

Significance
Polyketide natural products represent a rich source for discovery of new bioactive compounds. However, the optimization of polyketide structure for medicinal purposes can be difficult using chemical methods given their complexity. Site-selective introduction of fluorine into polyketides and other natural products is a particularly interesting area of exploration given the demonstrated effectiveness of fluorine in modulating the behavior of small-molecule therapeutics. Here, we show that fluorine can be inserted site-selectively by an engineered polyketide synthase system via a fluorinated monomer that becomes covalently tethered to the enzyme to complete a canonical reaction cycle. By increasing the throughput of fluorinated extender units, we can produce multiply fluorinated polyketide products by chemoenzymatic synthesis and target the production of complex structures.
Keywords: polyketide synthase, fluorine, synthetic biology, natural products
Abstract
Polyketides are a large family of bioactive natural products synthesized by polyketide synthase (PKS) enzyme complexes predominantly from acetate and propionate. Given the structural diversity of compounds produced using these two simple building blocks, there has been longstanding interest in engineering the incorporation of alternative extender units. We have been investigating the mechanism of fluorinated monomer insertion by three of the six different modules of the PKS involved in erythromycin biosynthesis (6-deoxyerythronolide B synthase, DEBS) to begin understanding the contribution of different steps, such as enzyme acylation, transacylation, C–C bond formation, and chain transfer, to the overall selectivity and efficiency of this process. In these studies, we observe that inactivation of a cis-acyltransferase (AT) domain to circumvent its native extender unit preference leads concurrently to a change of mechanism in which chain extension with fluorine-substituted extender units switches largely to an acyl carrier protein (ACP)-independent mode. This result suggests that the covalent linkage between the growing polyketide chain and the enzyme is lost in these cases, which would limit efficient chain elongation after insertion of a fluorinated monomer. However, use of a standalone trans-acting AT to complement modules with catalytically deficient AT domains leads to enzyme acylation with the fluoromalonyl-CoA extender unit. Formation of the canonical ACP-linked intermediate with fluoromalonyl-CoA allows insertion of fluorinated extender units at 43% of the yield of the wild-type system while also amplifying product yield in single chain-extension experiments and enabling multiple chain extensions to form multiply fluorinated products.
Polyketide natural products represent a large class of bioactive compounds with a broad range of medicinal properties, including antibiotic and anticancer activity. Despite their diversity in structure, they are assembled with a common mechanism involving multiple Claisen condensations of acetate-derived extender units catalyzed by polyketide synthase (PKS) enzymes (1, 2). In addition to setting the oxidation state of the β-carbon at each polymerization step, modular PKSs can also select the α-substituent on the extender unit, which is predominantly derived from either acetate (R = H) or propionate (R = Me) (1, 2). The breadth of compounds (>10,000) that can be made from these two simple building blocks is quite considerable, and many efforts have thus centered on increasing the types of extender units that can be incorporated into polyketides (1, 3, 4).
We have focused specifically on the insertion of fluorinated monomers by PKSs as an approach to merge the advantages of complex natural product scaffolds with the effectiveness of fluorine medicinal chemistry (5). In this regard, the type I modular PKSs are an especially attractive target for engineering because their organization and structure lends itself to connecting sequence with product structure (1, 2, 6). Previous studies have shown that the acyltransferase (AT) domain within the PKS (cis-AT) is essential to determining extender unit identity and can be engineered to enable insertion of alternative extender units (7–9). Alternatively, the intrinsic selectivity of the cis-AT can be bypassed by inactivation and complementation with a standalone AT enzyme (trans-AT) as another approach to introducing nonnative substituents (10–12). Although these methods are effective for altering product structure, product yield can often diminish in these engineered systems and, thereby, limit their scalability. As such, we set out to elucidate the mechanism by which alternative extender units are accepted or rejected in these systems to expand our understanding of the native process of extender unit selection and to define design principles for PKS engineering.
We initiated these studies by using the 6-deoxyerythronolide B synthase (DEBS), which is responsible for the synthesis of the core of the antibiotic erythromycin, because it has been well characterized and is amenable to dissection to its modular units (2). We observe that the fluoromalonyl-CoA extender unit can be accepted directly by the ketosynthase (KS) domain in modules where the cis-AT is inactivated, with a 2:1 ratio of C–C bond formation to nonproductive decarboxylation to form fluoroacetyl-CoA. Under these conditions, a single fluorine substituent can be introduced, but further chain extension cannot easily occur when ligation of the extender unit to the acyl carrier protein (ACP) domain is bypassed. In contrast, complementation with a trans-AT leads to quantitative C–C bond formation with a fluorinated extender unit and restores the canonical PKS mechanism with 43% of wild-type (WT) yield. In this system, we can amplify single-chain extensions with fluorinated monomers and enable further chain growth to produce polyketide products with multiple fluorine substitutions.
Results and Discussion
ACP-Independent Chain Extension with Methyl- and Fluoromalonyl-CoA Extender Units.
