [PubMed] [Google Scholar]
- 77.Bastid C, Sahel J, Choux R, Payan MJ, Sarles H. Eosinophilic pancreatitis: report of a case. Pancreas. 1990;5:104–107. doi: 10.1097/00006676-199001000-00016. [DOI] [PubMed] [Google Scholar]
- 78.Kakodkar S, Omar H, Cabrera J, Chi K. Eosinophilic Pancreatitis Diagnosed With Endoscopic Ultrasound. ACG Case Rep J. 2015;2:239–241. doi: 10.14309/crj.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barthet M, Hastier P, Buckley MJ, Bernard JP, Sastre B, Baroni JL, Salducci J, Delmont J. Eosinophilic pancreatitis mimicking pancreatic neoplasia: EUS and ERCP findings--is nonsurgical diagnosis possible? Pancreas. 1998;17:419–422. [PubMed] [Google Scholar]
- 80.Juniper K. Chronic relapsing pancreatitis with associated marked eosinophilia and pleural effusion. Am J Med. 1955;19:648–651. doi: 10.1016/0002-9343(55)90368-5. [DOI] [PubMed] [Google Scholar]
- 81.Tokoo M, Oguchi H, Kawa S, Homma T, Nagata A. Eosinophilia associated with chronic pancreatitis: an analysis of 122 patients with definite chronic pancreatitis. Am J Gastroenterol. 1992;87:455–460. [PubMed] [Google Scholar]
- 82.Wang Q, Lu CM, Guo T, Qian JM. Eosinophilia associated with chronic pancreatitis. Pancreas. 2009;38:149–153. doi: 10.1097/MPA.0b013e31818d8ecc. [DOI] [PubMed] [Google Scholar]
- 83.Kamisawa T, Anjiki H, Egawa N, Kubota N. Allergic manifestations in autoimmune pancreatitis. Eur J Gastroenterol Hepatol. 2009;21:1136–1139. doi: 10.1097/meg.0b013e3283297417. [DOI] [PubMed] [Google Scholar]
- 84.Sah RP, Pannala R, Zhang L, Graham RP, Sugumar A, Chari ST. Eosinophilia and allergic disorders in autoimmune pancreatitis. Am J Gastroenterol. 2010;105:2485–2491. doi: 10.1038/ajg.2010.236. [DOI] [PubMed] [Google Scholar]
- 85.Hyun JJ, Lee HS. Experimental models of pancreatitis. Clin Endosc. 2014;47:212–216. doi: 10.5946/ce.2014.47.3.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lampel M, Kern HF. Acute interstitial pancreatitis in the rat induced by excessive doses of a pancreatic secretagogue. Virchows Arch A Pathol Anat Histol. 1977;373:97–117. doi: 10.1007/BF00432156. [DOI] [PubMed] [Google Scholar]
- 87.Saluja A, Saito I, Saluja M, Houlihan MJ, Powers RE, Meldolesi J, Steer M. In vivo rat pancreatic acinar cell function during supramaximal stimulation with caerulein. Am J Physiol. 1985;249:G702–G710. doi: 10.1152/ajpgi.1985.249.6.G702. [DOI] [PubMed] [Google Scholar]
- 88.Klöppel G, Maillet B. Chronic pancreatitis: evolution of the disease. Hepatogastroenterology. 1991;38:408–412. [PubMed] [Google Scholar]
- 89.Yamaguchi T, Kihara Y, Taguchi M, Nagashio Y, Tashiro M, Nakamura H, Otsuki M. Persistent destruction of the basement membrane of the pancreatic duct contributes to progressive acinar atrophy in rats with experimentally induced pancreatitis. Pancreas. 2005;31:365–372. doi: 10.1097/01.mpa.0000179729.61457.e5. [DOI] [PubMed] [Google Scholar]
- 90.Kim H. Cerulein pancreatitis: oxidative stress, inflammation, and apoptosis. Gut Liver. 2008;2:74–80. doi: 10.5009/gnl.2008.2.2.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mizunuma T, Kawamura S, Kishino Y. Effects of injecting excess arginine on rat pancreas. J Nutr. 1984;114:467–471. doi: 10.1093/jn/114.3.467. [DOI] [PubMed] [Google Scholar]
- 92.Tani S, Itoh H, Okabayashi Y, Nakamura T, Fujii M, Fujisawa T, Koide M, Otsuki M. New model of acute necrotizing pancreatitis induced by excessive doses of arginine in rats. Dig Dis Sci. 1990;35:367–374. doi: 10.1007/BF01537416. [DOI] [PubMed] [Google Scholar]
