

Abstract
Recent NMR chemical shift measurements of the 99 residue C-terminal fragment of Amyloid Precursor Protein (APP-C99) in the presence of cholesterol provide evidence of binary complex formation between C99 and cholesterol in membrane mimetic environments. It has also been observed that the production of Aβ protein is enhanced under conditions of high cholesterol concentration. In this study, we investigated the impact of the charge state of C99 on the structure and stability of the C99-cholesterol complex. We observed that the binding of C99 to cholesterol depends critically on the charge state of Glu 693 (E22) and Asp 694 (D23). Evaluation of the pKa values of the Asp and Glu sidechains suggests that these residues may be predominantly neutral in existing experimental observations of a stable C99-cholesterol complex at lower pH (characteristic of the endosomal environment), while binding is destabilized near neutral pH (characteristic of the cytoplasm). These observations suggest that specific binding of cholesterol to C99 is a sensitive function of the pH encountered in vivo, with key E22 and D23 residues serving as a “pH switch” controlling C99-cholesterol binding.
Keywords: Amyloid Precursor Protein (APP), C99, Alzheimer’s disease (AD), Amyloid β protein, protonation, pH, charge state, complex formation, molecular dynamics
TOC image
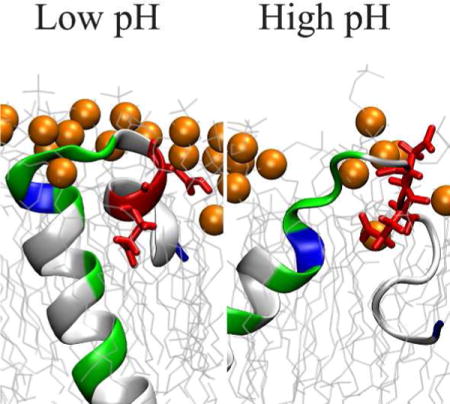
While the cause of Alzheimer’s disease (AD) remains under debate, the aggregation of Amyloid β (Aβ) peptides is widely thought to play a key role in the onset of senile dementia and progression of the disease1. The Aβ peptide is the product of processive cleavage of substrate C99, the 99 residue C-terminal fragment of Amyloid Precursor Protein (APP) by γ-secretase2,3, and is found in a variety of isoforms varying in length from 38 to 43 residues. The mechanistic details of formation of aggregates from Aβ monomers have been explored using a wide variety of experimental and computational techniques4–7. However, until recently the mechanism of production of Aβ peptide from the precursor protein C99 has received relatively little attention.
Cleavage of full length APP770 by β-secretase generates the membrane associated C99 (APP672–770). The transmembrane domain of C99 is subsequently processively cleaved by γ-secretase. Due to variation in points of initiation and termination of cleavage, a variety of isoforms of Aβ peptide is produced with Aβ40 being predominant and the more amyloidogenic Aβ42 occurring in a 10:1 Aβ40: Aβ42 ratio. These and a variety of other isoforms, varying from Aβ30 to Aβ43, are formed with absolute and relative quantities dependent on a number of factors including APP sequence and membrane lipid composition2,3. Recent studies of the structure of C99 monomer suggest that sequence and membrane environment hold the key to understanding why one cleavage site is favored over another in the genesis of Aβ peptide of varying lengths8–11. Nevertheless, there remain fundamental questions related to the mechanism of cleavage of C99 by γ-secretase, including whether the monomer or homodimer of C99 is the predominant cleavage substrate, how membrane lipid composition impacts the structure of C99 monomer and homodimer, and how the presence of cholesterol impacts the recognition of C99 by γ secretase and ultimately its cleavage. (Please refer to the recent detailed reviews on the impact of cholesterol on membrane proteins12,13)
In addition to nonspecific interactions8–11, NMR chemical shift measurements of C99 in bicelles of varying cholesterol concentration indicate that cholesterol forms a binary complex with C99 that competes with formation of C99 dimer11,14,15. This surprising observation is highly significant as γ-secretase localized in cholesterol rich environments such as lipid raft domains and its activity is known to be modulated by cholesterol concentration16–19. Therefore, binding to cholesterol may recruit C99 to cholesterol-rich raft domains and facilitate the cleavage process.
The most profound chemical shift variations in response to cholesterol binding were centered around the short juxtamembrane helix (JM) in the N terminal loop of C99 peptide and three Gly residues (Gly 29, Gly 33, Gly 37) that form a GXXXG repeat motif in the N-terminal transmembrane (TM) domain14. Mutation of Phe 20 or Glu 22 to Ala, from the JM helix and N terminal loop, or any Gly residues from the GXXXG motif substantially diminished cholesterol binding, suggesting that these residues are vital to C99-cholesterol complex formation14. This pattern of chemical shift variations suggests an induced fit model in which the flat α-face of cholesterol is kept in contact with the relatively smooth Gly face of the TM helix, created by the GXXXG repeat motif, while its head group is constrained by formation of hydrogen bonds with Asn27 and Glu2214 (see Figure 1).
Figure 1.