We were able to observe chain elongation by individual DEBS modules with a catalytically deficient AT domain (AT0), which we attributed to the greater number of enzyme turnovers possible when the extender unit pool is maintained by using an in situ regeneration system for methyl- and fluoromalonyl-CoA (10). However, end product analysis of single module systems does not necessarily distinguish between the two general mechanisms that could arise in this system. One possibility is that the extender unit could be loaded onto the ACP domain either by residual AT activity or by direct thiol-thioester chemical exchange to generate a canonical ACP-tethered intermediate. In this case, the polyketide product remains covalently attached to the enzyme after C–C bond formation and can be further processed effectively by the PKS (Scheme 1A). However, an alternative mechanism is that the extender unit could directly diffuse into the KS active site, bypassing both the AT and ACP domains to undergo a decarboxylative Claisen condensation to form a free polyketide product that is not covalently attached to the PKS (Scheme 1B). In the latter case, generation of full-length polyketides would be challenging once the covalent link between the PKS and the growing chain is lost because it amplifies substrate channeling from module to module by up to three orders of magnitude (13). Therefore, we set out to determine which mechanism was dominant under our assay conditions for both the native and fluorinated extender units.
Scheme 1.

Proposed mechanisms of extender unit incorporation in a generalized type I PKS module. For clarity, only the KS, AT, and ACP domains are shown. (A) Chemical transacylation of extender unit onto the ACP. (B) Direct diffusion of extender unit into the KS active site. ACP, acyl carrier protein; AT, acyltransferase; KS, ketosynthase; R1, growing polyketide chain; R2, H, Me, or F.
To distinguish between the two proposed mechanisms, the ACP domain was inactivated by mutation of the serine of the phosphopantetheine arm modification site (S1430) to alanine (ACP0). Because the canonical chain extension mechanism requires the phosphopantetheine arm to form the malonyl-ACP intermediate, we would expect that product formation in an AT0 ACP0 construct would be abolished if the malonyl-ACP intermediate is required for chain elongation (Scheme 1A) but should not be affected if the ACP can be bypassed (Scheme 1B). To generate these mutants, we started with DEBS module 3 constructs fused to a thioesterase domain (Mod3TE) and introduced the S1430A mutation into both a wild-type and AT0 background where the catalytic Ser651 is mutated to alanine (SI Appendix, Table S1). A construct encoding for a truncated Mod3 protein containing only the KS and AT domains (KSAT didomain) (14) was also used as a control to eliminate the contribution of any remaining interactions with the ACP (SI Appendix, Table S1).
These four protein constructs were heterologously expressed and purified as described (SI Appendix, Fig. S1) (10). They were first assayed for their ability to carry out chain elongation on the natural diketide N-acetylcysteamine (NDK-SNAC) substrate surrogate (SI Appendix, Fig. S2) with their native methylmalonyl-CoA extender unit to yield the corresponding triketide lactone (TKL) product (Fig. 1A). The samples were quantified under end-product and initial rate conditions using authentic standards by liquid chromatography monitored by UV-visible spectroscopy (LC-UV) and mass spectrometry (LC-MS) (Fig. 1 A and B and SI Appendix, Fig. S3). As consistent with previous studies (10, 15), the production of TKL in the AT0 module is dramatically decreased compared with production in the WT module, with a 110-fold drop in initial rate and a 16- to 24-fold decrease in yield. However, assays with the ACP0, AT0ACP0, and KSAT didomain yielded a similar catalytic defect as the AT0 construct with regard to yield and initial rate. This observation implies that the ACP domain is not required for the residual chain elongation activity that is observed when the AT domain is inactivated. As an additional control, the KS0, AT0, and KS0 AT0 mutations were also introduced into the KSAT didomain, verifying that TKL formation was only observed in the presence of an active KS and not a result of unexpected activity from another domain in these mutants (Fig. 1B). From these observations, we conclude that the observed TKL product arises from direct diffusion of the extender unit into the KS active site where C–C bond formation is catalyzed between the polyketide chain and methylmalonyl-CoA rather than methylmalonyl-ACP (Scheme 1B).
Fig. 1.

Analysis of TKL and F-TKL formation with constructs of DEBS Mod2, Mod3TE, and Mod6TE. (A) Reaction scheme for the TKL formation assay with Mod3TE. KR*, natively inactive ketoreductase domain. (B) Endpoint and initial rate quantification of TKL and F-TKL formation with 10 µM DEBS Mod3TE constructs and KSAT3. (C) Endpoint quantification of TKL and F-TKL formation with 10 µM DEBS Mod2 and Mod6TE constructs and KSAT6. Only KS, AT, and ACP domains are shown in the labels for Fig. 1 B and C. All data are mean ± SE (n = 3). n.d., not detected; –, data not collected.
Because different modules may display diverse behavior, similar experiments were carried out on WT, AT0, and KSAT didomain constructs of Mod2 and Mod6TE (SI Appendix, Table S1 and Fig. S1) to test whether the results with Mod3TE could be generalized. Indeed, we observed that ACP-independent chain extension can also occur with Mod2 and Mod6TE as well, based on the comparison of activities of the WT, AT0, and KSAT didomain constructs (Fig. 1C). The relatively high yield of TKL in the AT0 Mod2 construct compared with Mod3TE and Mod6TE may further indicate that the KS of Mod2 is more promiscuous with regard to nonnative substrates. Experiments with Mod2 constructs also yielded additional information with regard to ACP-independent chain extension. We found that product formation with WT Mod2 occurred at an order of magnitude lower efficiency than WT Mod3TE and Mod6TE, likely due to the absence of a thioesterase fusion. TKL is still formed by this construct because of the stability of the six-membered lactone ring but relies on uncatalyzed release from the ACP domain. However, the longer residence time of the polyketide on the ACP may create a bottleneck for formation of methylmalonyl-ACP. Under these conditions, the production of TKL by WT, AT0, and KSAT Mod2 constructs is essentially identical (Fig. 1C), suggesting that the ACP-independent mode of chain extension may be able to compete with canonical chain extension if the KS domain is saturated with methylmalonyl-CoA. The CoA pool was then reduced 10-fold to 0.1 mM to reduce saturation of the KS domain, resulting in reduction of the AT0 and KSAT-didomain yields by up to 5.2-fold, whereas WT only saw a twofold loss in TKL formation (Fig. 1C). These observations imply that saturation of the ACP-independent chain extension requires higher extender unit concentrations and that bias toward direct KS use could occur in the WT module at high methylmalonyl-CoA concentration.
