

Abstract
Human UDP-glucose dehydrogenase (hUGDH) is regulated by an atypical allosteric mechanism in which the feedback inhibitor UDP-xylose (UDP-Xyl) competes with substrate for the active site. Binding of UDP-Xyl triggers the T131-Loop/α6 allosteric switch, which converts the hexameric structure of hUGDH into an inactive, horseshoe-shaped complex (EΩ). This allosteric transition buries residue A136 in the protein core to produce a subunit interface that favors the EΩ structure. Here we use a methionine substitution to prevent the burial of A136 and trap the T131-Loop/α6 in the active conformation. We show that hUGDHA136M does not exhibit substrate cooperativity, which is strong evidence that the methionine substitution prevents the formation of the low UDP-Glc affinity EΩ state. In addition, the inhibitor affinity of hUGDHA136M is reduced 14 fold, which most likely represents the Ki for competitive inhibition in the absence of the allosteric transition to the higher affinity EΩ state. hUGDH also displays a lag in progress curves, which is caused by a slow, substrate-induced isomerization that activates the enzyme. Stopped flow analysis shows that hUGDHA136M does not exhibit hysteresis, which suggests that the T131-Loop/α6 switch is the source of the slow isomerization. This interpretation is supported by the 2.05 Å resolution crystal structure of hUGDHA136M, which shows that the A136M substitution has stabilized the active conformation of the T131-loop/α6 allosteric switch. This work shows that the T131-Loop/α6 allosteric switch couples allostery and hysteresis in hUGDH.
Introduction
Glucuronidation is a major component of the Phase II metabolism of drugs1–3. This pathway has been shown to be upregulated in some lung, breast and colorectal cancer cells, where it functions as an intrinsic drug resistance mechanism4–8. Thus, controlling glucuronidation is a promising strategy for sensitizing this class of tumors to existing chemotherapeutics. It is possible to inhibit glucuronidation by limiting the availability of the essential substrate, UDP-glucuronic acid9–11. UDP-glucose dehydrogenase (UGDH) catalyzes the NAD+ dependent oxidation of UDP-glucose (UDP-Glc) to produce UDP-glucuronic acid12–15. Understanding how UGDH activity is regulated is an important goal in developing new strategies to control glucuronidation-dependent drug resistance.
Human UGDH (hUGDH) forms a hexamer that is regulated by an atypical allosteric mechanism in which the feedback inhibitor UDP-xylose (UDP-Xyl) competes with substrate for the active site16–19. Here, allostery arises from the distinct conformational changes that are induced by the binding of substrate or inhibitor (Figure 1A). The binding of UDP-Glc favors the formation of an active 32 symmetry hexamer called the E state. In contrast, UDP-Xyl binding produces an inactive, horseshoe-shaped hexamer (EΩ). These effector specific transitions are controlled by a buried allosteric switch called the T131-loop/α6 helix (Figure 1A–C). To select for a specific hexamer conformation, the NAD+ binding domain (NB) of hUGDH must adopt an ‘open’ conformation (the E* or E*Bound state) which exposes the allosteric switch (Figure 1D). Once exposed, the T131-Loop and α6 helix are free to change conformation in response to UDP-Glc or UDP-Xyl binding to produce the E or EΩ hexamer, respectively (Figure 1D)16–18. Thus, the active site of hUGDH also functions as an allosteric site in that it controls the structure of the hexamer-building interface and the affinity between subunits16, 17. To our knowledge, the only other enzyme known to have a similar bifunctional active/allosteric site is dCTP-deaminase20, 21
Figure 1. hUDGH undergoes conformational changes during allostery and hysteresis.

(A) Semitransparent surface rendering showing the buried T131-Loop/α6 helix (red) in the E and EΩ states. UDP-Glc (green balls, UDG) binds to the active site and favors the 32 symmetry E state. UDP-Xyl (teal balls, UDX) competes for the active site and triggers the allosteric switch to rotate the α6 helix and favor the EΩ state. (B) The position of the T131-Loop/α6 helix in the E (green), E* (yellow) and EΩ (cyan) state. All of the unique chains in the crystal structures of E, E* and EΩ are superimposed (PDB entries 2Q3E, 4RJT and 3PTZ, respectively). (C) The hexamer-building interfaces of the E, E* and EΩ states are depicted as in panel B. Panels B & C are adapted from ref. 16. (D) The relationship between domain rotations (clamshell circles) and the α6 helix (black filled circle) conformation in the E (active), EΩ (inhibited), E* (inactive) and E*Bound (inactive, inhibitor/substrate bound) states. In the absence of ligand, the inactive E* state is favored. The binding of substrate and NAD+ to the E* induces a slow isomerization (hysteresis) to produce the active E state22. The E*Bound state forms as UDG and UDX compete to induce the transition between the E and EΩ states (the ligand stoichiometry that induces the transition is unknown). UDP-Xyl binding shifts the α6 helix to strengthen the packing interactions between adjacent subunits (semicircle cutouts) and form the EΩ hexamer-building interface16, 17. The EΩ conformation of the allosteric switch reduces the affinity for substrate. For simplicity, only one trimer of the hUGDH hexamer is depicted.
hUGDH also displays hysteresis, which can be observed as a lag in progress curves16–19, 22, 23. In the absence of any effector, hUGDH favors the inactive E* conformation(Figure 1D)16. Hysteresis is caused by coenzyme and substrate binding, which induces the E* state to slowly isomerize to the active E conformation22. Still, the molecular basis of the hysteretic transition was not known. The E* adopts an open domain structure that stabilizes the allosteric switch and the hexamer-building interfaces in an intermediate conformation between the E and EΩ states16, 18, 24(Figure 1B–D). When an effector binds to E*, the T131-Loop/α6-helix and eight surrounding residues repack into the appropriate E or EΩ conformation (Figure 1D)16. We have proposed that the repacking of the allosteric switch and adjacent residues from the E* to E conformation is the source of hysteresis observed in hUGDH22. Here we have used an amino acid substitution (A136M) to stabilize the E state and test our hypothesis.
