

Abstract
Objective
Diabetes mellitus causes vascular endothelial dysfunction and alters vascular microRNA expression. We investigated if endothelial microRNA-34a (miR-34a) leads to diabetic vascular dysfunction by targeting endothelial Sirtuin1 (Sirt1), and asked if the oxidative stress protein p66Shc governs miR-34a expression in the diabetic endothelium.
Approach and Results
MiR-34a is upregulated, and Sirt1down-regulated, in aortic endothelium of db/db and streptozotocin (STZ)-induced diabetic mice. Systemic administration of miR-34a inhibitor, or endothelium-specific knockout of miR-34a, prevents downregulation of aortic Sirt1 and rescues impaired endothelium-dependent aortic vasorelaxation induced by diabetes. Moreover, overexpression of Sirt1 mitigates impaired endothelium-dependent vasorelaxation caused by miR-34a mimic ex vivo. Systemic infusion of miR-34a inhibitor or genetic ablation of endothelial miR-34a prevents downregulation of endothelial Sirt1 by high glucose. MiR-34a is upregulated, Sirt1 is downregulated, and oxidative stress (H2O2) is induced in endothelial cells incubated with high glucose, or the free fatty acid palmitate in vitro. Increase of H2O2 and induction of endothelial miR-34a by high glucose or palmitate in vitro is suppressed by knockdown of p66shc. In addition, overexpression of wild-type but not redox-deficient p66Shc upregulates miR-34a in endothelial cells. P66Shc-stimulated upregulation of endothelial miR-34a is suppressed by cell-permeant antioxidants. Finally, mice with global knockdown of p66Shc are protected from diabetes-induced upregulation of miR-34a, and downregulation of Sirt1, in the endothelium.
Conclusions
These data show that hyperglycemia and elevated free fatty acids in the diabetic milieu recruit p66Shc to upregulate endothelial miR-34a via an oxidant-sensitive mechanism, which leads to endothelial dysfunction by targeting Sirt1.
Keywords: MicroRNA-34a, P66Shc, Sirtuin1, diabetic endothelial dysfunction
Diabetes mellitus (DM) is a global epidemic. Atherosclerotic vascular disease is the major cause of morbidity and mortality in diabetes. Vascular endothelial dysfunction precedes and promotes vascular inflammation and atherosclerosis. Hyperglycemia of diabetes impairs vascular endothelial function through many mechanisms. Recent evidence indicates that hyperglycemia alters the expression pattern of endothelial microRNAs (miRs)1,2.
MicroRNAs are small non-coding RNA molecule (~22 nucleotides) found in all cell types that lead to RNA silencing and post-transcriptional regulation of gene expression, thus affecting cellular function and determining cell fate. MicroRNAs play an important role in regulating vascular function and contribute to the development of diabetic vascular pathology1,2. MiR-34a, a p53-regulated microRNA3, has emerged as a mediator of several vascular pathologies. MiR-34a promotes senescence in the vascular endothelium4, causes endothelial inflammation by upregulating basal vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1)5, and impairs angiogenesis by inducing senescence of endothelial progenitor cells6, However, little is known about how miR-34a is regulated in the diabetic vasculature. Moreover, what genes are targeted by miR-34a, and whether their downregulation is responsible for diabetic endothelial dysfunction has not been experimentally verified.
The NAD+-dependent lysine deacetylase Sirtuin1 (Sirt1) has a vital part in governing vascular endothelial function. Sirt1 plays a salutary role in the vasculature via several mechanisms including deacetylation-induced activation of eNOS7, inhibition of macrophage foam cell formation, and prevention of hyperglycemia-induced vascular cell senescence8, Caloric restriction, a well-known stimulant of Sirt1, decreases arterial blood pressure in healthy individuals and improves endothelium-dependent vasodilation in obese and overweight individuals7. In addition, nutritional pharmaceuticals such as docosahexaenoic acid (DHA) act via Sirt1-dependent deacetylation of eNOS to increase endothelial NO production and relax blood vessels9. Importantly, Sirt1 is a bona fide target of several miRNAs, including miR-34a10. Moreover, serum miR-34a is upregulated in diabetics10, suggesting a role for it in diabetic end organ pathology.
P66shc is a master promoter of oxidative stress-related pathologies. Inactivation of p66Shc confers resistance to oxidative stress and protects mice from age-associated and diabetic vascular endothelial dysfunction11,12. Interestingly, p66Shc is regulated by Sirt1 in diabetes13. However, whether p66Shc governs expression of Sirt1 in the diabetic vasculature, and if this expression is regulated through miR-34a, is not known. Recognizing that miR-34a is an oxidative stress-induced microRNA, we asked if p66Shc promotes miR-34a expression in the endothelium, which then leads to diabetic endothelial dysfunction by targeting Sirt1.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Upregulation of miR-34a causes vascular endothelial dysfunction in db/db diabetic mice
To explore the role of miR-34a in diabetic vascular dysfunction, we first determined its expression in the vasculature of type 2 diabetic db/db mice. At 8 weeks of age, db/db mice were hyperglycemic in fed and fasted states (Supplemental Figure IA & B). MiR-34a was upregulated in aortas of db/db mice, as measured by in situ hybridization and real time qPCR (Figure 1A & B). Of note, upregulation of miR-34a in db/db mice was not exclusive to endothelium (Figure 1A), as it was evident in the vascular smooth muscle layer as well.
Figure 1. Vascular miR-34a is induced in diabetes.