With a basic understanding of extender unit incorporation in hand, we set out to compare behavior with fluoromalonyl-CoA. Based on the presence of the α-fluorine substituent, which activates this analog toward nucleophilic attack at the carbonyl, it is possible that fluoromalonyl-CoA would be more prone to undergo chemical transacylation to form the ACP-tethered intermediate (Scheme 1A). However, both the initial rate and end product analysis of fluorinated triketide lactones (F-TKLs) provide evidence that chain extensions carried out under these conditions occur independently of the ACP based on the similar activities measured for all five constructs (Fig. 1B and SI Appendix, Fig. S4). As such, endpoint F-TKL formation experiments with Mod2 and Mod6TE constructs are consistent with this conclusion that product is mostly formed in these AT0 variants without loading of the extender unit onto the ACP domain (Fig. 1C). These initial rate and endpoint experiments provide evidence that malonyl-ACP intermediates are not required for C–C bond formation. However, chain extension with the CoA-linked extender unit leads to early chain termination because the covalent tether between the PKS and the growing polyketide chain is lost.
Consequences of Chain Extension with CoA- Versus ACP-Linked Extender Units.
We were interested in further examining the nature of ACP-independent chain extension and its behavior compared with canonical ACP-dependent chain extension so that we could better understand the issues that may arise when this mode of reactivity occurs. For example, we reasoned that the coupling between extender unit decarboxylation and C–C bond formation may be affected, leading to release of propoionate or fluoroacetate, which cannot be regenerated under our current assay conditions. To address this question, we set out to characterize the partitioning between productive extender unit decarboxylation leading to TKL formation compared with nonproductive decarboxylation leading to propionate production (Fig. 2). To detect the products of the reactions with methylmalonyl-CoA, [4-13C]-methylmalonyl-CoA was chemoenzymatically synthesized from [4-13C]-methylmalonic acid (16) (SI Appendix, Fig. S5) and used as a substrate with the WT and AT0ACP0 Mod3TE constructs. Monitoring the reaction of WT Mod3TE by quantitative 13C-NMR spectroscopy allowed us to determine that for every 10 extender unit turnovers, 6 are hydrolyses, 3 are C–C bond formations, and 1 is a nonproductive decarboxylation (SI Appendix, Fig. S6). These results indicate that hydrolysis of extender units by the AT domain and nonproductive decarboxylation by the KS domain are both competitive with product formation under in vitro conditions. Along these lines, the measured rate constants for methylmalonyl-CoA hydrolysis (1.0 min−1; SI Appendix, Fig. S7) and triketide formation (1.0 min−1; Fig. 1B) are similar. In this particular case, the observation that extender unit hydrolysis is the predominant reaction observed could arise from the use of a nonnative polyketide starter substrate for this module (17).
Fig. 2.

13C-NMR quantitation of products formed in the TKL assay with WT and AT0 ACP0 Mod3TE using the native methylmalonyl-CoA extender unit. For clarity, only the KS, AT, and ACP domains are shown with the KR* and TE domains omitted. (A) With WT Mod3TE, extender unit decarboxylation leads to formation of TKL as the major C–C bond-coupled product rather than protonation to form propionate products. (B) With AT0 ACP0 Mod3TE AT0 ACP0, nonproductive decarboxylation becomes the major pathway. *, site of 13C label.
In WT Mod3TE, where chain extension presumably takes place with methylmalonyl-ACP, the ratio of productive to nonproductive decarboxylation was observed to be 3:1 (Fig. 2 and SI Appendix, Fig. S6). However, this ratio drastically shifted to 1:228 in AT0 ACP0 Mod3TE, where we have proposed that TKL formation occurs using methylmalonyl-CoA directly, suggesting that the ratio of productive decarboxylation is increased ∼685-fold when the methylmalonyl-ACP intermediate is formed (Fig. 3 and SI Appendix, Fig. S6). Thus, extender units that diffuse directly into the active site of the KS are lost to nonproductive decarboxylation, but the fidelity of the growing polyketide chain is maintained. In contrast, analysis of reactions of AT0ACP0 Mod3TE and fluoromalonyl-CoA by 19F-NMR using the in situ extender unit regeneration system unexpectedly revealed a significantly higher ratio (2:1) for C–C bond formation compared with nonproductive decarboxylation to form fluoroacetate (SI Appendix, Fig. S8). The observed difference in behavior between methylmalonyl-CoA and fluoromalonyl-CoA in this experiment could be related to the stabilizing effects of the α-fluoro group on the corresponding enolate or a difference in how native and nonnative extender units are treated by the enzyme. Given the inefficiency of ACP independent C–C bond formation and processivity with fluoromalonyl-CoA in DEBS modules bearing the AT0 mutation, we turned our attention to examining the changes in chain extension mechanism that occur when AT0 Mod3TE is complemented in trans with standalone ATs.
Fig. 3.

Complementation of AT0 Mod3TE constructs with DszAT. Reactions contain 10 μM DEBS Mod3TE and 5 mM NDK-SNAC with 30 μM DszAT. For clarity, only the KS, AT, and ACP domains are shown. (A) Endpoint quantification of F-TKL formation. Data are mean ± SD (n = 3). (B) Initial rate of F-TKL formation. Data are mean ± SE (n = 3).