Methods
Protein expression, crystallization and structure solution
hUGDHA136M and wild type hUDGH were recombinantly expressed in E. coli as previously described16–19, 22. Following purification, the His tags were removed from both proteins using TEV protease. The purified protein was dialyzed into storage buffer [25mM Tris pH8.0 and 50mM NaCl] and concentrated using a Millipore Amicon Ultra-15 10K centrifugal filter unit to ~20 mg/ml. Proteins were quantified using an Agilent 8453 UV/Vis with ε280 = 48360 M−1cm−1 and ε280 = 49850 M−1cm−1 for hUGDH and hUGDHA136M, respectively. The molar absorptivities for these proteins were calculated using the amino acid sequence and Protparam25. hUGDHA136M was crystallized at 20 °C using a hanging drop vapor diffusion method with a 2 μL drop mixed in a 1:1 ratio of protein to reservoir (final protein concentration 10mg/mL). The reservoir was comprised of 0.2M NaCl, 12% PEG 3350 and 0.1M Tris buffer pH 7.6. Crystals were cryoprotected using a solution matching the reservoir supplemented with 20% of a cryprotectant mixture (a 1:1:1 ratio of dimethyl sulfoxide, ethylene glycol and glycerol) and then cooled by plunging in liquid nitrogen. A 2.05 Å resolution data set was collected on the 21-ID beamline (SER-CAT) at the Argonne National Laboratory (Argonne, IL) using a Rayonix MX300HS CCD detector. The data set was processed using XDS26 and 5% of the data set aside for cross-validation27. The data collection statistics are in Table 1.
Table 1.
Data collection and refinement statistics
| Data collection | hUDGHA136M |
|---|---|
| Protein Data Bank entry | 5TJH |
| Space Group | P1 |
| Unit cell dimensions a, b, c, (Å) | 92.35, 104.59, 107.71 |
| α, β, γ (°) | 64.82, 68.35, 73.72 |
| Completeness (%) | 96.6 (97.1)a [91.3]b |
| Redundancy | 3.9 (4.0) [3.8] |
| No. of reflections | 987044 |
| I/σ(I) | 7.82 (1.80) [1.03] |
| CC1/2c | 99.6 (70.3) [45.6] |
| Rmeasd (%) | 11.7 (92.8) [154.1] |
| Refinement | |
| Resolution (Å) | 2.05 |
| Rwork/Rfree | 0.171/0.203 |
| Rfree in highest resolution shell | 0.345 |
| No. atoms Protein/Ligand/Water | 21736/89/799 |
| B-factors (Å2) Protein/Ligand/Water | 53.5/58.6/47.7 |
| Stereochemical Ideality | |
| Bond lengths (Å2) | 0.008 |
| Bond angles (°) | 0.89 |
| φ, ψPreferred region (%) | 98.3 |
| φ, ψAdditionally allowed (%) | 1.7 |
| φ, ψDisallowed region (%) | 0.0 |
Values in parentheses are for the highest-resolution shell (2.16–2.22) based on an I/σ cutoff of 1.8.
Values in brackets are for the highest-resolution shell [2.05 – 2.10] based on the CC1/2 cutoff defined by Diederichs and Karplus42
CC1/2 is the percentage of correlation between intensities from random half-data sets42
Rmeas is the redundancy independent merging R-factor43
The structure of hUGDHA136M was solved by molecular replacement using PDB id 4RJT as a search model in the PHENIX software suite28. The hUGDHA136M model was subjected to iterative cycles of manual rebuilding using COOT29 followed by automated refinement with NCS restraints. B-factors were refined using TLS as implemented in PHENIX28, 30. We used the following rationale in modeling the occupancy of the co-purified UDP-Glc. At resolutions lower than 1.5Å, B-factors and occupancy are strongly correlated, which makes it impossible to refine both of these parameters. Still, it is possible to approximate occupancy because ligands and interacting residues are similarly ordered. Briefly, the occupancy of the ligand is fixed at different values and only the B-factors are refined. The approximate occupancy is the value at which the ligand B-factors converge to those of the interacting residues. Final model refinement statistics are reported in Table 1.
Sedimentation velocity
hUGDH and hUGDHA136M were dialyzed into buffers containing 150 mM KCl, and 25 mM buffer [HEPES (pH 7.5) or TRIS (pH 8.5)], quantified (as above), and then diluted to a final protein concentration of 9 μM. Samples were loaded into 12 mm double-sector Epon centerpieces equipped with quartz windows and equilibrated for 1 hour at 20 °C in an An60 Ti rotor. Sedimentation velocity data were collected in an Optima XLA analytical ultracentrifuge using a rotor speed of 50,000 rpm at 20 °C. Sedimentation data were recorded at 280 nm in radial step sizes of 0.003 cm. SEDNTERP31 was used to estimate the partial specific volume of hUGDH (0.73840 mL/g) and hUGDHA136M (0.73842 mL/g), and the densities of both the pH 7.5 (1.00726 g/ml) and 8.5 (1.00603 g/ml) buffers. Viscosities for both buffers pH 7.5 and 8.5 were calculated to be 0.01018 and 0.01007 P, respectively. SEDFIT32 was used to analyze raw sedimentation data. Data were modeled as a continuous sedimentation coefficient c(s) distribution and were fit using the baseline, meniscus, frictional coefficient, and systematic time-invariant, and radial-invariant noise. Theoretical sedimentation coefficient (s) values were calculated from hUGDH atomic coordinates under standard conditions using HYDROPRO20 (www.bbri.org/RASMB).
Stopped-flow analysis of hysteresis
NAD+ and UDP-glucose were purchased from Sigma and UDP-xylose was purchased from Carbosource (University of Georgia, Complex Carbohydrate Research Center). Enzyme hysteresis was monitored at 25° C using an Olis RSM 1000 Rapid-scanning Absorbance and Fluorescence Spectrophotometer with a stopped-flow assembly and a 0.4 mm pathlength. The 480nM enzyme solution in 50 mM HEPEs pH 7.5, 50 mM NaCl and 5mM EDTA was rapidly mixed with an equal volume of 1mM UDP-Glc and 10mM NAD+ in the same buffer (final concentrations 240nM enzyme 0.5mM UDP-Glc, 5mM NAD+, 50 mM HEPEs (pH 7.5), 50 mM NaCl and 5mM EDTA). Absorbance readings at 340 nm were taken every 0.1 second for a total of 120 seconds to monitor NADH production. hUGDH progress curves were fit to Frieden’s equation33 describing enzyme hysteresis:
| (1) |
where P is the concentration of product at the time t and τ is equal to 1/kobs, where kobs is the apparent rate constant for the transition between the initial velocity (vi) and the steady state velocity (vss). Here, the initial velocity vi, in eq. 1 is the pre-steady state velocity, and does not obey the steady state approximation. The length of the lag in seconds is calculated as the product of Euler’s number and τ (eτ). Data was fit using PRISM (Graphpad Software Inc., San Diego, CA), and all fits were analyzed using residual plot analysis34.
