

Abstract
Altered DNA methylation in addiction-related genes may modify the susceptibility to alcohol or drug dependence (AD or ND). We profiled peripheral blood DNA methylation levels of 384 CpGs in promoter regions of 82 addiction-related genes in 256 African Americans (AAs) (117 cases with AD-ND codependence and 139 controls) and 196 European Americans (103 cases with AD-ND codependence and 93 controls) using Illumina’s GoldenGate DNA methylation array assays. AD-ND codependence-associated DNA methylation changes were analyzed using linear mixed-effects models with consideration of batch effects and covariates age, sex, and ancestry proportions. Seventy CpGs (in 41 genes) showed nominally significant associations (P < 0.05) with AD-ND codependence in both AAs and EAs. One CpG (HTR2B cg27531267) was hypomethylated in AA cases (P = 7.2 × 10−5), while 17 CpGs in 16 genes (including HTR2B cg27531267) were hypermethylated in EA cases (5.6 × 10−9 ≤ P ≤ 9.5 × 10−5). Nevertheless, 13 single nucleotide polymorphisms (SNPs) nearby HTR2B cg27531267 and the interaction of these SNPs and cg27531267 did not show significant effects on AD-ND codependence in either AAs or EAs. Our study demonstrated that DNA methylation changes in addiction-related genes could be potential biomarkers for AD-ND co-dependence. Future studies need to explore whether DNA methylation alterations influence the risk of AD-ND codependence or the other way around.
Alcohol and nicotine are the two most commonly used substances of abuse. In 2013, almost 17 million American adults had alcohol abuse or dependence1, and more than 42 million American adults were smokers2. Tobacco use and excessive alcohol consumption can result in substantial health problems. Tobacco use is the leading cause and excessive alcohol consumption is the third leading cause of preventable death in the United States, with more than 480,000 and 80,000 deaths, respectively, attributable to these behaviors in a one-year period2,3,4. Although alcohol and nicotine addiction are often viewed as separate disorders, they frequently co-occur. Individuals with alcohol dependence (AD) are four times more likely to be affected with nicotine dependence (ND), and nearly 23% of ND subjects met past-year criteria for alcohol use disorders5,6,7. Additionally, individuals with comorbid AD and ND had worse clinical outcomes than those with either AD or ND alone8.
AD and ND are both genetically-influenced complex disorders with both shared and specific genetic risk factors. The heritability of AD is estimated to be 50–70%9,10,11,12,13 and that of ND is estimated to be 40–60%11,14. The co-occurrence of AD and ND suggests a substantial genetic correlation between these two disorders and a possible common genetic vulnerability. The common genetic influence may partially explain the observations that smoking was a significant risk factor for promoting the progression of AD15,16, while AD was associated with greater nicotine withdrawal17. The reinforcing effects of alcohol and nicotine could be mediated by common reward pathways18,19. Genetic and epigenetic variants in genes involved in these reward pathways may confer vulnerability to AD-ND codependence.
Genome-wide association studies (GWAS) have been performed to identify genetic variants that are associated with AD and/or ND. At least eight original GWAS on AD have been conducted20,21,22,23,24,25,26,27. Findings from these AD GWAS were largely inconsistent, with the exception of variants in genes encoding the alcohol-metabolizing enzymes28. At least nine original GWAS on ND have been published29,30,31,32,33,34,35,36,37; three of them reported genome-wide significant results for single nucleotide polymorphisms (SNPs) in exon 5 of CHRNA3 on chromosome 1532, the upstream region of IL15 on chromosome 433, the intronic region of DLC1 on chromosome 837, or intergenic regions on chromosomes 7 (between CACNA2D1 and PCLO), 8 (flanked by INTS10), or 14 (from 45,307,535 to 45,613,093 base pairs or from rs146754986 to rs145624594)37. To increase the sample size and statistical power, several meta-analyses of GWAS on AD or ND have been conducted. For example, genome-wide meta-analyses identified a locus [located in the intronic region of the autism susceptibility candidate 2 gene (AUTS2)] that was associated with alcohol consumption and multiple loci (including CHRNA3 rs1051730) that were associated with smoking behaviors38. To date, no studies are known to have studied the pleiotropic effects of gene variants on both AD and ND. Only two published GWASs have studied the association of genetic variants with the co-occurrence of AD and ND (or AD-ND codependence): one identified genome-wide significant SNPs that are located near MARK1 on chromosome 1, proximal to DDX6 on chromosome 11, or in the intronic region of KIAA1409 on chromosome 1439, and another, based exclusively on publicly-available data, found genome-wide significant association signals between IPO11 and HTR1A on chromosome 540.
Although a number of AD and/or ND-associated genetic variants have been identified by GWAS or candidate gene studies, they explain only a small proportion of the genetic variance for these disorders38,41. Among the likely explanations for the “missing” genetic variance are epigenetic events (such as DNA methylation, histone modifications, chromatin remodeling, and noncoding RNA regulation), rare genetic variation, copy number variants, and the interaction among genes and between genes and environment. There is also evidence that environmental factors exert their effects on gene transcription through epigenetic mechanisms42. Candidate gene DNA methylation or epigenome-wide association studies (EWAS)43,44,45,46,47,48, including ours49,50, have shown altered DNA methylation in the peripheral blood of AD subjects or lymphoblastoid cell lines derived from AD subjects. We51 and others52,53 have also found altered DNA methylation in postmortem brains of AD subjects. Similarly, ND-associated DNA methylation changes have been found in the peripheral blood/lymphoblast cell lines46,54,55, lung56, or other tissues57. We are not aware of any published studies that have examined epigenetic changes contributing to AD-ND codependence. Given that AD and ND are genetically influenced complex disorders that exhibit a high degree of comorbidity, we examined AD-ND codependence-associated DNA methylation alterations in the promoter regions of 82 addiction-related genes in two populations: African Americans (AAs) and European Americans (EAs).
Results
AD-ND codependence-associated promoter DNA methylation changes in addiction-related genes
Methylation levels of 384 CpGs in the promoter region of 82 addiction-related genes were examined by the custom-designed Illumina GoldenGate assay for DNA methylation profiling, and AD-ND codependence-associated DNA methylation changes were identified in both AAs and EAs. The association analysis results are summarized in Supplementary Tables S1 and S2. In AAs, 103 (26.8%) CpGs in 54 genes showed nominally significant associations (P < 0.05) with AD-ND codependence (Supplementary Table S1). Only one CpG (cg27531267, located in the promoter region of HTR2B) remained significant (P = 7.2 × 10−5) after Bonferroni correction for multiple testing (P value ≤ 0.05/384 = 1.3 × 10−4 as the corrected statistical significance threshold), and it was hypomethylated in AAs with AD-ND codependence (Table 1). In EAs, 152 (39.6%) CpGs in 63 genes showed nominally significant associations (P < 0.05) with AD-ND codependence (Supplementary Table S2). The findings from 17 CpGs in 16 genes (MBD3 cg21372728, HTR2B cg27531267, PENK cg26106216, DRD2 cg05421426, NCAM1 cg14313206, NCAM1 cg21572351, HTR2C cg02156408, HTR3A cg08989585, GABRB2 cg02095443, OPRD1 cg01706569, DRD4 cg08079114, RGS17 cg12505522, SLC6A4 cg14534584, SLC6A3 cg00037218, OPRK1 cg07344165, GAD1 cg04123893, and GABRB1 cg21074850) survived Bonferroni correction (5.6 × 10−9 ≤ P ≤ 9.5 × 10−5, using P value ≤ 0.05/384 = 1.3 × 10−4 as the statistical significance threshold). All 17 CpGs were hypermethylated in EAs with AD-ND codependence (Table 1). As displayed in Fig. 1, the findings from HTR2B cg27531267 withstood multiple testing corrections in both AAs (AAs: P = 7.2 × 10−5) and EAs (P = 2.1 × 10−7). Additionally, 70 CpGs (in 41 genes) showed nominally significant associations (P < 0.05) with AD-ND codependence in both AAs and EAs (Supplementary Tables S1 and S2).