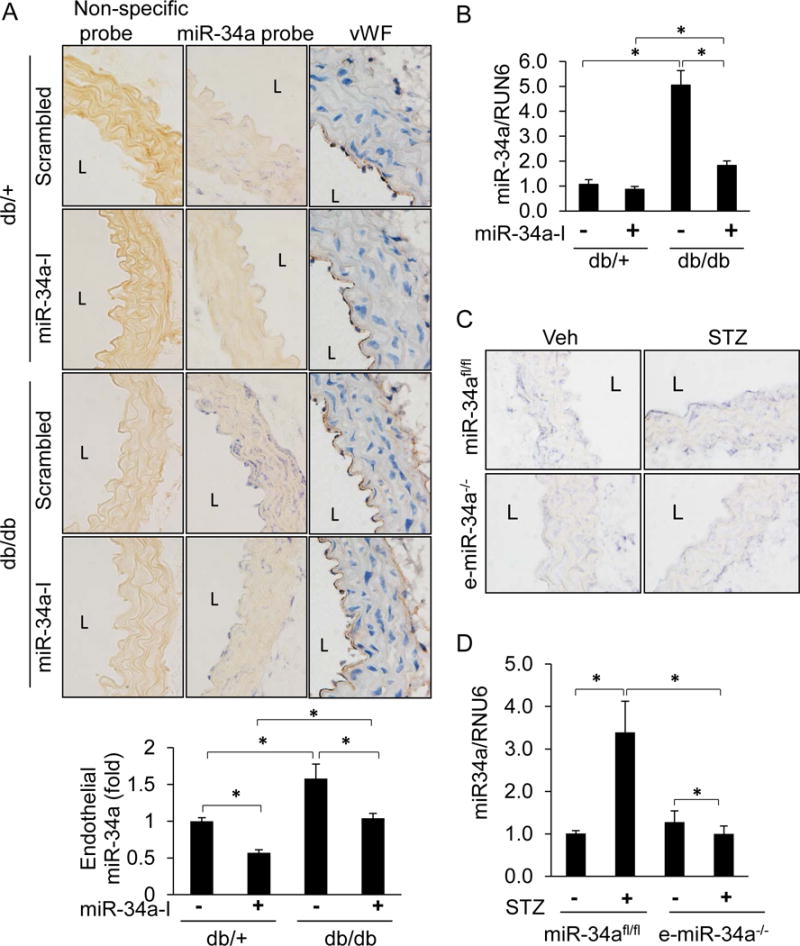
A–B, MiR-34a is upregulated in db/db diabetic mouse aortas, and suppressed by miR-34a inhibitor. A, Top: In situ hybridization (ISH) for miR-34a (purple-blue) in diabetic db/db and non-diabetic db/+ mouse aortas. Mice were systemically infused with a locked nucleic acid miR34a inhibitor (miR-34a-I) or a scrambled control locked nucleic acid (scrambled) for 4 weeks. Aortas were stained with miR-34a probe or a non-specific probe. Von Willebrand factor (vWF; brown) was stain by using DAB peroxidase substrate. Images were captured at 40 X, and representative images are shown. Bottom: Quantification for endothelial miR-34a was performed using ImageJ. B, Real time qPCR (q-RTPCR) for miR-34a in whole aortas of db/db and db/+ mice. C–D, Upregulation of endothelial miR-34a is suppressed in endothelium-specific knockout (e-miR-34a−/−) mice. C, ISH for miR-34a (purple-blue) in aortas of miR-34afl/fl and e-miR-34a−/− mice. Mice were made diabetic with STZ or given vehicle (Veh) control. D, Q-RTPCR for miR-34a in whole aortas of STZ-induced diabetic miR-34afl/fl and e-miR-34a−/− mice. n=4–5 per group. Data are shown as mean ± SEM. * P<0.05. L: lumen.
Db/db mouse aortas had impaired endothelium-dependent relaxation compared to db/+ controls (Figure 2A, left panel). To determine if miR-34a is responsible for this impairment, osmotic mini-pumps delivering a locked nucleic acid miR-34a inhibitor (miR-34a-I) or a scrambled control locked nucleic acid were implanted subcutaneously into mice. Systemic administration of miR-34a-I resulted in suppression of miR-34a expression in whole aortas of db/db mice (Figure 1A & B). The inhibitor did not selectively target endothelium, as its effect on miR-34a expression was seen in both the endothelial and medial layers of mouse aortas (Figure 1A & B). Hyperglycemic status was not altered in db/db mice that received miR-34a inhibitor (Supplemental Figure IA & B), indicating that miR-34a does not affect systemic glucose homeostasis. These findings indicate that systemic administration of miR-34a-I is an effective way to decrease miR-34a expression in mouse arteries in vivo.
Figure 2. MiR-34a mediates impairment of endothelium-dependent vasorelaxation in diabetes.
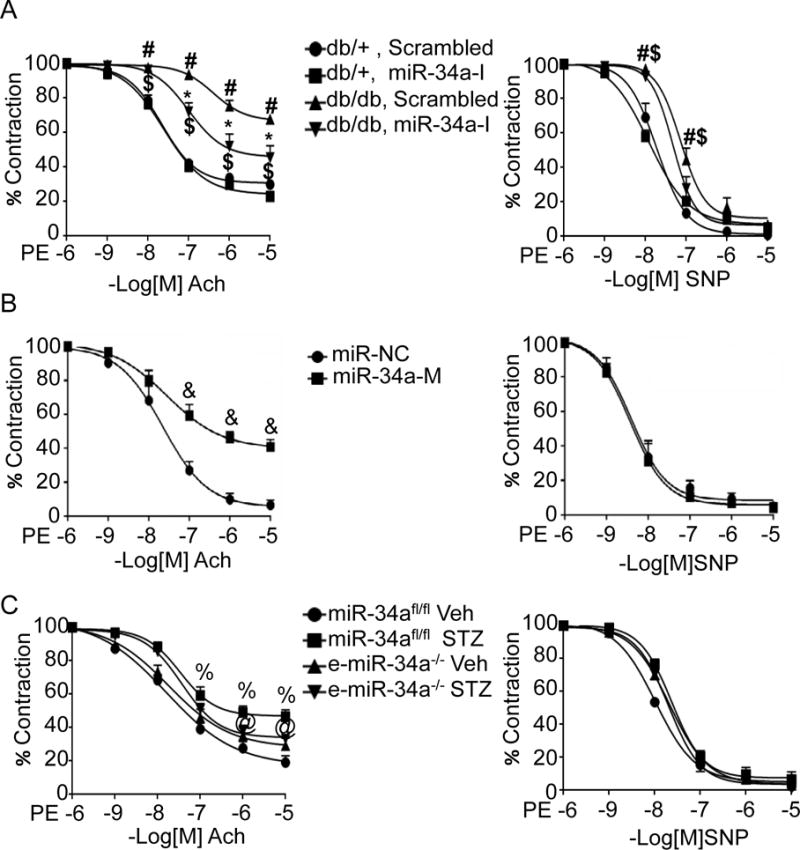
A, MiR-34a inhibitor protects against endothelial dysfunction in db/db diabetic mice. Acetylcholine (Ach)-stimulated endothelium-dependent (left), and sodium nitroprusside (SNP)-stimulated endothelium-independent (right), vasorelaxation in aortas of db/db and db/+ mice systemically administered locked nucleic acid miR-34a inhibitor (miR-34a-I) or scrambled control locked nucleic acid. # p<0.05 vs. db/+ scramble; * p<0.05 vs. db/db scrambled; $ p<0.05 vs. db/+ miR-34a-I. B, MiR-34a mimic promotes endothelial dysfunction. Ach-stimulated (left), and SNP-stimulated (right), vasorelaxation in wild-type aortas transduced ex vivo with miR-34a oligonucleotide mimic (miR-34a-M) or a scrambled control miR (miR-NC). & p<0.05 vs. miR-NC. C, Endothelium-specific deletion of miR-34a partially protects against endothelial dysfunction in STZ-induced diabetes. Ach-stimulated (left), and SNP-stimulated (right) vasorelaxation in STZ-induced diabetic and non-diabetic (Veh) miR-34afl/fl and e-miR-34a−/− mice; % p<0.05 vs. miR-34afl/fl Veh; @ p<0.05 vs. miR-34afl/fl STZ. n=8–10 aortic rings from 4–6 mice per group. Data are shown as mean ± SEM.
Systemic delivery of miR-34a-I preserved endothelium-dependent vasorelaxation in aortas of db/db mice (Figure 2A, left panel). Although miR-34a-I decreased miR-34a expression in the medial smooth muscle cell layer (Figure 1A), this did not result in an improvement in endothelium-independent vasorelaxation in the db/db mice (Figure 2A, right panel). These data indicate that in db/db mice, although miR-34a is upregulated throughout the vascular wall, its effect on vasoreactivity is primarily restricted to impairing endothelium-dependent vasorelaxation.