Efficient Complementation with Fluorinated Extender Units Using the trans-AT from the Disorazole Biosynthetic Cluster.
The formation of C–C bonds via ACP-independent reaction of the extender unit inhibits processive growth of the polyketide chain on the PKS to form the full-length product (13). Previous work with the trans-AT of the disorazole biosynthetic cluster (DszAT) has shown that it can acylate DEBS ACPs with malonyl-CoA (11, 18, 19) and also amplifies chain extension with fluoromalonyl-CoA to produce F-TKL (10). Given our observation that direct reaction of fluoromalonyl-CoA with the KS domain can lead to chain extension, we decided to further probe the behavior of the trans-AT in this system. We found that complementation of AT0 Mod3TE with DszAT results in a 10-fold increase in F-TKL formation, and a 40-fold improvement in the initial rate of fluorinated extender unit incorporation (Fig. 3 and SI Appendix, Fig. S9). This enhancement depends on the presence of a functional ACP domain, because no yield or rate enhancements were observed in the control when AT0ACP0 Mod3TE was complemented with DszAT (Fig. 3 and SI Appendix, Fig. S9). Moreover, no fluoroacetate was observed by 19F-NMR when Mod3TE AT0 was complemented with DszAT, implying that nonproductive decarboxylation does not occur when the extender unit is properly attached to the ACP (SI Appendix, Fig. S10). These results are consistent with a mechanism of DszAT complementation by transacylation of the ACP with fluoromalonyl-CoA to form a covalently tethered intermediate. As such, the restoration of the canonical PKS mechanism through this complementation can provide a successful solution to the challenge of chain transfer between upstream and downstream PKS modules.
To confirm that fluoromalonyl-CoA could be effectively transacylated onto the ACP domain, we adapted a MS/MS spectrometry-based assay that allows us to directly detect malonyl-ACP intermediates and calculate a semiquantitative occupancy of ACPs with different extender units (Fig. 4A and SI Appendix, Fig. S11) (20–22). The different DEBS modules were first incubated with the appropriate extender unit and then subjected to tryptic digestion of the protein. To simplify analysis, the in situ regeneration system for the extender unit was not included. The parent ion of the peptide containing the phosphopantetheine arm of the ACP was then selected and further fragmented to eject the phosphopantetheine arm, confirming its identity and allowing us to measure the ratio of acyl- to holo-ACP (Fig. 4B). When WT Mod3TE is incubated with methylmalonyl-CoA, the ACP is almost fully acylated (92%) as is expected (Fig. 4C). However, no detectable acyl-ACP intermediates can be observed under the same conditions with AT0 Mod3TE. Similarly, basal levels of acylation by nonnative extender units (malonyl-CoA and fluoromalonyl-CoA) can be observed in both the WT and AT0 constructs (Fig. 4C). We attribute this low level of acylation to an uncatalyzed thioester exchange between the CoA thioesters and the ACP thiol (23) because it is similar to the occupancy of acyl-ACPs for modules that are subjected to tryptic digestion before incubation with the various extender units, where acylation must occur chemically (Fig. 4C). The similarity of the malonyl- and fluoromalonyl-ACP occupancies under all three conditions further implies that the AT domain does not acylate unnatural extender units onto the ACP, or does so at levels lower than uncatalyzed or chemical transacylation. As such, these data are consistent with our hypothesis that formation of products in modules with inactive AT and/or ACP domains is the result of KS-catalyzed C–C bond formation between the growing polyketide chain and a CoA-linked extender unit.
Fig. 4.

Semiquantitative analysis of ACP acylation of Mod3TE constructs with malonyl-CoA, methylmalonyl-CoA, and fluoromalonyl-CoA by using LC-MS/MS. KR*, natively inactive ketoreductase domain. (A) Scheme for phosphopantetheine arm ejection from the ACP. (B) Extracted ion chromatograms (EIC) of the malonyl-ACP fragments and holo-ACP. (C) Semiquantitative ACP acylation occupancy calculated based on the MS peak area of the acylated and unacylated phosphopantetheine arm. Data are mean ± SD (n = 3).
When examining the reaction of AT0 Mod3TE in the presence of DszAT, we found that 89% of the ACPs were acylated after a 20-min incubation with malonyl-CoA, which is the native substrate of DszAT (Fig. 4C). Although 10% of ACPs were acylated when incubated with methylmalonyl-CoA, DszAT active sites have been shown to contain methylmalonyl-CoA acyl-enzyme intermediates (24), but the degree of acylation is sufficiently low that it does not appear to contribute to TKL production. Interestingly, only 28% occupancy of fluoromalonyl-ACP was observed (Fig. 4C) despite the robust yield increase in F-TKL formation observed in the presence of DszAT (Fig. 3A). However, the relatively low occupancy appears to be derived from rapid hydrolysis of fluoromalonyl-CoA by DszAT (SI Appendix, Fig. S12), which would not be yield-limiting under our in situ extender unit regeneration conditions where fluoromalonyl-CoA can be regenerated at a rate of 1.5 s−1.