Steady state kinetics
The assay conditions for the steady state analysis of hUGDH have been described previously16–19, 22. Briefly, assays contained 100nM hUDGH or 230nM hUGDHA136M in a standard reaction buffer of 50 mM HEPEs pH 7.5, 50 mM NaCl and 5mM EDTA with a saturating concentration of either 2.5 mM NAD+ or 1mM UDP-Glucose. Prior to the reaction, substrate and enzyme were separately pre-incubated at 25°C for 5 minutes, and the reaction was initiated by rapidly mixing the enzyme and substrate solutions. Progress curves were measured by monitoring NADH production at 340 nm (molar absorptivity coefficient of 6220 M−1 cm−1) with 0.5 s data points using an Agilent 8453 UV/Vis spectrometer at 25° C. All data were fit using nonlinear regression from PRISM. hUDGH steady state velocities were calculated by fitting progress curves to eq. 1, as we have previously described16–19, 22. For a hysteretic enzyme, the initial velocity does not satisfy the steady state approximation. Thus, the vss prior to the depletion of 10% of the substrate represents the initial steady state velocity. hUGDHA136M did not display hysteresis, thus the initial velocities satisfy the steady state approximation, and were determined using the linear portion of the progress curves. Initial steady state velocities were fit to a sigmoidal rate equation and analyzed with residual analysis34:
| (2) |
Negative cooperativity in the NAD+ saturation curve of hUGDH was evaluated using Kurganov’s analysis of a concave-up Eadie-Hofstee plot35. Briefly, data were fit to the equation:
| (3) |
where K0 is the estimated KM for the high affinity binding sites, and Klim is the average of the lower affinity binding sites.
We determined the Ki for the inhibitor UDP-Xyl as previously described22. Briefly, UDP-Xyl competes with UDP-Glc for the active site and induces hUGDH to form the EΩ state17, 22. Because the E and EΩ states have different affinities for UDP-Glc and UDP-Xyl, the inhibition studies display cooperativity. To determine the Ki, the substrate saturation curves from different concentrations of UDP-Xyl were fit simultaneously to the equation for competitive inhibition corrected for substrate cooperativity using global analysis in PRISM:
| (4) |
Results
Design and rational for A136M substitution
The A136M substitution was designed to prevent the isomerization of the T131-Loop/α6 allosteric switch. In the active conformation of hUGDH, the Cβ atom of A136 is solvent exposed (Figure 2A). To form the inhibited EΩ state, the α6-helix tilts ~10.6° then rotates about the helix axis ~12° burying A136 near a cavity in the core of the protein (Figure 2B)17. We hypothesized that an amino acid substitution could prevent the burial of A136 and effectively lock the α6-helix in the active conformation. We excluded polar and charged residues from consideration because A136 is located in the hexamer-building interface, and the burial of an unsatisfied electrostatic group might destabilize the hexamer. We also excluded β-branched amino acids based on a modeling experiment with valine at position 136; each preferred rotamer of valine introduced at least one steric clash between a γ -methyl and the surrounding residues of the A136 pocket (Figure 2C). Similarly, leucine, phenylalanine, and tryptophan are also expected to introduce steric clashes. The structure of the A136 surface pocket only permits an unbranched amino acid substitution with a χ1 torsion angle of 180° (Figure 2C). Based on these observations, we chose the A136M substitution; methionine not only satisfies the criteria above, but it is also flexible enough to conform to the local packing constraints in the A136 pocket.
Figure 2. Rational for design of A136M substitution.

(A) The left panel is a semitransparent surface rendering of the E state (PDB entry 2Q3E) showing the α6 helix (green) with the Cβ atom of A136 (yellow van der Waals dots) in a solvent accessible pocket. The right panel depicts a ‘cutaway’ of the protein surface looking down the axis of the α6 helix to show conformational changes that take place in the protein core; specifically, the position of the α6 helix and the solvent exposed A136 in the E state. Cavities buried in the protein core (red surfaces) were identified with a 1.2Å radius probe. The contribution of the A136 Cβ atom to the surface was removed prior to rendering. (B) The EΩ conformation (cyan) of hUGDH rotates the α6 helix to bury A136 in the protein core (PDB entry 3PTZ). The right panel depicts the rotation of the α6 helix to bury A136 in the EΩ state. The left and right panels are illustrated as in (A). Note that the cavities in the core change shape and number due to the repacking of allosteric switch. (C) To investigate the steric constraints (dotted van der Waals surfaces) of a β-branched amino acid substitution in the A136 pocket, we modeled A136V with its three preferred rotamers superimposed (purple sticks). Each rotamer introduces a bad contact (red dashed lines) between a γ-methyl and adjacent amino acids. Thus, the pocket will only accommodate an unbranched amino acid with a 180° χ1 torsion angle (60°,180°, and −60° χ1 torsion angles are labeled).
Structural analysis of hUGDHA136M
The crystal structure of hUDGHA136M was solved in the space group P1 and refined to a resolution of 2.05 Å (Table 1). The unit cell contains a single 32 symmetry hexamer (Figure 3A). The residues in loop 385–387 and the C-terminal residues 467–494 are disordered in each monomer and are not included in the final model. All 466 Cα atoms in each of the six monomers superimpose with RMSDs ranging from 0.344 to 0.834 Å. The largest structural differences between the chains involve variability in the amount of hinge-bending motion of the NB (residues 1–212) and SB domains (residues 323–466) about an axis located between residues 219–220 (Figure 3B). A DynDom36 comparison of hUDGHA136M to the closed conformation of hUGDH (represented by PDB entry 2Q3E) shows that the NB and SB domains of chains A, B, C, D, E and F are rotated open by 11.2°, 9.4°, 9.1°, 10°, 12.4° and 5.9°, respectively. In the SB domain of chain C we observed electron density consistent with a weakly ordered nucleotide sugar that has co-purified with the enzyme (Figure 3C). We have modeled the density as a UDP-Glc based on the observation that it binds in the same position and orientation as the substrate in the abortive ternary complex of UGDH (PDB entry 2Q3E). The occupancy of the UDP-Glc was adjusted to 50% to match the B-factors of the interacting residues in the active site (see Methods). In the other five chains of hUDGHA136M the corresponding density is weaker and only supports our modeling of the diphosphate in UDP-Glc (not shown). The quality of the nucleotide sugar electron density appears to correlate with the average B-factors of the chains; chain C has the lowest average B-factor (44 Å2) and the best ordered UDP-Glc, while the average B-factors for the remaining chains range from 51 Å2-60 Å2 with much weaker density for the nucleotide sugar. This is not the first observation of a nucleotide sugar co-purifying with UGDH. Dickinson reported that bovine UGDH purified from liver contained a nucleotide sugar that he tentatively identified as UDP-Glc or UDP-Xyl23. We chose to model UDP-Glc based on the fact that E. coli does not produce UDP-Xyl.
Figure 3. Crystal structure of hUGDHA136M in the open domain conformation.

(A) Side and top views of the UDGHA136M 32 symmetry hexamer. The surfaces of the six monomers are colored by domain: NB (blue), dimerization (light green), and SB (red). The allosteric switch is colored orange. The active sites and hexamer-building interfaces are identified with circles and arrows, respectively. (B) Stereoview of a Cα trace of hUDGHA136M chain C (colored as above) superimposed onto hUGDH in the closed conformation (grey, PDB entry 2Q3E) to illustrate the open domain conformation. The hinge-bending axis is depicted as a purple rod. (C) The difference (Fo − Fc) electron density map contoured at 3σ for the partially occupied UDP-Glc (sticks) calculated after the nucleotide sugar was omitted and the resulting model subjected to simulated annealing.