To confirm if miR-34a directly impairs endothelium-dependent vascular relaxation, a miR-34a oligonucleotide mimic was transfected into wild-type mouse aortas ex vivo (Supplemental Figure II). Transfection of the miR-34a mimic impaired endothelium-dependent vascular relaxation (Figure 2B, left panel), but had no effect on endothelium-independent vascular relaxation (Figure 2B, right panel). These findings further suggest that the effect of miR-34a on vasomotor function is principally by targeting the vascular endothelium.
STZ-induced diabetes causes vascular endothelial dysfunction through upregulation of endothelial miR-34a
The role of miR-34a upregulation in impairing vascular function was also evaluated in a streptozotocin (STZ)–induced type 1 diabetes mouse model. This model was established by administrating a single dose of STZ (100mg/kg) and resulted in robust increase in blood glucose over the course of 4 weeks (Supplemental Figure III). Although mice injected with STZ did not gain weight as their non-treated counterparts, they appeared healthy and were not emaciated (Supplemental Figure IV). Similar to db/db mouse aortas, miR-34a was upregulated in aortas, including the endothelium, of mice rendered diabetic by injection of STZ (Figure 1C &D). To tease out the role of endothelial miR-34a in diabetes-induced impairment of endothelial vasorelaxation, we turned to mice conditionally lacking endothelial miR-34a (e-miR-34a−/−). Knockout of miR-34a in the endothelium did not affect basal or STZ-induced blood glucose levels (Supplemental Figure V). E-miR-34a−/− mice were protected from STZ-induced impairment of endothelium-dependent vasorelaxation compared to mir34afl/fl control mice (Figure 2C, left panel). STZ did not impair, and endothelial knockout of miR-34a had no effect on, endothelium-independent vasorelaxation (Figure 2C, right panel). Taken together, these data show that upregulation of endothelial miR-34a is responsible for vascular endothelial dysfunction in the STZ model of diabetes.
Diabetes, high glucose, and free fatty acids downregulate endothelial Sirt1 via miR-34a
Previous studies have demonstrated that miR-34a targets Sirt114, although no studies have addressed the role of miR-34a-mediated Sirt1 downregulation in diabetic endothelial dysfunction. We therefore asked if miR-34a promotes diabetic vascular endothelial dysfunction by targeting Sirt1. We first examined if diabetes affects the expression of vascular Sirt1. Sirt1 was downregulated in aortas of db/db and STZ-induced diabetic mice (Figure 3A–3C). This downregulation was evident in both vascular smooth muscle cells as well as endothelial layers (Figure 3B & C). To determine if miR-34a induced by diabetes is responsible for downregulation of Sirt1, we assessed Sirt1 expression in aortas of db/db mice systemically infused with miR-34a inhibitor (miR-34a-I). MiR-34a-I rescued vascular Sirt1 expression in db/db mice (Figure 3A & B). This was particularly evident with regards to endothelial Sirt1 (Figure 3B). We also asked if the same is true in a STZ-induced model of diabetes. Vascular Sirt1 was downregulated throughout the aortic wall of miR-34afl/fl mice rendered diabetic with STZ (Figure 3C). In contrast, whole vascular, and endothelial, Sirt1 downregulation by STZ was mitigated in e-miR-34a−/− mice (Figure 3C). Thus, upregulation of miR-34a in endothelium results in downregulation of endothelial Sirt1.
Figure 3. MiR-34a is required for endothelial Sirt1 downregulation in diabetes.
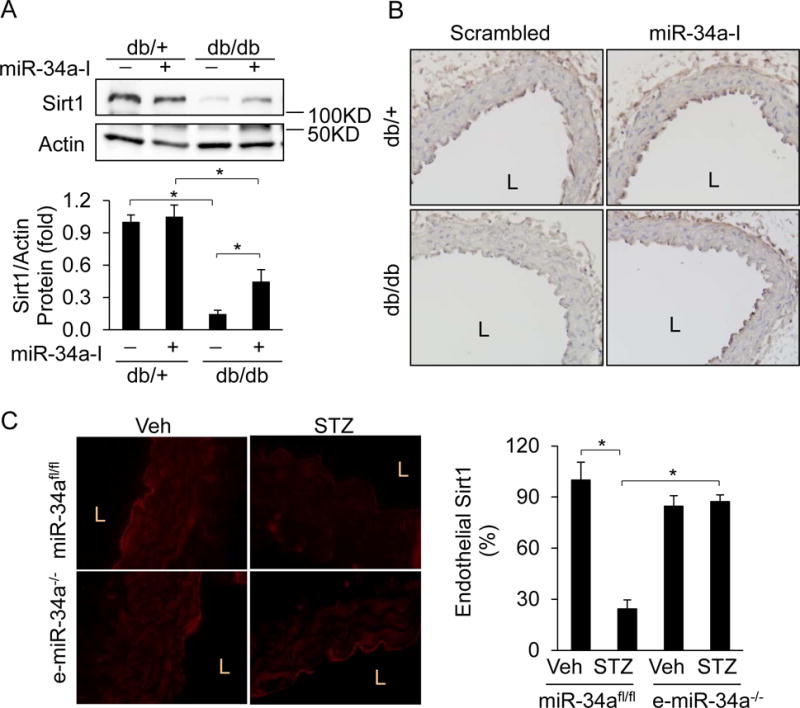
A–B, Sirt1 is downregulated in db/db diabetic mouse aortas, and is rescued by inhibition of miR-34a. Sirt1 expression by (A) immunoblotting and (B) IHC (brown color) in aortas of db/db and db/+ mice systemically administered locked nucleic acid miR-34a inhibitor (miR-34a-I) or scrambled control locked nucleic acid miR. C, Endothelium-specific deletion of miR-34a rescues downregulation of Sirt1 in STZ diabetes. Immunofluorescence images of Sirt1 (red) in aortas of STZ-induced diabetic and non-diabetic (Veh) miR-34afl/fl and e-miR-34a−/− mice. Representative immunoblots and IHC or immunofluorescence images are shown. Endothelial or total aortic Sirt1 was quantified and is expressed as percent or fold of Veh-treated miR-34afl/fl mice, or db/+ mice infused with scrambled miR. Data are shown as mean ± SEM. n=4–5, * P<0.05. L: lumen.
We next asked if hyperglycemia per se leads to miR-34a-mediated downregulation of Sirt1 in the endothelium. To answer this question, we performed in vitro studies in endothelial cells incubated with medium supplemented with high glucose. HUVECs and aortic endothelial cells isolated from mice (miR-34afl/fl mice) cultured in high glucose (HG; 30mM) medium showed upregulation of miR-34a and downregulation of Sirt1 (Figure 4A, B & C). Decrease of Sirt1 by HG was rescued when HUVEC were pre-treated with miR-34-I (Figure 4B). Complementary studies in aortic endothelial cells isolated from e-miR-34a−/− mice showed that HG failed to upregulate miR-34a (Figure 4C) and downregulate Sirt1 (Figure 4D). Conversely, miR-34a mimic (miR-34a-M) downregulated, and miR-34-I upregulated, basal Sirt1 in HUVECs (Figure 4E).
Figure 4. High glucose and palmitate downregulate Sirt1 in endothelial cells by inducing miR-34a.