- 93.Hegyi P, Rakonczay Z, Sári R, Góg C, Lonovics J, Takács T, Czakó L. L-arginine-induced experimental pancreatitis. World J Gastroenterol. 2004;10:2003–2009. doi: 10.3748/wjg.v10.i14.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kui B, Balla Z, Vasas B, Végh ET, Pallagi P, Kormányos ES, Venglovecz V, Iványi B, Takács T, Hegyi P, et al. New insights into the methodology of L-arginine-induced acute pancreatitis. PLoS One. 2015;10:e0117588. doi: 10.1371/journal.pone.0117588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bernard C. Leçons de physiologie expérimentale appliquée à la médecine, 1: faites au Collége de France. Paris: Chez J.-B. Baillière; 1855. [Google Scholar]
- 96.Sun W, Watanabe Y, Toki A, Wang ZQ. Beneficial effects of hydrocortisone in induced acute pancreatitis of rats. Chin Med J (Engl) 2007;120:1757–1761. [PubMed] [Google Scholar]
- 97.Terry TR, Grant DA, Hermon-Taylor J. Intraduct enterokinase is lethal in rats with experimental bile-salt pancreatitis. Br J Surg. 1987;74:40–43. doi: 10.1002/bjs.1800740113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wan MH, Huang W, Latawiec D, Jiang K, Booth DM, Elliott V, Mukherjee R, Xia Q. Review of experimental animal models of biliary acute pancreatitis and recent advances in basic research. HPB (Oxford) 2012;14:73–81. doi: 10.1111/j.1477-2574.2011.00408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Niederau C, Lüthen R, Niederau MC, Grendell JH, Ferrell LD. Acute experimental hemorrhagic-necrotizing pancreatitis induced by feeding a choline-deficient, ethionine-supplemented diet. Methodology and standards. Eur Surg Res. 1992;24 Suppl 1:40–54. doi: 10.1159/000129238. [DOI] [PubMed] [Google Scholar]
- 100.Ida S, Ohmuraya M, Hirota M, Ozaki N, Hiramatsu S, Uehara H, Takamori H, Araki K, Baba H, Yamamura K. Chronic pancreatitis in mice by treatment with choline-deficient ethionine-supplemented diet. Exp Anim. 2010;59:421–429. doi: 10.1538/expanim.59.421. [DOI] [PubMed] [Google Scholar]
- 101.Opie EL. On the relation of chronic interstitial pancreatitis to the islands of langerhans and to diabetes melutus. J Exp Med. 1901;5:397–428. doi: 10.1084/jem.5.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Opie EL. The relation oe diabetes mellitus to lesions of the pancreas. hyaline degeneration of the islands oe langerhans. J Exp Med. 1901;5:527–540. doi: 10.1084/jem.5.5.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ohshio G, Saluja A, Steer ML. Effects of short-term pancreatic duct obstruction in rats. Gastroenterology. 1991;100:196–202. doi: 10.1016/0016-5085(91)90601-g. [DOI] [PubMed] [Google Scholar]
- 104.Watanabe S, Abe K, Anbo Y, Katoh H. Changes in the mouse exocrine pancreas after pancreatic duct ligation: a qualitative and quantitative histological study. Arch Histol Cytol. 1995;58:365–374. doi: 10.1679/aohc.58.365. [DOI] [PubMed] [Google Scholar]
- 105.Aghdassi AA, Mayerle J, Christochowitz S, Weiss FU, Sendler M, Lerch MM. Animal models for investigating chronic pancreatitis. Fibrogenesis Tissue Repair. 2011;4:26. doi: 10.1186/1755-1536-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kono H, Nakagami M, Rusyn I, Connor HD, Stefanovic B, Brenner DA, Mason RP, Arteel GE, Thurman RG. Development of an animal model of chronic alcohol-induced pancreatitis in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;280:G1178–G1186. doi: 10.1152/ajpgi.2001.280.6.G1178. [DOI] [PubMed] [Google Scholar]
- 107.Lieber CS, DeCarli LM. Alcoholic liver injury: experimental models in rats and baboons. Adv Exp Med Biol. 1975;59:379–393. doi: 10.1007/978-1-4757-0632-1_27. [DOI] [PubMed] [Google Scholar]