The A136M substitution is well ordered and fills the A136 surface pocket in the hexamer-building interface without making any van der Waals contacts with the adjacent monomer in the hexamer (Figure 4 A, B). As expected, M136 adopts the trans rotamer with a χ1= 179.3, χ2= 73.4° and χ3= −111.0°. The A136M substitution does not interfere with the lattice contacts in either of the two UDP-Xyl bound EΩ crystal forms we have previously described17. Still, our attempts to obtain X-ray diffraction quality crystals of hUDGHA136M using both previously published conditions failed. The fact hUDGHA136M will not crystallize in the EΩ state suggests that the substitution prevents the allosteric transition.
Figure 4. The A136M substitution stabilizes the E state of hUGDH.
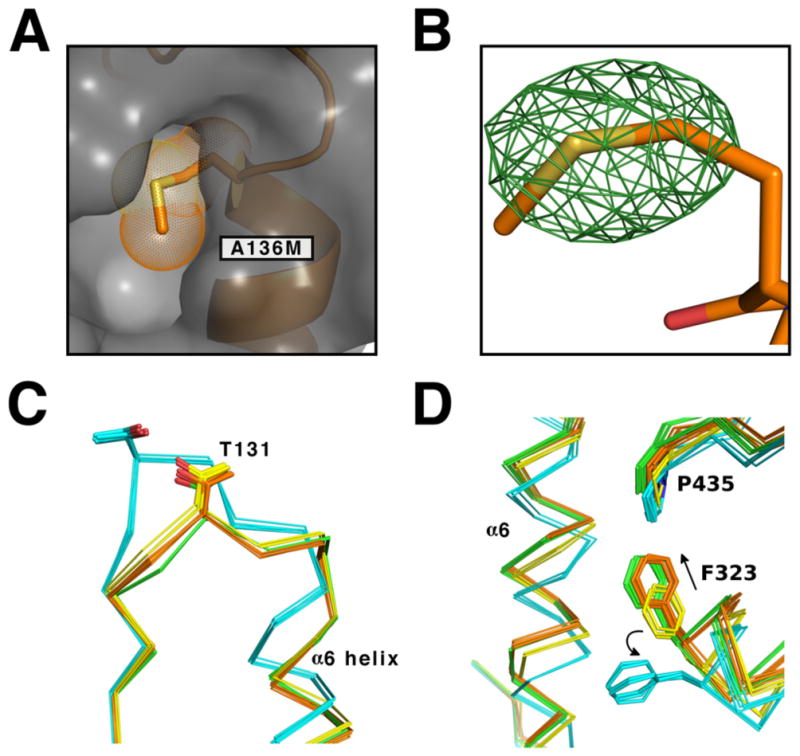
(A) The difference (Fo − Fc) electron density map contoured at 4σ for A136 calculated prior to modeling the methionine substitution. (B) A surface rendering (grey) of hUDGHA136M showing the A136M substitution (orange stick with van der Waals surface dots) and the α6 helix (orange). As expected, the A136M substitution fills the A136 pocket. (C) The position of the T131 loop in the inactive (cyan), active (green), E* (yellow) and hUDGHA136M (orange). (D) The hexamer building interfaces of the inactive (cyan), active (green), E*(yellow) hUDGHA136M (orange). Key residues are labeled with arrows used to depicted direction of motion between states.
As in previous studies16, we analyzed the conformation of the allosteric switch by superimposing the Cα atoms of residues 1–124 in hUDGHA136M onto to the active E, the E* and the UDP-Xyl inhibited EΩ structures of hUGDH (PDB entries 2Q3E, 4QEJ and 3PTZ, respectively (Figure 4C). The T131-Loops in all six monomers of hUDGHA136M are in a conformation between the active E and E* state, with the T131 Cα atom of hUGDHA136M being displaced 0.5–0.7 Å and 0.4–0.6 Å from the corresponding atoms in the E and E* structures, respectively. In contrast, the α6 helix and the hexamer building interface of hUGDHA136M are most similar to the E conformation of hUGDH (PDB 2Q3E) (Figure 4D). Specifically, F323 in the hexamer-building interface adopts the same rotamer observed in the active state, with the Cα atoms of F323 in all chains of hUDGHA136M positioned 0.4 – 0.8 Å from the E state compared to 1.5–1.9 Å from the E* state.
The A136M substitution stabilizes the hexamer
We have previously shown that the ligand-free hUGDH hexamer is relatively unstable in solution, and dissociates into a concentration-dependent distribution of dimers, tetramers and hexamers in rapid equilibrium19. Because of its location in the hexamer-building interface, it is possible that the A136M substitution could alter the stability of the hexamer. We used sedimentation velocity studies to analyze the oligomeric structure of hUGDHA136M in solution. The c(s) distribution of 9 μM hUGDHA136M at pH 7.5 is dominated (94.1%) by an 11.5 S species corresponding to the hexamer, and a smaller (5.9%) amount of the 5.2 S dimer (Figure 5A). For comparison, the c(s) distribution of 9 μM hUGDH reveals an 11.4 S hexamer (78.2 %), an 8.3 S tetramer (4.2%) and a 5.5 S dimer (13.1%) (Figure 5B). The distribution also contains a 3.2 S species that is most likely a small amount (4.5%) of misfolded monomer. The slight differences in S values for corresponding species in the hUDGH and hUGDHA136M distributions are not significant as S values obtained from a system in rapid equilibrium are inherently biased by the mean of the c(s) distribution32, 37.
Figure 5. The A136M substitution stabilizes the hexamer.
