

Abstract
AIM
To evaluate the effect of orally administered plecanatide on colorectal dysplasia in Apc+/Min-FCCC mice with dextran sodium sulfate (DSS)-induced inflammation.
METHODS
Inflammation driven colorectal carcinogenesis was induced in Apc+/Min-FCCC mice by administering DSS in their drinking water. Mice were fed a diet supplemented with plecanatide (0-20 ppm) and its effect on the multiplicity of histopathologically confirmed polypoid, flat and indeterminate dysplasia was evaluated. Plecanatide-mediated activation of guanylate cyclase-C (GC-C) signaling was assessed in colon tissues by measuring cyclic guanosine monophosphate (cGMP) by ELISA, protein kinase G-II and vasodilator stimulated phosphoprotein by immunoblotting. Ki-67, c-myc and cyclin D1 were used as markers of proliferation. Cellular levels and localization of β-catenin in colon tissues were assessed by immunoblotting and immunohistochemistry, respectively. Uroguanylin (UG) and GC-C transcript levels were measured by quantitative reverse transcription polymerase chain reaction (RT-PCR). A mouse cytokine array panel was used to detect cytokines in the supernatant of colon explant cultures.
RESULTS
Oral treatment of Apc+/MinFCCC mice with plecanatide produced a statistically significant reduction in the formation of inflammation-driven polypoid, flat and indeterminate dysplasias. This anti-carcinogenic activity of plecanatide was accompanied by activation of cGMP/GC-C signaling mediated inhibition of Wnt/β-catenin signaling and reduced proliferation. Plecanatide also decreased secretion of pro-inflammatory cytokines (IL-6, IL1 TNF), chemokines (MIP-1, IP-10) and growth factors (GCSF and GMCSF) from colon explants derived from mice with acute DSS-induced inflammation. The effect of plecanatide-mediated inhibition of inflammation/dysplasia on endogenous expression of UG and GC-C transcripts was measured in intestinal tissues. Although GC-C expression was not altered appreciably, a statistically significant increase in the level of UG transcripts was detected in the proximal small intestine and colon, potentially due to a reduction in intestinal inflammation and/or neoplasia. Taken together, these results suggest that reductions in endogenous UG, accompanied by dysregulation in GC-C signaling, may be an early event in inflammation-promoted colorectal neoplasia; an event that can potentially be ameliorated by prophylactic intervention with plecanatide.
CONCLUSION
This study provides the first evidence that orally administered plecanatide reduces the multiplicity of inflammation-driven colonic dysplasia in mice, demonstrating the utility for developing GC-C agonists as chemopreventive agents.
Keywords: Guanylate cyclase-C, Uroguanylin, Plecanatide, Inflammation, Colorectal cancer
Core tip: Plecanatide, an analog of human uroguanylin, binds and activates guanylate cyclase-C signaling to produce its anti-tumorigenic activity. This study provides the first evidence that oral treatment with plecanatide inhibits inflammation-driven colorectal carcinogenesis. The potential mechanism in Apc+/MinFCCC mice appears to be agonist-mediated activation of guanylate cyclase-C signaling, resulting in inhibition of Wnt/β-catenin pathway and downregulation of pro-inflammatory cytokines and growth factors.
INTRODUCTION
Colorectal cancer (CRC) ranks third among newly diagnosed cancers in the United States and is the third most common cause of cancer mortality[1]. Clinical studies indicate that patients with long-standing inflammatory bowel disease (IBD) have a 2 to 8-fold increased risk of developing CRC as compared to the general population[2]. Although the precise etiology underlying inflammation-promoted CRC remains unclear, emerging data suggest that chronic inflammation, oxidative stress and excessive production of cytokines, chemokines and growth factors by infiltrating immune cells, may eventually lead to the development of dysplasia[3-6]. Like sporadic CRC, development of IBD-promoted carcinogenesis follows a sequential progression of disease from low-grade to high-grade dysplasia, and eventually CRC[7]. Prophylactic intervention with 5-aminosalicylate (5-ASA) is widely considered as a promising chemopreventive strategy[8]. However, additional case-controlled studies with larger numbers of patients are needed to further elucidate the chemopreventive utility of 5-ASA in IBD-promoted CRC. Conceptually, chronic prophylactic intervention with an orally safe and mucosally active agent that not only suppresses inflammation but also regulates renewal of the gastrointestinal (GI) mucosa is desirable.
Uroguanylin (UG) and guanylin (GN), endogenous natriuretic peptides, are agonists of guanylate cyclase-C (GC-C) that are structurally similar to bacterial enterotoxin (ST), secreted by the pathogenic Escherichia coli (E. coli) responsible for traveler’s diarrhea[9]. Binding of these peptides to GC-C on the apical surface of epithelial cells lining the GI tract stimulates intracellular production of cyclic guanosine monophosphate (cGMP), resulting in activation of cGMP-dependent protein kinase G-II (PKG-II) and cystic fibrosis transmembrane conductance regulator. This leads to enhanced transepithelial efflux of Cl and HCO3-, inhibition of Na+ absorption and passive secretion of water into the intestinal lumen; a process essential for a normal bowel movement[10]. Thus, a major physiological function of UG and GN is to regulate ion and fluid homeostasis in the GI tract[10-12].
GC-C signaling also plays a key physiological role in regulating the proliferative index of epithelial cells and maintaining the integrity of the GI mucosa[10,13,14]. Thus, disruption of GC-C signaling, due to reduced production of UG and/or GN, could potentially lead to neoplastic transformation. Indeed, transcript levels of both UG and GN are reduced dramatically in colon polyps and adenocarcinomas[13]. Furthermore, dietary supplementation of Apc+/Min mice with human UG not only inhibits polyp formation but also delays tumor progression[13]. Recent studies performed using GC-C and UG knockout mice further illustrate the involvement of GC-C signaling in the maintenance of homeostatic intestinal barrier function, gut permeability and intestinal epithelial cell proliferation[15]. Recently, we demonstrated that oral treatment with GC-C agonists such as plecanatide or dolcanatide effectively ameliorated GI inflammation in acute and chronic models of experimental colitis in murine models[16]. Thus, treatment with UG to overcome this deficiency may represent a promising approach for the prevention of inflammation-driven CRC.
The present study provides the first evidence to demonstrate that oral treatment with plecanatide effectively suppresses the formation of inflammation-driven CRC in Apc+/Min-FCCC mice.
MATERIALS AND METHODS
Materials
DSS (molecular weight 30000-40000) was purchased from MP Biomedicals (Solon, OH). All other chemicals and reagents were obtained from commercial vendors. Plecanatide (H-Asn1-Asp2-Glu3-Cys4-Glu5-Leu6-Cys7-Val8-Asn9-Val10-Ala11-Cys12-Thr13-Gly14-Cys15-Leu16-OH; Disulfide bond between Cys4 and Cys12; Cys7 and Cys15) was synthesized for this study according to procedures described previously[17].
Animals
Female Apc+/Min-FCCC (C57BL/6J) mice (7-wk-old, n = 92) were obtained from the Laboratory Animal Facility at Fox Chase Cancer Center (FCCC)[18]. Mice were genotyped for a point mutation in codon 850 of the Apc gene[19]. Animals were maintained in a temperature- and humidity-controlled room and received Teklad Global 2018SX diet (Harlan Teklad, Madison, WI) and drinking water ad libitum. All animal protocols were approved by the Institutional Animal Care and Use Committee at FCCC (IACUC # 08-4). Drug-supplemented diet was prepared as described previously[13].