- 108.Neuschwander-Tetri BA, Bridle KR, Wells LD, Marcu M, Ramm GA. Repetitive acute pancreatic injury in the mouse induces procollagen alpha1(I) expression colocalized to pancreatic stellate cells. Lab Invest. 2000;80:143–150. doi: 10.1038/labinvest.3780018. [DOI] [PubMed] [Google Scholar]
- 109.Vonlaufen A, Xu Z, Daniel B, Kumar RK, Pirola R, Wilson J, Apte MV. Bacterial endotoxin: a trigger factor for alcoholic pancreatitis? Evidence from a novel, physiologically relevant animal model. Gastroenterology. 2007;133:1293–1303. doi: 10.1053/j.gastro.2007.06.062. [DOI] [PubMed] [Google Scholar]
- 110.Apte M, Pirola R, Wilson J. New insights into alcoholic pancreatitis and pancreatic cancer. J Gastroenterol Hepatol. 2009;24 Suppl 3:S51–S56. doi: 10.1111/j.1440-1746.2009.06071.x. [DOI] [PubMed] [Google Scholar]
- 111.Cosen-Binker LI, Lam PP, Binker MG, Reeve J, Pandol S, Gaisano HY. Alcohol/cholecystokinin-evoked pancreatic acinar basolateral exocytosis is mediated by protein kinase C alpha phosphorylation of Munc18c. J Biol Chem. 2007;282:13047–13058. doi: 10.1074/jbc.M611132200. [DOI] [PubMed] [Google Scholar]
- 112.Clemens DL, Wells MA, Schneider KJ, Singh S. Molecular mechanisms of alcohol associated pancreatitis. World J Gastrointest Pathophysiol. 2014;5:147–157. doi: 10.4291/wjgp.v5.i3.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gaisano HY, Sheu L, Whitcomb D. Alcoholic chronic pancreatitis involves displacement of Munc18c from the pancreatic acinar basal membrane surface. Pancreas. 2004;28:395–400. doi: 10.1097/00006676-200405000-00008. [DOI] [PubMed] [Google Scholar]
- 114.Whitcomb DC. Inflammation and Cancer V. Chronic pancreatitis and pancreatic cancer. Am J Physiol Gastrointest Liver Physiol. 2004;287:G315–G319. doi: 10.1152/ajpgi.00115.2004. [DOI] [PubMed] [Google Scholar]
- 115.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 116.Rockey DC, Bell PD, Hill JA. Fibrosis--a common pathway to organ injury and failure. N Engl J Med. 2015;372:1138–1149. doi: 10.1056/NEJMra1300575. [DOI] [PubMed] [Google Scholar]
- 117.Brenner DA, Kisseleva T, Scholten D, Paik YH, Iwaisako K, Inokuchi S, Schnabl B, Seki E, De Minicis S, Oesterreicher C, et al. Origin of myofibroblasts in liver fibrosis. Fibrogenesis Tissue Repair. 2012;5:S17. doi: 10.1186/1755-1536-5-S1-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sindrilaru A, Scharffetter-Kochanek K. Disclosure of the Culprits: Macrophages-Versatile Regulators of Wound Healing. Adv Wound Care (New Rochelle) 2013;2:357–368. doi: 10.1089/wound.2012.0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mahdavian Delavary B, van der Veer WM, van Egmond M, Niessen FB, Beelen RH. Macrophages in skin injury and repair. Immunobiology. 2011;216:753–762. doi: 10.1016/j.imbio.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 120.Longnecker DS. Anatomy and Histology of the Pancreas. Pancreapedia: Exocrine Pancreas Knowledge Base; 2014. p. Version 1.0. [Google Scholar]
- 121.Klöppel G, Detlefsen S, Feyerabend B. Fibrosis of the pancreas: the initial tissue damage and the resulting pattern. Virchows Arch. 2004;445:1–8. doi: 10.1007/s00428-004-1021-5. [DOI] [PubMed] [Google Scholar]
- 122.Jura N, Archer H, Bar-Sagi D. Chronic pancreatitis, pancreatic adenocarcinoma and the black box in-between. Cell Res. 2005;15:72–77. doi: 10.1038/sj.cr.7290269. [DOI] [PubMed] [Google Scholar]
- 123.Apte MV, Wilson JS, Lugea A, Pandol SJ. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology. 2013;144:1210–1219. doi: 10.1053/j.gastro.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Talukdar R, Saikia N, Singal DK, Tandon R. Chronic pancreatitis: evolving paradigms. Pancreatology. 2006;6:440–449. doi: 10.1159/000094561. [DOI] [PubMed] [Google Scholar]