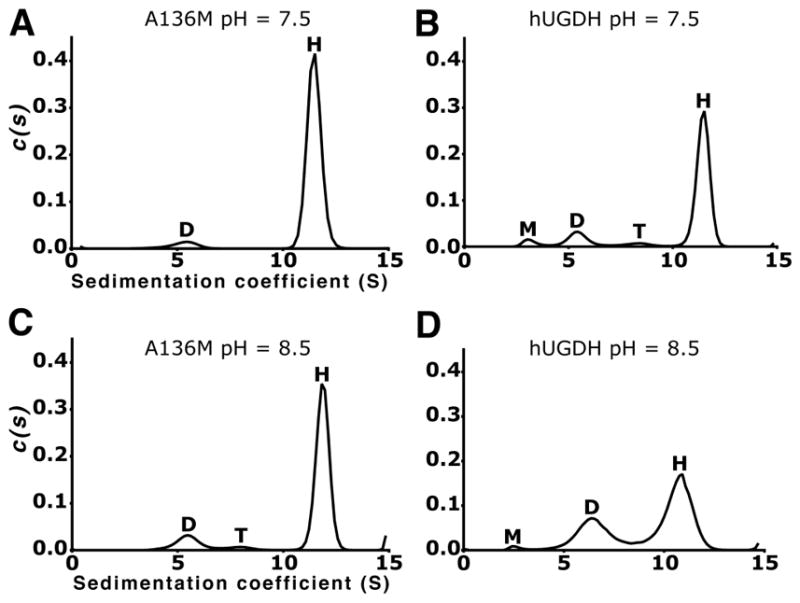
(A) Sedimentation velocity c(s) distribution of hUDGHA136M at pH 7.5 showing the hexamer (H), and dimer (D). (B) The hUDGH c(s) distribution at pH 7.5 shows hexamer (H), tetramer (T), dimer (D) and a peak that we interpret as misfolded monomer (M). (C) hUDGHA136M at pH 8.5. (D) hUDGH at pH 8.5.
Next, we examined the oligomeric state of hUGDHA136M at the more alkaline pH 8.5, which we have previously shown to weaken the hUGDH hexamer16. Again, the main sedimenting species in the hUGDHA136M c(s) distribution is an 11.85 S hexamer (83.7%), followed by a 5.49 S dimer (13.4%) and a 7.9 S tetramer (3.0%) (Figure 5C). In contrast, the c(s) distribution of hUGDH reveals significantly less 10.6 S hexamer (64.4%), and a complementary increase in the 6.5 S dimer concentration (34.0%) (Figure 5D). The broadening of the hexamer and dimer peaks in hUGDH is a characteristic of the rapid equilibrium between species16, 19, 38. This likely explains why we do not observe a quantifiable peak for the tetramer, which represents a transient in the formation and dissociation of the hexamer16. These results show that, A136M substitution favors the hexameric state of hUGDH.
Hysteresis is not observed in hUGDHA136M
Hysteresis in hUGDH and hUDGHA136M was analyzed using stopped-flow absorbance spectroscopy. hUGDH was injected into a rapid mixing cell with saturating substrate and NAD+, and the characteristic lag in enzyme activity was measured by fitting the data to eq. 1 as described in the Materials and Methods (Table 2). The relaxation of hUGDH has a τ of 16.30 ± 0.24 seconds, which corresponds to a lag of 44.3 ± 0.66 seconds before reaching the steady state velocity of 1.0± 0.01 nM NADH s−1 (Figure 6A). In contrast, we observed no lag for hUDGHA136M under these conditions (Figure 6B). In fact, hUGDHA136M does not display hysteresis even at saturating concentrations of the feedback inhibitor UDP-Xyl (Table 2). This is not true of hUGDH, where the lag has been shown16 to increase with UDP-Xyl concentrations (Table 2). Finally, the steady state velocity for hUDGHA136M appears to be 30% faster than hUGDH, but this is most likely due to negative cooperativity in the latter enzyme (discussed below) (Table 2).
Table 2.
Effect of UDP-Xyl on Hysteresis
| Enzyme | UDP-Xyl (μM) | τ (s−1) | Lag (s) | νi | νss |
|---|---|---|---|---|---|
| hUGDH | 0 | 16.3 ± 0.24 | 44.3 ± 0.66 | 0.43 ± 0.02a | 1.0 ± 0.01a |
| 3b | 38.3 ± 2.7 | 104 ± 7.3 | 0.35 ± 0.03 | 1.1 ± 0.01 | |
| hUGDHA136M | 0 | N.D.c | N.D. | N.D. | 1.3 ± 0.03 |
| 60b | N.D. | N.D. | N.D. | 1.4 ± 0.03 |
units = nM NADH per nM enzyme per second.
These concentrations of UDP-Xyl are ≥10× the Ki listed in Table 4
N.D.= not detected
Figure 6. The A136M substitution disrupts hysteresis and cooperativity.

(A) Stopped-flow analysis of hysteresis in the hUDGH progress curve under saturating concentrations of NAD+ (5mM) and UDP- Glc (0.5mM). The data points (black line) are fit (yellow line) to eq. 1 as described in the Materials and Methods. (B) The hUDGHA136M progress curve was analyzed as in panel A shows no lag, and is fit to linear equation (yellow line) to obtain the initial steady state velocity. The yellow line represents a linear fit for the first 20 seconds of the reaction. (C and D) The NAD+ saturation curves for hUGDH and hUGDHA136M, respectively, fit to eq. 3. The rates are normalized to turnover (nM NADH per nM enzyme per second). Dashed lines represent the 95% confidence interval of the fit in all panels. (E) An Eadie-Hofstee plot of the hUGDH NAD+ saturation data in panel C is concave up (negative cooperativity), and was fit to eq. 3 to determine the K0 and Klim values in Table 3. Velocity (v) was normalized to turnover (s−1) as in panels C & D. (F) hUGDH UDP-glucose saturation curves (black lines) with 0 μM (squares) and 3 μM (diamonds) UDP-xylose were globally fit to eq. 4 to determine Ki and the Hill coefficient. For comparison, data from a hUGDHA136M UDP-glucose saturation curve with 60 μM UDP-Xyl (red triangles) is plotted. (G) hUGDHA136M UDP-glucose saturation curves with 0 μM (black circles), 11 μM (blue squares) and 30 μM (red triangles) UDP-xylose were globally fit to eq. 6 to determine Ki.
The A136M substitution disrupts NAD+-induced negative cooperativity
The NAD+ saturation curve of hUGDH displays negative cooperativity (Hill = 0.73± 0.07), which we have previously shown22 to be an intrinsic property of the substrate bound complex (in the absence of UDP-Glc, the binding of NAD+ is noncooperative) (Figure 6C and Table 3). In contrast, the NAD+ saturation curve of hUGDHA136M is hyperbolic (Figure 6D and Table 3). Negative cooperativity indicates an asymmetry in an enzyme that results in a mixture of high affinity and low affinity binding sites. It is possible to estimate the KM for the high affinity and low affinity NAD+ binding (K0 and Klim respectively) using Kurgonov’s methodology35 (see Materials and Methods) (Figure 6E). This analysis of hUGDH NAD+ saturation curves yields a K0 of 88 ± 20 μM which is similar the KM (90 ± 6 μM) observed for hUGDHA136M (Figure 6D and Table 3).
Table 3.