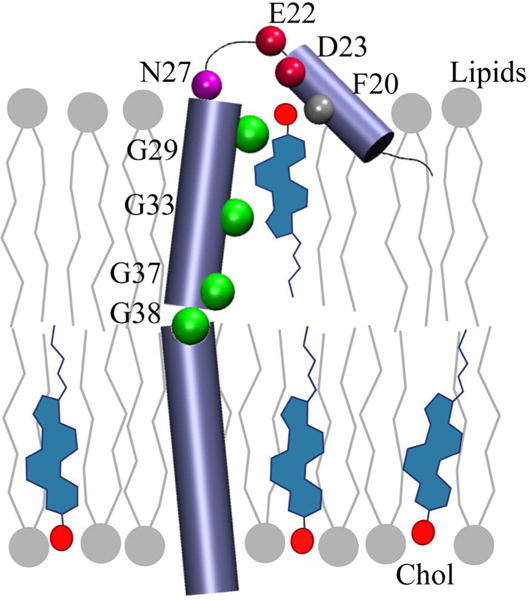
Cartoon representation of C99-cholesterol complex. Cholesterol molecules are shown in blue and their oxygen atoms with red circles. The lipid molecules are depicted in gray. The peptide is represented with purple cylinders and cholesterol contacting residues are marked with circles that are colored as following: Gly residues (green), Asp and Glu residues (red), Phe (light gray) and Asn (magenta).
While chemical shift and mutagenesis data on C99 in the presence of cholesterol has informed an initial model of the C99-cholesterol complex, little is known regarding the detailed interaction of key peptide residues with cholesterol and their specific role in stabilizing the putative C99-cholesterol complex. Specifically, the role of key ionizable residues, including E22 and D23, must be critically evaluated. The proposed binding model is based on chemical shift data collected at pH 4.5, close to the natural pKa values of Asp and Glu side chains (4.0 and 4.4, respectively). However, the true protonation state of these sidechains in the hydrophobic environment of a membrane bilayer is unclear and may play an essential role in modulating C99 structure and interaction with cholesterol.
In this study, we apply molecular dynamics (MD) simulation using all atom models from NMR chemical shift and mutagenesis studies14 to investigate the detailed interaction of C99 with cholesterol in DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) bilayers. The ionization states of residues E22 and D23 are found to be critically important to the structure of C99 monomer and its interaction with cholesterol, suggesting that C99-cholesterol interaction is a sensitive function of pH. These observations shed light on the role of environment in C99 processing and the potential mechanism of action of a number of known familial AD mutations.
Two replica simulations were initiated from a proposed C99-cholesterol complex structure primarily based on the results of Ala scanning data14 (see Figure S1) for both protonated E22/D23 (C9915–55[0]) and charged E22/D23 (C9915–55[−]) (for information on simulation details, see Computational Methods). Cholesterol was considered to be bound to C99 if the distance between its center-of-mass and that of a critical GXXXG repeat (residues 29 to 37) was less than or equal to 10Å. This cut-off value was chosen based on earlier binding free energy calculations by Nierzwicki and Czub20.
In order to probe the effect of interactions between individual residues and cholesterol in the complex, the average time each residue spends within 5Å of cholesterol was calculated over all simulations (See Figure 2). Contact between cholesterol and Gly 29 and Gly 32 is observed across all simulations of C9915–55[−] in agreement with NMR chemical shift measurments14. However, these contacts are short-lived (no more than several nanoseconds). In contrast, cholesterol forms persistent contacts with C9915–55[0] on the Gly-repeat face, as well as with the TM domain and short N terminal loop, including direct interaction with protonated E22. Comparison with NMR chemical shift variations14 (gray bars in Figure 2) supports the earlier conjecture that the GXXXG motif provides a favorable surface for cholesterol interaction in both C9915–55[−] and C9915–55[0]. However, long-lived contacts with the N terminal loop were only evident in C9915–55[0]. Importantly, overall agreement between simulation and NMR14 data is improved in the case of protonated side chains. It should be noted that the reported residence times are based on collected sampling over 5 independent simulations of 400ns. Longer simulations involving multiple binding and unbinding events would provide better statistics and a more complete estimate of the uncertainty in the observed residence times.
Figure 2.
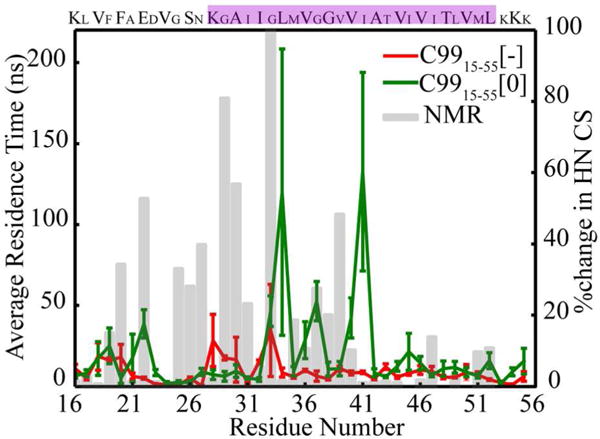
Average residence time of any heavy atom of cholesterol within 5Å of any heavy atom of the side chains of C99. The red (green) lines represent data for C9915–55[−] (C9915–55[0]). The gray bars represent contact probability inferred from NMR experiment14. The TM region is highlighted in magenta.