- 125.Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43:128–133. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Phillips PA, McCarroll JA, Park S, Wu MJ, Pirola R, Korsten M, Wilson JS, Apte MV. Rat pancreatic stellate cells secrete matrix metalloproteinases: implications for extracellular matrix turnover. Gut. 2003;52:275–282. doi: 10.1136/gut.52.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 128.Prud’homme GJ. Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest. 2007;87:1077–1091. doi: 10.1038/labinvest.3700669. [DOI] [PubMed] [Google Scholar]
- 129.Massagué J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 131.van Laethem JL, Deviere J, Resibois A, Rickaert F, Vertongen P, Ohtani H, Cremer M, Miyazono K, Robberecht P. Localization of transforming growth factor beta 1 and its latent binding protein in human chronic pancreatitis. Gastroenterology. 1995;108:1873–1881. doi: 10.1016/0016-5085(95)90152-3. [DOI] [PubMed] [Google Scholar]
- 132.Friess H, Büchler MW, Mueller C, Malfertheiner P. Immunopathogenesis of chronic pancreatitis. Gastroenterology. 1998;115:1018–1022. doi: 10.1016/s0016-5085(98)70278-1. [DOI] [PubMed] [Google Scholar]
- 133.Friess H, Lu Z, Riesle E, Uhl W, Bründler AM, Horvath L, Gold LI, Korc M, Büchler MW. Enhanced expression of TGF-betas and their receptors in human acute pancreatitis. Ann Surg. 1998;227:95–104. doi: 10.1097/00000658-199801000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Satoh K, Shimosegawa T, Hirota M, Koizumi M, Toyota T. Expression of transforming growth factor beta1 (TGFbeta1) and its receptors in pancreatic duct cell carcinoma and in chronic pancreatitis. Pancreas. 1998;16:468–474. doi: 10.1097/00006676-199805000-00002. [DOI] [PubMed] [Google Scholar]
- 135.Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999;44:534–541. doi: 10.1136/gut.44.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shek FW, Benyon RC, Walker FM, McCrudden PR, Pender SL, Williams EJ, Johnson PA, Johnson CD, Bateman AC, Fine DR, et al. Expression of transforming growth factor-beta 1 by pancreatic stellate cells and its implications for matrix secretion and turnover in chronic pancreatitis. Am J Pathol. 2002;160:1787–1798. doi: 10.1016/s0002-9440(10)61125-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ohnishi H, Miyata T, Yasuda H, Satoh Y, Hanatsuka K, Kita H, Ohashi A, Tamada K, Makita N, Iiri T, et al. Distinct roles of Smad2-, Smad3-, and ERK-dependent pathways in transforming growth factor-beta1 regulation of pancreatic stellate cellular functions. J Biol Chem. 2004;279:8873–8878. doi: 10.1074/jbc.M309698200. [DOI] [PubMed] [Google Scholar]
- 138.Aoki H, Ohnishi H, Hama K, Ishijima T, Satoh Y, Hanatsuka K, Ohashi A, Wada S, Miyata T, Kita H, et al. Autocrine loop between TGF-beta1 and IL-1beta through Smad3- and ERK-dependent pathways in rat pancreatic stellate cells. Am J Physiol Cell Physiol. 2006;290:C1100–C1108. doi: 10.1152/ajpcell.00465.2005. [DOI] [PubMed] [Google Scholar]
- 139.Yoo BM, Yeo M, Oh TY, Choi JH, Kim WW, Kim JH, Cho SW, Kim SJ, Hahm KB. Amelioration of pancreatic fibrosis in mice with defective TGF-beta signaling. Pancreas. 2005;30:e71–e79. doi: 10.1097/01.mpa.0000157388.54016.0a. [DOI] [PubMed] [Google Scholar]