Methods
Experimental design: Inflammation-promoted colorectal neoplasia was induced by administering 2% dextran sodium sulfate (DSS) to female Apc+/Min-FCCC mice in the drinking water as outlined in Figure 1A. At treatment week 0 (8 wk of age), mice were randomized to one of four experimental groups: DSS treatment alone (no plecanatide treatment (control)) or DSS plus 2, 10 or 20 ppm plecanatide in the diet (n = 23/group). All animals were administered DSS for 4 d beginning at treatment week 1 and received regular water for the remainder of the study (Figure 1A). At the end of the study, the entire small intestine and colon were excised, cut longitudinally and rinsed with saline. An equivalent strip of the small intestine and colon from each animal was snap frozen for molecular analysis of UG and GC-C transcript levels by quantitative reverse-transcription polymerase chain reaction (RT-PCR). The remainder of the colon was fixed in 10% formalin overnight, cross-sectioned at 2 mm intervals and processed for histopathological review.
Figure 1.
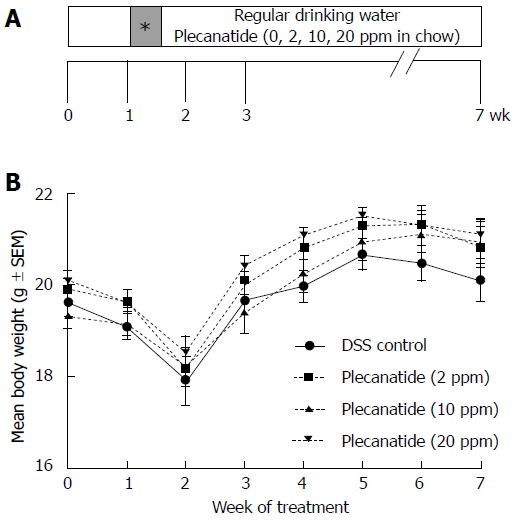
Inflammation-driven colorectal carcinogenesis in Apc+/Min-FCCC mice. A: Outline depicting the experimental design of the animal study. Female Apc+/Min-FCCC mice (n = 23/group) were randomized into four treatment groups: DSS alone (vehicle control) or DSS plus diet supplemented with 2, 10 or 20 ppm plecanatide. One week later, all animals were administered 2% DSS in the drinking water for 4 d (shaded box with asterisk) and regular water for the remainder of the study. At the time of euthanasia, (7 wk of study), the entire colon were fixed in formalin for histopathological evaluation; B: Body weights of Apc+/Min-FCCC mice treated with either DSS alone or DSS plus a diet supplemented with indicated concentrations of plecanatide (n = 20-23/group). Body weights were obtained weekly, and DSS was administered to all animals on days 7-10 of study. DSS: Dextran sodium sulfate.
Histopathological analyses: Sections stained with H and E were histopathologically evaluated for neoplasia in a blinded manner, as described previously[20]. All classifications were based on standardized morphology and nomenclature for the human pathology of inflammation-promoted colorectal neoplasia[21]. A diagnosis of carcinoma was assigned when neoplastic glands had invaded into the muscularis mucosae or beyond. Any dysplasia or cancer exhibiting an elevated growth pattern was considered polypoid. Non-polypoid (flat) lesions were elevated less than 2-fold above the adjacent non-neoplastic colorectal mucosa. Lesions that could not be readily classified as either polypoid or non-polypoid were categorized as indeterminate.
Immunohistochemistry: Ki-67 was selected as a biomarker of cell proliferation. Antigen retrieval was performed prior to staining in a Ventana Benchmark XT automated stainer (Tucson, AZ). All buffers and washes were per standard XT protocol. For Ki-67 staining, sections were incubated with Ki-67 primary antibody (1:1500 dilution; Vector Laboratories, Inc., Burlingame, CA) for one hour at room temperature. Negative controls were processed with rabbit IgG at approximately the same protein concentration as the primary antibody. Staining was detected using a rabbit secondary antibody kit (Vector Laboratories, Inc.) according to the manufacturer’s instructions. All sections were counterstained with lite hematoxylin. Only cells with nuclear staining of Ki67 were considered positive. The number of Ki-67 positive cells in dysplasias (2 fields/dysplasia, 600 ×) and non-neoplastic colonic crypts (20 crypt columns/animal, 600 ×) were counted and recorded as a Ki-67 labeling index (number of positive cells/total number of cells evaluated). The rate of proliferation of each tumor was established and the mean rate of all tumors in the treatment group was calculated.
The cellular localization of β-catenin was determined using specific polyclonal antibodies (1:4000 dilution; Sigma, St Louis, MO). Negative controls were processed with rabbit IgG. Staining was detected using a goat anti-rabbit secondary antibody kit (Vector Laboratories, Inc.) as per the manufacturer’s instructions. Sections were counterstained with lite hematoxylin. The number of tumor cells with nuclear localization of β-catenin was counted and expressed as a percentage of the total number of tumor cells per field (400 ×).
Immunoblotting: One centimeter long colon tissues from 6 animals per group were pooled and homogenized in RIPA buffer (10 mmol/L Tris, pH 7.2; 150 mmol/L NaCl, 1% sodium deoxycholate, 1% triton × 100, 0.1% SDS, 0.1 mmol/L Na3VO4; 50 mmol/L NaF), containing a protease inhibitor cocktail (Boehringer Mannheim, GmbH, Germany). The homogenate was centrifuged at 12000 g for 15 min at 4 °C and the supernatant was used as crude tissue lysate. The crude lysates (approximately 50 g protein) were subjected to 10% SDS-PAGE under reducing conditions, followed by immunoblotting with antibodies specific for β-catenin (1:4000 dilution, Sigma), PKG-II (1:200, dilution Santa Cruz Biotechnology, Inc., CA), glyceraldehyde 3phosphate dehydrogenase (GAPDH; 1:10000 dilution, Ambion), GC-C (1:500, Santa Cruz Biotechnology Inc., CA), phospho-VASP and c-myc (1:1000, Cell Signaling, MA, United States), cyclin D1 (1:1000, Abcam, MA, United States), and β-actin (1:1000, Chemicon, CA). Blots were developed using the ECL plus Western Blotting Detection System (GE Healthcare, Buckinghamshire, United Kingdom) or LiCor blotting system. The resulting images were analyzed using the FluorChem E system (Cell Biosciences, Santa Clara, CA).
Measurement of cyclic GMP in tissue lysates: Colon tissues (1 cm) from 6 animals per group were pooled to prepare crude tissue lysates. The levels of cGMP were determined using an ELISA kit (Cayman Chemical Co., Ann Arbor, MI)[13]. The protein concentration of the lysates was determined using the Pierce BCA protein assay (Thermo Fisher Scientific, Rockford, IL). Results were normalized per mg protein and expressed as mean pmol ± SEM.
Quantification of UG and GC-C transcript levels by RT-PCR: Representative areas of the proximal and distal small intestine and colon were randomly selected from DSS control and DSS + plecanatide-treated mice (n = 45/group) for analysis. Total RNA was extracted from 5-10 mg of tissue using the RNAqueous®-4PCR Kit (Ambion, Austin, TX) and quantified using a Nanodrop2000 (Thermo Fisher, Waltham, MA). Total RNA (1 μg) was reverse-transcribed to cDNA using a High Capacity RNA-to-cDNA Kit (Applied Biosystems, Carlsbad, CA), according to the manufacturer’s instructions. Quantitative RT-PCR amplification and analysis were carried out using a LightCycler480 (Roche, Basel, Switzerland), UG and GC-C specific TaqMan reagents (Integrated DNA Technologies, Coralville, IA) and RT-PCR Master Mix (Roche, Basel, Switzerland). All amplification reactions (20 L total volume) were performed in duplicate with 10 ng cDNA (based on input RNA) and subjected to 35 PCR cycles using parameters set by the manufacturer. GAPDH was used as an endogenous control. The data generated were analyzed and expressed as target gene expression relative to endogenous control, using the comparative Ct method and the 2ΔΔCt formula. Results are expressed as fold change in relative levels of UG or GC-C transcripts per segment of small intestine or colon of plecanatide-treated mice, as compared to those of control mice treated with only DSS.