An earlier simulation study assessing the stability of the C99-cholesterol complex in C9915–55[−] showed stable binding of cholesterol, on the order of a few kcal/mol. However, no significant contacts were observed between Asp and Glu residues and cholesterol20. This further supports the importance of protonation of key residues E22 and D23 in the formation of a stable C99-cholesterol complex DMPC bilayers.
The observed increased contact of cholesterol and E22 can be explained by the critical impact of protonation on E22 and D23 on the structure of C99, including the orientation and insertion of Asp and Glu residues in the membrane bilayer. The density distributions of F19, E22 and D23 are superimposed on the density distribution of P atoms in the head groups of the lipids (Figure 3. Figure S2 for snapshots from the simulations). For C9915–55[0] (Figure 3 top panel, Figure S2 left panel) all three residues are inserted in the lipid bilayers while D23 is localized at the watermembrane interface. In contrast, for C9915–55[−] (Figure 3, bottom panel, Figure S2, right panel) a clear shift toward the membrane exterior is observed for all three residues.
Figure 3.
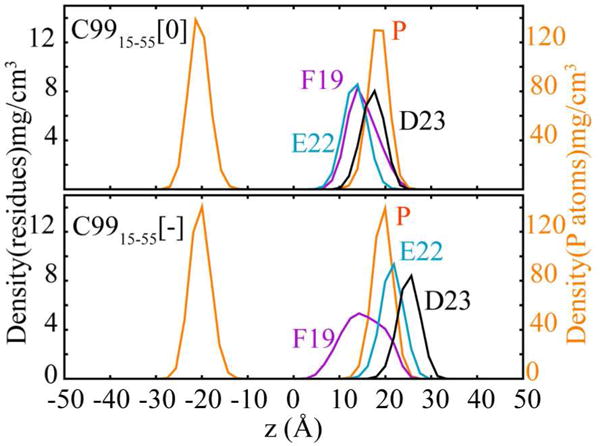
Density distributions of key residues across the membrane normal (z-axis). The scale for P atom (orange) density distribution is shown on the right, while the scale for densities of side chains is marked on the left. F19, E22 and D23 are colored as magenta, teal and black, respectively. Deeper penetration of E22 and D23 is observed in C9915–55[0] (upper) as opposed to C9915–55[−] (lower).
A clear enhancement in interaction between water and E22 and D23 is observed in C9915–55[−], whereas in C9915–55[0] the residues are membrane inserted and shielded from water (See radial distribution function (RDF) in Figure S3). An increase in insertion depth of Glu and Asp in their neutral states and the increased exposure to the membrane exterior in the charged state supports the conjecture that Glu and Asp are unavailable for direct interaction with cholesterol in their charged state and that stable binding of cholesterol with C99 is observed only in the neutral state of these key residues.
The average insertion depth of each residue was calculated as the distance of each residue center-of-mass relative to the average center of mass of all lipid head groups along the membrane normal (z-axis). Computed insertion depths of individual residues was compared with relative insertion depths obtained from EPR measurements in POPC/POPG vesicles14, as well as NMR chemical shift variations in the presence of hydrophobic (DSA) and hydrophilic (Gd) probes21 (Figure 4). Results suggest that the average insertion depth obtained from both C9915–55[0] and C9915–55[−] is in qualitative agreement with these experiments. However, the agreement is improved for C9915–55[0] with neutral E22 and D23, especially for the JM helix in the neighborhood of these two residues. This suggests that the presence of negatively charged lipids (POPG) in the vesicle may have increased the pKa of E22 and D23 and enhanced the probability of their protonation. Notably, the number of water molecules within 5 Å of each side chain (red bars) shows the elevated solvent exposure of the JM helix region in C9915–55[−].
Figure 4.
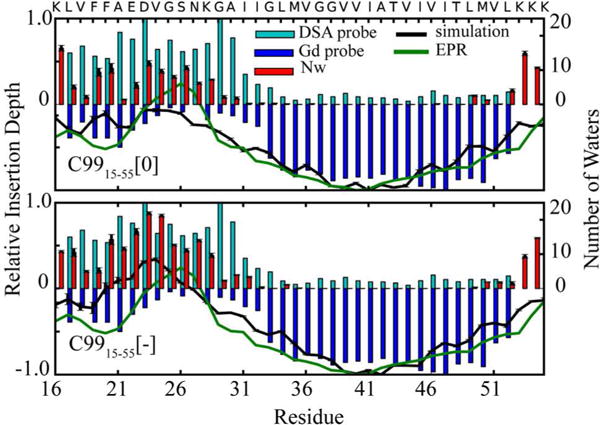
Insertion depth of each residue in the DMPC+20% cholesterol bilayer (black curve) compared with relative insertion depth from EPR (green)14 and relative variation in NMR chemical shift in the presence of hydrophilic (cyan bars) and hydrophobic (blue bars) probes21. Shown for comparison is the number of water molecules observed within 5Å of each residue in simulation (red bars).