- 140.Wildi S, Kleeff J, Mayerle J, Zimmermann A, Böttinger EP, Wakefield L, Büchler MW, Friess H, Korc M. Suppression of transforming growth factor beta signalling aborts caerulein induced pancreatitis and eliminates restricted stimulation at high caerulein concentrations. Gut. 2007;56:685–692. doi: 10.1136/gut.2006.105833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hahm KB, Im YH, Lee C, Parks WT, Bang YJ, Green JE, Kim SJ. Loss of TGF-beta signaling contributes to autoimmune pancreatitis. J Clin Invest. 2000;105:1057–1065. doi: 10.1172/JCI8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Boomershine CS, Chamberlain A, Kendall P, Afshar-Sharif AR, Huang H, Washington MK, Lawson WE, Thomas JW, Blackwell TS, Bhowmick NA. Autoimmune pancreatitis results from loss of TGFbeta signalling in S100A4-positive dendritic cells. Gut. 2009;58:1267–1274. doi: 10.1136/gut.2008.170779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tahara H, Sato K, Yamazaki Y, Ohyama T, Horiguchi N, Hashizume H, Kakizaki S, Takagi H, Ozaki I, Arai H, et al. Transforming growth factor-α activates pancreatic stellate cells and may be involved in matrix metalloproteinase-1 upregulation. Lab Invest. 2013;93:720–732. doi: 10.1038/labinvest.2013.59. [DOI] [PubMed] [Google Scholar]
- 144.Garcia-Carracedo D, Yu CC, Akhavan N, Fine SA, Schönleben F, Maehara N, Karg DC, Xie C, Qiu W, Fine RL, et al. Smad4 loss synergizes with TGFα overexpression in promoting pancreatic metaplasia, PanIN development, and fibrosis. PLoS One. 2015;10:e0120851. doi: 10.1371/journal.pone.0120851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Deng S, Zhu S, Wang B, Li X, Liu Y, Qin Q, Gong Q, Niu Y, Xiang C, Chen J, et al. Chronic pancreatitis and pancreatic cancer demonstrate active epithelial-mesenchymal transition profile, regulated by miR-217-SIRT1 pathway. Cancer Lett. 2014;355:184–191. doi: 10.1016/j.canlet.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 146.Wakefield LM, Roberts AB. TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12:22–29. doi: 10.1016/s0959-437x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- 147.Nicolás FJ, Hill CS. Attenuation of the TGF-beta-Smad signaling pathway in pancreatic tumor cells confers resistance to TGF-beta-induced growth arrest. Oncogene. 2003;22:3698–3711. doi: 10.1038/sj.onc.1206420. [DOI] [PubMed] [Google Scholar]
- 148.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 149.Masamune A, Kikuta K, Satoh M, Satoh A, Shimosegawa T. Alcohol activates activator protein-1 and mitogen-activated protein kinases in rat pancreatic stellate cells. J Pharmacol Exp Ther. 2002;302:36–42. doi: 10.1124/jpet.302.1.36. [DOI] [PubMed] [Google Scholar]
- 150.McCarroll JA, Phillips PA, Park S, Doherty E, Pirola RC, Wilson JS, Apte MV. Pancreatic stellate cell activation by ethanol and acetaldehyde: is it mediated by the mitogen-activated protein kinase signaling pathway? Pancreas. 2003;27:150–160. doi: 10.1097/00006676-200308000-00008. [DOI] [PubMed] [Google Scholar]
- 151.Yasuda M, Ito T, Oono T, Kawabe K, Kaku T, Igarashi H, Nakamura T, Takayanagi R. Fractalkine and TGF-beta1 levels reflect the severity of chronic pancreatitis in humans. World J Gastroenterol. 2008;14:6488–6495. doi: 10.3748/wjg.14.6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Uchida M, Ito T, Nakamura T, Igarashi H, Oono T, Fujimori N, Kawabe K, Suzuki K, Jensen RT, Takayanagi R. ERK pathway and sheddases play an essential role in ethanol-induced CX3CL1 release in pancreatic stellate cells. Lab Invest. 2013;93:41–53. doi: 10.1038/labinvest.2012.156. [DOI] [PubMed] [Google Scholar]
- 153.Kikuta K, Masamune A, Satoh M, Suzuki N, Satoh K, Shimosegawa T. Hydrogen peroxide activates activator protein-1 and mitogen-activated protein kinases in pancreatic stellate cells. Mol Cell Biochem. 2006;291:11–20. doi: 10.1007/s11010-006-9189-4. [DOI] [PubMed] [Google Scholar]
