

Abstract
Mucopolysaccharidosis type I (MPS I) is due to deficiency of α-L-iduronidase (IDUA) and subsequent storage of undegraded glycosaminoglycans (GAG). The severe form of the disease, known as Hurler syndrome, is characterized by mental retardation and neurodegeneration of unknown etiology. To identify potential biomarkers and unveil the neuropathology mechanism of MPS I disease, two-dimensional polyacrylamide gel electrophoresis (PAGE) and nanoliquid chromatography-tandem mass spectrometry (nanoLC-MS/MS) were applied to compare proteome profiling of brains from MPS I and control mice (5-month old). A total of 2,055 spots were compared, and 25 spots (corresponding to 50 different proteins) with a fold change ≥3.5 and a p value <0.05 between MPS I and control mice were further analyzed by nanoLC-MS/MS. These altered proteins could be divided into three major groups based on Gene Ontology (GO) terms: proteins involved in metabolism, neurotransmission and cytoskeleton. Cytoskeletal proteins including ACTA1, ACTN4, TUBB4B and DNM1 were significantly downregulated. STXBP1, a regulator of synaptic vesicle fusion and docking was also downregulated, indicating impaired synaptic transmission. Additionally, proteins regulating Ca2+ and H+ homeostasis including ATP6V1B2 and RYR3 were downregulated, which may be related to disrupted autophagic and endocytotic pathways. Notably, there is no altered expression in proteins associated with cell death, ubiquitin or inflammation. These results for the first time highlight the important role of alterations in metabolism pathways, intracellular ionic homeostasis and the cytoskeleton in the neuropathology of MPS I disease. The proteins identified in this study would provide potential biomarkers for diagnostic and therapeutic studies of MPS I.
Keywords: mucopolysaccharidosis, proteomics, 2D-PAGE
1. Introduction
Mucopolysaccharidosis type I (MPS I) is an autosomal recessive disease that results from deficiency of α-L-iduronidase (IDUA, E.C.3.2.1.76), which degrades the glycosaminoglycans (GAG) heparan sulfate and dermatan sulfate. The widespread accumulation of GAG leads to progressive cellular damage and organ dysfunction, with the central nervous system (CNS) being one of the primary sites of pathology. The CNS pathology in MPS I patients is manifested by learning delays, dementia, hydrocephalus and mental retardation. The etiology of neurological dysfunction in MPS I is unclear. It has been reported that neurons and glial cells accumulate GAG (1) and gangliosides (2). Activation of glial cells (3) and alterations in oxidative status (4) in the cortex and cerebellum has been reported. It has been previously shown that MPS I mice had difficulty to habituate in the repeated open field test (5) and impaired long-term memory for aversive training (6). Interestingly, although one study (7) reported abnormal performance of MPS I mice in Morris water maze tests, another study found inconclusive results (8). In more recent studies, MPS I mice showed impaired learning behaviors in water T maze test (9) and spatial memory skills in the Barnes maze test (10). These findings describe abnormal cognitive and neuropathology in MPS I, but the mechanisms are likely to be complex and remain to be elucidated.
Development of proteomics technology includes two-dimensional polyacrylamide gel electrophoresis (2D-PAGE), which has provided a powerful tool to study the complicated biological processes in cells and rapidly profiling the global protein expression alterations. 2D-PAGE is powerful in identifying proteins and protein isoforms that may be neglected by other methods such as in-solution digestion and nanoLC-MS/MS or 1D-PAGE and nanoLC-MS/MS. As a mass spectrometric screening method, 2D-PAGE offers many advantages: high throughput, broad dynamic range, good reproducibility and adequate sensitivity. Although combing 2D-PAGE with mass spectrometry is a common proteomic approach for high throughput screening of putative biomarkers in several disorders (11,12), it has not been used in lysosomal diseases.
In this study, a comparative analysis of the proteome of MPS I and wildtype mouse brains using two dimensional gel electrophoresis (2D-GE) and nanoLC-ESI-MS/MS were performed. We identified 50 proteins that were differentially expressed in brains of MPS I versus wildtype mice. Bioinformatics analyses using Database for Annotation, Visualization, and Integrated Discovery (DAVID) (13), Protein ANalysis THrough Evolutionary Relationships (PANTHER) (14) and Search Tool for the Retrieval of Interacting genes (STRING) (15) databases allowed for functional classification of the detected proteins and highlighted MPS I-relevant biological pathways. This approach of screening identified potential biomarkers of MPS I that may reveal specific protein signatures indicating MPS I risk. These might also prove useful to assess prognosis, and provide outcome measures for assessing response to therapies.
diagnosis and subtype.
2. Materials and Methods
2.1 Animals and sample collection
MPS I knockout mice (idua−/−), a kind gift from Dr. Elizabeth Neufeld, UCLA, has been generated by insertion of neomycin resistance gene into exon 6 of the 14-exon IDUA gene on the C57BL/6 background. MPS I mice (idua−/−) and wildtype were genotyped by PCR. All mouse care and handling procedures were in compliance with the rules of the Institutional Animal Care and Use Committee (IACUC) of the University of Minnesota. The whole mouse brains were collected from one MPS I and one wildtype mouse (5-month old) for further analysis.
2.2 2D-PAGE
The samples were homogenized on ice using glass/glass dounces each with 1 mL osmotic lysis buffer (containing 10 X nuclease stock, phosphatase inhibitor stock and protease inhibitor stock) and 1 mL SDS boiling buffer without reducing agents. The samples were treated with 3 μL Omnicleave and heated in a boiling water bath for 5 minutes before the protein concentrations were determined using the BCA assay (Pierce Chemical Co., Rockford, IL, USA) (16). The samples were diluted to 3.33 and 0.67 mg/mL in 1:1 diluted SDS boiling buffer: Urea sample buffer with reducing agents before loading. 2D-PAGE was performed with the carrier ampholine method of isoelectric focusing as previously reported (17). Isoelectric focusing was conducted in a glass tube of inner diameter 3.3 mm using 2% pH 3–10 Isodalt Servalytes (Serva, Heidelberg, Germany) for 20,000 volt-hrs. Then, 100 ng of an IEF internal standard, tropomyosin, was added to the sample. This protein migrates as a doublet with lower polypeptide spot of MW 33,000 and pI 5.2. The enclosed tube gel pH gradient plot for this set of Servalytes was determined with a surface pH electrode. After equilibration for 10 minutes in Buffer ‘O’ (50 mM dithiothreitol, 10% glycerol, 2.3% SDS and 0.0625 M Tris, pH 6.8), each tube gel was sealed to the top of a stacking gel that overlaid a 10% acrylamide slab gel (1.00 mm thick). SDS slab gel electrophoresis was conducted for approximately 5 hours at 25 mA/gel. The following proteins (Sigma Chemical Co., St. Louis, MO, USA and EMD Millipore, Billerica, MA, USA) were used as molecular weight standards: lysozyme (14,000), carbonic anhydrase (29,000), actin (43,000), catalase (60,000), phosphorylase A (94,000) and myosin (220,000). These standards appear along the basic edge of the silver-stained 10% acrylamide slab gel. The silver-stained gels were dried between sheets of cellophane with the acid edge to the left (18). The gels were dried between sheets of cellophane paper with the acid edge to the left.