Strong correlation is observed between cholesterol concentration and chemical shift variations in the GXXXG motif region within the TM domain of the peptide14. It has been proposed that the α-face of cholesterol provides a favorable surface for interaction with the Gly face of the TM helix14,22. In order to investigate the configuration of cholesterol in complex with the peptide, we introduce two Crick angles, ψ1 and ψ2, that describe the relative orientation of cholesterol, once in complex with the peptide, and the GXXXG motif of the TM helix (Figure 5). These angles were defined in terms of an intermolecular vector passing through (1) the center-of-mass of the sterol ring system of cholesterol and (2) center-of-mass of residues 29 to 37 of C99. ψ1 was defined as the angle between the central vector and a vector connecting the center-of-mass of residues 29 to 37 and the Cα of Gly33. Similarly, ψ2 was defined as the angle between the central vector and a vector connecting the center-of-mass of the sterol ring system and C19 of cholesterol (methyl carbon in CHARMM nomenclature). Cα of Gly 29, Gly 33, Gly 37 and C19 are depicted as spheres in Figure 5. In calculating ψ1 and ψ2, we considered the projection of all vectors on the x-y plane. MDAnalysis toolkit23 was used to perform the analysis.
Figure 5.
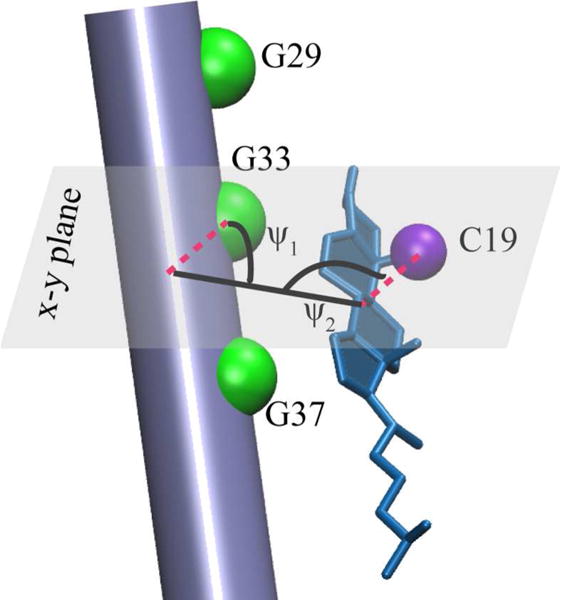
Definition of ψ1 and ψ2 Crick angles with Cα of Gly 29, 33 and 37 residues (green) and C19 carbon of cholesterol (purple). Cholesterol is shown in blue and the peptide is represented as a cylinder.
Small ψ1 values correspond to TM helix Gly-face oriented toward cholesterol, while small ψ2 values correspond to the β-face of cholesterol oriented toward the peptide. The distribution of Crick angles was compared for all cholesterol-C99 complexes in both C9915–55[0] and C9915–55[−]. The potential of mean force (PMF) projected along these two angles is depicted in Figure 6. Representative structures from different basins are marked on the PMF.
Figure 6.
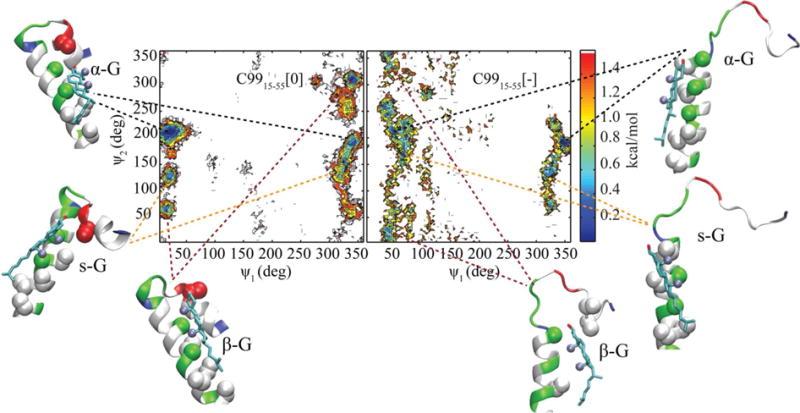
PMF projected along the ψ1 and ψ2 Crick angles for C9915–55[0] (left) and C9915–55[−] (right). Color bar is in kcal/mol. Representative structures for each basin are illustrated. Residues are colored based on type: hydrophobic (gray), hydrophilic including Gly (green), acidic Asp and Glu (red), and basic Lys (blue). Cα and Cβ atoms of residues within 5Å of cholesterol are depicted with spheres. Cholesterol is colored based on atom types: oxygens (red) and carbons (cyan). Methyl groups of cholesterol are marked by purple spheres. Structures are rendered in VMD24.
Our calculations show that in addition to complexes in which cholesterol α face is oriented toward peptide GXXXG motif (α-G), there are two additional basins. One basin corresponds to small ψ2 values and represents complexes in which the cholesterol β-face is oriented towards the peptide G-face (β-G) stabilized by a favorable “knob-in-hole” interaction of the cholesterol methyl group and C99. An additional basin represents complexes in which cholesterol is oriented sideways with respect to the peptide Gly-face (s-G). Similar sideways structures were observed in earlier computational studies of cholesterol-C99 interaction20. Importantly, in C9915–55[0] ψ1 angles are smaller (0°–20°) compared with C9915–55[−] (20°–60°) suggesting a strong preference for the dominant α-G configuration in the neutral state.