DSS treatment to induce acute inflammation in Apc+/Min-FCCC mice: An independent study was designed to evaluate the effect of plecanatide on GC-C signaling and cytokine expression during the acute phase of colonic inflammation. Apc+/Min-FCCC mice (n = 12; 6-8 wk old) were administered 2% DSS in the drinking water for 4 d followed by 3 d of regular water. Starting on day 1, six animals received an oral gavage of plecanatide (2.5 mg/kg body weight) daily while the other six were administered vehicle (0.9% sodium chloride solution, Sigma, St. Louis, MO). Vehicle control animals (n = 6) received regular drinking water (no DSS) and were administered an oral gavage of saline daily. It should be noted that the amount of plecanatide delivered by a single oral gavage at this dose is similar to that ingested daily when animals were administered a diet supplemented with 10 ppm plecanatide in the main tumorigenesis study. Mice were euthanized on day 7, and the entire colon was excised. Part of the colon tissue from each animal was snap frozen for analysis of intracellular cGMP and determination of the expression of GC-C, PKG-II, p-VASP and β-actin by immunoblot. The remaining tissue was used immediately for explant cultures as described below.
Explant culture: Tissues were washed in PBS containing 100 units of penicillin, 0.1 mg streptomycin and 0.25 μg amphotericin B per milliliter (1 x antibiotic and anti-mycotic solution; Sigma, St. Louis, MO) and cut into 1 cm pieces. Tissue explants were cultured in a 24-well plate overnight in RPMI 1640 media (Mediatech, Manassas, VA) in the absence or presence of 1 μmol/L plecanatide at 37 °C in a CO2 incubator. After the incubation period, explants were snap frozen and stored for analysis of cyclin D1, c-myc and β-actin expression by immunoblot.
Cytokine analysis: Expression of select mouse cytokines was detected in the spent media of pooled explant cultures (n = 6/pool) using a mouse Proteome Profiler Panel A kit array panel (R and D Systems, Minneapolis, MN). The mean intensities of the dot blots were calculated using ImageJ software (NIH).
Statistical analysis
Wilcoxon 2-sample test and analysis of variance (ANOVA) were used to compare differences between groups such as body weight, lesion multiplicity, and lesion type. Student’s t test was used to evaluate differences in cGMP, Ki-67 labeling index, expression of total and nuclear β-catenin, cytokines, PKG-II protein, UG and GC-C transcript levels.
RESULTS
Plecanatide reduced inflammation-promoted dysplasia in Apc+/Min-FCCC mice
All experimental treatments were well tolerated. The rate of survival was: 91% for mice in the control group, 83% for mice administered 2 ppm plecanatide, and 96% for mice receiving diet supplemented with either 10 or 20 ppm plecanatide. As observed previously in this model[22], body weights declined immediately following DSS treatment and increased gradually thereafter in all groups (Figure 1B). The body weights of mice treated with DSS + plecanatide increased more rapidly than did those of mice receiving DSS + vehicle. Tissues were also scored for the degree of inflammation (data not shown). Consistent with our previous experience in this animal model, the inflammation scores were generally low, most likely due to anticipated resolution of the colonic inflammation by the end of the study.
The effect of plecanatide on the multiplicity of polypoid, flat, indeterminate and total colonic dysplasias was determined (Figure 2). While the multiplicity of all morphological subtypes of colon lesions in mice treated with 2 ppm plecanatide did not differ from that of controls, a reduction in the multiplicity of each of the subtypes of dysplasia was observed in mice treated with higher doses of the drug. For example, 10 and 20 ppm plecanatide reduced polypoid dysplasias (Figure 2A), with a statistically significant reduction (approximately 40%) observed at a dose of 20 ppm, as compared to the control group (P = 0.05). A similar reduction in the multiplicity of non-polypoid/flat (P = 0.041) and indeterminate (P = 0.05) dysplasias was observed post treatment, but only in mice receiving 10 ppm plecanatide (Figure 2B and C). Surprisingly, the higher dose of plecanatide (20 ppm) did not produce an appreciable reduction in the multiplicity of either flat or indeterminate colonic dysplasias. A statistically significant reduction (P = 0.028) in total colonic dysplasia was observed in mice administered 10 ppm of plecanatide in the diet (Figure 2D). As previously observed with oral UG treatment[13], plecanatide also reduced the multiplicity of small intestinal tumors (data not shown). Since suppression of polyp formation by oral treatment with GC-C agonists has now been well-established[13,23], this study focused only the effect of orally administered plecanatide on the multiplicity of colonic dysplasias. Therefore, subsequent analyses were performed on colon tissues from mice treated with 10 ppm plecanatide.
Figure 2.
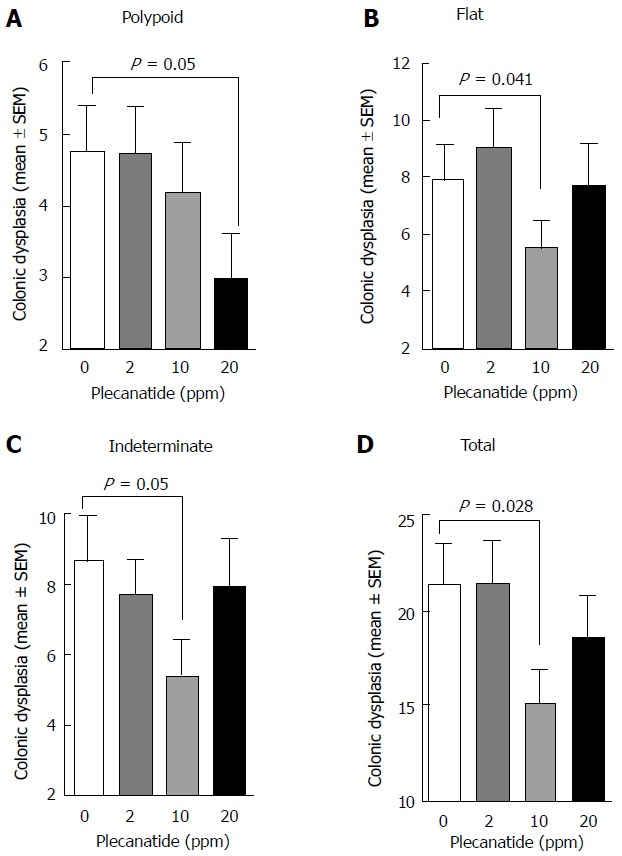
Treatment with plecanatide inhibits inflammation-associated colonic dysplasia in dextran sodium sulfate-treated Apc+/Min-FCCC mice. Analyses revealed the number of pathologically confirmed polypoid (A), flat (B), indeterminate (C) and total (D) dysplasias within the colon of DSS-treated mice following administration (7 wk) of either control diet or diet supplemented with varying doses of plecanatide (n = 23/group). Wilcoxon 2-sample test and analysis of variance (ANOVA) were used to compare the multiplicity of dysplasias in independent groups. A P value ≤ 0.05 was considered statistically significant. DSS: Dextran sodium sulfate
Plecanatide mediated activation of GC-C signaling
Although UG, GN, and other GC-C related agonists are known to stimulate production of cGMP via activation of GC-C, in epithelial cells lining the GI tract, and cultured T84 and Caco-2 cells[12,24,25], it was of interest to evaluate if orally administered plecanatide could stimulate cGMP production within the murine colon. Acute inflammation was induced in mice as described in the Materials and Methods section. Colon tissues (1 cm piece) from 6 mice within a treatment group were pooled, homogenized, and crude lysates were used to measure cGMP. A significant reduction in the cGMP levels was observed in colon tissues from DSS-treated animals as compared to vehicle controls (P = 0.01). Oral treatment with plecanatide (2.5 mg/kg) completely restored the DSS-mediated reduction in cGMP levels (Figure 3A).
Figure 3.