Steady State Parameters
| Enzyme | Ligand | KM (μM) | Hill | kcat (s−1)a | K0b | Klimb | kcatb |
|---|---|---|---|---|---|---|---|
| hUGDH | UDP-Glc | 9.7±0.8 | 1 | 0.55±0.01 | |||
| NAD+ | 780±180 | 0.73 ± 0.07 | 0.76±0.05 | 88 ± 20 | 1800 ± 400 | 0.79± 0.06 | |
| UDP-Glc | 7.26±0.67 | 1 | 0.73±0.02 | ||||
| hUGDHA136M | NAD+ | 90±6 | 1 | 0.83±0.02 |
One complete catalytic turnover produces 2 NADH,
results from Kurganov analysis of negatively cooperative enzyme
With respect to turnover, the kcat for the NAD+ saturation curves of both hUGDH and hUGDHA136M are comparable at 0.76± 0.05 s−1 and 0.83± 0.02 s−1, respectively (Table 3). The UDP-Glc saturation curves for hUGDH and hUGDHA136M are both hyperbolic, with similar KM’s of 9.7 ± 0.8 μM and 7.26 ± 0.67 μM, respectively (Table 3) However, there is a significant discrepancy in the kcat’s observed in the UDP-Glc saturation curves; while the hUGDHA136M kcat is similar to that measured in the NAD+ saturation curves, the kcat for hUGDH is significantly lower (0.76± 0.02s−1 and 0.55± 0.01s−1, respectively) (Table 3). The difference in turnover numbers is due to the coenzyme-dependent negative cooperativity of hUGDH, which makes it difficult to saturate the enzyme with NAD+ (Klim = 1800 μM) in order to achieve the pseudo-first order conditions necessary for steady state analysis of UDP-Glc kinetics.
hUDGHA136M binding of UDP-xylose is non-cooperative
Next we examined the impact of the A136M substitution on the allosteric inhibition mechanism of hUGDH. UDP-Xyl binding stabilizes the EΩ conformation of hUGDH (Figure 1)16–18. Competition with UDP-Glc induces the low substrate affinity EΩ conformation of hUGDH to undergo a cooperative conformational change to the high affinity E state16, 39. The allosteric transition can be observed as sigmoidicity in the UDP-Glc saturation curves during steady state analysis of UDP-Xyl inhibition (Figure 6F). Global analysis of the hUGDH steady state inhibition curves yields a Ki of 0.32 ± 0.05 μM for UDP-Xyl, which is consistent with our earlier work16. The UDP-Glc saturation curve in the presence of UDP-Xyl at a concentration that is ~10x Ki is strongly cooperative, with a Hill coefficient of 2.1 ± 0.2 (Figure 6F and Table 4). This agrees with our previous work, which showed that sigmoidicity increases with UDP-Xyl concentration16. In contrast, hUGDHA136M has a much lower affinity for UDP-Xyl (Ki of 4.2 ± 0.4 μM), and displays no cooperativity (Figure 6G and Table 4). In fact, the UDP-Glc saturation curves are hyperbolic even at inhibitor concentrations 14x Ki (60 μM UDP-Xyl), which is strong evidence that hUGDHA136M does not undergo a conformational change between inhibited and uninhibited states (Figure 6F).
Table 4.
Global analysis of competitive inhibition
| Enzyme | UDP-Xyl (μM) | UDP-Glc KM (μM) | kcat (s−1)a | Ki UDP-Xyl (μM) | Hill |
|---|---|---|---|---|---|
| hUGDH | 0 | 9.50±1.01 b | 0.55±0.02 b | 0.30±0.03 | 1 |
| 3 | 2.1±0.2 | ||||
| hUGDHA136M | 0 | 7.34±0.80b | 0.74±0.02 b | 4.2±0.4 | 1 |
| 11 | 1 | ||||
| 30 | 1 | ||||
| hUGDHA136Mc | 60 | 6.3±0.5 | 0.85±0.02 | 1 |
Turnover produces 2 NADH per cycle.
For global analysis, this parameter was refined as a shared value for all UDP-Xyl concentrations
The 60 μM UDP-Xyl experiment was conducted separately and excluded from global fit.
Discussion
We have previously shown that binding of the feedback inhibitor UDP-Xyl to hUGDH induces the T131-Loop/α6 allosteric switch to repack and form the inactive EΩ state (Figure 1)17. This remarkable allosteric transition requires the α6 helix to rotate, which buries A136 in the protein core (Figure 2 A, B). Here we have tested the allosteric mechanism by trapping the α6 helix in the active conformation using the A136M substitution (Figure 4). The burial of the methionine would be unlikely, as it would require a significant local unfolding of the protein and repacking of the core to accommodate the bulky side chain (Figure 2). This rationale is supported by the substrate saturation kinetics of the UDP-Xyl inhibited enzyme (Table 4). The EΩ state has a lower affinity for UDP-Glc, which results in positive cooperativity in substrate saturation curves of hUGDH (Figure 6F)16. The fact that UDP-Xyl saturated hUGDHA136M does not display substrate cooperativity is strong evidence that the A136M substitution prevents the formation of the EΩ state (Figure 6F, Table 4). This interpretation implies that the lower affinity for UDP-Xyl (Ki =4.2 μM) observed in hUGDHA136M corresponds to the competitive inhibition constant for the E state only (Figure 6G, Table 4). This is the first report of the E state affinity constant for UDP-Xyl, and suggests the higher affinity (Ki =0.32 μM) observed for hUGDH is a property of the EΩ state (Table 4).
The specific activity of hUGDH shows a hyperbolic dependency on protein concentration that can be modeled as three low activity dimers associating to form a higher activity hexamer16. It is possible for enzyme association to result in hysteresis40, 41 , but this is not the case for hUGDH. Instead, hysteresis in hUGDH is caused by the slow isomerization from the inactive E* state to the active E state upon binding of NAD+ and substrate 19, 22. While the increased stability of the hUGDHA136M hexamer does not explain the absence of a lag in progress curves, it does focus attention on the conformational flexibility of the allosteric switch in the E* state (Figure 5, 6A).The intermediate conformation T131-Loop/α6 allosteric switch and the hexamer-building interface in E* led us to hypothesize that the hysteresis was the result of NAD+ and substrate inducing the allosteric switch and surrounding residues to slowly repack into the E conformation16, 18 (Figure 1B,C). The A136M substitution allows us to test this model. The crystal structure of unliganded hUGDHA136M shows that both the allosteric switch and the hexamer-building interface favor the E state, despite the open-domain conformation of the enzyme (Figure 4C, D). By stabilizing the allosteric switch in the E state, the A136M substitution would prevent the core and interface repacking. This model accurately predicts the absence of a lag in the hUGDHA136M progress curves (Figure 6B). Additional evidence comes from UDP-Xyl inhibition studies. We have previously shown16 that the lag in hUGDH increases with UDP-Xyl concentration (Table 2). This increase is most likely caused by the inhibitor stabilizing the EΩ conformation, which must first convert to the E*Bound before it can slowly isomerize into the active E state (Figure 1D). Thus, the absence of hysteresis in UDP-Xyl saturated hUGDHA136M is strong evidence that the A136M substitution prevents the formation of both the E* and EΩ states (Table 2). Together, these results identify the isomerization of the allosteric switch as the source of the hysteresis observed in hUGDH.