For C9915–55[0] residues in the vicinity of cholesterol (Figure 6 and Figure S4), cholesterol molecules are observed in the bound state in three different conformations with respect to the peptide Gly-face. Stable complexes are facilitated by favorable van der Waals contacts between cholesterol tails and hydrophobic residues on either side of the GXXXG motifs (Figure 6 and Figure S4) and insertion of methyl groups on the cholesterol β-face into empty grooves between hydrophobic side chains. The same trend is observed with favorable C9915–55[−] cholesterol tail interactions occasionally involving Phe residues in the JM region. However, significant differences between cholesterol complexes with C9915–55[0] and C9915–55[−] are also observed (Figure 2), as direct interactions with Asp and Glu stabilizing the C9915–55[0] complex are absent in C9915–55[−]. Cholesterol oxygen forms a hydrogen bond with the hydrogen of the protonated E22 of C9915–55[0] 12% of the time compared to 1.5% with E22 and D23 in C9915–55[−]. We did not observe any long lasting hydrogen bonding between cholesterol’s polar hydrogen and residues in its vicinity. In addition, the orientation of the JM helix of C9915–55[−] disrupts the “binding pocket” such that cholesterol is only loosely associated with the peptide Gly-face. As a result, cholesterol moves with more freedom in the binding pocket, characterized by large ψ1 values (Figure 6).
The structural heterogeneity observed among the C99-cholesterol complexes indicates that C99 provides a flexible pocket that could be compatible with ligands of varying geometries. Recall that the chemical shift variation in residues of C99 was measured for varying cholesterol concentration at pH 4.5, close to the intrinsic pKa values of Asp and Glu sidechains (4.0 and 4.4, respectively). Similar binding affinity was observed for C99-cholesterol complex at pH of 6.5 in vesicles containing negatively charged lipids15 (POPC:POPG 3:1). Given the observation above, it is likely that the resulting cholesterol binding is a function of pH, raising the interesting question of the impact of pH variation on the production of Aβ. To explore this question, we performed constant pH molecular dynamics simulations to calculate pKa values of Asp and Glu side chains in DMPC+20% cholesterol. The pKa values obtained from our simulations are 6.5±0.1 (D23) and 7.4±0.1 (E22) (see Figure S5 for titration curves). These values are in excellent agreement with recently reported pKa of 6.825 for a Glu side chain in a pentapeptide localized near a membrane surface25. Our computed pKas, although within the error bars of the study mentioned above25, are likely elevated by the adjacency of E22 and D23, and the solvent exposure of F19 in C9915–55[−] (Figure 3 and Figure S2), leading to a higher population of neutral versus charged states. In this study, we focus on variations in pKa with respect to the reference value of the side chain in solution. Longer simulations, and more complete conformational sampling, would provide a better estimate of the true uncertainty in the calculated pKa values.
Our computed pKa values suggest that in a biologically relevant pH range (5.0–7.4), Asp and Glu will be found in both ionized and neutral states. At pH 4.5 employed in the recent NMR study14, the neutral state is expected to have a significantly higher population enhancing cholesterol binding.
Importantly, production and release of Aβ from APP involves endocytosis18,26–29. The low pH of the environment of the endosome (with estimated pH 6.5–6.0 and 5.5–4.5 for early and late endosomes, respectively) increases the population of the protonated state of Glu 22 and Asp 23, which according to our simulation is more compatible with cholesterol binding. Therefore, C99 may be more prone to binding to cholesterol in the endosome (pH≈5.0), as opposed to the extracellular environment near the plasma membrane (pH≈7.4), which could subsequently promote its cleavage by γ-secretase. Familial AD mutations are observed at both Glu 22 and Asp 23. In all known cases these residues are substituted with a neutral side chain (E22G, E22Q and D23N)30, deleted (E22Δ)30, or substituted with Lys (E22K)30. In the latter case, Lys possesses a long aliphatic sidechain and is known to insert into membranes, adopting a neutral state in a biologically relevant pH range31,32. Given this pattern, it is possible that these familial AD mutations enhance favorable interaction with cholesterol thereby promoting C99-chol complex formation. In addition, it is possible that small perturbations of cellular pH, such as defects in the pH control pathway, lead to a decrease in cellular pH or increase in neutral Asp and Glu populations that stabilize C99-cholesterol interaction.
In conclusion, our study suggests that C99 complexation with cholesterol, observed in recent NMR studies, depends sensitively on the protonation states of key ionizable residues (E22 and D23) in the JM domain of C99. For ionized (negatively charged) states of Asp and Glu, expected at high pH, the sidechains are observed to be solvated, disrupting formation of the JM helix as well as contacts between cholesterol and residues in the JM region of C99. In contrast, for neutral states of Glu and Asp, expected at lower pH, E22 and D23 sidechains are inserted into the membrane, stabilizing the JM helix and facilitating formation of a cholesterol binding pocket.