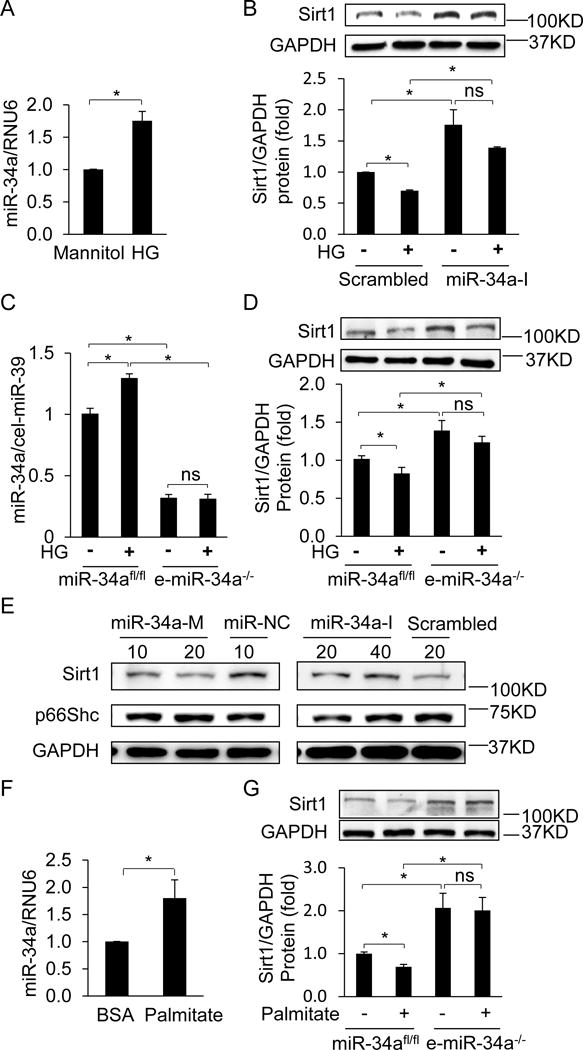
A, MiR-34a in HUVECs is upregulated by high glucose (HG, 30 mM glucose). HUVECs were incubated in HG for 24 h. MiR-34a expression is expressed relative to cells incubated in mannitol (30 mM) control. B, Inhibition of miR-34a rescues HG-induced downregulation of Sirt1. HUVECs transfected with miR-34a inhibitor (miR-34a-I) or scrambled miR were treated with mannitol or HG for 24 h. Data are expressed relative to cells transfected with scrambled miR and treated with mannitol control. C, HG incubation upregulates miR-34a in miR-34afl/fl, but not e-miR-34a−/−, aortic endothelial cells. Aortic endothelial cells isolated from miR-34afl/fl and e-miR-34a−/− mice were treated with mannitol or HG for 24 h. Data is expressed relative to miR-34afl/fl cells treated with mannitol. D, MiR-34a knockout inhibits HG-induced downregulation of Sirt1. Aortic endothelial cells were incubated in HG for 24 h as above. E, MiR-34a regulates basal Sirt1 expression in endothelial cells. HUVECs were transfected with miR-34a mimic (miR-34a-M), miR-34-I, or scrambled miR. Representative immunoblots are shown. F, Palmitate upregulates miR-34a in miR-34afl/fl aortic endothelial cells. Aortic endothelial cells were incubated in 500 uM palmitate for 12 h. MiR-34a expression is shown relative to miR-34afl/fl endothelial cells incubated in BSA vehicle. G, MiR-34a knockout inhibits palmitate-induced downregulation of Sirt1. MiR-34afl/fl and e-miR-34a−/− aortic endothelial cells were treated with 500 uM palmitate for 12 h. Data are expressed relative to miR-34afl/fl endothelial cells treated with BSA vehicle. Representative Immunoblots are shown. Data are shown as mean ± SEM. * P<0.05. n = 3–7.
In addition to increase in serum glucose, diabetes also results in increase of circulating free fatty acids (FFA). We therefore tested the effect of palmitate, a saturated FFA, on miR-34a and Sirt1 expression in endothelial cells. Palmitate (500 μM) stimulated miR-34a expression in mouse (miR-34afl/fl mice) aortic endothelial cells (Figure 4F). In addition, palmitate decreased Sirt1 expression in these cells (Figure 4G; Supplemental Figure VI). In contrast, Sirt1 expression was preserved in mouse endothelial cells derived from e-miR-34a−/− mice (Figure 4G). These findings underscore that 1) miR-34a targets Sirt1 in endothelial cells, 2) high glucose and FFA induce miR-34a in endothelial cells, and 3) downregulation of endothelial Sirt1 by high glucose and FFA is mediated by miR-34a.
Reconstitution of Sirt1 restores endothelium-dependent vasorelaxation in miR-34a-transfected vessels
Next, we probed for a causal relationship between downregulation of Sirt1 and endothelial dysfunction induced by miR-34a. MiR-34a mimic transfected into mouse aortas ex vivo led to marked upregulation of miR-34a expression (Figure 5A), and impairment of endothelium-dependent vasorelaxation (Figure 5B, left panel), but not endothelium-independent vasorelaxation (Figure 5B, right panel). This upregulation of miR-34a was accompanied by downregulation of vascular Sirt1 (Figure 5C & D). Reconstitution of Sirt1 expression with an adenovirus encoding Sirt1 lacking the 3′-UTR (AdSirt1) rescued impaired endothelium-dependent vasorelaxation triggered by miR34-a (Figure 5B, left panel). These data show that downregulation of Sirt1 is the proximate cause of endothelial dysfunction induced by miR-34a.
Figure 5. Sirt1 rescues vascular endothelial dysfunction induced by miR-34a.
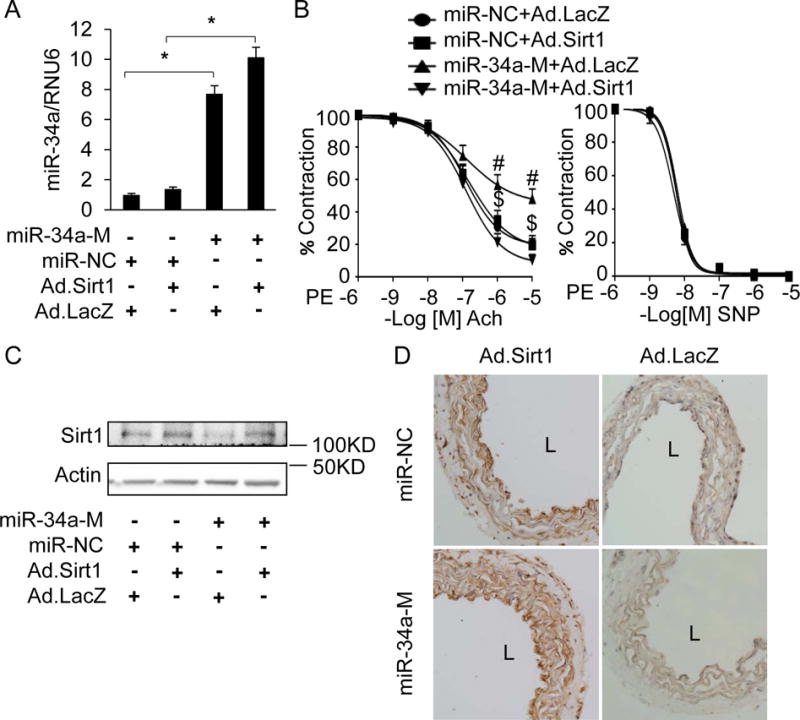
A–D, Adenoviral overexpression of Sirt1 ex vivo rescues downregulation of Sirt1 and mitigates endothelial dysfunction in aortas induced by miR-34a. (A) Real time qPCR for miR-34a, (B) endothelium-dependent (left) and –independent (right) vasorelaxation (C) immunoblot for Sirt1, and (D) IHC for Sirt1(dark brown) in wild-type mouse aortas transfected with miR-34a mimic (mirR-34a-M) or negative control microRNA with scrambled sequence (miR-NC), and simultaneously infected with adenovirus encoding human Sirt1 (Ad.Sirt1) or a control adenovirus encoding LacZ (Ad.LacZ). Representative IHC images and immunoblots are shown. n=3, * p<0.05 for q-RTPCR. # p<0.05, miR-34a-M+Ad.LacZ vs. miR-NC + Ad.LacZ; $ p<0.05, miR-34a-M+Ad.Sirt1 vs. miR-34a-M+Ad.LacZ, n=6–8 rings from 3–4 mice per group. Data are shown as mean ± SEM. L: lumen.