2.3 Computerized comparisons
Duplicate gels were obtained from each sample to reduce sources of variability and to detect differences with real statistical significance. The gels were scanned with a laser densitometer (Model PDSI; Molecular Dynamics Inc, Sunnyvale, CA, USA). The scanner was checked for linearity prior to scanning with a calibrated Neutral Density Filter Set (Melles Griot, Irvine, CA, USA). The images were analyzed using Progenesis Same Spots software (version 4.5, 2011; Nonlinear Dynamics, Durham, NC, USA) and Progenesis PG240 software (version 2006; Nonlinear Dynamics). The general method of computerized analysis for these pairs included image warping followed by spot finding, background subtraction (average on boundary), matching, and quantification in conjunction with detailed manual checking. A p value (Student’s t-test, n = 2 gels/sample) is calculated to help assess whether corresponding spots are different. As background is a factor, spot differences are checked by eye. Spot % is equal to spot integrated density above background (volume) expressed as a percentage of total density above background of all spots measured. Difference is defined as fold change of spot percentages. For instance, if corresponding protein spots from different samples (e.g. MPS I versus wildtype) have the same spot %, the difference field will show 1.0; if the spot % from MPS I is twice as large as wildtype, the difference field will display 2.0 indicating two fold up-regulation. If the spot % from MPS I has a value half as large, the difference field will display − 2.0 indicating a twofold downregulation.
2.4 Spot picking and in-gel digestion
Protein spots of interest were selected based on a fold increase or decrease of ≥3.5 and P-value <0.05. Picked spots were excised and subjected to in-gel tryptic digestion and peptide extraction for protein identification by nanoLC-MS/MS analysis, as described before (19). Briefly, spots were washed in HPLC grade water, 50 mM ammonium bicarbonate (ABC), 50% acetonitrile (ACN)/50% ABC for each 15 minutes under moderate shaking at room temperature. Next, the gel pieces were dehydrated with 100% ACN and dried under speed vac. Reduction and alkylation were carried out with 10 mM dithiothreitol (DTT) in 25 mM ABC for 30 minutes at 60°C and with 100 mM iodoacetamide in 25 mM ABC for 45 minutes in the dark respectively. Gel pieces were then dehydrated again, dried and rehydrated in 20 μL of a trypsin solution (10 ng/μL) overnight at 37°C. After incubation, peptide extraction was carried out with 5% formic acid (FA)/50 mM ABC/50% ACN and with 5% FA/100% ACN (20 minutes each). Extracted peptides were dried and submitted to a zip tip step for purification (EMD Millipore).
2.5 LC-MS/MS
The extracted peptides mixture was analyzed by reversed phase liquid chromatography (LC) and MS (LC-MS/MS) using a NanoAcuity UPLC (Micromass/Waters, Milford, MA) coupled to a Q-TOF Ultima API MS (Micromass/Waters, Milford, MA), according to published procedures (20). Briefly, the peptides were loaded onto a 100 μm × 10 mm NanoAquity BEH130 C18 1.7 μm UPLC column (Waters, Milford, MA) and eluted over a 150-minute gradient of 2–80% organic solvent (ACN containing 0.1% FA) at a flow rate of 400 nL/min. The aqueous solvent was 0.1% FA in HPLC water. The column was coupled to a Picotip Emitter Silicatip nano-electrospray needle (New Objective, Woburn, MA). MS data acquisition involved survey MS scans and automatic data dependent analysis (DDA) of the top three ions with the highest intensity ions with the charge of 2+, 3+ or 4+ ions. The MS/MS was triggered when the MS signal intensity exceeded 10 counts/second. In survey MS scans, the three most intense peaks were selected for collision-induced dissociation (CID) and fragmented until the total MS/MS ion counts reached 10,000 or for up to 6 seconds each. Calibration was performed for both precursor and product ions using 1 pmol GluFib (Glu1-Fibrinopeptide B) standard peptide with the sequence EGVNDNEEGFFSAR and the monoisotopic doubly-charged peak with m/z of 785.84.
2.6 Data processing and protein identification
The raw data were processed using ProteinLynx Global Server (PLGS, version 2.4) software as previously described (21). The following parameters were used: background subtraction of polynomial order 5 adaptive with a threshold of 30%, two smoothings with a window of three channels in Savitzky-Golay mode and centroid calculation of top 80% of peaks based on a minimum peak width of 4 channels at half height. The resulting pkl files were submitted for database search and protein identification to the public Mascot database search (www.matrixscience.com, Matrix Science, London, UK) using the following parameters: databases from NCBI, parent mass error of 1.3 Da, product ion error of 0.8 Da, enzyme used: trypsin, one missed cleavage, propionamide as cysteine fixed modification and methionine oxidized as variable modification. To identify the false negative results, we used additional parameters such as different databases or organisms, a narrower error window for the parent mass error (1.2 and then 0.2 Da) and for the product ion error (0.6 Da), and up to two missed cleavage sites for trypsin. In addition, the pkl files were also searched against in-house PLGS database version 2.4 (www.waters.com) using searching parameters similar to the ones used for Mascot search. The Mascot and PLGS database search provided a list of proteins for each gel band. To eliminate false positive results, for the proteins identified by either one peptide or a mascot score lower than 30, we verified the MS/MS spectra that led to identification of a protein.