Our findings also suggest that in addition to the α-G complex, originally proposed based on existing NMR data, there are β-G and s-Gly complexes that contribute to the overall cholesterol-C99 complex ensemble. Computed pKas of Asp and Glu side chains in membrane indicate that for biologically relevant pH values there will be a balance between the neutral state of the side chain of E22 and D23. Based on these results, we propose that known familial mutations of these key residues (E22G, E22Q and D23N) should enhance the stability of the cholesterol-C99 complex. Taken as a whole, our results are consistent with the proposal that enhanced cholesterol-C99 complex formation may lead to enhanced cleavage of C99 by γ-secretase and elevated production of Aβ.
Computational Methods
The residues that are shown to be critical both for peptide homo-dimerization and cholesterol complex formation are located in the TM and JM regions of C9911,14,15; therefore, in this study, we focus on residues 15–55 of C99, including both of these domains. The peptide congener C9915–5510 (PDBID 2LLM) was used as a model of full length APP672–725 (C99). The peptide was prepared with charged (C9915–55[−]) or neutral (C9915–55[0]) E22 and D23 residues using patches at both N and C termini (using CHARMM ACE, CT3 patches, respectively) and inserted into a membrane bilayer composed of DMPC and 20% cholesterol using CHARMM-GUI33. The composition of the bilayer was chosen to match recent bicelle compositions employed in the experimental study of Sanders and coworkers14. The bilayer was composed of 140 lipid molecules with 0.150 M concentration of Na+ and Cl− ions. One cholesterol molecule was positioned in the vicinity of the GXXXG motif (and Glu22 and Asp23) in agreement with Ala scanning results14. The initial model is depicted in Figure S1. The complex was energy minimized and equilibrated using the NVT ensemble. Initially, cholesterol was restrained to the binding pocket during the minimization and equilibration using harmonic restraints with force constant of 3500 kJ/(mol nm2). The cholesterol-C99 force constant was reduced to zero during 100 ns of NPT simulation. Subsequently, the complex was simulated for 400 ns of NPT dynamics using the Nose-Hoover thermostat and Parrinello-Rahman semi-isotropic barostat. Five replicates of each system were simulated and the first 40 ns was discarded. All simulations were performed using the GROMACS 5.0 simulation package34–38, the CHARMM36 all-atom force-field for proteins39 and lipids40, and the TIP341 water model. During all production runs, the CHARMM-GUI33 simulation protocol was followed. The temperature was maintained at 310 K and the pressure at 1bar.
Constant pH molecular dynamics simulations (CPHMD) were performed using the CHARMM multi-site λ dynamics frame work (MSλD)42,43 within the BLOCK facility. Details of this method, including the setup of each residue and its parameterization for the CHARMM 36 protein force-field, can be found elsewhere32,44–46. Structures of peptides with both charged and neutral states of E22 and D23 in the unbound configuration were used as initial structures of the CPHMD simulations. Following equilibration of each state in DMPC+20% cholesterol lipid bilayer according to the CHARMM-GUI33 default protocol, each structure was subjected to 10 ns of NPT dynamics followed by 10 ns of NVT dynamics at 298K. The resulting structures were subjected to 10 ns of CPHMD simulation from which the last 3 ns was used for analysis. The pH range was chosen to be 2 to 9 and λ values were saved every 10 steps. Three replicates were simulated. λ ≥0.8 was chosen as the cut-off for physical states. The fraction of unprotonated states for either E22 or D23 at each pH (Sunprot(pH)) was calculated as
| (1) |
In Eq. 1, Nunprot and Nprot are the populations of physical states (λ ≥0.8) at any given pH. These values were fitted to the Henderson-Hasselbalch equation (Eq. 2) to calculate pKa values:
| (2) |
All equilibration and production runs of CPHMD were performed using the CHARMM simulation package47.
Supplementary Material
Figures S1 Initial structure of cholesterol-C99 complex employed in the simulations. Figure S2 Snapshots from the simulations depicting different orientations of E22 and D23 in their charged and neutral states. Figure S3 Radial distribution function of water around E22 and D23. Figure S4 Molecular representation of C99-cholesterol complexes. Figure S5 Titration curves of E22 and D23.
Acknowledgments
We are grateful to the National Institutes of Health (R01 GM107703) and the National Science Foundation (CHE-1362524) for the generous support of our research. GAP is supported by a National Science Foundation Graduate Research Fellowship (DGE-1545957). The Center of Computational Science and the Shared Computer Cluster at Boston University are acknowledged for providing computational support. We are grateful to Dr. Charles Sanders for valuable comments and advice. AP wishes to thank Dr. Tetsuro Nagai for scientific discussions and Dr. Garret Goh and Dr. Elena Laricheva for their assistance in performing constant pH simulations.
Footnotes
Supporting Information. The following files are available free of charge.
The authors declare no competing financial interests.