Table 1. Differentially methylated CpGs in subjects with alcohol-nicotine codependence.
| CpGs | Genes | β (Mean ± S.D.) | Effect size | P values |
|---|---|---|---|---|
| African Americans (n = 256) | ||||
| cg27531267 | HTR2B | 0.043 ± 0.013 | −0.005 | 7.2 × 10−5 |
| European Americans (n = 196) | ||||
| cg27531267 | HTR2B | 0.038 ± 0.012 | 0.006 | 2.1 × 10−7 |
| cg02156408 | HTR2C | 0.088 ± 0.023 | 0.009 | 8.0 × 10−6 |
| cg08989585 | HTR3A | 0.170 ± 0.016 | 0.007 | 1.5 × 10−5 |
| cg14534584 | SLC6A4 | 0.054 ± 0.017 | 0.007 | 8.1 × 10−5 |
| cg05421426 | DRD2 | 0.090 ± 0.022 | 0.008 | 4.0 × 10−6 |
| cg08079114 | DRD4 | 0.054 ± 0.013 | 0.006 | 4.5 × 10−5 |
| cg00037218 | SLC6A3 | 0.033 ± 0.014 | 0.006 | 8.2 × 10−5 |
| cg21074850 | GABRB1 | 0.059 ± 0.019 | 0.007 | 1.3 × 10−4 |
| cg02095443 | GABRB2 | 0.061 ± 0.016 | 0.006 | 3.8 × 10−5 |
| cg04123893 | GAD1 | 0.296 ± 0.032 | 0.012 | 9.5 × 10−5 |
| cg01706569 | OPRD1 | 0.045 ± 0.016 | 0.007 | 4.3 × 10−5 |
| cg07344165 | OPRK1 | 0.034 ± 0.015 | 0.006 | 8.9 × 10−5 |
| cg26106216 | PENK | 0.028 ± 0.014 | 0.007 | 5.7 × 10−7 |
| cg12505522 | RGS17 | 0.056 ± 0.017 | 0.007 | 6.7 × 10−5 |
| cg14313206 | NCAM1 | 0.049 ± 0.012 | 0.005 | 7.7 × 10−6 |
| cg21572351 | NCAM1 | 0.141 ± 0.019 | 0.008 | 8.2 × 10−6 |
| cg21372728 | MBD3 | 0.051 ± 0.014 | 0.008 | 5.6 × 10−9 |
Figure 1. Distribution of P values of 384 CpGs in 82 addiction-related genes in African Americans and European Americans.
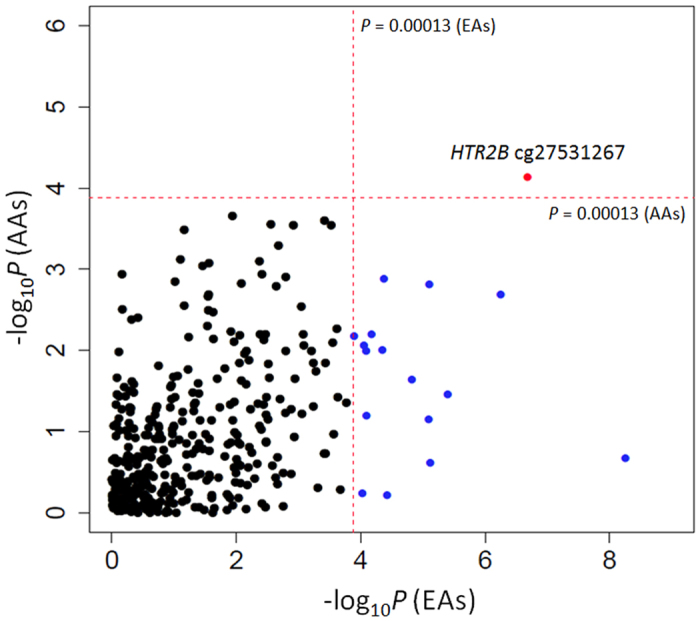
The association between 384 CpGs in 82 addiction-related genes and alcohol and nicotine codependence was analyzed using the liner mixed-effect model. There were 17 CpGs survived the Bonferroni adjustment (P < = 0.05/384 = 0.00013; the red dashed-line) in European Americans. Only one CpGs (cg27531267 in the promoter region of HTR2B) survived the Bonferroni correction in both African Americans (AAs) and European Americans (EAs).
No significant interactive effects of HTR2B cg27531267 and nearby SNPs on AD-ND codependence
Considering that genetic variants may either have a direct or an indirect (e.g., via altering DNA methylation patterns) influence on disease risk, we further analyzed the effects of 13 SNPs around HTR2B cg27531267 (50 kb up- or downstream of cg27531267) and the interaction of these SNPs with HTR2B cg27531267 on the susceptibility to AD-ND codependence. All 13 SNPs were located in a tight linkage disequilibrium (LD) block with average R2 of 0.48 in EAs, while the LD pattern was similar but less tight in AAs with average R2 of 0.28 (Figure S1). As shown in Table 2, none of the 13 SNPs was significantly associated with AD-ND codependence in either AAs or EAs. Moreover, the interactive effect of HTR2B cg27531267 and nearby SNPs on AD-ND codependence was not significant in either AAs or EAs. Additionally, the methylation level of HTR2B cg27531267 was not significantly correlated with the genotypes of 13 nearby SNPs in either AAs or EAs (Pcorrelation > 0.05).