2.7 Bioinformatics analysis
A series of analyses were undertaken with the differentially expressed proteins identified by 2D-PAGE. Interaction analysis was performed with STRING (http://string-db.org/newstring_cgi/show_input_page.pl). Further, PANTHER v9.0 and DAVID v6.7 (Database for Annotation, Visualization and Integrated Discovery) were employed for functional annotation analysis to understand the biological significance (e.g. physiological pathways) associated with large lists of genes or proteins. Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7 was used to functionally annotate the genes that clustered together. DAVID’s functional annotation tool allows for pathway analysis using the Kyoto Encyclopedia of Genes and Genomes (KEGG), gene ontology annotation for biological processes, molecular function, and cellular components, assessment of transcription factor binding sites, and identification of tissues matching the gene clusters. All relationships determined had corresponding statistics within DAVID. The functional annotations presented here had a p value <0.05 with Bonferroni correction.
3. Results
3.1 2D-PAGE of brain samples from MPS I and wildtype mice
Brain samples from MPS I and wildtype mice (5-month old) were collected for identifying putative differentially expressed proteins using 2D-PAGE combined with nanoLC-LC- MS/MS. In all, 2 Coomassie and 4 silver-stained (Figure S1) gels of 2D- PAGE of the brain samples (250 μg protein in each gel) were run for reasons of reproducibility. A total of 2,055 spots were compared (Figure 1) and only those spots with a fold change ≥3.5 and a p value <0.05 between MPS I and wildtype mice were picked for in-gel tryptic digestion. A list of all the picked spots with their pI, MW, spot percentage, fold change and p value is listed in Table S1.
Figure 1. 2D Gel difference image of brain samples from MPS I versus wildtype mice.

Polypeptide spots increased in MPS I versus control are outlined in blue, while spots decreased in MPS I versus control are outlined in red. See Table S1 for spot data and measurements.
3.2 Identification of differentially expressed proteins
Only proteins that were found in spots with a p value <0.05 and a fold change ≥3.5 were considered significant. This stringent criterion allows for true changes to be considered and for artifacts to be ignored. NanoLC-MS/MS analysis of the differentially expressed spots led to identification of 50 proteins that are up- or down-regulated in MPS I mouse brain (Table 1). Table 1 includes proteins that were identified by submitting the pkl files generated from the MS raw data to the MASCOT database. The average values of MOWSE score was 54.5 while the number of unique peptides identified by MS/MS was 5.5. In total, 27 spots were found to be corresponding to 50 unique proteins, of which, 36 proteins were down-regulated in MPS I mouse brain, while 14 proteins were up-regulated. Several representative gel images with some of the dysregulated protein spots identified in this study between MPS I and wildtype mice, along with their relative quantitative value is shown in Figure 2.
Table 1. Summary of proteins identified by LC-MS/MS from the picked 2D gel spots which are differentially expressed between MPS I and wildtype mice.
Positive fold changes represent an up-regulation while negative values signify a down-regulation of protein expression in MPS I versus wildtype.
| Spot# | Protein name | Mascot score | # of peptides | NCBI accession | Protein MW (kD) | Fold change (MPS vs WT) | T-test (p) |
|---|---|---|---|---|---|---|---|
| 81 | murinoglobulin-1 precursor | 29 | 1 | gi|31982171 | 166968 | −5.1 | 0.019 |
| oxidation resistance protein 1 isoform E | 28 | 10 | gi|194328708 | 96421 | |||
| ryanodine receptor 3 | 27 | 5 | gi|125628627 | 554439 | |||
| 149 | drebrin A | 37 | 8 | gi|6694227 | 78136 | 4.1 | 0.009 |
| 254 | alpha-actinin-4 isoform X2 | 76 | 4 | gi|568946338 | 104880 | −3.7 | 0.013 |
| dynamin/mKIAA4093 protein | 68 | 9 | gi|60360130 | 98009 | |||
| 285 | glucosidase 2 subunit beta isoform 2 precursor | 39 | 2 | gi|6679465 | 59963 | 3.8 | 0.023 |
| unnamed protein product | 27 | 1 | gi|74193788 | 27642 | |||
| phosphatidylcholine-specific phospholipase D1b | 27 | 2 | gi|2541940 | 120066 | |||
| 358 | dual specificity phosphatase-like 15, isoform CRA_b, partial | 29 | 2 | gi|148674056 | 10754 | 3.8 | 0.022 |
| Col4a6 protein | 20 | 2 | gi|34785873 | 107153 | |||
| epsin-1 | 29 | 1 | gi|46195711 | 60317 | |||
| 583 | unc-18 homologue/Munc18-1 | 83 | 3 | gi|1944322 | 68156 | −6.6 | 0.043 |
| dihydropyrimidinase-related protein 2 | 65 | 7 | gi|40254595 | 62736 | |||
| 591 | dihydropyrimidinase-related protein 2/Ulip2 protein | 276 | 24 | gi|40254595 | 62736 | −5.0 | 0.013 |
| unc-18 homologue/Munc18-1 | 95 | 10 | gi|3810884 | 68004 | |||
| FERM domain containing 3, isoform CRA_a, partial | 30 | 1 | gi|148699106 | 67284 | |||
| 609 | dihydrolipoyllysine-residue acetyltransferase component of pyruvate dehydrogenase complex, mitochondrial | 70 | 9 | gi|257796245 | 68609 | −5.5 | 0.014 |
| heat shock-related protein | 46 | 2 | gi|957195 | 71148 | |||
| dihydropyrimidinase-related protein 2/Ulip2 protein | 33 | 4 | gi|40254595 | 62736 | |||
| 613 | dihydropyrimidinase-related protein 2/Ulip2 protein | 106 | 23 | gi|40254595 | 62736 | −4.6 | 0.011 |
| unc-18 homologue/Munc18-1 | 58 | 3 | gi|1944322 | 68156 | |||
| 615 | dihydropyrimidinase-related protein 2/Ulip2 protein | 78 | 22 | gi|40254595 | 62736 | −4.2 | 0.001 |
| dihydropyrimidinase-related protein 4 isoform X2 | 48 | 4 | gi|568952370 | 34187 | |||
| glutaminase kidney isoform, mitochondrial isoform X2 | 36 | 2 | gi|568905794 | 50243 | |||