References
- 1.O’Brien RJ, Wong PC. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu Rev Neurosci. 2011;34:185–204. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S. Ihara, γ-Secretase: Successive Tripeptide and Tetrapeptide Release from the Transmembrane Domain of β-Carboxyl Terminal Fragment. J Neurosci. 2009;29 doi: 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kukar TL, Ladd TB, Robertson P, Pintchovski SA, Moore B, Bann MA, Ren Z, Jansen-West K, Malphrus K, Eggert S, et al. Lysine 624 of the Amyloid Precursor Protein (APP) Is a Critical Determinant of Amyloid Peptide Length: Support for a Sequential Model of γ-Secretase Intramembrane Proteolysis and Regulation by the Amyloid Precursor Protein (APP) Juxtamembrane Region. J Biol Chem. 2011;286:39804–39812. doi: 10.1074/jbc.M111.274696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Straub JE, Thirumalai D. Toward a Molecular Theory of Early and Late Events in Monomer to Amyloid Fibril Formation. Annu Rev Phys Chem. 2011;62:437–463. doi: 10.1146/annurev-physchem-032210-103526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldschmidt L, Teng PK, Riek R, Eisenberg D. Identifying the Amylome, Proteins Capable of Forming Amyloid-like Fibrils. Proc Natl Acad Sci. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nelson R, Sawaya MR, Balbirnie M, Madsen AØ, Riekel C, Grothe R, Eisenberg D. Structure of the Cross-β Spine of Amyloid-like Fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A Structural Model for Alzheimer’s β-Amyloid Fibrils Based on Experimental Constraints from Solid State NMR. Proc Natl Acad Sci. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dominguez L, Meredith SC, Straub JE, Thirumalai D. Transmembrane Fragment Structures of Amyloid Precursor Protein Depend on Membrane Surface Curvature. J Am Chem Soc. 2014;136:854–857. doi: 10.1021/ja410958j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dominguez L, Foster L, Meredith SC, Straub JE, Thirumalai D. Structural Heterogeneity in Transmembrane Amyloid Precursor Protein Homodimer Is a Consequence of Environmental Selection. J Am Chem Soc. 2014;136:9619–9626. doi: 10.1021/ja503150x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nadezhdin KD, Bocharova OV, Bocharov EV, Arseniev AS. Structural and Dynamic Study of the Transmembrane Domain of the Amyloid Precursor Protein. Acta Naturae. 2011;3:69–76. [PMC free article] [PubMed] [Google Scholar]
- 11.Beel AJ, Mobley CK, Kim HJ, Tian F, Hadziselimovic A, Jap B, Prestegard JH, Sanders CR. Structural Studies of the Transmembrane C-Terminal Domain of the Amyloid Precursor Protein (APP): Does APP Function as a Cholesterol Sensor? Biochemistry. 2008;47:9428–9446. doi: 10.1021/bi800993c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fantini J, Barrantes FJ. How Cholesterol Interacts with Membrane Proteins: An Exploration of Cholesterol-Binding Sites Including CRAC, CARC, and Tilted Domains. Front Physiol. 2013;4 doi: 10.3389/fphys.2013.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song Y, Kenworthy AK, Sanders CR. Cholesterol as a Co-Solvent and a Ligand for Membrane Proteins. Protein Sci. 2014;23:1–22. doi: 10.1002/pro.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barrett PJ, Song Y, Van Horn WD, Hustedt EJ, Schafer JM, Hadziselimovic A, Beel AJ, Sanders CR. The Amyloid Precursor Protein Has a Flexible Transmembrane Domain and Binds Cholesterol. Science. 2012;336:1168–1171. doi: 10.1126/science.1219988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song Y, Hustedt EJ, Brandon S, Sanders CR. Competition Between Homodimerization and Cholesterol Binding to the C99 Domain of the Amyloid Precursor Protein. Biochemistry. 2013;52:5051–5064. doi: 10.1021/bi400735x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol Depletion Inhibits the Generation of β-Amyloid in Hippocampal Neurons. Proc Natl Acad Sci. 1998;95:6460–6464. doi: 10.1073/pnas.95.11.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE. Cholesterol-Dependent γ-Secretase Activity in Buoyant Cholesterol-Rich Membrane Microdomains. Neurobiol Dis. 2002;9:11–23. doi: 10.1006/nbdi.2001.0470. [DOI] [PubMed] [Google Scholar]
- 18.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic Processing of the Alzheimer β-Amyloid Precursor Protein Depends on Lipid Rafts. J Cell Biol. 2003;160:113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT, Kosik KS. A Detergent-Insoluble Membrance Compartment Contains Aβ in Vivo. Nat Med. 1998;4:730–734. doi: 10.1038/nm0698-730. [DOI] [PubMed] [Google Scholar]
- 20.Nierzwicki Ł, Czub J. Specific Binding of Cholesterol to the Amyloid Precursor Protein: Structure of the Complex and Driving Forces Characterized in Molecular Detail. J Phys Chem Lett. 2015;6:784–790. doi: 10.1021/acs.jpclett.5b00197. [DOI] [PubMed] [Google Scholar]