- 154.Jaster R, Sparmann G, Emmrich J, Liebe S. Extracellular signal regulated kinases are key mediators of mitogenic signals in rat pancreatic stellate cells. Gut. 2002;51:579–584. doi: 10.1136/gut.51.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Masamune A, Kikuta K, Satoh M, Suzuki N, Shimosegawa T. Protease-activated receptor-2-mediated proliferation and collagen production of rat pancreatic stellate cells. J Pharmacol Exp Ther. 2005;312:651–658. doi: 10.1124/jpet.104.076232. [DOI] [PubMed] [Google Scholar]
- 156.Mazzon E, Impellizzeri D, Di Paola R, Paterniti I, Esposito E, Cappellani A, Bramanti P, Cuzzocrea S. Effects of mitogen-activated protein kinase signaling pathway inhibition on the development of cerulein-induced acute pancreatitis in mice. Pancreas. 2012;41:560–570. doi: 10.1097/MPA.0b013e31823acd56. [DOI] [PubMed] [Google Scholar]
- 157.Reinehr R, Zoller S, Klonowski-Stumpe H, Kordes C, Häussinger D. Effects of angiotensin II on rat pancreatic stellate cells. Pancreas. 2004;28:129–137. doi: 10.1097/00006676-200403000-00003. [DOI] [PubMed] [Google Scholar]
- 158.Hama K, Ohnishi H, Yasuda H, Ueda N, Mashima H, Satoh Y, Hanatsuka K, Kita H, Ohashi A, Tamada K, et al. Angiotensin II stimulates DNA synthesis of rat pancreatic stellate cells by activating ERK through EGF receptor transactivation. Biochem Biophys Res Commun. 2004;315:905–911. doi: 10.1016/j.bbrc.2004.01.155. [DOI] [PubMed] [Google Scholar]
- 159.Masamune A, Kikuta K, Satoh M, Satoh K, Shimosegawa T. Rho kinase inhibitors block activation of pancreatic stellate cells. Br J Pharmacol. 2003;140:1292–1302. doi: 10.1038/sj.bjp.0705551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Huang Y, Xiao S, Jiang Q. Role of Rho kinase signal pathway in inflammatory bowel disease. Int J Clin Exp Med. 2015;8:3089–3097. [PMC free article] [PubMed] [Google Scholar]
- 161.Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 162.Du XG, Chen XM, Gan H, Li ZR, Wen YJ, Wang XC. Continuous blood purification ameliorates RhoA-mediated endothelial permeability in severe acute pancreatitis patients with lung injury. Int J Artif Organs. 2011;34:348–356. doi: 10.5301/IJAO.2011.7742. [DOI] [PubMed] [Google Scholar]
- 163.Masamune A, Satoh M, Kikuta K, Sakai Y, Satoh A, Shimosegawa T. Inhibition of p38 mitogen-activated protein kinase blocks activation of rat pancreatic stellate cells. J Pharmacol Exp Ther. 2003;304:8–14. doi: 10.1124/jpet.102.040287. [DOI] [PubMed] [Google Scholar]
- 164.Kusama K, Nozu F, Awai T, Tanaka S, Honma I, Tsunoda Y, Mitamura K. Deactivation of ROCK-II by Y-27632 enhances basolateral pancreatic enzyme secretion and acute pancreatitis induced by CCK analogues. Biochem Biophys Res Commun. 2003;305:339–344. doi: 10.1016/s0006-291x(03)00758-7. [DOI] [PubMed] [Google Scholar]
- 165.Awla D, Hartman H, Abdulla A, Zhang S, Rahman M, Regnér S, Thorlacius H. Rho-kinase signalling regulates trypsinogen activation and tissue damage in severe acute pancreatitis. Br J Pharmacol. 2011;162:648–658. doi: 10.1111/j.1476-5381.2010.01060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ihle JN, Kerr IM. Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet. 1995;11:69–74. doi: 10.1016/s0168-9525(00)89000-9. [DOI] [PubMed] [Google Scholar]
- 167.Ihle JN, Witthuhn BA, Quelle FW, Yamamoto K, Thierfelder WE, Kreider B, Silvennoinen O. Signaling by the cytokine receptor superfamily: JAKs and STATs. Trends Biochem Sci. 1994;19:222–227. doi: 10.1016/0968-0004(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 168.Yu JH, Kim KH, Kim H. Suppression of IL-1beta expression by the Jak 2 inhibitor AG490 in cerulein-stimulated pancreatic acinar cells. Biochem Pharmacol. 2006;72:1555–1562. doi: 10.1016/j.bcp.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 169.Luttenberger T, Schmid-Kotsas A, Menke A, Siech M, Beger H, Adler G, Grünert A, Bachem MG. Platelet-derived growth factors stimulate proliferation and extracellular matrix synthesis of pancreatic stellate cells: implications in pathogenesis of pancreas fibrosis. Lab Invest. 2000;80:47–55. doi: 10.1038/labinvest.3780007. [DOI] [PubMed] [Google Scholar]