| 644 | dihydropyrimidinase-related protein 2/Ulip2 protein | 44 | 11 | gi|40254595 | 62736 | −5.0 | 0.017 |
| chaperonin containing TCP-1 theta subunit | 34 | 2 | gi|5295992 | 60170 | |||
| 720 | dihydropyrimidinase-related protein 2/Ulip2 protein | 51 | 10 | gi|40254595 | 62736 | −3.8 | 0.026 |
| mKIAA3028 protein | 23 | 4 | gi|50511271 | 189949 | |||
| 906 | actin, alpha skeletal muscle | 43 | 4 | gi|4501881 | 42450 | −3.8 | 0.018 |
| perinuclear binding protein | 41 | 1 | gi|577078 | 47127 | |||
| tubulin beta-4B chain | 35 | 4 | gi|5174735 | 50367 | |||
| 919 | V-type proton ATPase subunit B, brain isoform | 132 | 12 | gi|17105370 | 56941 | −4.2 | 0.007 |
| MNCb-1930 protein/cytosolic non-specific dipeptidase | 53 | 7 | gi|12697592 | 53274 | |||
| PHD finger protein 24 | 48 | 2 | gi|27369996 | 42227 | |||
| spliceosome RNA helicase Ddx39b | 41 | 2 | gi|9790069 | 49572 | |||
| solute carrier family 9 (sodium/hydrogen exchanger), isoform 3 regulator 1, isoform CRA_b, partial | 36 | 10 | gi|148702516 | 32049 | |||
| 947 | PHD finger protein 24 isoform X2 | 40 | 4 | gi|1039767264 | 47318 | −8.7 | 0.012 |
| Na(+)/H(+) exchange regulatory cofactor NHE-RF1 | 30 | 3 | gi|6755566 | 38932 | |||
| 1088 | NADP-dependent isocitrate dehydrogenase | 37 | 1 | gi|3641400 | 47128 | 3.6 | 0.004 |
| erythrocyte protein band 4.9, isoform CRA_a | 30 | 1 | gi|148703936 | 36082 | |||
| SEC14-like protein 2 | 31 | 3 | gi|21362309 | 46839 | |||
| 1162 | Melanocyte proliferating gene 1 | 36 | 3 | gi|20306404 | 43166 | −4.0 | 0.027 |
| UPF0160 protein MYG1, mitochondrial precursor | 35 | 5 | gi|11096332 | 43193 | |||
| heterogeneous nuclear ribonucleoprotein A/B isoform 2 | 32 | 4 | gi|6754222 | 30954 | |||
| 1206 | leukocyte elastase inhibitor A | 131 | 6 | gi|114158675 | 42761 | −4.2 | 0.042 |
| 1226 | fructose-bisphosphate aldolase A isoform 2 | 40 | 5 | gi|6671539 | 39900 | −14.1 | 0.021 |
| guanine nucleotide-binding protein G(o) subunit alpha isoform A | 24 | 6 | gi|6754012 | 40769 | |||
| 1351 | SIR2L2 | 119 | 8 | gi|11141704 | 44026 | −5.7 | 0.013 |
| poly(rC)-binding protein 1 | 3 | 32 | gi|6754994 | 38113 | |||
| acyl-CoA hydrolase | 30 | 1 | gi|14587839 | 38028 | |||
| fructose-bisphosphate aldolase A isoform 2 | 29 | 2 | gi|6671539 | 39900 | |||
| 1575 | pyridoxal phosphate phosphatase | 344 | 19 | gi|47059486 | 31989 | −3.6 | 0.042 |
| immunoglobulin variable region | 27 | 2 | gi|288821 | 13708 | |||
| 1613 | 14-3-3 protein theta | 80 | 2 | gi|6756039 | 28116 | 4.7 | 0.033 |
| 14-3-3 zeta | 79 | 3 | gi|1526539 | 27950 | |||
| isoamyl acetate-hydrolyzing esterase 1 homolog ] | 38 | 1 | gi|27754071 | 28524 | |||
| 1636 | mCG145112, partial | 28 | 1 | gi|148676777 | 12544 | 4.9 | 0.019 |
| glutamate receptor channel subunit epsilon 4 | 28 | 1 | gi|2160438 | 144523 | |||
| 1669 | MRJ | 68 | 2 | gi|3142372 | 26995 | −4.1 | 0.017 |
| glutathione S-transferase Mu 2 | 35 | 1 | gi|6680121 | 25913 | |||
| triosephosphate isomerase | 29 | 3 | gi|54855 | 27105 | |||
| plexin 2 | 34 | 2 | gi|1655432 | 214098 | |||
| triosephosphate isomerase | 30 | 5 | gi|54855 | 27105 | |||
| 1889 | alpha-crystallin B chain | 36 | 2 | gi|6753530 | 20056 | −6.8 | 0.007 |
Figure 2. Montage images of example protein spots differentially regulated between MPS I and wildtype mouse brain samples.

Column charts show the ratio in percentage for each dysregulated protein. See Table S1 for the whole spot data and measurements.
3.3 Bioinformatics analysis of dysregulated proteins
Dysregulated proteins were investigated with regard to their functional categories and their biological pathways. To this end, these proteins were imported into the PANTHER database, which allows classification and identification of the function of gene products. PANTHER analysis of the dysregulated proteins with regard to molecular function, biological process, cellular component, protein class and cellular pathway is represented in Figure 3. Most dysregulated proteins are catalytically active (40%), whereas the predominant biological process is the cellular process (26%) and metabolic process (18%). As to cellular component, cell part and organelle are 46% and 30%, respectively. The majority of the proteins belong to the protein class of hydrolase (21%), enzyme modulator (17%) and cytoskeletal protein (17%). Interestingly, a variety of pathways are involved, but no predominant pathways are identified. It indicates the complexity of pathways involved in MPS I neuropathology.
Figure 3. Functional classifications of the dysregulated proteins.

Dysregulated proteins are classified according to cellular component (A), biological process (B), protein class (C) and molecular function (D) by PANTHER.
To extract additional information from our dataset, the same list was submitted to the DAVID. DAVID renders functional annotation and interpretation of lists of protein identifications. According to DAVID functional chart, the most frequent biological process is generation of precursor metabolites and energy, indicating role of altered energy metabolic pathways in MPS I neuropathology. Another interesting finding is that 22% of proteins are in mitochondrion, which is also associated with energy metabolism. Additionally, the two most frequent post-translational modifications are phosphorylation (65%) and acetylation (35%). With DAVID functional annotation clustering, three major clusters were identified: energy metabolism, neurotransmission and cytoskeletal system (summarized in Table 2). Collectively, these results suggested that altered metabolism plays a major role in neuropathology of MPS I disease.
Table 2. DAVID functional annotation clustering analysis of all dysregulated proteins.