Table 2. Interactive effects of HTR2B cg27531267 with 13 nearby SNPs on alcohol and nicotine codependence.
| SNP | CpG-SNP distance (bp) | Effects of SNPs on AD-ND codependence |
cg27531267-SNP interactions on AD-ND codependence |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Estimate | Std. error | Z | P | Estimate | Std. error | Z | P | ||
| Interactions of cg27531267 and nearby SNPs in African Americans (AAs): | |||||||||
| rs6436999 | −49250 | −0.396 | 0.913 | −0.433 | 0.665 | −1.053 | 20.899 | −0.050 | 0.960 |
| rs2303357 | −41796 | −0.160 | 0.735 | −0.218 | 0.828 | 6.543 | 16.433 | 0.398 | 0.691 |
| rs16827801 | 22590 | 0.500 | 0.775 | 0.645 | 0.519 | −10.499 | 17.543 | −0.598 | 0.550 |
| rs10194776 | −11723 | −0.467 | 0.964 | −0.484 | 0.628 | 0.907 | 22.385 | 0.041 | 0.968 |
| rs17619588 | −19567 | 0.468 | 0.841 | 0.557 | 0.578 | −8.995 | 19.191 | −0.469 | 0.639 |
| kgp14695769 | −18326 | −1.186 | 1.350 | −0.878 | 0.380 | 25.791 | 29.761 | 0.867 | 0.386 |
| rs16827784 | −10965 | −1.203 | 1.352 | −0.890 | 0.374 | 26.079 | 29.803 | 0.875 | 0.382 |
| rs10199752 | −22188 | 0.552 | 0.916 | 0.603 | 0.547 | −1.156 | 20.849 | −0.055 | 0.956 |
| rs1549339 | −8916 | −0.297 | 0.776 | −0.383 | 0.702 | 9.857 | 17.497 | 0.563 | 0.573 |
| rs10187149 | −1371 | 0.496 | 0.778 | 0.637 | 0.524 | −10.489 | 17.673 | −0.594 | 0.553 |
| rs4635521 | 29520 | −0.539 | 0.848 | −0.635 | 0.525 | 4.375 | 19.022 | 0.230 | 0.818 |
| rs13430407 | 43687 | −0.552 | 0.916 | −0.603 | 0.547 | 1.156 | 20.849 | 0.055 | 0.956 |
| rs12468767 | 45101 | 2.926 | 1.655 | 1.768 | 0.077 | −68.597 | 39.852 | −1.721 | 0.085 |
| Interactions of cg27531267 and nearby SNPs in European Americans (EAs): | |||||||||
| rs6436999 | −49250 | −5.551 | 7.974 | −0.696 | 0.486 | 177.230 | 238.254 | 0.744 | 0.457 |
| rs2303357 | −41796 | −4.473 | 7.842 | −0.570 | 0.568 | 143.319 | 232.680 | 0.616 | 0.538 |
| rs16827801 | 22590 | 5.283 | 6.727 | 0.785 | 0.430 | −173.067 | 200.339 | −0.864 | 0.380 |
| rs10194776 | −11723 | −2.252 | 8.112 | −0.278 | 0.781 | 85.800 | 241.817 | 0.355 | 0.722 |
| rs17619588 | −19567 | 1.276 | 9.212 | 0.138 | 0.890 | −89.607 | 270.476 | −0.331 | 0.740 |
| kgp14695769 | −18326 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| rs16827784 | −10965 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| rs10199752 | −22188 | 4.366 | 7.833 | 0.557 | 0.577 | −140.073 | 232.332 | −0.603 | 0.547 |
| rs1549339 | −8916 | −4.472 | 7.838 | −0.571 | 0.568 | 143.368 | 232.103 | 0.618 | 0.537 |
| rs10187149 | −1371 | 5.182 | 7.409 | 0.699 | 0.484 | −169.876 | 221.410 | −0.767 | 0.443 |
| rs4635521 | 29520 | −5.112 | 7.429 | −0.688 | 0.491 | 167.447 | 222.060 | 0.754 | 0.451 |
| rs13430407 | 43687 | −4.366 | 7.833 | −0.557 | 0.577 | 140.073 | 232.333 | 0.603 | 0.547 |
| rs12468767 | 45101 | −0.250 | 9.311 | −0.027 | 0.979 | −12.922 | 279.219 | −0.046 | 0.963 |
Potential function of AD-ND codependence-associated CpGs
The program PROMO was used to predict putative transcription factor binding sites (TFBS) in DNA sequences harboring differentially methylated CpGs. Among the 17 differentially methylated CpGs (Table 1), 11 were predicted to be located in the core binding site of one or more transcription factors (Table 3). The UCSC Genome Browser was used to query DNase hypersensitivity sites (DHSs) and H3K27Ac marks in DNA sequences harboring differentially methylated CpGs. Among the 17 differentially methylated CpGs, 14 were located in DHSs and five were situated in DNA sequences that are associated with the H3K27Ac histone mark (Table 3). Although the most significant HTR2B cg27531267 was not predicted to be located in TFBSs, it was mapped to DHSs and DNA sequences associated with histone protein modification mark H3K27Ac (Fig. 2).
Table 3. Function prediction of alcohol-nicotine codependence-associated CpGs.
| CpGs | Genes | Chr. | Coordinate | DNA Sequences around CpGs | aTF core binding sites | bDHSs | cH3K27Ac |
|---|---|---|---|---|---|---|---|
| cg01706569 | OPRD1 | 1 | 29011768 | GTGGGGATCA[CG]AACTTGAGAC | Yes | No | |
| cg04123893 | GAD1 | 2 | 171380892 | AACCTTCAAA[CG]TGATTAATCA | Yes | Yes | |
| cg27531267 | HTR2B | 2 | 231699986 | CACACATACA[CG]CACACACACG | Yes | Yes | |
| cg21074850 | GABRB1 | 4 | 46729246 | ACATGGTCTC[CG]AAGTGAATAT | TFII-I (GTCTCC) | No | No |
| cg00037218 | SLC6A3 | 5 | 1498390 | CCCCGGCCCC[CG]CCCCTGCGCC | E2F-1 (GCCCCCGC); | No | No |
| Sp1 (CCCCCGCCCC) | |||||||
| cg02095443 | GABRB2 | 5 | 160908531 | TGGCGGCAGG[CG]GCGGAAGTAG | AP-2alphaA (GCAGGC); | Yes | No |
| Elk-1 (CGGCGGAAG) | |||||||
| cg12505522 | RGS17 | 6 | 153494537 | CTGCCTGCTC[CG]GGTCCCGGAG | Yes | Yes | |
| cg07344165 | OPRK1 | 8 | 54326330 | ACAGGGAGAA[CG]GACTTCTCGC | FOXP3 (GAGAAC) | Yes | No |
| cg26106216 | PENK | 8 | 57521167 | AGGGGATCGT[CG]AGCAAAAGCC | NF-kappaB1 (GGGGATCGTCG) | Yes | No |
| cg08079114 | DRD4 | 11 | 627981 | CCCTGGCTGC[CG]TGGGACACAC | ENKTF-1 (TGGCTGCC) | Yes | No |
| cg21572351 | NCAM1 | 11 | 112335975 | GGGCAGATCA[CG]AGGTCAGGAG | GR-alpha (CGAGGTCAGG); | No | Yes |
| COUP-TF1 (CACGAGGTCAGGA) | |||||||
| cg14313206 | NCAM1 | 11 | 112337308 | CCCGGGCCAG[CG]CAAGGATCTC | GCF (GGCCAGCGC) | Yes | Yes |
| cg05421426 | DRD2 | 11 | 112850463 | ACCGTGGGAG[CG]GGAGAATCCA | E2F-1 (GCGGGAGA) | Yes | No |
| cg08989585 | HTR3A | 11 | 113350239 | GCAGTGATTG[CG]CCACTGCACT | C/EBPbeta (TTGC); | Yes | No |
| C/EBPalpha (GATTGCG); | |||||||
| ENKTF-1 (TTGCGCCA) | |||||||
| cg14534584 | SLC6A4 | 17 | 25587232 | CCTGCCGCCC[CG]CGCCCACAGG | Pax-5 (GCCGCCCCGCGCCC); | Yes | No |
| p53 (GCCGCCCCGCGCCC); | |||||||
| Sp1 (CTGCCGCCCC); | |||||||
| GCF (GCCCCGCGC); | |||||||
| ETF (GCCCCGCGCCC) | |||||||
| cg21372728 | MBD3 | 19 | 1543111 | CCCTGCTCCC[CG]AAATCCCGGC | Yes | Yes | |
| cg02156408 | HTR2C | X | 113725033 | GGCCTTCGTC[CG]TTTAGAGTAG | Yes | No |
aTranscription factor (TF) binding sites predicted by PROMO.