Negative cooperativity can arise from enzyme heterogeneity that results in a mixed population with different affinity constants, but this model is not supported by ligand binding studies in hUGDH22. Briefly, the binding of UDP-Glc or NAD+ to hUGDH is noncooperative, indicating that the enzyme is homogeneous, with single affinity constants for substrate and coenzyme 22. However, the binding of NAD+ to the binary hUGDH:UDP-Glc complex displays negativity cooperativity22. The simplest interpretation of these results is that the binding of UDP-Glc induces an asymmetry in the enzyme that produces two distinct affinities for the coenzyme. The molecular basis of negative cooperativity in hUGDH is not known, but the fact that the T131-Loop contributes to both substrate and cofactor binding17 suggests that it may play a role. This assumption is supported by the observation that hUGDHA136M constrains the T131-Loop and does not display negative cooperativity (Figure 6C–D, Table 3). A comparison of hUGDH and hUGDHA136M suggests that the A136M substitution selects for the high affinity conformation of the NAD+ binding sites. Using Kurgonov’s approach35, we estimated the high and low affinity KM’s for the negative cooperativity in coenzyme binding to hUGDH (Figure 6E, Table 3). The apparent high affinity KM (K0) for hUGDH is very similar to the noncooperative KM we observe for hUGDHA136M (Table 3). This suggests that the A136M substitution prevents the asymmetry induced by UDP-Glc binding. This suggests that the flexibility of the allosteric switch is linked to the negative cooperativity observed in NAD+ substrate saturation curves.
The work presented here shows that targeting the allosteric switch is a practical method of selecting specific conformations of hUGDH, and by extension, regulating enzyme activity. Regulating hUGDH is an important milestone in our long term goal of controlling the pharmacokinetics of chemotherapeutics. Future work will focus on inhibiting hUGDH by trapping the allosteric switch in the EΩ conformation.
Acknowledgments
We thank Dr. Renuka Kadirvelraj for helpful discussions and assistance with designing the A136M substitution. We also thank the SER-CAT beamline personnel (Advanced Photon Source, Argonne National Lab, Argonne, Illinois) for assistance with X-ray data collection.
Funding
Funding from NIH (grant R01GM114298) to Z.A.W. is gratefully acknowledged.
Footnotes
Accession Codes
The atomic coordinates and structure factors have been deposited in the Protein Data Bank. (PDB entry 5TJH)
References
- 1.Guillemette C. Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics Journal. 2003;3:136–158. doi: 10.1038/sj.tpj.6500171. [DOI] [PubMed] [Google Scholar]
- 2.Tukey RH, Strassburg CP. Human UDP-glucuronosyltransferases: Metabolism, expression, and disease. Annual Review of Pharmacology and Toxicology. 2000;40:581–616. doi: 10.1146/annurev.pharmtox.40.1.581. [DOI] [PubMed] [Google Scholar]
- 3.Lin JH, Lu AYH. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacological Reviews. 1997;49:403–449. [PubMed] [Google Scholar]
- 4.Cummings J, Boyd G, Ethell BT, Macpherson JS, Burchell B, Smyth JF, Jodrell DI. Enhanced clearance of topoisomerase I inhibitors from human colon cancer cells by glucuronidation. Biochemical Pharmacology. 2002;63:607–613. doi: 10.1016/s0006-2952(01)00812-7. [DOI] [PubMed] [Google Scholar]
- 5.Cummings J, Ethell BT, Jardine L, Boyd G, Macpherson JS, Burchell B, Smyth JF, Jodrell DI. Glucuronidation as a mechanism of intrinsic drug resistance in human colon cancer: Reversal of resistance by food additives. Cancer Research. 2003;63:8443–8450. [PubMed] [Google Scholar]
- 6.Cummings J, Zelcer N, Allen JD, Yao DG, Boyd G, Maliepaard M, Friedberg TH, Smyth JF, Jodrell DI. Glucuronidation as a mechanism of intrinsic drug resistance in colon cancer cells: contribution of drug transport proteins. Biochemical Pharmacology. 2004;67:31–39. doi: 10.1016/j.bcp.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 7.de Almagro MC, Selga E, Thibaut R, Porte C, Noe V, Ciudad CJ. UDP-glucuronosyltransferase 1A6 overexpression in breast cancer cells resistant to methotrexate. Biochemical Pharmacology. 2011;81:60–70. doi: 10.1016/j.bcp.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 8.Oguri T, Takahashi T, Miyazaki M, Isobe T, Kohn N, Mackenzie PI, Fujiwara Y. UGT1A10 is responsible for SN-38 glucuronidation and its expression in human lung cancers. Anticancer Research. 2004;24:2893–2896. [PubMed] [Google Scholar]
- 9.Wei Q, Galbenus R, Raza A, Cerny RL, Simpson MA. Androgen-stimulated UDP-glucose dehydrogenase expression limits prostate androgen availability without impacting hyaluronan levels. Cancer Res. 2009;69:2332–2339. doi: 10.1158/0008-5472.CAN-08-3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kultti A, Pasonen-Seppanen S, Jauhiainen M, Rilla KJ, Karna R, Pyoria E, Tammi RH, Tammi MI. 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Exp Cell Res. 2009;315:1914–1923. doi: 10.1016/j.yexcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 11.Kakizaki I, Kojima K, Takagaki K, Endo M, Kannagi R, Ito M, Maruo Y, Sato H, Yasuda T, Mita S, Kimata K, Itano N. A novel mechanism for the inhibition of hyaluronan biosynthesis by 4-methylumbelliferone. J Biol Chem. 2004;279:33281–33289. doi: 10.1074/jbc.M405918200. [DOI] [PubMed] [Google Scholar]
- 12.Balduini C, Brovelli A, Deluca G, Galligan L, Castella Aa. Uridine Diphosphate Glucose Dehydrogenase from Cornea and Epiphyseal-Plate Cartilage. Biochemical Journal. 1973;133:243–249. doi: 10.1042/bj1330243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Egger S, Chaikuad A, Kavanagh KL, Oppermann U, Nidetzky B. UDP-glucose dehydrogenase: structure and function of a potential drug target. Biochemical Society Transactions. 2010;38:1378–1385. doi: 10.1042/BST0381378. [DOI] [PubMed] [Google Scholar]
- 14.Gainey PA, Phelps CF. Uridine Diphophate Glucuronic Acid Production and Utilization in Various Tissues Actively Synthesizing Glycosaminoglycans. Biochemical Journal. 1972;128:215. doi: 10.1042/bj1280215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maxwell ES, Kalckar HM, Strominger JL. Some Properties of Uridine Diphosphoglucose Dehydrogenase. Archives of Biochemistry and Biophysics. 1956;65:2–10. doi: 10.1016/0003-9861(56)90171-0. [DOI] [PubMed] [Google Scholar]