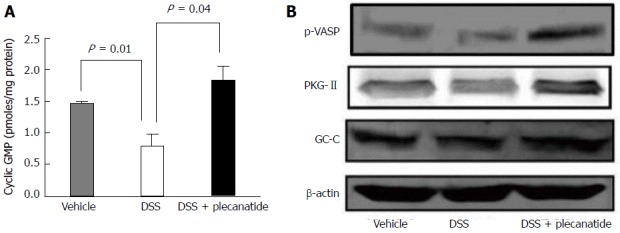
Orally administered plecanatide activates guanylate cyclase-C signaling within the colon. Effect of plecanatide on (A) stimulation of cGMP production and (B) expression of phosphorylated VASP, PKG-II and GC-C in colon tissues from Apc+/Min-FCCC mice with DSS-induced acute inflammation (n = 6/group). Mice with acute inflammation were administered plecanatide (2.5 mg/kg) by oral gavage; a dose equivalent to that ingested daily by animals fed a diet supplemented with 10 ppm plecanatide in the main tumorigenesis study. Colon tissue (1 cm) from 6 animals per group was pooled to prepare cell lysates. Intracellular cGMP levels depicted in (A) are expressed as pmoles/mg protein ± SEM. Student t test was used to evaluate differences in cGMP between treatment groups. P values ≤ 0.05 were considered statistically significant. Representative Western blot analyses of phospho-VASP, PKG-II and GC-C were performed using appropriate antibodies. To demonstrate equivalent protein loading for each condition, membranes were probed subsequently with β-actin antibody. GC-C: Guanylate cyclase-C; GMP: Guanosine monophosphate; p-VASP: Phospho-vasodilator-stimulated phosphoprotein; PKG-II: Protein kinase G-II.
PKG-II, a cGMP-dependent protein kinase, is activated by cGMP following activation of GC-C by its agonists. PKG-II is expressed on epithelial cells lining the GI tract and undergoes auto-phosphorylation upon activation of GC-C signaling[26]. Colon tissues from mice with DSS-induced inflammation were homogenized and lysates were subjected to immunoblotting with antibodies specific for phosphorylated-vasodilatorstimulated phosphoprotein (p-VASP), PKG-II and GC-C (Figure 3B). The p-VASP antibody detects only Ser239 phosphorylated VASP. Activated PKG-II expression, seen on blots as a partially resolved doublet, was considerably higher in colon tissues from mice treated with both DSS and plecanatide as compared to those treated with only DSS. The level of p-VASP was also much higher in colon tissues from DSS + plecanatide treated mice as compared to that observed in tissues from DSS treated mice. Colonic expression of GC-C was comparable among all cohorts, irrespective of the treatment group (Figure 3B). Collectively, these results suggest that GC-C signaling is activated in the colon by orally administered plecanatide.
Plecanatide reduces proliferation of colonic epithelial cells
Immunohistochemical staining of Ki-67 was performed using non-neoplastic (normal) and neoplastic mucosa from the colons of mice treated with DSS or DSS + plecanatide (10 ppm). As depicted in Figure 4A, plecanatide reduced the number of Ki-67 positive epithelial cells in both the non-neoplastic and neoplastic mucosa. However, the reduction in Ki-67 labeling only achieved significance in the neoplastic mucosa (P < 0.001). Similarly, although the number of caspase-3 positive cells was increased in both non-neoplastic and neoplastic colon tissue, the elevation was significant (P = 0.05) only in plecanatide treated non-neoplastic tissue (data not shown). It should be noted that endogenous UG expression and GC-C signaling-mediated regulation of epithelial cell homeostasis is not altered in the normal epithelium. In addition, the rate of cell proliferation is also much higher in tumor tissue as compared to the normal epithelium. Thus, it is possible that oral treatment with plecanatide has a more pronounced inhibitory effect on proliferation in tumor tissue than in the normal epithelium. To further confirm the anti-proliferative activity, crude lysates of colon tissues from mice treated with vehicle, DSS and DSS + plecanatide (2.5 mg/kg) were examined by immunoblotting with antibodies specific for c-myc and cyclin D1 (markers of proliferation) (Figure 4B). Plecanatide reduced levels of c-myc and phosphorylated cyclin D1 (slower moving band) in colon tissues as compared to treatment with either vehicle or DSS alone. Taken together, these results suggest that plecanatide reduces proliferation of epithelial cells lining the GI mucosa.
Figure 4.
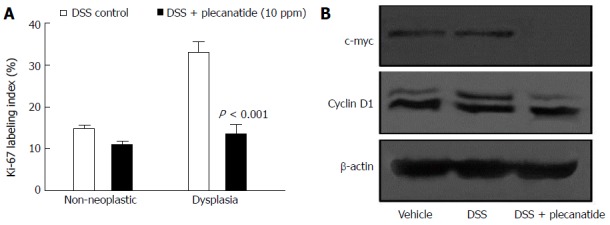
Effect of plecanatide on markers of proliferation in colonic epithelial cells from Apc+/Min-FCCC mice with dextran sodium sulfate-induced inflammation. A: Normal (non-neoplastic) and neoplastic colon tissues from mice treated with DSS only and DSS + plecanatide (10 ppm) were stained with antibodies specific for Ki-67. Nuclear staining of Ki-67 (positive) was recorded as a labeling index (number of positive cells/total number of cells evaluated; mean ± SEM). Statistical comparisons between DSS control and DSS plus plecanatide-treated groups (n = 7-9 mice per group) were performed using the Student t test. A P value ≤ 0.05 was considered significant; B: Colon tissue (1 cm) from 6 animals per group was pooled to prepare cell lysates. A representative immunoblot demonstrating the effect of plecanatide on expression of c-Myc and cyclin D1 is shown. β-actin was used to normalize protein loading. DSS: Dextran sodium sulfate.
A statistically significant reduction in the levels of total βcatenin was observed in colon tissues from mice administered 10 ppm plecanatide in the diet as compared to control DSS-treated mice. The densitometry of the blot is shown in Figure 5A. Consistent with our previous report[18], immunohistochemical staining revealed membranous localization of β-catenin within the non-neoplastic colonic mucosa. Strong cytoplasmic and nuclear staining of βcatenin staining was observed in colonic dysplasias from DSS-treated mice (panel I, Figure 5B). Treatment with plecanatide (10 ppm) reduced nuclear staining of βcatenin, with a concomitant increase in its localization to the membrane of neoplastic cells (Figure 5B panel II). The densitometric analysis revealed that plecanatide treatment reduced β-catenin levels in the nucleus by 41% (P = 0.02) as compared to analogous neoplastic regions within the colons of DSS-treated mice (Figure 5B, panel III).
Figure 5.
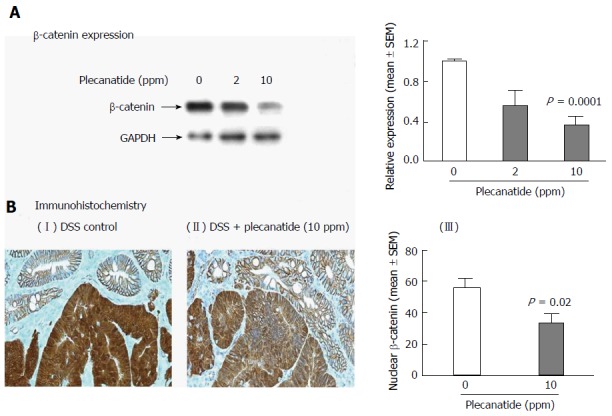
Total β-catenin expression is reduced within the colon of Apc+/Min-FCCC mice treated with dextran sodium sulfate plus plecanatide. A: Western blot analysis and the associated densitometric quantification of levels of total β-catenin expression (mean ± SEM) within the colon; B: Immunohistochemical localization of β-catenin within the colonic mucosa. Membranous localization of β-catenin was observed within the normal colonic mucosa irrespective of the treatment group, while cytoplasmic and nuclear β-catenin staining predominant in adenomas from DSS-treated mice (panel I). Plecanatide treatment caused a significant reduction in nuclear staining of β-catenin in dysplasias, while the cell membranes exhibited enhanced protein localization (panel II). The number of tumor cells with nuclear localization of β-catenin was counted in distal colon tumors (n = 7-9 mice/group) and expressed as a percentage of the total number of tumor cells per 400 X field (panel III). Statistical comparisons between DSS control and DSS plus plecanatide-treated groups were performed using the Student’s t test. A P value of ≤ 0.05 was considered significant. DSS: Dextran sodium sulfate; GAPDH: Glyceraldehyde 3-phosphate dehydrogenase; .