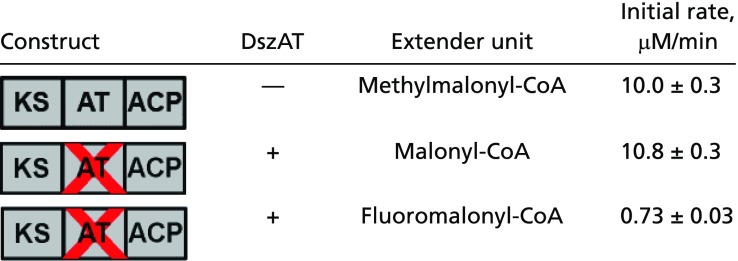
Because the direct observation of DszAT-catalyzed ACP acylation provides compelling evidence that F-TKL was being produced through the fluoromalonyl-ACP intermediate, we sought to assess the overall efficiency of this process by using both the initial rate and yield of chain extension as markers. We thus characterized DszAT complementation of AT0 Mod3TE with fluoromalonyl-CoA in comparison with its native substrate, malonyl-CoA. With malonyl-CoA as the extender unit, the initial rate of desmethyl triketide lactone observed was 10.8 ± 0.3 μM⋅min−1 (Table 1 and SI Appendix, Fig. S13). This value is within error of the initial rate of TKL formation measured with WT Mod3TE (10.0 ± 0.3 μM⋅min−1) with its native methylmalonyl-CoA extender unit (Table 1), which indicates that DszAT performs well in complementing the AT0 mutation in trans. In comparison, the initial rate of F-TKL formation for AT0 Mod3TE complemented with DszAT using fluoromalonyl-CoA had been determined to be 0.73 ± 0.03 μM⋅min−1 (Table 1), which is 7.3% of the rate of the WT Mod3TE system.
Table 1.
Comparison of initial rate of TKL formation with different extender units
 |
We also characterized the product yield, which could be more relevant to engineering the production of fluorinated polyketides. Because the single modular systems have been found to produce other compounds in addition to the expected one, we quantified other major products resulting from C–C bond formation to better assess the stoichiometry of chain extension reactions. With fluoromalonyl-CoA, these products arise after the expected chain extension reaction when the TE releases the free acid rather than the lactone and still represent a positive C–C bond-forming event (SI Appendix, Fig. S14). When the data are represented in terms of chain extension stoichiometry, we find that AT0 Mod3TE can incorporate fluorinated extender units at 43% efficiency when complemented with DszAT, compared with WT Mod3TE incubated with its native methylmalonyl-CoA extender unit (SI Appendix, Figs. S14 and S15). The endpoint yield relative to the native system is higher than expected based on the initial rate data, even taking into account other products that are formed. However, it is potentially consistent with the higher level of observed fluoromalonyl-ACP (Fig. 4). This difference could result from the initial rate being limited by a slower rate of KS-catalyzed C–C bond formation with the α-fluoro substituent that is separate from the effectiveness of the ACP acylation reaction carried out by DszAT. Taken together, this data demonstrates that DszAT is sufficiently active for catalyzing ACP acylation with fluoromalonyl-CoA to produce a significant yield of fluorinated polyketide product compared with the wild-type system.
Site-Specific Incorporation of Fluorinated Extender Units into Polyketide Synthases.
One major challenge for fluorine incorporation into full-length polyketides is the transfer of the growing chain from module to module. We have shown that a fluorinated extender unit can be incorporated into a polyketide nascent chain and undergo an additional chain extension with methylmalonyl-CoA to produce a monofluorinated product by using a noncovalent dimodular “mini-PKS” system containing Mod2 and Mod3TE (10). However, this system requires a 5:1 ratio of Mod2 AT0 to Mod3TE to overcome the barrier to chain transfer, which is a stoichiometry that would be difficult to control under in vivo conditions. Given our ability to efficiently complement AT0 Mod3TE with DszAT and fluoromalonyl-CoA, we decided to approach both the production of bis-fluorinated products and the improvement of the ratio of PKS modules to a more physiologically relevant regime (1:1).
We chose to use propionyl-SNAC (SI Appendix, Fig. S16) as the starter unit to produce F-TKLs to avoid the offloading that results from the propensity of triketidyl-thioesters to undergo cyclization of the polyketide chain after a single extension from NDK-SNAC (Fig. 5A) (10). Additionally, the CoA-pool concentration was lowered from 1 mM to 0.1 mM to lower competition between fluoromalonyl-CoA and fluoromalonyl-ACP for the KS active site that we observed with saturating concentrations of extender unit. Under these conditions, the complementation with the different AT0 variants of the mini-PKS under regenerative methylmalonyl-CoA and fluoromalonyl-CoA conditions led to the formation of the expected site-specifically fluorinated products with a 1:1 ratio of Mod2 to Mod3TE (Fig. 5B). To further substantiate the identity of these products, [1-13C]-propionyl-SNAC was used as the starter unit, leading to the expected 1 amu shift in mass of the products. The two monofluorinated regioisomers were determined to be unique by different retention times, as well as distinctive fragmentation patterns (Fig. 5B and SI Appendix, Figs. S17 and S18). Control reactions with no added DszAT showed a greatly diminished F-TKL yield (up to 75- to 93-fold for monofluorinated products, whereas the bisfluorinated product could not be detected), indicating that DszAT complementation is crucial to achieve efficient production of an F-TKL formed from subsequent chain extensions (Fig. 5B).
Fig. 5.