Endothelial p66Shc is activated by diabetes, high glucose, and free fatty acids, and promotes vascular oxidative stress and diabetic endothelial dysfunction
P66shc is a master regulator of the redox state and promotes endothelial dysfunction by inducing oxidative stress. We determined the status of p66Shc, and its role in endothelial dysfunction, in our models of diabetes. Because p66Shc is activated by serine phosphorylation15, we first assessed the phosphorylation state of p66Shc in diabetic vasculature. Phosphorylation on serine 36, as well as expression, of p66Shc was increased in db/db diabetic mouse aortas compared to db/+ non-diabetic controls (Figure 6A). Similarly, endothelial p66Shc was phosphorylated on serine 36 in the STZ model of diabetes (Figure 6B top panels; p66-N mice). To assess the role of p66Shc in diabetic oxidative stress and endothelial dysfunction, we used mice expressing a shRNA targeting p66Shc (p66-T) in which p66Shc is globally knocked down16. Phosphorylation of p66Shc on serine 36 was significantly reduced in p66-T mice compared to p66-N non-transgenic littermates (Figure 6B). Consistent with the phosphorylation status of p66Shc, staining for 8-hydroxy-deoxyguanosine (8-OH-dG), a marker of oxidative DNA damage, was significantly lower throughout the aorta, including the endothelium, of diabetic p66-T mice, compared to their p66-N non-transgenic controls (Figure 6C). P66-T mice were partially protected from STZ-induced impairment of endothelium-dependent vasorelaxation (Figure 6D, left panel), but not endothelium-independent vasorelaxation (Figure 6D, right panel). In HUVECs and mouse aortic endothelial cells, knockdown of p66Shc mitigated high glucose-induced hydrogen peroxide (H2O2) (Figure 6E & F), while overexpression of p66Shc stimulated H2O2 production (Figure 6G). Additionally, palmitate-induced H2O2 was significantly higher in aortic endothelial cells isolated from p66-N mice than p66-T mice (Figure 6H). These findings highlight the vital role p66Shc plays in diabetic vascular/endothelial oxidative stress and endothelial dysfunction, and indicate that both hyperglycemia and free fatty acids act via p66shc to induce endothelial oxidative stress.
Figure 6. Endothelial p66Shc is upregulated and phosphorylated by diabetes and promotes vascular oxidative stress and endothelial dysfunction.
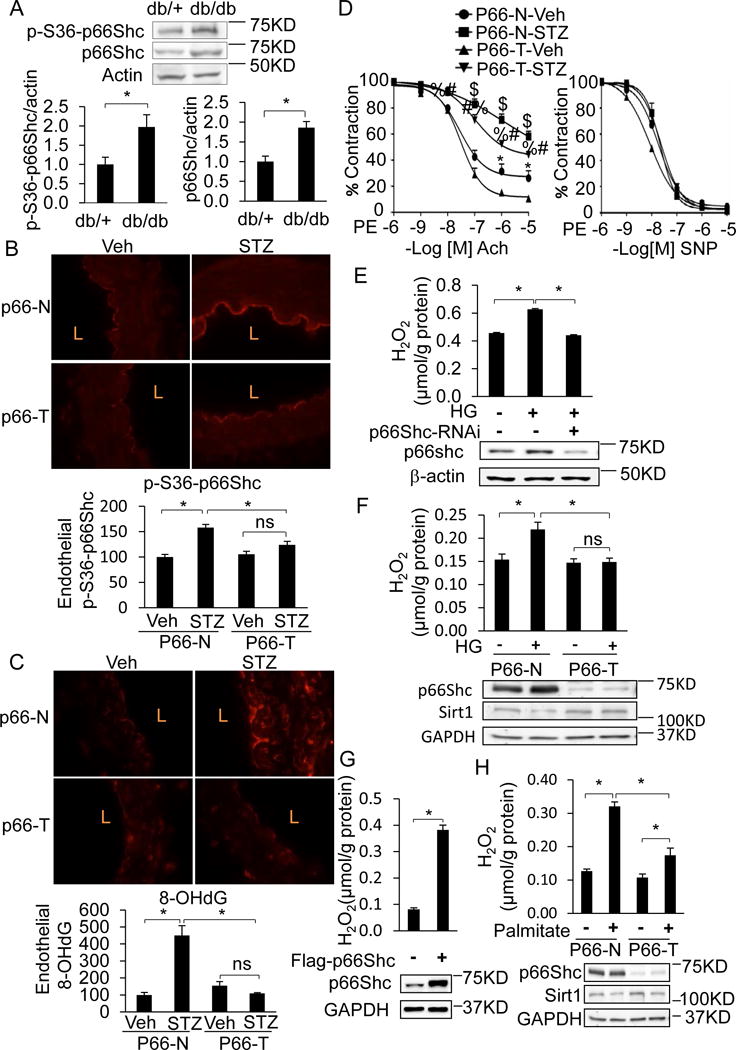
A–D, Endothelial p66Shc is phosphorylated on serine 36 in diabetes and promotes vascular oxidative stress and endothelial dysfunction. (A) Immunoblots for p66Shc and phospho-S36 p66Shc (P-S36-p66Shc) in whole aortas of db/db diabetic and db/+ non-diabetic mice. IHC for (B) P-S36-p66Shc (red) and (C) the oxidative stress marker 8-hydroxy-deoxyguanosine (8-OHdG; red) in aortas of transgenic mice expressing shRNA targeting p66Shc (p66-T) or their wild-type non-transgenic littermates (p66-N), treated with vehicle control (Veh) or rendered diabetic with STZ (STZ). L: lumen. Graphs at bottom show quantification of endothelial P-S36-p66Shc and 8-OHdG expressed relative to non-diabetic (Veh-treated) p66-N mice. (D) P66-T mice are protected from diabetic endothelial dysfunction. Endothelium-dependent (left) and –independent (right) vasorelaxation in aortas of p66-T or p66-N mice treated with vehicle (Veh) or rendered diabetic with STZ. $ p<0.05, p66-N-Veh vs. P66-T-STZ; # p<0.05, p66-T-STZ vs. P66-N-STZ; % p<0.05, p66-T-STZ vs P66-T-Veh; * p<0.05, p66-T-Veh vs P66-N-Veh. n=8–10 from 4–5 mice per group. E, p66Shc silencing in HUVECs abrogates HG-induced oxidative stress (H2O2). H2O2 was quantified by Amplex red fluorescence. F, p66Shc knockdown in aortic endothelial cells abrogates HG-induced oxidative stress. Aortic endothelial cell isolated from p66-T mice or p66-N littermates were treated with HG for 24 h. H2O2 was quantified as above. G, Overexpression of p66Shc in HUVECs induces oxidative stress (H2O2). H, p66Shc knockdown in aortic endothelial cells abrogates palmitate-induced oxidative stress. Aortic endothelial cell isolated from p66-T mice or their p66-N littermates were treated with 500 uM for 12 h. H2O2 was quantified as above. In all panels, representative immunoblots, or IHC images are shown. Data are shown as mean ± SEM. n=3–5 for all data except vasorelaxation, and * P<0.05.