Plecanatide treatment restores UG expression
Consistent with previous reports that expression of UG is reduced dramatically in inflamed tissues from colitic mice and in colon biopsies from IBD patients[13,27], UG transcript levels were also reduced significantly in intestinal tissues following DSS treatment as compared to those of vehicle-treated mice (data not shown). Since administration of plecanatide reduces GI inflammation[16] and the multiplicity of colonic dysplasias in colitic mice, it was important to determine if plecanatide treatment increased transcript levels of UG and GCC following amelioration of GI inflammation in intestinal tissues from Apc+/Min-FCCC mice. UG and GCC expression was determined in the proximal and distal segments of the small intestine and colon by quantitative RT-PCR (Figure 6A). UG expression was increased significantly within the proximal small intestine and proximal colon of animals receiving oral plecanatide. A similar increase in UG expression was observed in the distal small intestine, but did not achieve statistical significance. It should be noted that the relative expression of UG is known to be extremely low in the distal colon as compared to that in the proximal region of the small intestine[24]. Thus, accurate quantitative measurement of UG expression in the distal colon segment may be compromised by low endogenous levels. No appreciable change in relative levels of GC-C transcripts was observed in either the small intestine or colon following plecanatide treatment (Figure 6B).
Figure 6.
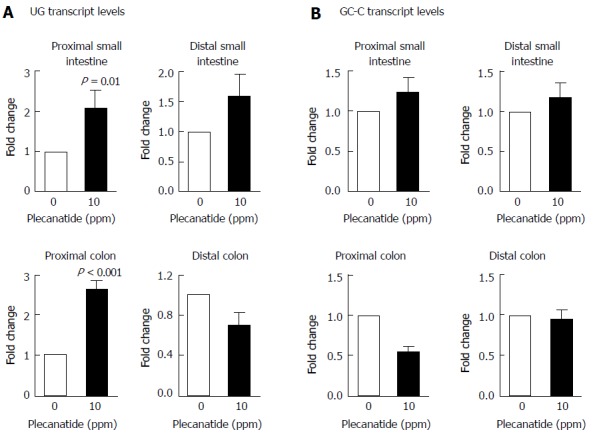
Effect of plecanatide on expression of uroguanylin and guanylate cyclase-C. The relative levels of UG (A) and GC-C (B) transcripts in the small intestine and colon of DSS-treated and DSS + plecanatide treated Apc+/Min-FCCC mice (n = 5-6/group) were determined by quantitative RT-PCR and normalized to those of GAPDH in the same sample. Transcript levels are expressed as fold change (mean ± SEM) as compared to control samples treated with only DSS. Student’s t-test was used to evaluate statistical differences between DSS control and plecanatide-treated mice. A P value of ≤ 0.05 was considered significant. GC-C: Guanylate cyclase-C; UG: Uroguanylin; DSS: Dextran sodium sulfate.
Plecanatide downregulates pro-inflammatory cytokines in colon explants
Induction of colonic inflammation with DSS is known to increase the production of proinflammatory cytokines and growth factors in mice[28]. Thus the effect of plecanatide treatment on activation of GC-C signaling and on secretion of select pro-inflammatory cytokines, chemokines and growth factors was examined in colon explants from Apc+/Min-FCCC mice with acute inflammation. Consistent with the in vivo results shown in Figure 3, plecanatide treatment activated GC-C signaling, increased cGMP production and restored levels of PKG-II in colon explants (data not shown), confirming that orally administered plecanatide is able to activate GC-C signaling within the colon. Next, we determined the effect of plecanatide treatment on the secretion of cytokines and growth factors in explant cultures of colon tissues from mice in the above study. Analysis of the supernatant from pooled explant cultures using a mouse cytokine array revealed a significant reduction in secretion of pro-inflammatory cytokines (IL-1α, IL-6 and TNFalpha), chemokines (IP-10, MIP-1α, KC and CXCL9) and growth factors (GCSF and GMCSF) in DSS + plecanatide treated mice as compared to mice treated with DSS alone (Figure 7). These data are consistent with the anti-inflammatory activity of plecanatide in experimental models of murine colitis[16].
Figure 7.
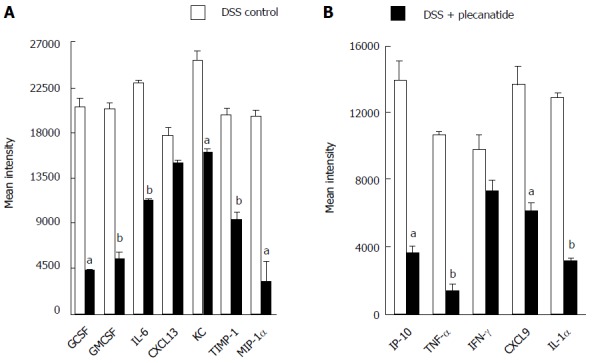
Effect of plecanatide on cytokine expression. Expression of cytokines, chemokines and growth factors was measured in the spent media of colonic explant cultures derived from mice treated with DSS only or DSS + plecanatide. Murine cytokines, chemokines and growth factors were measured in pooled supernatants (n = 6/pool) using membranes coated with specific capture antibodies (Proteome Profiler Panel A kit; R and D Systems, Minneapolis, MN). Immunoblots were scanned using Image J software. The data represent the mean intensity of cytokine/chemokine levels in colonic samples derived from untreated and plecanatide-treated mice. Student’s t test was used to evaluate statistical differences in the secretion of cytokines/chemokines between DSS control and DSS + plecanatide treated samples. aP value ≤ 0.05, bP ≤ 0.01; vs DSS control.
DISCUSSION
This is the first study to demonstrate that oral treatment with plecanatide reduces the multiplicity of DSS-promoted colonic dysplasias in mice; a well-established model for studying inflammation-associated colorectal carcinogenesis[29]. Results presented here demonstrate that Apc+/Min-FCCC mice treated with DSS exhibit an increased multiplicity of colonic dysplasias and that oral treatment with plecanatide produces a statistically significant reduction in inflammation-induced dysplasia in these mice. Although the multiplicity of all morphological subtypes of colonic dysplasias was reduced in mice exposed to plecanatide, the dose required to achieve tumor inhibition varied. For example, plecanatide reduced the formation of polypoid dysplasias in a dose-dependent manner, with an approximate 40% reduction observed at 20 ppm. On the other hand, a similar magnitude of reduction in the multiplicity of flat and indeterminate colonic dysplasias was observed at 10 ppm, with no appreciable inhibition recorded at a higher dose (20 ppm). The chemopreventive effect of NSAIDs and cyclooxygenase-2 inhibitors against inflammation-driven colorectal carcinogenesis has been evaluated in several animal models including Apc+/Min mice. While the reduction in colonic tumors in these studies was approximately 50%[29], it is noteworthy that the observed approximately 40% reduction in dysplasia following plecanatide treatment, is comparable to the result obtained from a meta-analysis of 5-ASA in a clinical setting (37%-49% reduction in neoplasia)[30]. Our results, indicating a differential response of lesion types to plecanatide, are consistent with previous observations suggesting that flat and polypoid dysplasias arise via distinct genetic mechanisms[31,32] and respond differently to prophylactic therapies[8]. In addition, a differential response of polypoid and flat dysplasias to celecoxib (Celebrex®) has also been observed[32].