Formation of TKL products from propionyl-SNAC by using a dimodular PKS system consisting of Mod2 and Mod3TE. (A) Reaction scheme for TKL formation (R = Me and F). *, natively inactive KR domain. (B) EICs of the two monofluorinated TKL isomers (natural abundance, 173.0619 m/z; 13C-labeled product, 174.0653 m/z) and the bis-fluorinated TKL (natural abundance, 177.0369 m/z; 13C-labeled product, 178.0403 m/z) corresponding to the insertion of one and two fluoromalonyl-CoA monomers, respectively (propionyl-SNAC + DszAT, black; propionyl-SNAC – DszAT, blue; 13C-propionyl-SNAC + DszAT, red). The splitting of the peak observed for the bis-fluorinated TKL is likely related to the existence of tautomeric forms.
Conclusions
We set out to explore the molecular mechanism of fluorinated monomer incorporation by modular PKSs using DEBS as a model system with the goal of identifying limitations to be addressed to develop a robust system for production of site-selectively fluorinated polyketides. In this study, we found that inactivation of the AT domain, which is a strategy used to eliminate one of the major selectivity filters for choosing the extender unit, initiates a mode of C–C bond formation that is independent of the ACP (Scheme 2). Although we had previously hypothesized that the more activated fluoromalonyl-CoA extender unit might be able to achieve ACP acylation with a catalytically-deficient AT domain, we observed instead that the KS domain can compete with the AT domain for both the native methylmalonyl-CoA unit and fluoromalonyl-CoA to carry out direct C–C bond formation while bypassing the canonical ACP-linked intermediate. This observation is consistent with previous studies demonstrating direct KS reactivity with nonhydrolyzable malonyl-SNAC mimics (25, 26) and the in vitro detection of unnatural TKLs using Mod6TE heterologously expressed in Escherichia coli without a phosphopantetheinyl transferase enzyme (15). Although this mode of reactivity leads to a single chain extension, the covalent tether between the ACP and growing polyketide chain that ensures its transfer to the downstream module is lost, which leads to loss of processivity and ability to efficiently produce full-length polyketide products. Interestingly, we found that reaction partitioning between productive substrate decarboxylation to form the C–C bond and nonproductive pathways leading to release of propionate or fluoroacetate was also altered. For the native methylmalonyl-CoA, an ∼685-fold improvement in this partition was observed when C–C bond formation proceeded through the proper ACP-linked rather than CoA-linked extender unit. Although less dramatic, the partition for C–C bond formation with the fluoromalonyl-CoA extender also improves from 2:1 to quantitative.
Scheme 2.

Possible reaction pathways for PKS-catalyzed chain extension when fluoromalonyl-CoA is used. Use of a PKS module with the AT inactivated (Ser → Ala) allows bypassing the main selectivity filter for native extender unit selection and enabling the use of fluoromalonyl-CoA. In the absence of a trans-AT, the extender unit can diffuse directly into the KS active site and undergo decarboxylation. In this mode of reactivity, decarboxylation is decoupled from the chain extension and enolate protonation can lead to release of fluoroacetyl-CoA and degradation of the extender unit pool. Even when C–C bond formation occurs, the lack of an ACP-bound intermediate leads to early chain termination and release of the growing polyketide. In the presence of a trans-AT (DszAT), the AT0 mutation can be complemented, leading to ACP acylation with fluoromalonyl-CoA. When the canonical intermediate is formed, decarboxylation of fluoromalonyl-ACP is coupled to C–C bond formation with no loss of extender unit, resulting ultimately in chain growth and formation of the full-length polyketide product.
Based on the results of these experiments, we carried out a detailed study of trans-AT complementation with DEBS modules in which we characterized the molecular mechanism of fluoromalonyl-CoA incorporation and rate enhancement upon the complementation of DszAT with DEBS modules. Using end product analysis, enzyme kinetics, and direct observation by MS with various DEBS constructs, we are able to show that complementation of AT0 modules with a trans-AT, DszAT, enables formation of the canonical ACP-linked intermediate with fluoromalonyl-CoA. Under these conditions, C–C bond formation with a fluorinated monomer occurs at a 43% yield compared with the wild-type system with the native methylmalonyl-CoA extender unit. Formation of fluoromalonyl-ACP has key implications for production of full-length polyketides, because the covalent linkage between the growing chain and PKS is maintained after the fluorinated monomer is incorporated. Furthermore, C–C bond formation is fully coupled to fluoromalonyl-ACP decarboxylation with no observed nonproductive loss to fluoroacetate.
We then sought to test these findings in a dimodular mini-PKS system, where we had required the use of a 5:1 ratio of the two modules to achieve two rounds of chain extension without DszAT complementation (10). It is in these multiple turnover systems where the identification of the two modes of C–C bond formation in AT0 systems has dramatic implications on product yield and regiochemistry. Given that the ACP-independent mode of chain extension can be activated with high levels of extender unit, decreasing the concentration of CoA extender units below this critical amount should increase chain transfer efficiency by reducing the amount of chain termination occurring from direct diffusion of the extender unit into the KS active site. Using this information, we are able to show that mono- and bis-fluorinated products can be synthesized from the dimodular system at a 1:1 ratio of DEBS modules, indicating that channeling fluorinated growing chain from an upstream to a downstream module is possible. We further show that DszAT complementation increases titers by up to ∼93-fold in this dimodular system. The ability to complement DszAT with two sequential AT0 modules to insert two fluorinated extender units suggests that trans-AT complementation with PKS modules shows promise as a method for achieving robust production of site-specifically fluorinated full-length polyketide products.
Materials and Methods
Construction of Vectors for Protein Expression and Isolation of His-Tagged and MBP-Tagged Proteins.
Expression plasmids were constructed by amplification of target genes from genomic or synthetic DNA and insertion into the appropriate pET vector (SI Appendix, Table S1). His-tagged and MBP-tagged proteins were expressed in E. coli BL21(de3) or BAP1 at 16 °C and isolated by using standard procedures.