bDNase I Hypersensitivity sites (DHSs) from ENCODE (95 cell types) by the UCSC Genome Browser.
cH3K27Ac mark (often found near active regulatory elements) on 7 cell lines from ENCODE by the UCSC Genome Browser.
Figure 2. Functional annotation of three HTR2B promoter CpGs.

The top panel contains the -log P values for the association between DNA methylation of three HTR2B promoter CpGs and alcohol-nicotine codependence. The other panels indicate the presence of coding exons (boxes in dark red) and noncoding introns (grey lines) of HTR2B (the second panel), DNase hypersensitivity cluster sites (the third panel), the location of a H3K27ac histone modification (the fourth panel), the conservative area (the fifth panel), the percentage of G (guanine) and C (cytosine) bases (the 6th panel), and the location of CpGs in transcription factor binding sites (TFBSs) (the seventh panel). The function of the most significant CpG (i.e., cg27531267 in the promoter region of HTR2B) is annotated by a brown line across all panels.
Discussion
Prior evidence supports the involvement of many of the genes included in the present study in neurobiologic processes underlying drug reward and addiction. However, with a few exceptions (such as those coding for nicotinic acetylcholine receptors or alcohol metabolizing enzymes), most of these genes have not been previously found to harbor genetic variants that are associated at a genome-wide level with AD and/or ND. There are other mechanisms by which these loci could exert more substantial effects on AD and/or ND: namely, DNA methylation changes in addiction-related genes could confer vulnerability to AD and/or ND. In the present study, we identified methylation alterations in the promoter regions of a number of addiction-related genes in African Americans (AAs) and European Americans (EAs) with AD-ND codependence.
We found that differentially methylated genes are involved in several critical pathways for AD and/or ND. A post hoc power analysis demonstrated that 196 EAs samples provided 80% statistical power for an effect size greater than 0.004, while 256 AA samples provided 80% statistical power to detect an effect size greater than 0.003, indicating that our sample size provided adequate statistical power to detect methylation changes. As summarized in Table 1, four of the differentially methylated genes were serotonergic (HTR2B, HTR2C, HTR3A, and SLC6A4), three were dopaminergic (DRD2, DRD4, and SLC6A3), two were GABAergic (GABRB1 and GABRB2), one was glutamatergic (GAD1), and three (OPRD1, OPRK1, and PENK) were opioidergic. Three other genes (RGS17, NCAM1, and MBD3), which do not belong to the above-listed neurotransmitter systems, also showed promoter DNA methylation changes in subjects with AD-ND codependence. RGS17 encodes a regulator of G-protein signaling (RGS)58, which can inactivate the G protein and rapidly switch off G-protein-coupled receptor signaling pathways59. Our candidate gene studies have demonstrated that variation in RGS17 is associated with a variety of different substance dependence disorders60. NCAM1 encodes a neural cell adhesion protein, a member of the immunoglobulin superfamily61. It is located in the DRD2-ANKK1-TTC12-NCAM1 gene cluster region, and SNPs or haplotypic variants in this region were found to be associated with dependence on alcohol, nicotine, or other drugs of abuse62,63,64. MBD3 encodes methyl-CpG binding domain protein 3, a nuclear protein that is potentially involved in chromatin remodeling and histone modifications65,66. Because genetic association studies did not reveal a strong effect (i.e., with genome-wide significance) of variants in the above genes on AD and/or ND, we postulate that either inherent DNA methylation of these genes results in AD and/or ND or long-term alcohol misuse or smoking leads to AD and/or ND through epigenetic modifications. It should be noted that these epigenetic changes may not contribute to the risk of AD and ND simultaneously or to the same extent. Significant CpGs identified in the present study are associated with AD-ND codependence, which was considered to be a new phenotype, i.e., the co-occurring risk of AD and ND.
Our findings suggest that the impact of DNA methylation of addiction-related genes on AD-ND codependence risk may be larger than that of genetic variants carried by these genes. Considering the possible correlation of methylation levels of CpGs and genotypes of nearby SNPs as reported in our previous studies67, we further investigated the effect of 13 SNPs within ± 50 kb of HTR2B cg27531267, as well as the interaction of cg27531267 with these SNPs on AD-ND codependence risk. Although HTR2B cg27531267 was differentially methylated in both AAs and EAs (albeit in opposite directions) with AD-ND codependence, no interactive effect of HTR2B cg27531267 with proximal SNPs on AD-ND codependence was observed in either population. Additionally, none of the 13 nearby SNPs was significantly associated with AD-ND codependence (Table 2). This finding is consistent with the results from our previous GWAS research that variants in HTR2B were not significantly associated with either AD26 or ND37.
The present study provides further evidence that DNA methylation changes within the regulatory (promoter) regions of addiction-related genes are associated with AD and/or ND, presumably because they may result in altered gene transcription. The rationale is that promoter DNA methylation may directly interfere with the binding of transcription factors (TFs) to the regulatory regions. Among the top 17 CpGs located in promoter regions of 16 genes (Table 1), 11 were predicted to be located in the core binding site of one or more TFs (Table 3). It is well known that chromatin structure mediates the interaction of TFs and DNA68,69. Our findings suggest that promoter DNA methylation may also modulate TF-DNA interactions and subsequently influence gene transcription. Moreover, methylated CpG falling within DNase hypersensitivity sites (DHSs) may impede the association of TFs to DNA, thus inhibiting the accessibility of chromatin. Because we did not extract RNA from blood samples (which were used only for genomic DNA extraction), we were unable to perform RT-qPCR to confirm gene expression changes caused by differentially methylated CpGs. Additionally, methylation of CpGs mapped to DNA sequences that are associated with histone marks (e.g., H3K27Ac mark, which is often found near active regulatory elements) may change histone protein epigenetic status, thus influencing the compact structure of chromatins. We also noticed that some of the 17 differentially methylated CpGs (including the most significant HTR2B cg27531267) were located in DHSs or DNA sequences that are associated with histone mark H3K27Ac. Taken together, subjects with altered DNA methylation in promoter regions of addiction-related genes may have an increased risk of AD and/or ND. Note that, further studies are needed to replicate the above findings in independent samples and extend the findings to other racial/ethnic groups.