P66Shc regulates miR-34a expression - role of p53
The mechanisms that regulate endothelial miR-34a expression, especially in the context of diabetes, are not well understood. We sought to determine if p66Shc is responsible for upregulation of miR-34a in diabetes. Induction of aortic miR-34a in STZ-induced diabetes was blunted in p66-T mice compared to p66-N controls (Figure 7A, Supplemental Figure VII). In addition, increase of miR-34a in HUVECs by HG was abrogated by siRNA-mediated knockdown of p66Shc (Figure 7B). Similarly, HG and palmitate upregulated miR-34a expression in aortic endothelial cells from p66-N mice but not p66-T mice (Figure 7C & D). P66Shc overexpression alone, in the absence of high glucose, also increased endothelial miR-34a (Figure 7E & F) and promoter activity (Figure 7G). The non-phosphorylatable redox-deficient mutant of p66Shc (S36A) on the other hand, led to significantly less induction of miR-34a expression and promoter activity (Figure 7F & G). Moreover, upregulation of endothelial miR-34a by p66Shc was blunted by two cell-permeable antioxidants: N-acetylcysteine (NAC) and polyethylene glycol-catalase (PEG-C) (Figure 7H & I). Finally, miR-34a was induced by treatment of endothelial cells with the cell-permeable oxidant H2O2 (Figure 7J). Taken together, these findings indicate that endothelial miR-34a expression is governed by p66Shc in an oxidant-dependent manner.
Figure 7. P66Shc stimulates endothelial miR-34a, and suppresses endothelial Sirt1, in diabetes.
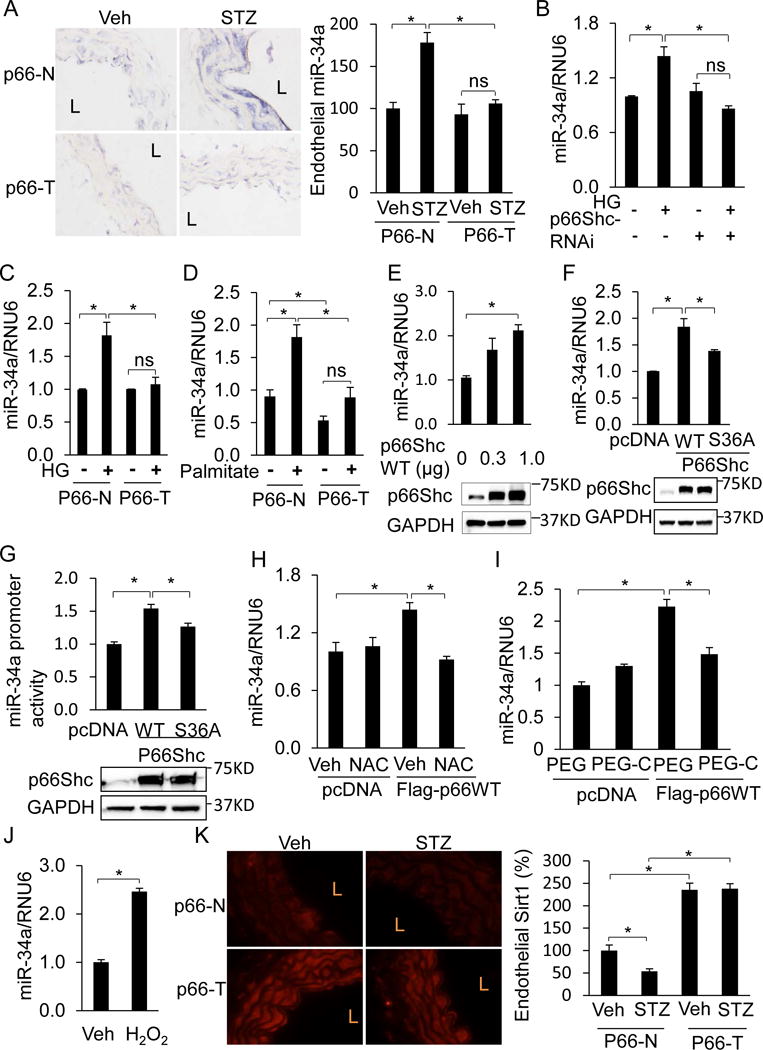
A, MiR-34a is not induced in diabetic p66-T mice. Left, ISH for miR-34a (purple-blue) in aortas of p66-T or p66-N mice treated with Veh or STZ. L: lumen. Right, graphs showing quantification of endothelial miR-34a relative to non-diabetic Veh-treated p66-N aortas. B, C, E, P66Shc promotes miR-34a expression in endothelial cells. (B) P66Shc Silencing in HUVECs prevents HG-induced upregulation of miR-34a. q-RTPCR for miR-34a in HUVECs transfected with p66Shc siRNA and treated with HG. (C) p66Shc knockdown in aortic endothelial cells prevents HG-induced upregulation of miR-34a. Aortic endothelial cell isolated from p66-T and p66-N mice were treated with HG for 24 h. miR-34a levels were quantified using q-RTPCR. (D) p66Shc knockdown in aortic endothelial cells prevents palmitate-induced upregulation of miR-34a. Aortic endothelial cell isolated from p66-T and p66-N mice were treated with 500 uM palmitate for 12 h. miR-34a levels were quantified using q-RTPCR. (E) Overexpression of wild-type p66Shc induces miR-34a. Top, q-RTPCR for miR-34a in HUVECs transfected with plasmid encoding wild-type p66Shc. Bottom, representative immunoblots for p66Shc. F–G, The non-phosphorylatable redox-deficient S36A mutant of p66Shc is impaired in its capacity to induce miR-34a. (F) Top, q-RTPCR and (G) Top, promoter activity for miR-34a in HUVECs overexpressing wild-type (WT) or redox-deficient (S36A) p66Shc. F-G bottom, representative immunoblots for p66Shc. H–I, P66Shc induces endothelial miR-34a through oxidative stress. Q-RTPCR for miR-34a in HUVECs expressing. WT p66Shc treated with the cell-permeable antioxidants (H) N-acetylcysteine (NAC, 500 μM, 24 h) or (I) polyethylene glycol-catalase (PEG-C, 500 U/ml, 24 h). Control HUVECs were treated with Veh or polyethylene glycol (PEG). J, MiR-34a is induced by exogenous oxidants. Q-RTPCR for miR-34a in HUVECs treated with H2O2 (50 μM, 24 h). K, P66Shc suppresses endothelial Sirt1 expression in diabetes. Left, immunofluorescence for Sirt1 (red) in aortas of p66-T or p66-N mice treated with Veh or STZ. L: lumen. Right, Quantification of Sirt1 immunofluorescence in endothelium relative to non-diabetic Veh-treated p66-N mice. All values are shown as mean ± SEM. n=3–5, * P<0.05.