Assays for Triketide Lactone Production and Initial Rate Determination.
TKL assays were performed in 400 mM sodium phosphate, pH 7.5 with phosphoenolpyruvate (50 mM), TCEP (5 mM), magnesium chloride (10 mM), ATP (2.5 mM), pyruvate kinase/lactate dehydrogenase (15 U/mL), myokinase (10 U/mL), methylmalonyl-CoA epimerase (5 µM), CoA (1 mM), MatB (20 µM), and fluoromalonate (5 mM). All reactions were preincubated at 37 °C for 30–45 min and initiated by the addition of NDK-SNAC (5 mM) and DEBS Mod3TE, Mod2, or Mod6TE (10 µM). DszAT (30 µM) was also added to the reaction mixtures when appropriate. The end product analysis reactions (50 µL) were incubated for 16–24 h at 37 °C. For reaction mixtures (500 µL) used to determine initial rates, aliquots (9 × 50 µL) were removed from the assay over the course of 24 h. Each aliquot was quenched with 0.05V 70% (vol/vol) perchloric acid (2.5 µL). Quenched samples were centrifuged at 18,000 × g for 10 min to pellet the precipitated protein. The supernatant (50 µL) was removed and flash frozen and recentrifuged at 18,000 × g for 5 min. The supernatant was then removed and analyzed by LC-MS and/or LC-UV.
Determination of Malonyl-ACP Occupancy in AT0 Mod3TE with DszAT.
Assays were performed in 50 mM sodium phosphate, pH 7.5 with TCEP (5 mM), magnesium chloride (10 mM), methylmalonyl-CoA epimerase (1 µM), and either methylmalonyl-CoA (1 mM), malonyl-CoA (1 mM), or fluoromalonyl-CoA (1 mM). The reaction was initiated by the addition of DEBS Mod3TE (1 µM). DszAT (1 µM) was also added to the reaction mixture, when appropriate. The reaction was incubated at 37 °C for 20 min before adding trypsin (1.25 µL, 2.5 µg/µL). The reaction was incubated at 37 °C for another 90 min, quenched with liquid N2, and stored at −80 °C until analysis. Excess salts were removed by centrifugation at 18,000 × g for 5 min. The supernatant was then removed and analyzed by LC-MS/MS. Products were characterized using both MS1 (exact mass <2 ppm) and MS2 (phosphopantetheine ejection).
Triketide Lactone Production Using Propionyl-SNAC with Mod2/AT0 and Mod3TE/AT0.
All reactions contained 400 mM sodium phosphate (pH 7.5), phosphoenolpyruvate (50 mM), TCEP (10 mM), magnesium chloride (5 mM), ATP (2.5 mM), pyruvate kinase (18 U/mL), myokinase (10 U/mL), methylmalonyl-CoA epimerase (5 µM), MatB (20 µM), CoA (0.1 mM), methylmalonate (5 mM, omitted from reaction with both AT0s) and fluoromalonate (30 mM), propionyl-SNAC (5 mM) and reduced nicotinamide adenine dinucleotide phosphate (NADPH; 5 mM). Reactions were initiated by addition of Mod2/AT0 (10 µM), Mod3TE/AT0 (10 µM), and DszAT (30 µM) and incubated overnight at 37 °C. Aliquots were removed, quenched, and processed as described above. The supernatant was then removed and analyzed by LC-MS and/or LC-UV.
Complete Materials and Methods.
Detailed procedures for the methods are described above, and additional experiments can be found in the SI Appendix, SI Materials and Methods.
A full description of materials and methods, sequences of oligonucleotides, SDS/PAGE gels, NMR spectra, kinetics and endpoint assays for enzymes, and mass spectrometry is available in SI Appendix, SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank T. Privalsky for cloning the Rhodopseudomonas palustris matB gene and O. Sokolovskaya for cloning the KSAT2 didomain construct; A. Aron for help in the synthesis of [4-13C]-methylmalonyl-CoA; and J. Pelton (QB3 Central California 900 MHz NMR Facility) for assistance in analysis and development of the 13C NMR stoichiometry experiments. This work was funded by the generous support of a National Institutes of Health New Innovator Award 1 DP2 OD008696. O.A. and B.W.T. also acknowledge the support of a National Institutes of Health Training Grant T32 GMO66698. The College of Chemistry NMR Grants at the University of California, Berkeley is supported in part by National Institutes of Health Grants 1S10RR023679-01 and S10 RR16634-01.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1614196114/-/DCSupplemental.