The major limitation of the present study is that we only investigated the association of CpGs in promoter regions of a number of preselected addiction-related genes. For a more complete understanding of the epigenetic mechanism of AD-ND codependence, it will be necessary to use a high-resolution DNA methylation array (such as the Illumina MethylationEPIC BeadChip) assays or whole genome bisulfite sequencing (WGBS) to identify DNA methylomic changes. Another limitation is that we did not consider the relative proportion of different types of blood cells in the DNA methylation data analysis. Inter-individual differences in DNA methylation levels due to different blood cell composition may confound the findings. In future, when we have the high-density DNA methylation data (such as those generated by the Illumina MethylationEPIC BeadChip), we could use the method developed by Jaffe and Irizarry70 to estimate the relative proportions of CD4+ and CD8+ T-cells, natural killer cells, monocytes, granulocytes, and B-cells in blood samples and then incorporate the cell proportion estimation into the data analysis. Although DNA methylation patterns in the peripheral blood may not reflect those in the brain, peripheral blood samples are easier to collect than brain tissue samples, and blood DNA methylation changes in regulatory regions of genes could be accessible biomarkers.
We are aware that there are more significant CpGs in EAs than AAs, even though the EA sample was smaller than the AA sample. Additionally, it is unknown why HTR2B cg27531267 showed an opposite methylation direction in AAs (hypomethylation) and EAs (hypermethylation) with AD-ND codependence. One possible explanation is that the DNA methylation status of CpGs can be influenced by genetic variation. Similar to many disease-associated SNPs that are specific to a certain population, CpG methylation levels may also be population-specific, leading to inconsistent results between EAs and AAs. For example, the top CpG cg27531267 showed a mean methylation level of 0.045 in AA controls, which was significantly higher than that of EA controls (β = 0.035, P = 1.8 × 10−8). Among the 13 SNPs that are proximal to HTR2B cg27531267 and included in CpG-SNP interaction analysis, two were either not existent or rare in EAs (Table 2).
In summary, the present study examined DNA methylation alterations in promoter regions of addiction-related genes among individuals with AD-ND codependence. We identified both specific and shared DNA methylation changes in the two populations. The overlap of the differentially methylated promoter CpGs and TFBSs or DHSs and the location of differentially methylated CpGs in DNA sequences that are associated with specific histone marks (e.g., H3K27Ac) imply that promoter CpG methylation may modulate gene transcription and influence an individual’s susceptibility to AD-ND codependence. Considering the reversibility of DNA methylation, the findings from the present study could provide the basis for effective pharmacotherapies for AD-ND co-dependence that target specific epigenetic marks in promoter regions of addiction-related genes.
Methods
Subjects
Two hundred fifty-six African Americans (AAs) [including 117 cases (71 males and 46 females) with AD-ND codependence and 139 controls (31 males and 108 females)] and 196 European Americans [including 103 cases (59 males and 44 females) with AD-ND codependence and 93 controls (49 males and 44 females)] were recruited from substance abuse treatment centers or through advertisements at the University of Connecticut Health Center (n = 200), Yale University (n = 126), and the Medical University of South Carolina (n = 126). The mean age of AA cases and AA controls was 42 ± 8 and 37 ± 14 years, respectively (P < 0.05). The mean age of EA cases and EA controls was 43 ± 12 and 37 ± 16 years, respectively (P > 0.05). Both cases and controls were chosen from a large sample of subjects recruited for studies of the genetics of substance dependence. Subjects were interviewed using an electronic version of the Semi-Structured Assessment for Drug Dependence and Alcoholism (SSADDA)71. Lifetime diagnoses for AD and ND codependence were made according to the criteria of the Diagnostic and Statistical Manual of Mental Disorders, 4th edition (DSM-IV) [American Psychiatric Association, 1994]. None of the subjects had a lifetime major psychotic disorder such as schizophrenia and bipolar disorder. Additionally, no control subjects (139 AAs and 93 EAs) were affected with alcohol or drug abuse or dependence. Subjects gave informed consent as approved by the institutional review board at each clinical site, and certificates of confidentiality were obtained from the National Institute on Drug Abuse and the National Institute on Alcohol Abuse and Alcoholism. All methods were carried out in accordance with relevant guidelines and regulations. All experimental protocols were approved by the Human Investigation Committee of the above three institutes.
The self-reported genetic background of all subjects included in the present study was verified using a set of 41 ancestry informative markers (AIMs), including 36 short tandem repeat markers and five SNPs, implemented in the program STRUCTURE72. Subjects were defined as EAs if their ancestry proportion scores were less than 0.5; otherwise, they were considered AAs. To minimize the influence of subjects’ genetic background on the results of the DNA methylation analysis, subjects’ ancestry proportions were considered as a covariate in the differential CpG analysis.
DNA extraction and bisulfite modification
Genomic DNA was extracted from the peripheral blood of cases and controls using the PAXgene Blood DNA Kit (PreAnalytiX, Hombrechtikon, Switzerland). One microgram of genomic DNA was treated with the bisulfite reagent included in the EZ DNA Methylation Kit (Zymo Research, Orange, CA, USA). Unmethylated cytosines were converted to uracils while methylated cytosines remained unchanged73. Bisulfite-converted DNA samples were then used in the custom-designed Illumina GoldenGate DNA methylation assay.
Illumina GoldenGate DNA methylation assay
The design of the customized Illumina GoldenGate DNA Methylation array was described in our previous publication50. Supplementary Table SI contains information on 384 CpGs in the promoter regions of 82 candidate genes involved in the opioidergic system (8 genes/51 CpGs), the serotonergic system (11 genes/52 CpGs), the dopaminergic system (8 genes/45 CpGs), the GABAergic system (13 genes/42 CpGs), the glutamatergic system (7 genes/45 CpGs), the cannabinoid system (CNR1: 3 CpGs), and the cholinergic system (6 genes/34 CpGs), as well as alcohol metabolism (5 genes/14 CpGs), DNA methylation (8 genes/34 CpGs), signal transduction (12 genes/53 CpGs), and several others (ANKK1: 4 CpGs; NCAM1: 4 CpGs; TTC12: 3 CpGs). The hybridization probes were highly specific for these 384 CpGs [GGMAScore (mean ± S.D.): 0.85 ± 0.06].
After bisulfite conversion of genomic DNA, the remaining methylation assay steps were the same as those previously described50. Image processing and intensity data extraction were performed using the Illumina GenomeStudioTM Methylation Module v.1.0 Software. The background normalization algorithm was used to minimize background variation within the array by using built-in negative control signals. The methylation level (defined as β) of each individual CpG site was estimated as the ratio of intensities between methylated and unmethylated alleles. The β value was calculated as β = [Max(Cy5,0)]/[Max (Cy3,0) + Max(Cy5,0) + α]. A constant offset α (by default, α = 100) was added to the denominator to adjust β values when both methylated and unmethylated probe intensities were low. The β value ranges from 0 (i.e., completely unmethylated) to 1 (i.e., completely methylated).