- 21.Song Y, Mittendorf KF, Lu Z, Sanders CR. Impact of Bilayer Lipid Composition on the Structure and Topology of the Transmembrane Amyloid Precursor C99 Protein. J Am Chem Soc. 2014;136:4093–4096. doi: 10.1021/ja4114374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fantini J, Yahi N, Garmy N. Cholesterol Accelerates the Binding of Alzheimer’s β-Amyloid Peptide to Ganglioside GM1 through a Universal Hydrogen-Bond-Dependent Sterol Tuning of Glycolipid Conformation. Front Physiol. 2013;4:1–10. doi: 10.3389/fphys.2013.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michaud-Agrawal N, Denning EJ, Woolf TB, Beckstein O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J Comput Chem. 2011;32:2319–2327. doi: 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Humphrey W, Dalke A, Schulten K. VMD: Visual Molecular Dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 25.Teixeira VH, Vila-Viçosa D, Reis PBPS, Machuqueiro M. pKa Values of Titrable Amino Acids at the Water/Membrane Interface. J Chem Theory Comput. 2016;12:930–934. doi: 10.1021/acs.jctc.5b01114. [DOI] [PubMed] [Google Scholar]
- 26.Koo EH, Squazzo SL. Evidence That Production and Release of Amyloid β-Protein Involves the Endocytic Pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- 27.Marquer C, Devauges V, Cossec J-C, Liot G, Lecart S, Saudou F, Duyckaerts C, Leveque-Fort S, Potier M-C. Local Cholesterol Increase Triggers Amyloid Precursor Protein-Bace1 Clustering in Lipid Rafts and Rapid Endocytosis. FASEB J. 2011;25:1295–1305. doi: 10.1096/fj.10-168633. [DOI] [PubMed] [Google Scholar]
- 28.Perez RG, Soriano S, Hayes JD, Ostaszewski B, Xia W, Selkoe DJ, Chen X, Stokin GB, Koo EH. Mutagenesis Identifies New Signals for β-Amyloid Precursor Protein Endocytosis, Turnover, and the Generation of Secreted Fragments, Including Aβ 42. J Biol Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- 29.Grbovic OM, Mathews PM, Jiang Y, Schmidt SD, Dinakar R, Summers-Terio NB, Ceresa BP, Nixon RA, Cataldo AM. Rab5-Stimulated Up-Regulation of the Endocytic Pathway Increases Intracellular β-Cleaved Amyloid Precursor Protein Carboxyl-Terminal Fragment Levels and Aβ Production. J Biol Chem. 2003;278:31261–31268. doi: 10.1074/jbc.M304122200. [DOI] [PubMed] [Google Scholar]
- 30.Tomidokoro Y, Rostagno A, Neubert TA, Lu Y, Rebeck GW, Frangione B, Greenberg SM, Ghiso J. Iowa Variant of Familial Alzheimer’s Disease. Am J Pathol. 2010;176:1841–1854. doi: 10.2353/ajpath.2010.090636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gleason NJ, Vostrikov VV, Greathouse DV, Koeppe RE. Buried Lysine, but Not Arginine, Titrates and Alters Transmembrane Helix Tilt. Proc Natl Acad Sci USA. 2013;110:1692–1695. doi: 10.1073/pnas.1215400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Panahi A, Brooks CL. Membrane Environment Modulates the pKa Values of Transmembrane Helices. J Phys Chem B. 2015;119:4601–4607. doi: 10.1021/acs.jpcb.5b00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jo S, Kim T, Im W. Automated Builder and Database of Protein/membrane Complexes for Molecular Dynamics Simulations. PLoS One. 2007;2:e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindahl E, Hess B, van der Spoel D. GROMACS 3.0: A Package for Molecular Simulation and Trajectory Analysis. Mol Model Annu. 7:306–317. [Google Scholar]
- 35.Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput Phys Commun. 1995;91:43–56. [Google Scholar]
- 36.Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: Fast, Flexible, and Free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 37.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 38.Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX. 2015:1–2. [Google Scholar]
- 39.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RW. Update of the CHARMM AllAtom Additive Force Field for Lipids: Validation on Six Lipid Types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of Simple Potential Functions for Simulating Liquid Water. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 42.Knight JL, Brooks CL. Applying Efficient Implicit Nongeometric Constraints in Alchemical Free Energy Simulations. J Comput Chem. 2011;32:3423–3432. doi: 10.1002/jcc.21921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knight JL, Brooks CL. Multi-Site λ-Dynamics for Simulated Structure-Activity Relationship Studies. J Chem Theory Comput. 2011;7:2728–2739. doi: 10.1021/ct200444f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goh GB, Hulbert BS, Zhou H, Brooks CL. Constant pH Molecular Dynamics of Proteins in Explicit Solvent with Proton Tautomerism. Proteins Struct Funct Bioinforma. 2014;82:1319–1331. doi: 10.1002/prot.24499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goh GB, Knight JL, Brooks CL. Constant pH Molecular Dynamics Simulations of Nucleic Acids in Explicit Solvent. J Chem Theory Comput. 2012;8:36–46. doi: 10.1021/ct2006314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goh GB, Laricheva EN, Brooks CL. Uncovering pH-Dependent Transient States of Proteins with Buried Ionizable Residues. J Am Chem Soc. 2014;136:8496–8499. doi: 10.1021/ja5012564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, et al. CHARMM: The Biomolecular Simulation Program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1 Initial structure of cholesterol-C99 complex employed in the simulations. Figure S2 Snapshots from the simulations depicting different orientations of E22 and D23 in their charged and neutral states. Figure S3 Radial distribution function of water around E22 and D23. Figure S4 Molecular representation of C99-cholesterol complexes. Figure S5 Titration curves of E22 and D23.