The lack of an observed plecanatide dose response is consistent with the results of several animal studies examining the effect of plecanatide and dolcanatide (another UG analog) on amelioration of colitis in mice[16]. Orally administered plecanatide or dolcanatide activates GC-C receptors and produces fluid distention only in the duodenum and jejunum[33], suggesting that they act primarily in the proximal intestine to stimulate fluid secretion. Therefore, a higher dose of plecanatide may lead to excess fluid production in the proximal intestine, resulting in dilution of the orally administered plecanatide prior to reaching the colon segments. It should also be noted that plecanatide acts locally by binding to GC-C expressed on epithelial cells lining the luminal surface of the GI tract and its systemic absorption is not needed to produce a pharmacological effect. Thus, lack of a dose response could also be due to saturation of the GC-C that is available on the luminal surface of the GI mucosa. In addition, results from animal studies conducted by this group with orally administered 5-ASA demonstrate that higher doses do not confer greater protection from the formation of colitis-associated tumors[8]. Thus, development and optimization of a new formulation of plecanatide that bypasses the proximal intestine is warranted to not only decrease fluid secretion, but also enhance its chemoprotective activity. A longer duration of exposure to plecanatide would also allow more time for the colonic mucosa to heal and could potentially enhance chemopreventive response to treatment.
Activation of GC-C signaling by its ligands is associated with an increase in cGMP, a decrease in cyclin D1, delayed cell cycle progression and reduced DNA synthesis[34-36]. Importantly, oral administration of cGMP restored crypt proliferative homeostasis and reduced proliferation (Ki-67 positive cells) in the crypts, but not in the villi of Gucy2c-/- mice[36]. In addition, normal functioning of GC-C signaling appears to also regulate the balance between proliferation and differentiation in the intestinal epithelium[37]. In this context, GC-C signaling plays a key role in organizing the crypt-surface axis, restricting the depth of the crypts and the number of proliferating cells and regulating the rate of cell cycle progression through the G1-S transition[23]. Consistent with these findings, UG and E. coli ST inhibit the proliferation of T84 and Caco-2 colon carcinoma cells[13,14]. Data from the present study demonstrating that plecanatide reduced levels of β-catenin, c-myc and cyclin D1 as well as nuclear and total β-catenin provide further support for these findings. Of relevance, treatment with dolcanatide also reduced transcript levels of c-myc, cyclin D1 and Birc5 (survivin) in T84 cells[38]. This study suggests that orally administered plecanatide may act via cGMP/GC-C signaling to mediate downregulation of Wnt/β-catenin signaling within the colon.
The ability of plecanatide to retard the progression of inflammation-associated colorectal neoplasia represents an extension of our prior findings, demonstrating that oral treatment with UG suppressed polyp formation in Apc+/Min mice[13] and plecanatide ameliorated colitis in mice[16]. It should be noted that the expression of UG is reduced significantly in inflamed tissue from IBD patients as well as in colon tissue from mice with colitis[13,27,39]. These findings suggest that loss of UG expression, in the presence of key mutations in the APC gene, results in dysregulation of GC-C signaling and in downstream activation of the Wnt/β-catenin pathway. The resulting transactivation of genes responsible for hyperproliferation and anti-apoptotic mechanisms may be the quintessential events during the early stages of neoplastic transformation in colonocytes. In this context, silencing of GC-C signaling is typically associated with early loss of APC heterozygosity and subsequent AKT-mediated inhibition of apoptosis; a potential trigger for neoplastic transformation in colonocytes[23,36]. These reports also suggest that loss of GC-C signaling could be associated with increased susceptibility to intestinal carcinogenesis in mice. However, the possibility that the observed reduction in polyp formation occurs via a non-GC-C mechanism[40] cannot be completely ruled out. Transcription of the GC-C gene can be regulated by β-catenin/TCF signaling[40]. Interestingly, treatment with E.coli ST peptide stimulated duodenal HCO3- secretion, albeit at a reduced level, in GC-C-/- mice[41], suggesting the existence of a non-GC-C mechanism, possibly involving a UG/ST receptor yet to be identified.
A known human kindred mutation that causes altered expression of GC-C and presents clinically as bowel dysfunction in Norwegian families has been reported[42]. This GC-C “gain-of-function” mutation in infants of these families leads to chronic diarrheal diseases, often accompanied by electrolyte imbalance, dehydration, metabolic acidosis and ileal inflammation. Two additional kindred mutations in GUCY2C were reported in two unrelated Bedouin families, where the “loss-of-function” of GC-C was associated with meconium ileus[43]. Although there is no mention of an association of these kindred mutations with increased susceptibility to colon cancer, deregulated GC-C signaling early in life may be the key event that increases susceptibility to intestinal inflammation and eventually colon cancer.
The precise cause for downregulation of UG and GN and early dysregulation of GC-C signaling during inflammation and colon carcinogenesis remains largely unexplored. Nevertheless, it is known that the genes encoding endogenous GCC ligands UG and GN are located on chromosome 4 in mice and 1p34-35 in humans, a region lost frequently during human colon carcinogenesis[44-46]. Our results, albeit preliminary, suggest that the level of UG transcripts in the small intestine and proximal colon increase following treatment with plecanatide. One possible explanation for the restoration of UG expression is the mucosal healing that eventually follows inhibition of inflammation and/or colorectal dysplasia. It should be noted that UG transcripts levels were measured in intestinal tissue samples comprised of both neoplastic and adjacent normal tissue. Since the expression of UG is lost in colon polyps and tumors but not in the surrounding normal colonic mucosa[13], measurement of UG and GC-C transcript levels could be influenced by the number of tumors present, the severity of inflammation, and the proportion of normal tissue in the sample. A more accurate comparative analysis of UG and GC-C expression in microdissected inflamed, normal vs tumor tissue will be needed in the future. It is known that UG is predominantly expressed in the small intestine and proximal colon, whereas GN expression is more abundant in the colon[24]. An analysis of GN transcript levels in colon tissue would be useful to better understand the cooperative functions of UG and GN within the colon. Another limitation in this study is that expression of UG was examined only at the transcriptional level and not at the protein level. Antibodies specific for UG are being generated currently for immunohistochemical analysis of UG expression in normal, inflamed and tumor tissue; studies that are anticipated to provide new insight into the molecular basis for the disruption of GC-C signaling during colon carcinogenesis.
In summary, results from the present study suggest that administering plecanatide to overcome a deficiency in endogenous GC-C ligands ameliorates inflammation/colitis and delays progression to CRC. These findings represent a new role for GC-C agonists in the prevention of inflammation-associated CRC in humans. Recent clinical studies suggest that plecanatide is a safe and orally active drug candidate, with promising potential for use in the treatment of various GI disorders and diseases[47].
ACKNOWLEDGMENTS
We wish to thank Maureen Climaldi and Susan Nagele for their excellent assistance in preparing this manuscript for publication. This work was performed with the technical support of the Laboratory Animal Facility and the Genomics Facility at Fox Chase Cancer Center.
COMMENTS
Background
Patients with long-standing inflammatory bowel disease (IBD) have a 2 to 8-fold increased relative risk of developing colorectal cancer as compared to the general population. Although prophylactic intervention with 5-aminosalicylate (5-ASA) is considered to be a promising chemopreventive strategy, additional studies are needed to elucidate its utility in IBD-promoted colorectal cancer. Plecanatide is a synthetic analog of the endogenous peptide uroguanylin (UG) and, like UG, is an activator of receptor guanylate cyclase-C (GC-C) signaling cascade that regulates fluid/ion secretion and epithelial cell homeostasis in the gastrointestinal (GI) tract. Oral treatment with plecanatide ameliorates GI inflammation in animal models of experimental colitis. Conceptually, chronic prophylactic intervention with an orally safe and locally-acting agent that not only suppresses inflammation but also regulates renewal of the GI mucosa is desirable.
Research frontiers
Therapeutic intervention with locally acting, minimally absorbed analogs of UG, represents a novel and safe approach for delaying the transition from IBD to colon carcinogenesis.
Innovations and breakthroughs
This is the first report highlighting the therapeutic potential of an orally administered and mucosally active GC-C agonist for delaying the progression of ulcerative colitis to colorectal cancer in humans.