- 170.Chen P, Huang L, Zhang Y, Qiao M, Yao W, Yuan Y. The antagonist of the JAK-1/STAT-1 signaling pathway improves the severity of cerulein-stimulated pancreatic injury via inhibition of NF-κB activity. Int J Mol Med. 2011;27:731–738. doi: 10.3892/ijmm.2011.632. [DOI] [PubMed] [Google Scholar]
- 171.Martelli AM, Nyåkern M, Tabellini G, Bortul R, Tazzari PL, Evangelisti C, Cocco L. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia. 2006;20:911–928. doi: 10.1038/sj.leu.2404245. [DOI] [PubMed] [Google Scholar]
- 172.Lupia E, Pigozzi L, Goffi A, Hirsch E, Montrucchio G. Role of phosphoinositide 3-kinase in the pathogenesis of acute pancreatitis. World J Gastroenterol. 2014;20:15190–15199. doi: 10.3748/wjg.v20.i41.15190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Faes S, Dormond O. PI3K and AKT: Unfaithful Partners in Cancer. Int J Mol Sci. 2015;16:21138–21152. doi: 10.3390/ijms160921138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Singh VP, Saluja AK, Bhagat L, van Acker GJ, Song AM, Soltoff SP, Cantley LC, Steer ML. Phosphatidylinositol 3-kinase-dependent activation of trypsinogen modulates the severity of acute pancreatitis. J Clin Invest. 2001;108:1387–1395. doi: 10.1172/JCI12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Xu P, Wang J, Yang ZW, Lou XL, Chen C. Regulatory roles of the PI3K/Akt signaling pathway in rats with severe acute pancreatitis. PLoS One. 2013;8:e81767. doi: 10.1371/journal.pone.0081767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gukovsky I, Cheng JH, Nam KJ, Lee OT, Lugea A, Fischer L, Penninger JM, Pandol SJ, Gukovskaya AS. Phosphatidylinositide 3-kinase gamma regulates key pathologic responses to cholecystokinin in pancreatic acinar cells. Gastroenterology. 2004;126:554–566. doi: 10.1053/j.gastro.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 177.Lupia E, Goffi A, De Giuli P, Azzolino O, Bosco O, Patrucco E, Vivaldo MC, Ricca M, Wymann MP, Hirsch E, et al. Ablation of phosphoinositide 3-kinase-gamma reduces the severity of acute pancreatitis. Am J Pathol. 2004;165:2003–2011. doi: 10.1016/s0002-9440(10)63251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Affronti J. Chronic pancreatitis and exocrine insufficiency. Prim Care. 2011;38:515–537; ix. doi: 10.1016/j.pop.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 179.Stevens T. aCDL. Chronic pancreatitis, W. Carey (Ed.), Current clinical medicine (2nd edition) Philadelphia: Saunders Elsevier; 2009. pp. 451–456. [Google Scholar]
- 180.Ammann RW, Muellhaupt B. The natural history of pain in alcoholic chronic pancreatitis. Gastroenterology. 1999;116:1132–1140. doi: 10.1016/s0016-5085(99)70016-8. [DOI] [PubMed] [Google Scholar]
- 181.Meher S, Mishra TS, Sasmal PK, Rath S, Sharma R, Rout B, Sahu MK. Role of Biomarkers in Diagnosis and Prognostic Evaluation of Acute Pancreatitis. J Biomark. 2015;2015:519534. doi: 10.1155/2015/519534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Rau BM, Krüger CM, Schilling MK. Anti-cytokine strategies in acute pancreatitis: pathophysiological insights and clinical implications. Rocz Akad Med Bialymst. 2005;50:106–115. [PubMed] [Google Scholar]