Statistical and bioinformatics analyses
The statistical analysis of DNA methylation data was implemented in R (version 3.1.3) (http://www.r-project.org/). To identify differentially methylated CpGs in subjects with AD and ND codependence, we used the linear mixed-effects model handled by the R package CpGassoc, which was designed for analyzing the association between methylation at CpG sites across the genome and a phenotype of interest74. In the linear mixed-effect model built-in CpGassoc, the methylation level of CpGs was the response variable, the status of AD-ND codependence and other covariates (age, sex, and ancestry proportions) were fitted via fixed effects, and batch factor (referring to methylation chips) was fitted as a random effect. A post hoc power analysis by R package SIMR75 served to calculate the power for a linear mixed model based on the observed data structure. For both EAs and AAs, we evaluated the sample power by varying effect sizes and using 100 simulations.
The interactive effect of differentially methylated CpGs and nearby single nucleotide polymorphisms (SNPs) on AD-ND codependence risk was examined using the generalized linear mixed-effects model by the lme4 package in R (http://CRAN.R-project.org/package=lme4). Although the correlation of CpGs and SNPs can be detected when they are within one million bases apart, the strength of correlation between CpGs and SNPs is dramatically decreased when their distance is increased67. Here, we just included those SNPs that are 50 kb away from the CpG site in the SNP-CpG interaction analysis. SNP genotype data were generated via the Illumina HumanOmni1-Quad v1.0 microarray, as described in our previous GWAS on AD26 and ND37. Genotype data were cleaned with a commonly used procedure, that is, SNPs were excluded if they met any of following criteria: 1) minor allele frequency (MAF) was <0.05; 2) missing genotyping rate was >10%; or 3) P value of Hardy-Weinberg disequilibrium test was <1.0 × 10−3. The association of SNP genotypes (in an additive model) and CpG methylation levels was analyzed using PLINK76. For both EAs and AAs, linkage disequilibrium (LD) plots of SNPs near CpGs were generated by R package LDheatmap77.
Additionally, the function of differentially methylated promoter CpGs was predicted by bioinformatics programs. The online program PROMO78,79 was applied to predict whether differentially methylated promoter CpGs were located in transcription factor binding sites (TFBS) as defined in the TRANSFAC database80. The USCS Genome Browser (http://genome.ucsc.edu) was used to query whether differentially methylated promoter CpGs were located in DNase hypersensitivity sites (DHSs) or in chromosomal regions that were associated with H3K27Ac mark (which are often found near active regulatory elements).
Additional Information
How to cite this article: Xu, H. et al. Alcohol and nicotine codependence-associated DNA methylation changes in promoter regions of addiction-related genes. Sci. Rep. 7, 41816; doi: 10.1038/srep41816 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This study was supported by the National Institutes of Health (NIH) grants K99/R00 DA022891 (HZ), R21 AA023068 (HZ), R01 AA025080 (HZ), R01 AA017535 (JG), R01 DA012690 (JG), and P50 AA12870 (JG).
Footnotes
Although unrelated to this research, Dr. Kranzler has been a consultant, advisory board member, or CME speaker for Indivior, Lundbeck, and Otsuka. He is also a member of the Alcohol Clinical Trials Initiative of the American Society of Clinical Psychopharmacology, which is supported by AbbVie, Ethypharm, Lilly, Lundbeck, and Pfizer. Other authors declare that they have no competing interests.
Author Contributions All authors contributed to the study design, data acquisition, or data analysis and interpretation. Specifically, H.Z. contributed to the study design as well as data acquisition, analysis, and interpretation. H.X., F.W., H.R.K., and J.G. performed data acquisition, analysis, and interpretation. H.Z., H.X., and F.W. drafted the manuscript. All authors were involved in either drafting or revising the manuscript. All authors have approved the final version of the manuscript.
References
- SAMHSA. Substance Abuse and Mental Health Services Administration, Results from the 2013 National Survey on Drug Use and Health: Mental Health Findings, NSDUH Series H-49, HHS Publication No. (SMA) 14–4887. Rockville, MD: Substance Abuse and Mental Health Services Administration, 2014 (2014).
- Jamal A. et al. Current cigarette smoking among adults--United States, 2005–2013. MMWR Morb Mortal Wkly Rep 63, 1108–1112 (2014). [PMC free article] [PubMed] [Google Scholar]
- Sacks J. J. et al. State costs of excessive alcohol consumption, 2006. Am J Prev Med 45, 474–485, doi: 10.1016/j.amepre.2013.06.004 (2013). [DOI] [PubMed] [Google Scholar]
- Higgins S. T. Behavior change, health, and health disparities: an introduction. Prev Med 68, 1–4, doi: 10.1016/j.ypmed.2014.10.007 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B. F., Hasin D. S., Chou S. P., Stinson F. S. & Dawson D. A. Nicotine dependence and psychiatric disorders in the United States: results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry 61, 1107–1115, doi: 10.1001/archpsyc.61.11.1107 (2004). [DOI] [PubMed] [Google Scholar]
- Hasin D. S. & Grant B. F. The National Epidemiologic Survey on Alcohol and Related Conditions (NESARC) Waves 1 and 2: review and summary of findings. Soc Psychiatry Psychiatr Epidemiol 50, 1609–1640, doi: 10.1007/s00127-015-1088-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant B. F. et al. Epidemiology of DSM-5 Alcohol Use Disorder: Results From the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry 72, 757–766, doi: 10.1001/jamapsychiatry.2015.0584 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez E., Quijano-Carde N. & De Biasi M. Nicotinic Mechanisms Modulate Ethanol Withdrawal and Modify Time Course and Symptoms Severity of Simultaneous Withdrawal from Alcohol and Nicotine. Neuropsychopharmacology 40, 2327–2336, doi: 10.1038/npp.2015.80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath A. C. et al. Genetic and environmental contributions to alcohol dependence risk in a national twin sample: consistency of findings in women and men. Psychol Med 27, 1381–1396 (1997). [DOI] [PubMed] [Google Scholar]
- Hiroi N. & Agatsuma S. Genetic susceptibility to substance dependence. Mol Psychiatry 10, 336–344, doi: 10.1038/sj.mp.4001622 (2005). [DOI] [PubMed] [Google Scholar]
- Goldman D., Oroszi G. & Ducci F. The genetics of addictions: uncovering the genes. Nat Rev Genet 6, 521–532, doi: 10.1038/nrg1635 (2005). [DOI] [PubMed] [Google Scholar]
- Ystrom E., Reichborn-Kjennerud T., Aggen S. H. & Kendler K. S. Alcohol dependence in men: reliability and heritability. Alcohol Clin Exp Res 35, 1716–1722, doi: 10.1111/j.1530-0277.2011.01518.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young-Wolff K. C., Chereji E. & Prescott C. A. Heritability of alcohol dependence is similar in women and men. Evid Based Ment Health 15, 57, doi: 10.1136/ebmental-2012-100670 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGue M., Elkins I. & Iacono W. G. Genetic and environmental influences on adolescent substance use and abuse. Am J Med Genet 96, 671–677 (2000). [DOI] [PubMed] [Google Scholar]