Count stands for number of genes involved in individual term. P value stands for EASE score (the modified Fisher exact p value). The overall enrichment score based on the EASE scores of each term member. The higher, the more enriched. A cluster is a group of terms having similar biological meaning due to sharing similar gene members. For clustering analysis, classification stringency is set at medium, while enrichment threshold at 0.05.
| Cluster 1 (Enrichment score: 2.14) | |||
|---|---|---|---|
| Functional annotation | Count | P value | Bonferroni |
| Generation of precursor metabolites and energy | 5 | 2.20E-03 | 5.70E-01 |
| Glycolysis | 3 | 3.10E-03 | 3.10E-01 |
| Glucose catabolic process | 3 | 5.50E-03 | 8.80E-01 |
| Hexose catabolic process | 3 | 5.50E-03 | 8.80E-01 |
| Monosaccharide catabolic process | 3 | 5.90E-03 | 9.00E-01 |
| Cellular carbohydrate catabolic process | 3 | 7.20E-03 | 9.40E-01 |
| Alcohol catabolic process | 3 | 8.40E-03 | 9.60E-01 |
| Carbohydrate catabolic process | 3 | 1.30E-02 | 9.90E-01 |
| Glycolysis / Gluconeogenesis | 3 | 2.10E-02 | 6.10E-01 |
| Glucose metabolic process | 3 | 3.60E-02 | 1.00E+00 |
| Cluster 2 (Enrichment score: 1.95) | |||
| Synaptic transmission | 4 | 6.30E-03 | 9.20E-01 |
| Regulation of system process | 4 | 8.80E-03 | 9.70E-01 |
| Transmission of nerve impulse | 4 | 1.20E-02 | 9.90E-01 |
| Cell-cell signaling | 4 | 2.30E-02 | 1.00E+00 |
| Cluster 3 (Enrichment score: 1.64) | |||
| Actin filament organization | 3 | 6.30E-03 | 9.20E-01 |
| Cytoskeleton | 7 | 4.10E-02 | 9.90E-01 |
| Actin cytoskeleton organization | 3 | 4.80E-02 | 1.00E+00 |
Protein – protein interaction (PPI) between the dysregulated proteins was also performed with the STRING. In general, ACTA1 and other cytoskeletal proteins (TUBB4B, DNM1, and ACTN4) seem to be at the heart of most PPIs suggesting that alterations of cytoskeleton might play a major role in MPS I neuropathogenesis (Figure 4). These findings suggest that cytoskeletal proteins are implicated in the neuropathology of MPS I disease.
Figure 4. Interaction network of dysregulated proteins by STRING.
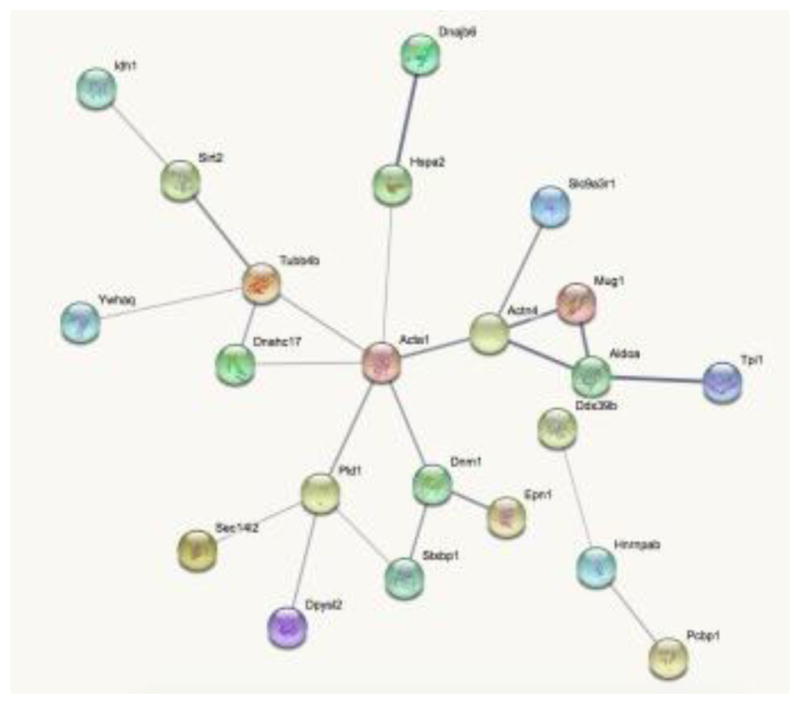
Line thickness indicates the strength of data support for protein-protein interactions. Disconnected proteins are hided from the network. Small nodes indicate unknown 3D structure, while large nodes have known or predicted 3D structure within nodes. Associations are meant to be specific and meaningful, i.e. proteins jointly contribute to a shared function; this does not necessarily mean they are physically binding each other.
4. Discussion
4.1 Functional classification of dysregulated proteins
In this study, 50 proteins were revealed as differentially expressed between MPS I and wildtype mouse brains. These proteins function in diverse biological processes, and some functional groups are formed through cluster and pathway analysis. We discuss below the different pathways and their interdependency, as well as some interesting proteins.
4.1.1 Cytoskeleton
A major group of dysregulated proteins in MPS I mouse brain are cytoskeletal proteins. Specifically, ACTA1 (actin), DNM1 (dynamin), ACTN4 (actinin), TUBB4B (tubulin) and DNAH17 (dynein) are major components of cytoskeletal system, while PDXP (pyridoxal phosphatase), DBN1 and SLC9A3R1 regulate actin cytoskeleton reorganization. Besides, it has been shown that SIRT2 modulates the acetylation status of α-tubulin (22). Cytoskeletal proteins are important for vesicular trafficking (reviewed in 23), which is involved in multiple lysosomal membrane trafficking pathways including endocytosis, autophagy and exocytosis. Lysosomes receive inputs from both endocytotic and autophagic pathways, and release degraded products to Golgi apparatus through retrograde trafficking or to the extracellular space through exocytosis (reviewed in 24). Additionally, EPN1 (epsin 1), which regulates receptor-mediated endocytosis through facilitating formation of clathrin-coated invaginations, is upregulated. Upregulation of EPN1 also suggests abnormality of lysosomal membrane trafficking pathways. Therefore, dysregulation in cytoskeleton related proteins may indicate impaired lysosomal membrane trafficking pathways as lysosomal storage accumulates.
4.1.2 Metabolism pathways
Another group of dysregulated proteins in MPS I mouse brain are metabolism related proteins. GLS (glutaminase) catalyzes the first reaction in the primary pathway for the renal catabolism of glutamine and also regulates the neurotransmitter glutamate in the CNS. Downregulation of GLS indicates impairment of neurotransmission mediated by glutamate in MPS I disease. Further, other downregulated metabolism related proteins include DLAT (dihydrolipoamide S-acetyltransferase), TPI1 (triosephosphate isomerase 1) and ALDOA (aldolase A, fructose-bisphosphate), SERPINB1A and ACOT7 (acyl-CoA thioesterase 7). Besides, IDH1 (isocitrate dehydrogenase 1), involved in the TCA cycle, is upregulated. These results further highlight involvement of altered energy production in MPS I disease, consistent with DAVID clustering analysis.