P66Shc plays an important part in p53-induced oxidative stress and DNA damage17. In addition, miR-34a is a p53-inducible microRNA18. Therefore we attempted to clarify the role of p53 in regulation of miR-34a in endothelial cells. Silencing of p53 in HUVEC (Supplemental Figure VIIIA & B) curtailed HG-induced miR-34a (Supplemental Figure VIIIC). Interestingly, the converse was also true – p53 upregulation by HG was suppressed in endothelial cells lacking miR-34a (Supplemental Figure VIIID). These findings reveal bi-directional talk between miR-34a and p53 in each other’s induction by high glucose.
P66Shc downregulates vascular Sirt1
Finally, we asked if vascular Sirt1 downregulation in diabetes is mediated by p66Shc. Under basal conditions, aortic Sirt1 was higher in p66-T mice compared to non-transgenic p66-N controls (Figure 7K). Increase in aortic Sirt1 was evident in the tunica media as well as the endothelium (Figure 7K). In addition, while STZ-induced diabetes downregulated endothelial Sirt1 in aortas of p66-N mice, endothelial Sirt1 expression was essentially unchanged in diabetic p66-T mice (Figure 7K). These data show that p66Shc promotes downregulation of vascular Sirt1 expression in diabetes.
Discussion
Sirt1 was first described as an important determinant of vascular endothelial function by targeting endothelial nitric oxide synthase for deacetylation, and increasing endothelium-derived nitric oxide7. Since then, numerous studies have shown that Sirt1 plays a protective role in the vasculature19–21. Sirt1 exerts its vasoprotective effect not only through direct actions on the vasculature, but also indirectly such as through its stimulation of reverse cholesterol transport mechanisms22.
By using two models of diabetes (db/db and STZ), our present data show the vital part miR-34a plays in impairing endothelial function in diabetes in vivo. Further, these data corroborate the previously appreciated relationship between miR-34a and Sirt1in the context of diabetic endothelial dysfunction: Sirt1 is the principal functionally relevant target of miR-34a in the diabetic endothelium, as rescue of Sirt1 restores endothelium-dependent vasorelaxation. While we did not look at other metrics of endothelial dysfunction such as vascular inflammation and eventual atherosclerosis, given that impairment of endothelium-dependent vasorelaxation is one of the earliest manifestations of endothelial dysfunction that predicts the development of atherosclerotic disease, it would be safe to say that the p66shc-miR-34a-Sirt1 axis is also likely to be relevant to the development of accelerated atherosclerosis in diabetes.
MiR-34a was induced both in the endothelium and media of aortas from both db/db and STZ-treated mice. Moreover, knockdown of p66Shc (which, in the p66-T mice, is global, and therefore would be expected to be present in both the endothelial and smooth muscle layers) suppressed miR-34a expression throughout the thickness of diabetic aortas. This finding suggests that expression of miR-34a in diabetes, at least in the vasculature, is governed by molecular mechanisms that are common to all major cell types comprising the vascular wall. However, despite upregulation of miR-34a in the entire vascular wall, the fact that miR-34a deletion exclusively in the endothelium was sufficient to rescue endothelial function in STZ mice indicates that it is endothelial miR-34a that is responsible for diabetic vascular dysfunction, at least as it pertains to endothelium-dependent vasorelaxation. This conclusion is corroborated by the finding that miR-34a mimic impaired endothelium-dependent, but not endothelium-independent vasorelaxation. Further supporting this conclusion is data that although endothelium-independent relaxation was impaired in db/db mice, downregulation of miR-34a with miR-34a inhibitor in the whole vessel did not improve vasorelaxation. However, one cannot completely exclude the possibility that miR-34a upregulation in the medial smooth muscle layer also contributes to impaired vascular function of diabetes. Such function may take other forms not captured by vasorelaxation studies. For example, prior work has shown that miR-34a causes senescence and upregulation of pro-inflammatory markers in vascular smooth muscle cells23.
While our studies show that overexpression of Sirt1 rescues miR-34a-induced endothelial dysfunction, it would be naïve to conclude that miR-34a is acting exclusively via Sirt1. MicroRNAs target a network of genes, and it is likely that miR-34a’s effect on vascular function is mediated by many up- and down-regulated genes. The full spectrum of gene targets of miR-34a in the endothelium is presently unknown. In addition, by suppressing Sirt1, an epigenetic modifier, miR-34a may also indirectly regulate endothelial gene expression.
Our data indicate that endothelial miR-34a expression in diabetes is induced by p66Shc, that the redox property of p66Shc is essential for this induction, and reducing oxidative stress in endothelial cells suppresses miR-34a expression. Further, they show that endothelial p53 is upregulated by high glucose, and p53 participates in miR-34a induction. These findings are consistent with miR-34a being a tumor suppressor microRNA induced by p533. They are also consistent with the part p66Shc plays in p53-induced oxidative stress and DNA damage17. It is notable that p66shc is also transcriptionally upregulated by p53 in endothelial cells16. Thus, p53 may conspire with p66shc to induce miR-34a expression in a redox dependent fashion. It is also noteworthy that Sirt1 targets p53 for deacetylation, thereby suppressing its transcriptional activity24, and this feedback loop may provide a means in diabetic vasculature to mitigate miR-34a expression. Finally, discussion of p53 and miR-34a would be incomplete without pointing out that in certain tumors miR-34a forms a positive feedback loop with p53 by targeting Mdm4, a major negative regulator of p5325. This positive feedback mechanism, which amplifies p53 expression, may be operative in endothelial cells in the context of hyperglycemia as well, because lack of miR-34a negated induction of p53 by high glucose (Supplemental Figure VIII D).
Supplementary Material
Highlight.
Vascular endothelial microRNA-34a (miR-34a) is upregulated in Type 1 and 2 diabetes
miR-34a mediates diabetic vascular endothelial dysfunction
miR-34a promotes diabetic endothelial dysfunction by downregulating Sirt1
activation of p66shc in diabetes stimulates endothelial miR-34a expression
elevated glucose and free fatty acids in diabetes stimulate endothelial miR-34a via increase of p66shc-mediated reactive oxygen species
tumor suppressor p53 is obligatory for endothelial miR-34a induction by elevated glucose
Acknowledgments
Q. Li was supported by NIH grant T32 HL007344, M. Kassan by NIH grant T32 HL007121, and K. Irani by University of Iowa Endowed Professorship in Cardiovascular Medicine.
Footnotes
Author contributions
Q.L, Y-R. K, and K.I. conceived the study, Q.L., Y-R. K., A.V., S-K.L., M.K., M.G., and J.S.J. performed the experiments, Q.L., and Y-R. K. analyzed the data, and Q.L. and K.I. wrote the paper.