- Madden P. A., Heath A. C., Starmer G. A., Whitfield J. B. & Martin N. G. Alcohol sensitivity and smoking history in men and women. Alcohol Clin Exp Res 19, 1111–1120 (1995). [DOI] [PubMed] [Google Scholar]
- Bucholz K. K., Heath A. C. & Madden P. A. Transitions in drinking in adolescent females: evidence from the Missouri adolescent female twin study. Alcohol Clin Exp Res 24, 914–923 (2000). [PubMed] [Google Scholar]
- Madden P. A. et al. Nicotine withdrawal in women. Addiction 92, 889–902 (1997). [PubMed] [Google Scholar]
- Kapusta N. D. et al. Multiple substance use among young males. Pharmacol Biochem Behav 86, 306–311, doi: 10.1016/j.pbb.2006.10.007 (2007). [DOI] [PubMed] [Google Scholar]
- Locker A. R., Marks M. J., Kamens H. M. & Klein L. C. Exposure to nicotine increases nicotinic acetylcholine receptor density in the reward pathway and binge ethanol consumption in C57BL/6J adolescent female mice. Brain Res Bull, doi: 10.1016/j.brainresbull.2015.09.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein J. et al. Genome-wide association study of alcohol dependence. Arch Gen Psychiatry 66, 773–784, doi: 10.1001/archgenpsychiatry.2009.83 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut L. J. et al. A genome-wide association study of alcohol dependence. Proc Natl Acad Sci USA 107, 5082–5087, doi: 10.1073/pnas.0911109107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg H. J. et al. Genome-wide association study of alcohol dependence implicates a region on chromosome 11. Alcohol Clin Exp Res 34, 840–852, doi: 10.1111/j.1530-0277.2010.01156.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlojutro M. et al. Genome-wide association study of theta band event-related oscillations identifies serotonin receptor gene HTR7 influencing risk of alcohol dependence. Am J Med Genet B Neuropsychiatr Genet 156B, 44–58, doi: 10.1002/ajmg.b.31136 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler K. S. et al. Genomewide association analysis of symptoms of alcohol dependence in the molecular genetics of schizophrenia (MGS2) control sample. Alcohol Clin Exp Res 35, 963–975, doi: 10.1111/j.1530-0277.2010.01427.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath A. C. et al. A quantitative-trait genome-wide association study of alcoholism risk in the community: findings and implications. Biol Psychiatry 70, 513–518, doi: 10.1016/j.biopsych.2011.02.028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J. et al. Genome-wide association study of alcohol dependence:significant findings in African- and European-Americans including novel risk loci. Mol Psychiatry 19, 41–49, doi: 10.1038/mp.2013.145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillen E. E. et al. ALDH2 is associated to alcohol dependence and is the major genetic determinant of “daily maximum drinks” in a GWAS study of an isolated rural Chinese sample. Am J Med Genet B Neuropsychiatr Genet 165B, 103–110, doi: 10.1002/ajmg.b.32213 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart A. B. & Kranzler H. R. Alcohol Dependence Genetics: Lessons Learned From Genome-Wide Association Studies (GWAS) and Post-GWAS Analyses. Alcohol Clin Exp Res 39, 1312–1327, doi: 10.1111/acer.12792 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut L. J. et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Hum Mol Genet 16, 24–35, doi: 10.1093/hmg/ddl441 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl G. R. et al. Molecular genetics of nicotine dependence and abstinence: whole genome association using 520,000 SNPs. BMC Genet 8, 10, doi: 10.1186/1471-2156-8-10 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl G. R. et al. Molecular genetics of successful smoking cessation: convergent genome-wide association study results. Arch Gen Psychiatry 65, 683–693, doi: 10.1001/archpsyc.65.6.683 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorgeirsson T. E. et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature 452, 638–642, doi: 10.1038/nature06846 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Z. et al. Genome-wide association analyses suggested a novel mechanism for smoking behavior regulated by IL15. Mol Psychiatry 14, 668–680, doi: 10.1038/mp.2009.3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink J. M. et al. Genome-wide association study of smoking initiation and current smoking. Am J Hum Genet 84, 367–379, doi: 10.1016/j.ajhg.2009.02.001 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drgon T. et al. Genome-wide association for nicotine dependence and smoking cessation success in NIH research volunteers. Mol Med 15, 21–27, doi: 10.2119/molmed.2008.00096 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso N. et al. Genome-wide and candidate gene association study of cigarette smoking behaviors. PLoS One 4, e4653, doi: 10.1371/journal.pone.0004653 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J. et al. Genome-wide association study of nicotine dependence in American populations: identification of novel risk loci in both African-Americans and European-Americans. Biol Psychiatry 77, 493–503, doi: 10.1016/j.biopsych.2014.08.025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culverhouse R. C. et al. Uncovering hidden variance: pair-wise SNP analysis accounts for additional variance in nicotine dependence. Hum Genet 129, 177–188, doi: 10.1007/s00439-010-0911-7 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind P. A. et al. A genomewide association study of nicotine and alcohol dependence in Australian and Dutch populations. Twin Res Hum Genet 13, 10–29, doi: 10.1375/twin.13.1.10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo L. et al. Genome-wide significant association signals in IPO11-HTR1A region specific for alcohol and nicotine codependence. Alcohol Clin Exp Res 37, 730–739, doi: 10.1111/acer.12032 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal A. & Bierut L. J. Identifying genetic variation for alcohol dependence. Alcohol Res 34, 274–281 (2012). [PMC free article] [PubMed] [Google Scholar]
- Weaver I. C. et al. Epigenetic programming by maternal behavior. Nat Neurosci 7, 847–854, doi: 10.1038/nn1276 (2004). [DOI] [PubMed] [Google Scholar]
- Bonsch D., Lenz B., Kornhuber J. & Bleich S. DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport 16, 167–170, doi: 00001756-200502080-00020 [pii] (2005). [DOI] [PubMed] [Google Scholar]
- Bonsch D. et al. Lowered DNA methyltransferase (DNMT-3b) mRNA expression is associated with genomic DNA hypermethylation in patients with chronic alcoholism. J Neural Transm 113, 1299–1304, doi: 10.1007/s00702-005-0413-2 (2006). [DOI] [PubMed] [Google Scholar]
- Bleich S. et al. Epigenetic DNA hypermethylation of the HERP gene promoter induces down-regulation of its mRNA expression in patients with alcohol dependence. Alcoholism, clinical and experimental research 30, 587–591, doi: 10.1111/j.1530-0277.2006.00068.x (2006). [DOI] [PubMed] [Google Scholar]