4.1.3 Neurotransmission
Moreover, expression of several neurotransmission-related proteins is found to be downregulated. DPYSL2 (dihydropyrimidinase-like 2) plays a role in neuronal development and polarity, as well as in neuron projection morphogenesis, axon growth and guidance, neuronal growth cone collapse and cell migration. PICK1 (protein interacting with C kinase 1) plays a role in synaptic plasticity by regulating the trafficking and internalization of AMPA receptors. More importantly, SXTBP1 (syntaxin binding protein 1) is a regulator of synaptic vesicle docking and fusion, which has also been found to interact with cytoskeletal proteins (25). Further, synaptic plasticity requires the dynamic actin filaments, which is the major structural component of synapses (reviewed in 26). These findings indicate the downregulation of cytoskeletal proteins may affect synaptic transmission directly or indirectly through SXTBP1.
4.1.4 Ionic homeostasis
More interestingly, several proteins involved in intracellular ionic homeostasis are also found to be dysregulated. ATP6V1B2, a V-type proton ATPase, which is responsible for H+ transporting, is downregulated. It has been shown that deficiency of VMA21, which is responsible for V-ATPase assembly, leads to decreased V-ATPase, increased lysosomal pH, and thereby a genetic disorder known as X-linked myopathy with excessive autophagy (XMEA) (27). Besides, MFSD7C, GRIN2D, RYR3 (ryanodine receptor 3) and PRKCSH, four proteins involved in calcium homeostasis, are also dysregulated. More interestingly, a lower content of Ca2+ and H+ has been found to be elevated in MPS I mouse cells (28). An elevation of the pH in lysosome may severely compromise the activity of different lysosomal hydrolases and vesicle trafficking mechanisms (29). As a side effect, substrates and molecules not directly related to the primary enzyme deficiency may accumulate, as is the case with most lysosomal diseases (reviewed in 30). Further, elevation in pH and lower Ca2+ concentration in lysosomes can inhibit the fusion between endosomes, autophagosomes and lysosomes, resulting in impaired endocytotic and autophagic pathways (31).
4.1.5 Chaperone
Several proteins involved in protein folding are also downregulated in MPS I mouse brain. HSPA2 (heat shock protein 2), in cooperation with other chaperones, stabilize preexistent proteins against aggregation and mediate the folding of newly translated polypeptides in the cytosol and within organelles. DNAJB6 is an endogenous molecular chaperone for neuronal proteins including huntingtin, and acts as a co-chaperone of HSP70. CRYAB (crystallin, alpha B) has chaperone-like activity, preventing aggregation of various proteins under a wide range of stress conditions.
4.1.6 Other proteins
Additionally, we also identified some dysregulated proteins involved in oxidative stress response (OXR1, GSTM1), RNA splicing (DBX39B, HNRNPAB and PCBP1), signal transduction (GNAO1, YWHAQ, PLD1), hydrolase activity (IAH1, DUSP15), extracellular matrix (COL4A6) and other functions. Of particular interest is MYG1, which was downregulated in MPS I mouse brain. MYG1 is associated with locomotory exploration behaviors, anxiety and stress related responses (32). There is no report so far regarding of the possible roles of MYG1 in cognitive abilities of MPS I mice. However, it has been shown that MPS I mice had impaired locomotory exploration behaviors (5). Thus, it will be beneficial to investigate the role of MYG1 in the neuropathology of MPS I. More interestingly, YWHAQ, which was upregulated in MPS I mouse brain, has been shown to be responsible for cytosolic retention of transcription factor EB (TFEB) (33). After translocation into nucleus, TFEB can activate genes regulating most lysosome-based processes including macromolecule degradation, autophagy, lysosomal exocytosis, and proteostasis (summarized in 34). Downregulation of TFEB contributes to disease pathogenesis, and overexpression of TFEB can reduce storage accumulation, tissue inflammation and apoptosis (35). Therefore, upregulation of YWHAQ may impair translocation of TFEB into nucleus and thereby downregulate TFEB signaling, feeding a negative loop that ultimately inhibits lysosomal degradative pathways.
4.2 Validity of this study
In this study, we compared the proteomic profiling of brain samples from 5-month old MPS I and wildtype mice, and picked 25 spots with a fold change ≥3.5 (p value <0.05). For better reproducibility, 2D-PAGE was run in duplicate and gel images were analyzed through Progenesis Same Spots software and Progenesis PG240 software. Further, the brain sample from 1-month old MPS I mouse was also compared with wildtype mouse, yielding similar fold change patterns. Specifically, out of 6 upregulated spots in 5-month old MPS I vs wildtype mice, 5 spots were also upregulated in 1-month old MPS I mouse. Out of 19 downregulated spots in 5-month old MPS I mouse, 17 spots were also downregulated in 1-month old MPS I mouse (data not shown). Considering the 4-month age difference, it showed good reproducibility of our results.
The dysregulated proteins can be classified into several major groups: metabolism, cytoskeleton, neurotransmission, protein folding and intracellular ion homeostasis. A previous study using MS/MS to analyze proteomic profiling of MPS I mouse (8-month old) hippocampus identified inflammation and synaptic plasticity related proteins, but no cell death or ubiquitin related proteins (36). Consistent with their results, we did not observe any differences in cell death related proteins, indicating that massive cell death may not be the primary cause of neuropathology in MPS I disease. In addition, we also found alteration in synaptic transmission related proteins including SXTBP1, PICK1 and DPYSL2. Although we observed dysregulation of several chaperons including HSP2A, DNAJB6 and CRYAB, we did not find any differences in ubiquitin related proteins, indicating that protein folding and ubiquitin system may be functioning properly. Unlike Baldo et al, we did not identify any inflammation related proteins (e.g. GFAP). Admittedly, the stringent criteria (a fold change ≥3.5 and a p value <0.05) used in this study may neglect some proteins that were modestly upregulated or downregulated. Besides, we are investigating the whole brain while the previous study was using hippocampus. However, this discrepancy is more likely due to the fact that we are using 5-month old MPS I mice while they were using 8-month old mice. It has been found that 9-month old MPS I mice had significantly increased astrocyte and microglia activation than 4-month old MPS I mice (37). Another study showed that microglia activation occurred between 6 to 9 months of age in MPS I mice (3), suggesting a late onset of neuroinflammation. Therefore, since MPS I mice showed abnormality in behavior tests as early as 4 months of age (5, 9), neuroinflammation may not be an initiator or major contributor of impaired cognitive abilities of MPS I mice. It will be interesting to investigate proteomic profiling of older MPS I mice to better understand the role of neuroinflammation.