- 183.Matull WR, Pereira SP, O’Donohue JW. Biochemical markers of acute pancreatitis. J Clin Pathol. 2006;59:340–344. doi: 10.1136/jcp.2002.002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Shah AM, Eddi R, Kothari ST, Maksoud C, DiGiacomo WS, Baddoura W. Acute pancreatitis with normal serum lipase: a case series. JOP. 2010;11:369–372. [PubMed] [Google Scholar]
- 185.Lippi G, Valentino M, Cervellin G. Laboratory diagnosis of acute pancreatitis: in search of the Holy Grail. Crit Rev Clin Lab Sci. 2012;49:18–31. doi: 10.3109/10408363.2012.658354. [DOI] [PubMed] [Google Scholar]
- 186.Chang JWY, Chung CH. Diagnosing acute pancreatitis: amylase or lipase? Hong Kong J Emerg Med. 2011;18:20–25. [Google Scholar]
- 187.Lott JA, Patel ST, Sawhney AK, Kazmierczak SC, Love JE. Assays of serum lipase: analytical and clinical considerations. Clin Chem. 1986;32:1290–1302. [PubMed] [Google Scholar]
- 188.Tietz NW, Shuey DF. Lipase in serum--the elusive enzyme: an overview. Clin Chem. 1993;39:746–756. [PubMed] [Google Scholar]
- 189.Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG, Tsiotos GG, Vege SS. Classification of acute pancreatitis--2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62:102–111. doi: 10.1136/gutjnl-2012-302779. [DOI] [PubMed] [Google Scholar]
- 190.Petersson U, Appelros S, Borgström A. Different patterns in immunoreactive anionic and cationic trypsinogen in urine and serum in human acute pancreatitis. Int J Pancreatol. 1999;25:165–170. doi: 10.1007/BF02925965. [DOI] [PubMed] [Google Scholar]
- 191.Jun Song TK, Myung-Hwan K. Steroid Therapy in the Management of Autoimmune Pancreatitis. Pancreapedia: Exocrine Pancreas Knowledge Base; 2013. [Google Scholar]
- 192.Kamisawa T, Shimosegawa T, Okazaki K, Nishino T, Watanabe H, Kanno A, Okumura F, Nishikawa T, Kobayashi K, Ichiya T, et al. Standard steroid treatment for autoimmune pancreatitis. Gut. 2009;58:1504–1507. doi: 10.1136/gut.2008.172908. [DOI] [PubMed] [Google Scholar]
- 193.Bhardwaj P, Garg PK, Maulik SK, Saraya A, Tandon RK, Acharya SK. A randomized controlled trial of antioxidant supplementation for pain relief in patients with chronic pancreatitis. Gastroenterology. 2009;136:149–159.e2. doi: 10.1053/j.gastro.2008.09.028. [DOI] [PubMed] [Google Scholar]
- 194.De las Heras Castaño G, García de la Paz A, Fernández MD, Fernández Forcelledo JL. Use of antioxidants to treat pain in chronic pancreatitis. Rev Esp Enferm Dig. 2000;92:375–385. [PubMed] [Google Scholar]
- 195.Dite P, Precechtelova M, Novotny I, Soska V, Zakova A, Lata J. Changes of reactive oxidative substances in patients with morphologically diff erent degrees of chronic pancreatitis and effects of long-term therapy with natural antioxidants. Gastroenterologia Polska. 2003;10:379–383. [Google Scholar]
- 196.Kirk GR, White JS, McKie L, Stevenson M, Young I, Clements WD, Rowlands BJ. Combined antioxidant therapy reduces pain and improves quality of life in chronic pancreatitis. J Gastrointest Surg. 2006;10:499–503. doi: 10.1016/j.gassur.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 197.Uden S, Bilton D, Nathan L, Hunt LP, Main C, Braganza JM. Antioxidant therapy for recurrent pancreatitis: placebo-controlled trial. Aliment Pharmacol Ther. 1990;4:357–371. doi: 10.1111/j.1365-2036.1990.tb00482.x. [DOI] [PubMed] [Google Scholar]
- 198.Uden S, Schofield D, Miller PF, Day JP, Bottiglier T, Braganza JM. Antioxidant therapy for recurrent pancreatitis: biochemical profiles in a placebo-controlled trial. Aliment Pharmacol Ther. 1992;6:229–240. doi: 10.1111/j.1365-2036.1992.tb00266.x. [DOI] [PubMed] [Google Scholar]
- 199.Uomo G, Talamini G, Rabitti PG. Antioxidant treatment in hereditary pancreatitis. A pilot study on three young patients. Dig Liver Dis. 2001;33:58–62. doi: 10.1016/s1590-8658(01)80136-5. [DOI] [PubMed] [Google Scholar]