References
- 1.Staunton J, Weissman KJ. Polyketide biosynthesis: A millennium review. Nat Prod Rep. 2001;18(4):380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 2.Khosla C, Tang Y, Chen AY, Schnarr NA, Cane DE. Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu Rev Biochem. 2007;76:195–221. doi: 10.1146/annurev.biochem.76.053105.093515. [DOI] [PubMed] [Google Scholar]
- 3.Dunn BJ, Khosla C. Engineering the acyltransferase substrate specificity of assembly line polyketide synthases. J R Soc Interface. 2013;10(85):20130297. doi: 10.1098/rsif.2013.0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ray L, Moore BS. Recent advances in the biosynthesis of unusual polyketide synthase substrates. Nat Prod Rep. 2016;33(2):150–161. doi: 10.1039/c5np00112a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hagmann WK. The many roles for fluorine in medicinal chemistry. J Med Chem. 2008;51(15):4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 6.Keatinge-Clay AT. The structures of type I polyketide synthases. Nat Prod Rep. 2012;29(10):1050–1073. doi: 10.1039/c2np20019h. [DOI] [PubMed] [Google Scholar]
- 7.Bravo-Rodriguez K, et al. Substrate flexibility of a mutated acyltransferase domain and implications for polyketide biosynthesis. Chem Biol. 2015;22(11):1425–1430. doi: 10.1016/j.chembiol.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Sundermann U, et al. Enzyme-directed mutasynthesis: A combined experimental and theoretical approach to substrate recognition of a polyketide synthase. ACS Chem Biol. 2013;8(2):443–450. doi: 10.1021/cb300505w. [DOI] [PubMed] [Google Scholar]
- 9.McDaniel R, et al. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel “unnatural” natural products. Proc Natl Acad Sci USA. 1999;96(5):1846–1851. doi: 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walker MC, et al. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways. Science. 2013;341(6150):1089–1094. doi: 10.1126/science.1242345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunn BJ, Watts KR, Robbins T, Cane DE, Khosla C. Comparative analysis of the substrate specificity of trans- versus cis-acyltransferases of assembly line polyketide synthases. Biochemistry. 2014;53(23):3796–3806. doi: 10.1021/bi5004316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koryakina I, et al. Poly specific trans-acyltransferase machinery revealed via engineered acyl-CoA synthetases. ACS Chem Biol. 2013;8(1):200–208. doi: 10.1021/cb3003489. [DOI] [PubMed] [Google Scholar]
- 13.Wu N, Tsuji SY, Cane DE, Khosla C. Assessing the balance between protein-protein interactions and enzyme-substrate interactions in the channeling of intermediates between polyketide synthase modules. J Am Chem Soc. 2001;123(27):6465–6474. doi: 10.1021/ja010219t. [DOI] [PubMed] [Google Scholar]
- 14.Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. Extender unit and acyl carrier protein specificity of ketosynthase domains of the 6-deoxyerythronolide B synthase. J Am Chem Soc. 2006;128(9):3067–3074. doi: 10.1021/ja058093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koryakina I, McArthur JB, Draelos MM, Williams GJ. Promiscuity of a modular polyketide synthase towards natural and non-natural extender units. Org Biomol Chem. 2013;11(27):4449–4458. doi: 10.1039/c3ob40633d. [DOI] [PubMed] [Google Scholar]
- 16.Rocha DF, Wouters FC, Machado G, Marsaioli AJ. First biosynthetic pathway of 1-hepten-3-one in Iporangaia pustulosa (Opiliones) Sci Rep. 2013;3:3156. doi: 10.1038/srep03156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jenner M, et al. Acyl-chain elongation drives ketosynthase substrate selectivity in trans-acyltransferase polyketide synthases. Angew Chem Int Ed Engl. 2015;54(6):1817–1821. doi: 10.1002/anie.201410219. [DOI] [PubMed] [Google Scholar]
- 18.Wong FT, Jin X, Mathews II, Cane DE, Khosla C. Structure and mechanism of the trans-acting acyltransferase from the disorazole synthase. Biochemistry. 2011;50(30):6539–6548. doi: 10.1021/bi200632j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wong FT, Chen AY, Cane DE, Khosla C. Protein-protein recognition between acyltransferases and acyl carrier proteins in multimodular polyketide synthases. Biochemistry. 2010;49(1):95–102. doi: 10.1021/bi901826g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meluzzi D, Zheng WH, Hensler M, Nizet V, Dorrestein PC. Top-down mass spectrometry on low-resolution instruments: Characterization of phosphopantetheinylated carrier domains in polyketide and non-ribosomal biosynthetic pathways. Bioorg Med Chem Lett. 2008;18(10):3107–3111. doi: 10.1016/j.bmcl.2007.10.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagen A, et al. In vitro analysis of carboxyacyl substrate tolerance in the loading and first extension modules of borrelidin polyketide synthase. Biochemistry. 2014;53(38):5975–5977. doi: 10.1021/bi500951c. [DOI] [PubMed] [Google Scholar]
- 22.Poust S, et al. Divergent mechanistic routes for the formation of gem-dimethyl groups in the biosynthesis of complex polyketides. Angew Chem Int Ed Engl. 2015;54(8):2370–2373. doi: 10.1002/anie.201410124. [DOI] [PubMed] [Google Scholar]
- 23.Bracher PJ, Snyder PW, Bohall BR, Whitesides GM. The relative rates of thiol-thioester exchange and hydrolysis for alkyl and aryl thioalkanoates in water. Orig Life Evol Biosph. 2011;41(5):399–412. doi: 10.1007/s11084-011-9243-4. [DOI] [PubMed] [Google Scholar]
- 24.Randall SM, Koryakina I, Williams GJ, Muddiman DC. Evaluating nonpolar surface area and liquid chromatography/mass spectrometry response: An application for site occupancy measurements for enzyme intermediates in polyketide biosynthesis. Rapid Commun Mass Spectrom. 2014;28(23):2511–2522. doi: 10.1002/rcm.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tosin M, et al. In vivo trapping of polyketide intermediates from an assembly line synthase using malonyl carba(dethia)-N-acetyl cysteamines. Chem Commun (Camb) 2011;47(12):3460–3462. doi: 10.1039/c0cc05077f. [DOI] [PubMed] [Google Scholar]
- 26.Riva E, et al. Chemical probes for the functionalization of polyketide intermediates. Angew Chem Int Ed Engl. 2014;53(44):11944–11949. doi: 10.1002/anie.201407448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.