4.3 Perspectives and conclusions
Collectively, this study identifies potential biomarkers for MPS I, and, for the first time, highlights that alterations in metabolism pathways, intracellular ionic homeostasis and cytoskeletal systems may play an important role in neuropathology of MPS I disease. Based on detailed analysis of these 50 dysregulated proteins, a hypothetical model of MPS I pathology mechanism is proposed (summarized in Figure 5). Further studies are necessary to investigate specific pathways that could lead to the CNS pathology seen in patients and animal models of MPS I. Additionally, it would be interesting to investigate proteomic profiling of brains from other lysosomal disease animals.
Figure 5. Hypothetic model of MPS I neuropathology.

Energy production provide ATPs for cytoskeletal system, vesicle transport and ionic homeostasis. Ionic homeostasis and vesicle transport are important for lysosomal membrane trafficking pathways including endocytosis, autophagy, exocytosis and retrograde pathway. Cytoskeletal proteins and vesicle transport are important for synaptic transmission. Genes in red represent proteins downregulated in MPS I and genes in green are upregulated.
Supplementary Material
Highlights.
We conducted 2D PAGE analysis of MPS I mice brain.
The proteins identified in this study would provide potential biomarkers for diagnostic and therapeutic studies of MPS I.
These results for the first time highlight the important role of alterations in metabolism pathways, intracellular ionic homeostasis and the cytoskeleton in the neuropathology of MPS I disease.
Acknowledgments
The authors thank Kendrick Labs (Madison, WI, http://www.kendricklabs.com/) for 2D-PAGE and quantitative analyses. All authors declare that they have no conflicts of interest related to this work. This work is supported by NIH grant P01HD032652. Dr. Li Ou is a fellow of the Lysosomal Disease Network (U54NS065768). The Lysosomal Disease Network is a part of the Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), and NxCATS. This consortium is funded through a collaboration between NCATS, the National Institute of Neurological Disorders and Stroke (NINDS), and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Baldo G, Mayer FQ, Martinelli B, Dilda A, Meyer F, Ponder KP, Giugliani R, Matte U. Evidence of a progressive motor dysfunction in Mucopolysaccharidosis type I mice. Behav Brain Res. 2012 Jul 15;233(1):169–75. doi: 10.1016/j.bbr.2012.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walkley SU. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin Cell Dev Biol. 2004 Aug;15(4):433–44. doi: 10.1016/j.semcdb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Ohmi K, Greenberg DS, Rajavel KS, Ryazantsev S, Li HH, Neufeld EF. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc Natl Acad Sci U S A. 2003 Feb 18;100(4):1902–7. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reolon GK, Reinke A, de Oliveira MR, Braga LM, Camassola M, Andrades ME, Moreira JC, Nardi NB, Roesler R, Dal-Pizzol F. Alterations in oxidative markers in the cerebellum and peripheral organs in MPS I mice. Cell Mol Neurobiol. 2009 Jun;29(4):443–8. doi: 10.1007/s10571-008-9335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartung SD, Frandsen JL, Pan D, Koniar BL, Graupman P, Gunther R, Low WC, Whitley CB, McIvor RS. Correction of metabolic, craniofacial, and neurologic abnormalities in MPS I mice treated at birth with adeno-associated virus vector transducing the human alpha-L-iduronidase gene. Mol Ther. 2004 Jun;9(6):866–75. doi: 10.1016/j.ymthe.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 6.Reolon GK, Braga LM, Camassola M, Luft T, Henriques JA, Nardi NB, Roesler R. Long-term memory for aversive training is impaired in Idua(−/−) mice, a genetic model of mucopolysaccharidosis type I. Brain Res. 2006 Mar 3;1076(1):225–30. doi: 10.1016/j.brainres.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Wolf DA, Lenander AW, Nan Z, Belur LR, Whitley CB, Gupta P, Low WC, McIvor RS. Direct gene transfer to the CNS prevents emergence of neurologic disease in a murine model of mucopolysaccharidosis type I. Neurobiol Dis. 2011 Jul;43(1):123–33. doi: 10.1016/j.nbd.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan D, Sciascia A, 2nd, Vorhees CV, Williams MT. Progression of multiple behavioral deficits with various ages of onset in a murine model of Hurler syndrome. Brain Res. 2008 Jan 10;1188:241–53. doi: 10.1016/j.brainres.2007.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ou L, Herzog T, Koniar BL, Gunther R, Whitley CB. High-dose enzyme replacement therapy in murine Hurler syndrome. Mol Genet Metab. 2014 Feb;111(2):116–22. doi: 10.1016/j.ymgme.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ou L, DeKelver R, Tom S, Radeke R, Rohde M, Manning-Bog A, Sproul S, Przybilla MJ, Koniar BL, Podetz-Pedersen K, Laoharawee K, Cooksley RD, Holmes MC, Wechsler T, McIvor RS, Whitley CB. ZFN-mediated liver-targeting gene therapy corrects systemic and neurological disease of mucopolysaccharidosis type I. Mol Ther. 2016 May;24:S192–S193. [Google Scholar]
- 11.Chen Y, Azman SN, Kerishnan JP, Zain RB, Chen YN, Wong YL, Gopinath SC. Identification of host-immune response protein candidates in the sera of human oral squamous cell carcinoma patients. PLoS One. 2014 Oct 1;9(10):e109012. doi: 10.1371/journal.pone.0109012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Costa O, Schneider P, Coquet L, Chan P, Penther D, Legrand E, Jouenne T, Vasse M, Vannier JP. Proteomic profile of pre - B2 lymphoblasts from children with acute lymphoblastic leukemia (ALL) in relation with the translocation (12; 21) Clin Proteomics. 2014 Aug 1;11(1):31. doi: 10.1186/1559-0275-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(5):P3. Epub 2003 Apr 3. [PubMed] [Google Scholar]