- Philibert R. A., Gunter T. D., Beach S. R., Brody G. H. & Madan A. MAOA methylation is associated with nicotine and alcohol dependence in women. Am J Med Genet B Neuropsychiatr Genet 147B, 565–570, doi: 10.1002/ajmg.b.30778 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert R. A. et al. The relationship of 5HTT (SLC6A4) methylation and genotype on mRNA expression and liability to major depression and alcohol dependence in subjects from the Iowa Adoption Studies. American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 147B, 543–549, doi: 10.1002/ajmg.b.30657 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philibert R. A., Plume J. M., Gibbons F. X., Brody G. H. & Beach S. R. The impact of recent alcohol use on genome wide DNA methylation signatures. Front Genet 3, 54, doi: 10.3389/fgene.2012.00054 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. et al. Hypermethylation of OPRM1 promoter region in European Americans with alcohol dependence. J Hum Genet 57, 670–675, doi: jhg201298 [pii]10.1038/jhg.2012.98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. et al. Array-based profiling of DNA methylation changes associated with alcohol dependence. Alcohol Clin Exp Res 37 Suppl 1, E108–115, doi: 10.1111/j.1530-0277.2012.01928.x (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Xu H., Zhao H., Gelernter J. & Zhang H. DNA co-methylation modules in postmortem prefrontal cortex tissues of European Australians with alcohol use disorders. Sci Rep 6, 19430, doi: 10.1038/srep19430 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taqi M. M. et al. Prodynorphin CpG-SNPs associated with alcohol dependence: elevated methylation in the brain of human alcoholics. Addict Biol 16, 499–509, doi: 10.1111/j.1369-1600.2011.00323.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzardo A. M., Henkhaus R. S. & Butler M. G. Global DNA promoter methylation in frontal cortex of alcoholics and controls. Gene 498, 5–12, doi: 10.1016/j.gene.2012.01.096 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Launay J. M. et al. Smoking induces long-lasting effects through a monoamine-oxidase epigenetic regulation. PLoS One 4, e7959, doi: 10.1371/journal.pone.0007959 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan E. S. et al. Cigarette smoking behaviors and time since quitting are associated with differential DNA methylation across the human genome. Hum Mol Genet 21, 3073–3082, doi: 10.1093/hmg/dds135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenker N. S. et al. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet 22, 843–851, doi: 10.1093/hmg/dds488 (2013). [DOI] [PubMed] [Google Scholar]
- Ma Y. T., Collins S. I., Young L. S., Murray P. G. & Woodman C. B. Smoking initiation is followed by the early acquisition of epigenetic change in cervical epithelium: a longitudinal study. Br J Cancer 104, 1500–1504, doi: 10.1038/bjc.2011.113 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H. et al. RGS17/RGSZ2, a novel regulator of Gi/o, Gz, and Gq signaling. J Biol Chem 279, 26314–26322, doi: 10.1074/jbc.M401800200 (2004). [DOI] [PubMed] [Google Scholar]
- De Vries L., Zheng B., Fischer T., Elenko E. & Farquhar M. G. The regulator of G protein signaling family. Annu Rev Pharmacol Toxicol 40, 235–271, doi: 10.1146/annurev.pharmtox.40.1.235 (2000). [DOI] [PubMed] [Google Scholar]
- Zhang H., Wang F., Kranzler H. R., Anton R. F. & Gelernter J. Variation in regulator of G-protein signaling 17 gene (RGS17) is associated with multiple substance dependence diagnoses. Behav Brain Funct 8, 23, doi: 10.1186/1744-9081-8-23 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier L. L. et al. Molecular and functional analysis of human natural killer cell-associated neural cell adhesion molecule (N-CAM/CD56). J Immunol 146, 4421–4426 (1991). [PubMed] [Google Scholar]
- Yang B. Z. et al. Haplotypic variants in DRD2, ANKK1, TTC12, and NCAM1 are associated with comorbid alcohol and drug dependence. Alcohol Clin Exp Res 32, 2117–2127, doi: 10.1111/j.1530-0277.2008.00800.x (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson E. C. et al. ANKK1, TTC12, and NCAM1 polymorphisms and heroin dependence: importance of considering drug exposure. JAMA Psychiatry 70, 325–333, doi: 10.1001/jamapsychiatry.2013.282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidwell L. C. et al. NCAM1-TTC12-ANKK1-DRD2 variants and smoking motives as intermediate phenotypes for nicotine dependence. Psychopharmacology (Berl) 232, 1177–1186, doi: 10.1007/s00213-014-3748-2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich B. et al. Genomic structure and chromosomal mapping of the murine and human Mbd1, Mbd2, Mbd3, and Mbd4 genes. Mamm Genome 10, 906–912 (1999). [DOI] [PubMed] [Google Scholar]
- Jin S. G., Jiang C. L., Rauch T., Li H. & Pfeifer G. P. MBD3L2 interacts with MBD3 and components of the NuRD complex and can oppose MBD2-MeCP1-mediated methylation silencing. J Biol Chem 280, 12700–12709, doi: 10.1074/jbc.M413492200 (2005). [DOI] [PubMed] [Google Scholar]
- Zhang H. et al. Identification of methylation quantitative trait loci (mQTLs) influencing promoter DNA methylation of alcohol dependence risk genes. Hum Genet 133, 1093–1104, doi: 10.1007/s00439-014-1452-2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truss M., Bartsch J., Hache R. S. & Beato M. Chromatin structure modulates transcription factor binding to the mouse mammary tumor virus (MMTV) promoter. J Steroid Biochem Mol Biol 47, 1–10 (1993). [DOI] [PubMed] [Google Scholar]
- Felsenfeld G., Boyes J., Chung J., Clark D. & Studitsky V. Chromatin structure and gene expression. Proc Natl Acad Sci USA 93, 9384–9388 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe A. E. & Irizarry R. A. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol 15, R31, doi: 10.1186/gb-2014-15-2-r31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierucci-Lagha A. et al. Diagnostic reliability of the Semi-structured Assessment for Drug Dependence and Alcoholism (SSADDA). Drug Alcohol Depend 80, 303–312, doi: S0376-8716(05)00132-8 [pii]10.1016/j.drugalcdep.2005.04.005 (2005). [DOI] [PubMed] [Google Scholar]
- Pritchard J. K., Stephens M. & Donnelly P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Y., Gehrke C. W. & Ehrlich M. Comparison of bisulfite modification of 5-methyldeoxycytidine and deoxycytidine residues. Nucleic Acids Res 8, 4777–4790 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barfield R. T., Kilaru V., Smith A. K. & Conneely K. N. CpGassoc: an R function for analysis of DNA methylation microarray data. Bioinformatics 28, 1280–1281, doi: 10.1093/bioinformatics/bts124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green P. & MacLeod C. J. SIMR: an R package for power analysis of generalized linear mixed models by simulation. Methods in Ecology and Evolution (2016). [Google Scholar]
- Purcell S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575, doi: 10.1086/519795 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J.-H., Blay S., McNeney B. & Graham J. LDheatmap: an R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphisms. Journal of Statistical Software 16, 1–10 (2006). [Google Scholar]
- Messeguer X. et al. PROMO: detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18, 333–334 (2002). [DOI] [PubMed] [Google Scholar]
- Farre D. et al. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucleic Acids Res 31, 3651–3653 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V. et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 34, D108–110, doi: 10.1093/nar/gkj143 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.