Applications
UG is an endogenous peptide hormone that regulates fluid/ion homeostasis and epithelial cell homeostasis and maintains the barrier function within the GI tract. Several studies have demonstrated that transcript levels of UG and its related peptide guanylin are markedly reduced in inflamed colonic tissues from patients with ulcerative colitis and Crohn’s disease, as well as in human colonic polyps and tumors. It can be implied from these findings that the pathogenesis of these diseases might be associated with a deficiency of UG and GN. Oral therapy with plecanatide and other UG analogs could be considered as a replacement therapy to overcome the deficiency underlying the etiology of IBD and delay its progression to colorectal cancer.
Peer-review
This manuscript is well written and illustrated.
Footnotes
Supported by National Cancer Institute (CA133689 to Kunwar Shailubhai, CA006927 to Fox Chase Cancer Center); and an appropriation from the Commonwealth of Pennsylvania (Fox Chase Cancer Center).
Institutional review board statement: Experiments reported in this manuscript did not involve human samples and hence institutional review is not applicable.
Conflict-of-interest statement: Shailubhai K and Palejwala V are employees of Synergy Pharmaceuticals; remaining authors have no conflict of interest.
Data sharing statement: No additional data are available.
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: United States
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: June 24, 2016
First decision: August 11, 2016
Article in press: October 27, 2016
P- Reviewer: Broutier L, Garcia-Olmo DC, Hunt DM S- Editor: Qiu S L- Editor: A E- Editor: Wu HL
References
- 1.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Hardy RG, Meltzer SJ, Jankowski JA. ABC of colorectal cancer. Molecular basis for risk factors. BMJ. 2000;321:886–889. doi: 10.1136/bmj.321.7265.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itzkowitz SH, Yio X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: the role of inflammation. Am J Physiol Gastrointest Liver Physiol. 2004;287:G7–17. doi: 10.1152/ajpgi.00079.2004. [DOI] [PubMed] [Google Scholar]
- 4.Clevers H. At the crossroads of inflammation and cancer. Cell. 2004;118:671–674. doi: 10.1016/j.cell.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Kraus S, Arber N. Inflammation and colorectal cancer. Curr Opin Pharmacol. 2009;9:405–410. doi: 10.1016/j.coph.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 6.Saxena A, Baliga MS, Ponemone V, Kaur K, Larsen B, Fletcher E, Greene J, Fayad R. Mucus and adiponectin deficiency: role in chronic inflammation-induced colon cancer. Int J Colorectal Dis. 2013;28:1267–1279. doi: 10.1007/s00384-013-1664-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Itzkowitz SH, Harpaz N. Diagnosis and management of dysplasia in patients with inflammatory bowel diseases. Gastroenterology. 2004;126:1634–1648. doi: 10.1053/j.gastro.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 8.Clapper ML, Gary MA, Coudry RA, Litwin S, Chang WC, Devarajan K, Lubet RA, Cooper HS. 5-aminosalicylic acid inhibits colitis-associated colorectal dysplasias in the mouse model of azoxymethane/dextran sulfate sodium-induced colitis. Inflamm Bowel Dis. 2008;14:1341–1347. doi: 10.1002/ibd.20489. [DOI] [PubMed] [Google Scholar]
- 9.Forte LR. Guanylin regulatory peptides: structures, biological activities mediated by cyclic GMP and pathobiology. Regul Pept. 1999;81:25–39. doi: 10.1016/s0167-0115(99)00033-6. [DOI] [PubMed] [Google Scholar]
- 10.Shailubhai K. Therapeutic applications of guanylate cyclase-C receptor agonists. Curr Opin Drug Discov Devel. 2002;5:261–268. [PubMed] [Google Scholar]
- 11.Forte LR, Freeman RH, Krause WJ, London RM. Guanylin peptides: cyclic GMP signaling mechanisms. Braz J Med Biol Res. 1999;32:1329–1336. doi: 10.1590/s0100-879x1999001100002. [DOI] [PubMed] [Google Scholar]
- 12.Forte LR. Uroguanylin and guanylin peptides: pharmacology and experimental therapeutics. Pharmacol Ther. 2004;104:137–162. doi: 10.1016/j.pharmthera.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Shailubhai K, Yu HH, Karunanandaa K, Wang JY, Eber SL, Wang Y, Joo NS, Kim HD, Miedema BW, Abbas SZ, et al. Uroguanylin treatment suppresses polyp formation in the Apc(Min/+) mouse and induces apoptosis in human colon adenocarcinoma cells via cyclic GMP. Cancer Res. 2000;60:5151–5157. [PubMed] [Google Scholar]
- 14.Pitari GM, Di Guglielmo MD, Park J, Schulz S, Waldman SA. Guanylyl cyclase C agonists regulate progression through the cell cycle of human colon carcinoma cells. Proc Natl Acad Sci USA. 2001;98:7846–7851. doi: 10.1073/pnas.141124698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han X, Mann E, Gilbert S, Guan Y, Steinbrecher KA, Montrose MH, Cohen MB. Loss of guanylyl cyclase C (GCC) signaling leads to dysfunctional intestinal barrier. PLoS One. 2011;6:e16139. doi: 10.1371/journal.pone.0016139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shailubhai K, Palejwala V, Arjunan KP, Saykhedkar S, Nefsky B, Foss JA, Comiskey S, Jacob GS, Plevy SE. Plecanatide and dolcanatide, novel guanylate cyclase-C agonists, ameliorate gastrointestinal inflammation in experimental models of murine colitis. World J Gastrointest Pharmacol Ther. 2015;6:213–222. doi: 10.4292/wjgpt.v6.i4.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shailubhai K, Jacob G. Synergy Pharmaceuticals Inc, assignee. Agonists of guanylate cyclase useful for the treatment of gastrointestinal disorders patent. 2009
- 18.Cooper HS, Chang WC, Coudry R, Gary MA, Everley L, Spittle CS, Wang H, Litwin S, Clapper ML. Generation of a unique strain of multiple intestinal neoplasia (Apc(+/Min-FCCC)) mice with significantly increased numbers of colorectal adenomas. Mol Carcinog. 2005;44:31–41. doi: 10.1002/mc.20114. [DOI] [PubMed] [Google Scholar]
- 19.Gabriel A, Helman T, Wilks A. A PCR based assay for the murine multiple intestinal neoplasia (Min) mutation. Mouse Genome. 1993;91:326–328. [Google Scholar]
- 20.Cooper HS, Murthy S, Kido K, Yoshitake H, Flanigan A. Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-catenin and p53 expression and the role of inflammation. Carcinogenesis. 2000;21:757–768. doi: 10.1093/carcin/21.4.757. [DOI] [PubMed] [Google Scholar]
- 21.Riddell RH, Goldman H, Ransohoff DF, Appelman HD, Fenoglio CM, Haggitt RC, Ahren C, Correa P, Hamilton SR, Morson BC. Dysplasia in inflammatory bowel disease: standardized classification with provisional clinical applications. Hum Pathol. 1983;14:931–968. doi: 10.1016/s0046-8177(83)80175-0. [DOI] [PubMed] [Google Scholar]
- 22.Yan Y, Kolachala V, Dalmasso G, Nguyen H, Laroui H, Sitaraman SV, Merlin D. Temporal and spatial analysis of clinical and molecular parameters in dextran sodium sulfate induced colitis. PLoS One. 2009;4:e6073. doi: 10.1371/journal.pone.0006073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li P, Schulz S, Bombonati A, Palazzo JP, Hyslop TM, Xu Y, Baran AA, Siracusa LD, Pitari GM, Waldman SA. Guanylyl cyclase C suppresses intestinal tumorigenesis by restricting proliferation and maintaining genomic integrity. Gastroenterology. 2007;133:599–607. doi: 10.1053/j.gastro.2007.05.052. [DOI] [PubMed] [Google Scholar]