- 14.Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013 Jan;41(Database issue):D377–86. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ, von Mering C. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011 Jan;39(Database issue):D561–8. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985 Oct;150(1):76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 17.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975 May 25;250(10):4007–21. [PMC free article] [PubMed] [Google Scholar]
- 18.Oakley BR, Kirsch DR, Morris NR. A simplified ultrasensitive silver stain for detecting proteins in polyacrylamide gels. Anal Biochem. 1980 Jul 1;105(2):361–3. doi: 10.1016/0003-2697(80)90470-4. [DOI] [PubMed] [Google Scholar]
- 19.Ngounou Wetie AG, Wormwood K, Thome J, Dudley E, Taurines R, Gerlach M, Woods AG, Darie CC. A pilot proteomic study of protein markers in autism spectrum disorder. Electrophoresis. 2014 Jul;35(14):2046–54. doi: 10.1002/elps.201300370. [DOI] [PubMed] [Google Scholar]
- 20.Darie CC, Deinhardt K, Zhang G, Cardasis HS, Chao MV, Neubert TA. Identifying transient protein-protein interactions in EphB2 signaling by blue native PAGE and mass spectrometry. Proteomics. 2011 Dec;11(23):4514–28. doi: 10.1002/pmic.201000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spellman DS, Deinhardt K, Darie CC, Chao MV, Neubert TA. Stable isotopic labeling by amino acids in cultured primary neurons: application to brain-derived neurotrophic factor-dependent phosphotyrosine-associated signaling. Mol Cell Proteomics. 2008 Jun;7(6):1067–76. doi: 10.1074/mcp.M700387-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang L, Hou X, Ma R, Moley K, Schedl T, Wang Q. Sirt2 functions in spindle organization and chromosome alignment in mouse oocyte meiosis. FASEB J. 2014 Mar;28(3):1435–45. doi: 10.1096/fj.13-244111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamal A, Goldstein LS. Connecting vesicle transport to the cytoskeleton. Curr Opin Cell Biol. 2000 Aug;12(4):503–8. doi: 10.1016/s0955-0674(00)00123-x. [DOI] [PubMed] [Google Scholar]
- 24.Samie MA, Xu H. Lysosomal exocytosis and lipid storage disorders. J Lipid Res. 2014 Jun;55(6):995–1009. doi: 10.1194/jlr.R046896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhaskar K, Shareef MM, Sharma VM, Shetty AP, Ramamohan Y, Pant HC, Raju TR, Shetty KT. Co-purification and localization of Munc18-1 (p67) and Cdk5 with neuronal cytoskeletal proteins. Neurochem Int. 2004 Jan;44(1):35–44. doi: 10.1016/s0197-0186(03)00099-8. [DOI] [PubMed] [Google Scholar]
- 26.Spence EF, Soderling SH. Actin Out: Regulation of the Synaptic Cytoskeleton. J Biol Chem. 2015 Nov 27;290(48):28613–22. doi: 10.1074/jbc.R115.655118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramachandran N, Munteanu I, Wang P, Ruggieri A, Rilstone JJ, Israelian N, Naranian T, Paroutis P, Guo R, Ren ZP, Nishino I, Chabrol B, Pellissier JF, Minetti C, Udd B, Fardeau M, Tailor CS, Mahuran DJ, Kissel JT, Kalimo H, Levy N, Manolson MF, Ackerley CA, Minassian BA. VMA21 deficiency prevents vacuolar ATPase assembly and causes autophagic vacuolar myopathy. Acta Neuropathol. 2013 Mar;125(3):439–57. doi: 10.1007/s00401-012-1073-6. [DOI] [PubMed] [Google Scholar]
- 28.Pereira VG, Gazarini ML, Rodrigues LC, da Silva FH, Han SW, Martins AM, Tersariol IL, D’Almeida V. Evidence of lysosomal membrane permeabilization in mucopolysaccharidosis type I: rupture of calcium and proton homeostasis. J Cell Physiol. 2010 May;223(2):335–42. doi: 10.1002/jcp.22039. [DOI] [PubMed] [Google Scholar]
- 29.Jentsch TJ. Chloride and the endosomal-lysosomal pathway: emerging roles of CLC chloride transporters. J Physiol. 2007 Feb 1;578(Pt 3):633–40. doi: 10.1113/jphysiol.2006.124719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Campos D, Monaga M. Mucopolysaccharidosis type I: current knowledge on its pathophysiological mechanisms. Metab Brain Dis. 2012 Jun;27(2):121–9. doi: 10.1007/s11011-012-9302-1. [DOI] [PubMed] [Google Scholar]
- 31.Pryor PR, Mullock BM, Bright NA, Gray SR, Luzio JP. The role of intraorganellar Ca(2+) in late endosome-lysosome heterotypic fusion and in the reformation of lysosomes from hybrid organelles. J Cell Biol. 2000 May 29;149(5):1053–62. doi: 10.1083/jcb.149.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Philips MA, Abramov U, Lilleväli K, Luuk H, Kurrikoff K, Raud S, Plaas M, Innos J, Puussaar T, Kõks S, Vasar E. Myg1-deficient mice display alterations in stress-induced responses and reduction of sex-dependent behavioural differences. Behav Brain Res. 2010 Feb 11;207(1):182–95. doi: 10.1016/j.bbr.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Pena KA, Kiselyov K. Transition metals activate TFEB in overexpressing cells. Biochem J. 2015 Aug 15;470(1):65–76. doi: 10.1042/BJ20140645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sardiello M. Transcription factor EB: from master coordinator of lysosomal pathways to candidate therapeutic target in degenerative storage diseases. Ann N Y Acad Sci. 2016 May;1371(1):3–14. doi: 10.1111/nyas.13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Medina DL, Fraldi A, Bouche V, Annunziata F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA, Sardiello M, Palmieri M, Polishchuk R, Puertollano R, Ballabio A. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev Cell. 2011 Sep 13;21(3):421–30. doi: 10.1016/j.devcel.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baldo G, Lorenzini DM, Santos DS, Mayer FQ, Vitry S, Bigou S, Heard JM, Matte U, Giugliani R. Shotgun proteomics reveals possible mechanisms for cognitive impairment in Mucopolysaccharidosis I mice. Mol Genet Metab. 2015 Feb;114(2):138–45. doi: 10.1016/j.ymgme.2014.12.301. [DOI] [PubMed] [Google Scholar]
- 37.Wilkinson FL, Holley RJ, Langford-Smith KJ, Badrinath S, Liao A, Langford-Smith A, Cooper JD, Jones SA, Wraith JE, Wynn RF, Merry CL, Bigger BW. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS One. 2012;7(4):e35787. doi: 10.1371/journal.pone.0035787. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.