Disclosures
None
References
- 1.Floris I, Descamps B, Vardeu A, Mitic T, Posadino AM, Shantikumar S, Sala-Newby G, Capobianco G, Mangialardi G, Howard L, Dessole S, Urrutia R, Pintus G, Emanueli C. Gestational diabetes mellitus impairs fetal endothelial cell functions through a mechanism involving microRNA-101 and histone methyltransferase enhancer of zester homolog-2. Arterioscler Thromb Vasc Biol. 2015;35:664–674. doi: 10.1161/ATVBAHA.114.304730. [DOI] [PubMed] [Google Scholar]
- 2.Xiang Y, Cheng J, Wang D, et al. Hyperglycemia repression of miR-24 coordinately upregulates endothelial cell expression and secretion of von Willebrand factor. Blood. 2015;125:3377–3387. doi: 10.1182/blood-2015-01-620278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, Arking DE, Beer MA, Maitra A, Mendell JT. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26:745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito T, Yagi S, Yamakuchi M. MicroRNA-34a regulation of endothelial senescence. Biochem Biophys Res Commun. 2010;398:735–740. doi: 10.1016/j.bbrc.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Fan W, Fang R, Wu X, Liu J, Feng M, Dai G, Chen G, Wu G. Shear-sensitive microRNA-34a modulates flow-dependent regulation of endothelial inflammation. J Cell Sci. 2015;128:70–80. doi: 10.1242/jcs.154252. [DOI] [PubMed] [Google Scholar]
- 6.Zhao T, Li J, Chen AF. MicroRNA-34a induces endothelial progenitor cell senescence and impedes its angiogenesis via suppressing silent information regulator 1. Am J Physiol Endocrinol Metab. 2010;299:E110–E116. doi: 10.1152/ajpendo.00192.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orimo M, Minamino T, Miyauchi H, Tateno K, Okada S, Moriya J, Komuro I. Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler Thromb Vasc Biol. 2009;29:889–894. doi: 10.1161/ATVBAHA.109.185694. [DOI] [PubMed] [Google Scholar]
- 9.Jung SB, Kwon SK, Kwon M, Nagar H, Jeon BH, Irani K, Yoon SH, Kim CS. Docosahexaenoic acid improves vascular function via up-regulation of SIRT1 expression in endothelial cells. Biochem Biophys Res Commun. 2013;437:114–119. doi: 10.1016/j.bbrc.2013.06.049. [DOI] [PubMed] [Google Scholar]
- 10.Yamakuchi M. MicroRNA Regulation of SIRT1. Front Physiol. 2012;3:68. doi: 10.3389/fphys.2012.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Francia P, Delli GC, Bachschmid M, Martin-Padura I, Savoia C, Migliaccio E, Pelicci PG, Schiavoni M, Luscher TF, Volpe M, Cosentino F. Deletion of p66shc gene protects against age-related endothelial dysfunction. Circulation. 2004;110:2889–2895. doi: 10.1161/01.CIR.0000147731.24444.4D. [DOI] [PubMed] [Google Scholar]
- 12.Camici GG, Schiavoni M, Francia P, Bachschmid M, Martin-Padura I, Hersberger M, Tanner FC, Pelicci P, Volpe M, Anversa P, Luscher TF, Cosentino F. Genetic deletion of p66(Shc) adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci U S A. 2007;104:5217–5222. doi: 10.1073/pnas.0609656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou S, Chen HZ, Wan YZ, Zhang QJ, Wei YS, Huang S, Liu JJ, Lu YB, Zhang ZQ, Yang RF, Zhang R, Cai H, Liu DP, Liang CC. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ Res. 2011;109:639–648. doi: 10.1161/CIRCRESAHA.111.243592. [DOI] [PubMed] [Google Scholar]
- 14.Yamakuchi M, Ferlito M, Lowenstein CJ. miR-34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A. 2008;105:13421–13426. doi: 10.1073/pnas.0801613105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- 16.Kim CS, Jung SB, Naqvi A, Hoffman TA, DeRicco J, Yamamori T, Cole MP, Jeon BH, Irani K. p53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circ Res. 2008;103:1441–1450. doi: 10.1161/CIRCRESAHA.108.181644. [DOI] [PubMed] [Google Scholar]
- 17.Trinei M, Giorgio M, Cicalese A, Barozzi S, Ventura A, Migliaccio E, Milia E, Padura IM, Raker VA, Maccarana M, Petronilli V, Minucci S, Bernardi P, Lanfrancone L, Pelicci PG. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene. 2002;21:3872–3878. doi: 10.1038/sj.onc.1205513. [DOI] [PubMed] [Google Scholar]
- 18.Rokavec M, Li H, Jiang L, Hermeking H. The p53/miR-34 axis in development and disease. J Mol Cell Biol. 2014;6:214–230. doi: 10.1093/jmcb/mju003. [DOI] [PubMed] [Google Scholar]
- 19.Brandes RP. Activating SIRT1: a new strategy to prevent atherosclerosis? Cardiovasc Res. 2008;80:163–164. doi: 10.1093/cvr/cvn245. [DOI] [PubMed] [Google Scholar]
- 20.Bai B, Liang Y, Xu C, Lee MY, Xu A, Wu D, Vanhoutte PM, Wang Y. Cyclin-dependent kinase 5-mediated hyperphosphorylation of sirtuin-1 contributes to the development of endothelial senescence and atherosclerosis. Circulation. 2012;126:729–740. doi: 10.1161/CIRCULATIONAHA.112.118778. [DOI] [PubMed] [Google Scholar]
- 21.Wen L, Chen Z, Zhang F, Cui X, Sun W, Geary GG, Wang Y, Johnson DA, Zhu Y, Chien S, Shyy JY. Ca2+/calmodulin-dependent protein kinase kinase beta phosphorylation of Sirtuin 1 in endothelium is atheroprotective. Proc Natl Acad Sci U S A. 2013;110:E2420–E2427. doi: 10.1073/pnas.1309354110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, Zhang S, Blander G, Tse JG, Krieger M, Guarente L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol Cell. 2007;28:91–106. doi: 10.1016/j.molcel.2007.07.032. [DOI] [PubMed] [Google Scholar]
- 23.Badi I, Burba I, Ruggeri C, Zeni F, Bertolotti M, Scopece A, Pompilio G, Raucci A. MicroRNA-34a Induces Vascular Smooth Muscle Cells Senescence by SIRT1 Downregulation and Promotes the Expression of Age-Associated Pro-inflammatory Secretory Factors. J Gerontol A Biol Sci Med Sci. 2015;70:1304–1311. doi: 10.1093/gerona/glu180. [DOI] [PubMed] [Google Scholar]
- 24.Vaziri H, Dessain SK, Ng EE, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 25.Okada N, Lin CP, Ribeiro MC, Biton A, Lai G, He X, Bu P, Vogel H, Jablons DM, Keller AC, Wilkinson JE, He B, Speed TP, He L. A positive feedback between p53 and miR-34 miRNAs mediates tumor suppression. Genes Dev. 2014;28:438–450. doi: 10.1101/gad.233585.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.