- 16.Kadirvelraj R, Custer GS, Keul ND, Sennett NC, Sidlo AM, Walsh RM, Wood ZA. Hysteresis in Human UDP-Glucose Dehydrogenase Is Due to a Restrained Hexameric Structure That Favors Feedback Inhibition. Biochemistry. 2014;53:8043–8051. doi: 10.1021/bi500594x. [DOI] [PubMed] [Google Scholar]
- 17.Kadirvelraj R, Sennett NC, Polizzi SJ, Weitzel S, Wood ZA. Role of Packing Defects in the Evolution of Allostery and Induced Fit in Human UDP-Glucose Dehydrogenase. Biochemistry. 2011;50:5780–5789. doi: 10.1021/bi2005637. [DOI] [PubMed] [Google Scholar]
- 18.Sennett NC, Kadirvelraj R, Wood ZA. Conformational Flexibility in the Allosteric Regulation of Human UDP-alpha-D-Glucose 6-Dehydrogenase. Biochemistry. 2011;50:9651–9663. doi: 10.1021/bi201381e. [DOI] [PubMed] [Google Scholar]
- 19.Sennett NC, Kadirvelraj R, Wood ZA. Cofactor Binding Triggers a Molecular Switch To Allosterically Activate Human UDP-alpha-D-glucose 6-Dehydrogenase. Biochemistry. 2012;51:9364–9374. doi: 10.1021/bi301067w. [DOI] [PubMed] [Google Scholar]
- 20.Helt SS, Thymark M, Harris P, Aagaard C, Dietrich J, Larsen S, Willemoes M. Mechanism of dTTP inhibition of the bifunctional dCTP deaminase : dUTPase encoded by Mycobacterium tuberculosis. Journal of Molecular Biology. 2008;376:554–569. doi: 10.1016/j.jmb.2007.11.099. [DOI] [PubMed] [Google Scholar]
- 21.Johansson E, Thymark M, Bynckl JH, Fan M, Larsen S, Willemoes M. Regulation of dCTP deaminase from Escherichia coli by nonallosteric dTTP binding to an inactive form of the enzyme. Febs Journal. 2007;274:4188–4198. doi: 10.1111/j.1742-4658.2007.05945.x. [DOI] [PubMed] [Google Scholar]
- 22.Kadirvelraj R, Sennett NC, Custer GS, Phillips RS, Wood ZA. Hysteresis and Negative Cooperativity in Human UDP-Glucose Dehydrogenase. Biochemistry. 2013;52:1456–1465. doi: 10.1021/bi301593c. [DOI] [PubMed] [Google Scholar]
- 23.Dickinson FM. Studies on the Unusual Behavior of Bovine Liver UDP-Glucose Dehydrogenase in Assays at Acid and Neutral pH and on the Presence of Tightly Bound Nucleotide Material in Purified Preparations of this Enzyme. Biochemical Journal. 1988;255:775–780. doi: 10.1042/bj2550775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egger S, Chaikuad A, Kavanagh KL, Oppermann U, Nidetzky B. Structure and Mechanism of Human UDP-glucose 6-Dehydrogenase. Journal of Biological Chemistry. 2011;286:23877–23887. doi: 10.1074/jbc.M111.234682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gasteiger E, HC, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. The Proteomics Protocols Handbook. 2005:571–607. doi: 10.1385/1-59259-584-7:531. [DOI] [PubMed] [Google Scholar]
- 26.Kabsch W. XDS. Acta Crystallographica Section D-Biological Crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brunger AT. Free R value: Cross-validation in crystallography. In: Carter CW, Sweet RM, editors. Macromolecular Crystallography, Pt B. 1997. pp. 366–396. [DOI] [PubMed] [Google Scholar]
- 28.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D-Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallographica Section D-Biological Crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Urzhumtsev A, Afonine PV, Adams PD. TLS from fundamentals to practice. Crystallography Reviews. 2013;19:230–270. doi: 10.1080/0889311X.2013.835806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Analytical Ultracentrifugation in Biochemistry and Polymer Science. R Soc Chem. 1992:90–125. [Google Scholar]
- 32.Schuck P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Analytical Biochemistry. 2003;320:104–124. doi: 10.1016/s0003-2697(03)00289-6. [DOI] [PubMed] [Google Scholar]
- 33.Frieden C. Kinetic Aspects of Regulation of Metabolic Processes- Hysteretic Enzyme Concept. Journal of Biological Chemistry. 1970;245:5788. [PubMed] [Google Scholar]
- 34.Cornish-Bowden A. Detection of errors of interpretation in experiments in enzyme kinetics. Methods. 2001;24:181–190. doi: 10.1006/meth.2001.1179. [DOI] [PubMed] [Google Scholar]
- 35.Kurganov BI. Analysis of negative cooperativity for glutamate dehydrogenase. Biophysical Chemistry. 2000;87:185–199. doi: 10.1016/s0301-4622(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 36.Hayward S, Berendsen HJC. Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins-Structure Function and Genetics. 1998;30:144–154. [PubMed] [Google Scholar]
- 37.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophysical Journal. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao H, Balbo A, Brown PH, Schuck P. The boundary structure in the analysis of reversibly interacting systems by sedimentation velocity. Methods. 2011;54:16–30. doi: 10.1016/j.ymeth.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuo LC. Allosteric Cofactor-Mediated Enzyme Cooperativity- A Theoretical Treatment. Proceedings of the National Academy of Sciences of the United States of America-Biological Sciences. 1983;80:5243–5247. doi: 10.1073/pnas.80.17.5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurganov BI, Dorozhko AK, Kagan ZS, Yakovlev VA. The theoretical analysis of kinetic behaviour of kinetic behaviour of "hysteretic" allosteric enzymes. III. Dissociating and associating enzyme systems in which the rate of installation of equilibrium between the oligomeric forms in comparable to that of enzymatic reaction. J Theor Biol. 1976;60:287–299. doi: 10.1016/0022-5193(76)90061-8. [DOI] [PubMed] [Google Scholar]
- 41.Kurganov BI, Dorozhko AI, Kagan ZS, Yakovlev VA. The theoretical analysis of kinetic behaviour of "hysteretic" allosteric enzymes. II. The dissociating and associating enzymic systems in which the rate of installation of equilibrium between the oligomeric forms is small in comparison with that of enzymatic reaction. J Theor Biol. 1976;60:271–286. doi: 10.1016/0022-5193(76)90060-6. [DOI] [PubMed] [Google Scholar]
- 42.Karplus PA, Diederichs K. Linking Crystallographic Model and Data Quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nature Structural Biology. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]