- 24.Scheving LA, Jin WH. Circadian regulation of uroguanylin and guanylin in the rat intestine. Am J Physiol. 1999;277:C1177–C1183. doi: 10.1152/ajpcell.1999.277.6.C1177. [DOI] [PubMed] [Google Scholar]
- 25.Browning DD, Kwon IK, Wang R. cGMP-dependent protein kinases as potential targets for colon cancer prevention and treatment. Future Med Chem. 2010;2:65–80. doi: 10.4155/fmc.09.142. [DOI] [PubMed] [Google Scholar]
- 26.Selvaraj NG, Prasad R, Goldstein JL, Rao MC. Evidence for the presence of cGMP-dependent protein kinase-II in human distal colon and in T84, the colonic cell line. Biochim Biophys Acta. 2000;1498:32–43. doi: 10.1016/s0167-4889(00)00075-6. [DOI] [PubMed] [Google Scholar]
- 27.Brenna Ø, Bruland T, Furnes MW, Granlund Av, Drozdov I, Emgård J, Brønstad G, Kidd M, Sandvik AK, Gustafsson BI. The guanylate cyclase-C signaling pathway is down-regulated in inflammatory bowel disease. Scand J Gastroenterol. 2015;50:1241–1252. doi: 10.3109/00365521.2015.1038849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alex P, Zachos NC, Nguyen T, Gonzales L, Chen TE, Conklin LS, Centola M, Li X. Distinct cytokine patterns identified from multiplex profiles of murine DSS and TNBS-induced colitis. Inflamm Bowel Dis. 2009;15:341–352. doi: 10.1002/ibd.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clapper ML, Cooper HS, Chang WC. Dextran sulfate sodium-induced colitis-associated neoplasia: a promising model for the development of chemopreventive interventions. Acta Pharmacol Sin. 2007;28:1450–1459. doi: 10.1111/j.1745-7254.2007.00695.x. [DOI] [PubMed] [Google Scholar]
- 30.Zhao LN, Li JY, Yu T, Chen GC, Yuan YH, Chen QK. 5-Aminosalicylates reduce the risk of colorectal neoplasia in patients with ulcerative colitis: an updated meta-analysis. PLoS One. 2014;9:e94208. doi: 10.1371/journal.pone.0094208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chang WC, Coudry RA, Clapper ML, Zhang X, Williams KL, Spittle CS, Li T, Cooper HS. Loss of p53 enhances the induction of colitis-associated neoplasia by dextran sulfate sodium. Carcinogenesis. 2007;28:2375–2381. doi: 10.1093/carcin/bgm134. [DOI] [PubMed] [Google Scholar]
- 32.Chang WC, Zenser TV, Cooper HS, Clapper ML. Differential response of flat and polypoid colitis-associated colorectal neoplasias to chemopreventive agents and heterocyclic amines. Cancer Lett. 2013;334:62–68. doi: 10.1016/j.canlet.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Comiskey S, Foss J, Jacob G, Shailubhai K. Orally administered plecanatide, a guanylate cyclase-C agonist, acts in the lumen of the proximal intestine to facilitate normal bowel movement in mice and monkeys. Am J Gastroenterol. 2012;107:S700. [Google Scholar]
- 34.Lin JE, Li P, Pitari GM, Schulz S, Waldman SA. Guanylyl cyclase C in colorectal cancer: susceptibility gene and potential therapeutic target. Future Oncol. 2009;5:509–522. doi: 10.2217/fon.09.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basu N, Saha S, Khan I, Ramachandra SG, Visweswariah SS. Intestinal cell proliferation and senescence are regulated by receptor guanylyl cyclase C and p21. J Biol Chem. 2014;289:581–593. doi: 10.1074/jbc.M113.511311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin JE, Li P, Snook AE, Schulz S, Dasgupta A, Hyslop TM, Gibbons AV, Marszlowicz G, Pitari GM, Waldman SA. The hormone receptor GUCY2C suppresses intestinal tumor formation by inhibiting AKT signaling. Gastroenterology. 2010;138:241–254. doi: 10.1053/j.gastro.2009.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kazerounian S, Pitari GM, Shah FJ, Frick GS, Madesh M, Ruiz-Stewart I, Schulz S, Hajnóczky G, Waldman SA. Proliferative signaling by store-operated calcium channels opposes colon cancer cell cytostasis induced by bacterial enterotoxins. J Pharmacol Exp Ther. 2005;314:1013–1022. doi: 10.1124/jpet.105.089052. [DOI] [PubMed] [Google Scholar]
- 38.Arjunan K, Saykhedkar S, Zhang XC, Palejwala V, Shailubhai K. SP-333, a Guanylate Cyclase-C Agonist, Inhibits NF-kappa B Signaling and Modulates Related Genes and miRNAs Implicated in GI Inflammation and Carcinogenesis. Am J Gastroenterol. 2013;108:S517–S517. [Google Scholar]
- 39.Wu F, Dassopoulos T, Cope L, Maitra A, Brant SR, Harris ML, Bayless TM, Parmigiani G, Chakravarti S. Genome-wide gene expression differences in Crohn’s disease and ulcerative colitis from endoscopic pinch biopsies: insights into distinctive pathogenesis. Inflamm Bowel Dis. 2007;13:807–821. doi: 10.1002/ibd.20110. [DOI] [PubMed] [Google Scholar]
- 40.Giannella RA, Mann EA. E. coli heat-stable enterotoxin and guanylyl cyclase C: new functions and unsuspected actions. Trans Am Clin Climatol Assoc. 2003;114:67–85; discussion 85-86. [PMC free article] [PubMed] [Google Scholar]
- 41.Rao SP, Sellers Z, Crombie DL, Hogan DL, Mann EA, Childs D, Keely S, Sheil-Puopolo M, Giannella RA, Barrett KE, et al. A role for guanylate cyclase C in acid-stimulated duodenal mucosal bicarbonate secretion. Am J Physiol Gastrointest Liver Physiol. 2004;286:G95–G101. doi: 10.1152/ajpgi.00087.2003. [DOI] [PubMed] [Google Scholar]
- 42.Fiskerstrand T, Arshad N, Haukanes BI, Tronstad RR, Pham KD, Johansson S, Håvik B, Tønder SL, Levy SE, Brackman D, et al. Familial diarrhea syndrome caused by an activating GUCY2C mutation. N Engl J Med. 2012;366:1586–1595. doi: 10.1056/NEJMoa1110132. [DOI] [PubMed] [Google Scholar]
- 43.Romi H, Cohen I, Landau D, Alkrinawi S, Yerushalmi B, Hershkovitz R, Newman-Heiman N, Cutting GR, Ofir R, Sivan S, et al. Meconium ileus caused by mutations in GUCY2C, encoding the CFTR-activating guanylate cyclase 2C. Am J Hum Genet. 2012;90:893–899. doi: 10.1016/j.ajhg.2012.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sciaky D, Jenkins NA, Gilbert DJ, Copeland NG, Sonoda G, Testa JR, Cohen MB. Mapping of guanylin to murine chromosome 4 and human chromosome 1p34-p35. Genomics. 1995;26:427–429. doi: 10.1016/0888-7543(95)80238-h. [DOI] [PubMed] [Google Scholar]
- 45.Whitaker TL, Steinbrecher KA, Copeland NG, Gilbert DJ, Jenkins NA, Cohen MB. The uroguanylin gene (Guca1b) is linked to guanylin (Guca2) on mouse chromosome 4. Genomics. 1997;45:348–354. doi: 10.1006/geno.1997.4942. [DOI] [PubMed] [Google Scholar]
- 46.Leister I, Weith A, Brüderlein S, Cziepluch C, Kangwanpong D, Schlag P, Schwab M. Human colorectal cancer: high frequency of deletions at chromosome 1p35. Cancer Res. 1990;50:7232–7235. [PubMed] [Google Scholar]
- 47.Shailubhai K, Comiskey S, Foss JA, Feng R, Barrow L, Comer GM, Jacob GS. Plecanatide, an oral guanylate cyclase C agonist acting locally in the gastrointestinal tract, is safe and well-tolerated in single doses. Dig Dis Sci. 2013;58:2580–2586. doi: 10.1007/s10620-013-2684-z. [DOI] [PubMed] [Google Scholar]