

Abstract
Hypothermia is a key symptom of sepsis, but the mechanism(s) leading to hypothermia during sepsis is largely unknown and thus no effective therapy is available for hypothermia. Therefore, it is important to investigate the mechanisms and develop effective therapeutic methods. Lipopolysaccharide (LPS)-induced hypothermia accompanied by excess nitric oxide (NO) production, lead to a reduction in energy production in wild type mice. However, mice lacking inducible nitric oxide synthase did not suffer from LPS-induced hypothermia, suggesting that hypothermia is associated with excess NO production during sepsis. This observation is supported by the treatment of wild type mice with α-lipoic acid (LA) in that it effectively attenuates LPS-induced hypothermia with decreased NO production. We also found that LA partially restored ATP production, and activities of the mitochondrial enzymes involved in energy metabolism, which were inhibited during sepsis. These data suggest that hypothermia is related to mitochondrial dysfunction, which is likely compromised by excess NO production and that LA administration attenuates hypothermia mainly by protecting mitochondrial enzymes from NO damage.
Keywords: Lipoic acid, antioxidant, mitochondria, body energy
Introduction
Sepsis remains a critical problem resulting in high morbidity and mortality rates. In the United States alone, there are 751,000 estimated cases of sepsis every year with an overall mortality of 100,000 people [1]. The incidence of sepsis is expected to rise at a rate of 1.5% per year with total costs of $16.7 billion [2].
Both hyperthermia (body temperature >38 °C) and hypothermia (body temperature <36 °C) are key diagnostic criteria for sepsis [3]. Hypothermia results in an increased rate of wound infection, coagulopathy and other postoperative complications [4], and is particularly associated with severe infection and a higher mortality rate [5, 6]. The mechanism for hypothermia remains largely unknown. Many strategies have been proposed for the treatment of sepsis, but few noteworthy breakthroughs have been made during the past several decades [7]. Since there is no effective therapy to prevent hypothermia, it is also clinically important to develop effective methods for modulating hypothermia.
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are involved in sepsis [8-10]. One of the characteristic features of sepsis is markedly increased levels of plasma nitric oxide (NO) [11]. NO plays an important role in oxidative phosphorylation [12], and it is well-known that excess NO causes inhibition of complexes I and IV in the electron transport chain in several ways [13-15]. Antioxidant therapy is a plausible adjunctive method for treatment of sepsis. Among antioxidants, α-lipoic acid (1, 2-dithiolane-3-pentanoic acid; LA) is a well-known potent antioxidant with anti-inflammatory activity [16-18]. LA is also a cofactor for several enzymes including pyruvate dehydrogenase complex (PDH; EC1.2.4.1 + EC 1.6.4.3 + EC 2.3.1.2) and α-ketoglutarate dehydrogenase complex (KDH; EC 1.2.4.2 + EC 2.3.1.61 + EC 1.8.1.4), two enzymes located in mitochondria and involved in energy production. LA has a therapeutic potential in mitochondria-related disorders [19, 20]. We have previously reported that a mouse embryo cannot survive without endogenous LA production, strongly suggesting that LA is essential for maintenance of normal levels of energy metabolism in addition to its antioxidant activity [21].
Lipopolysaccharide (LPS) administration may produce hypothermia in rodents [22, 23]. In the current study, we hypothesize that excess production of NO during sepsis inhibits some proteins in the Krebs cycle and the mitochondrial respiratory chain, leading to overall energy deficiency. To demonstrate this, we treated mice with LPS to induce hypothermia and studied molecular and physiological responses associated with this treatment. In this study, we found that LA increased mitochondrial antioxidant capacity, partially restored enzymatic activities in two mitochondrial enzymes, and ATP yield. These changes may contribute to the modification of hypothermia-associated sepsis mechanisms.
Materials and Methods
Materials
R-(+)-α-lipoic acid was from Toronto Chemical Research (Toronto, Canada), and Cytochrome C from Sigma Aldrich (St. Louis, MO).
LPS-induced endotoxic shock in mice
Three-month-old female C57BL/6J mice, and female iNOS-null mice were given intraperitoneally (i.p.) a dose (4 mg/kg body weight) of LPS (E. Coli 055:B5, Sigma, St. Louis), or saline as a control [18, 24]. Half of each of the LPS-treated and control mice received i.p. a single injection (40 mg/kg body weight) of LA 30 min before or after the LPS injection [18]. To help distinguish the effect of LA on hypothermia from other antioxidants, four LPS-treated mice were i.p. injected with ascorbate (pH 7.0 previously adjusted with NaOH; 5 mg/g of body weight) [25]. The dosage of LA was based on our experience and that of other investigators [26-29]. Body temperature was monitored for 4 hours after LPS injection using rectal temperature sensors (Physitemp Instruments Inc. Clifton, NJ). The experimental protocols were approved by the Institutional Animal Care and Use Committee of the University of North Carolina and complied with the U. S. National Institutes of Health standards.
Nitrate/Nitrite (NOx) Assay
Plasma nitrate (NO3−) and nitrite (NO2−) levels, indicators of the production of NO, were measured after 4 hours of LPS injection using a colorimetric assay kit (Cayman Chemical, Ann Arbor, MI). The absorbance was read on a Biotek Synergy HT Multi-Mode Microplate Reader (Winooski, VT) using a wavelength of 540 nm.
Mitochondria isolation
Liver mitochondria were isolated following protocols already published [30, 31] with minor modifications. Mice were sacrificed by cervical dislocation and the liver was rapidly explanted from the peritoneal cavity. The liver was immersed in ice-cold isolation buffer [100 mM Tris– 3-(N-morpholino) propanesulfonic acid (MOPS, Sigma Aldrich), 10 mM of ethylene-bis (oxyethylenenitrilo) tetraacetic acid tetrasodium (EGTA, Sigma Aldrich)/Tris and 200 mM sucrose, pH 7.4] and chopped finely with scissors and then rinsed with the isolation buffer several times to remove traces of blood. Chopped tissue was homogenized in the buffer, with 12 strokes of a loose pestle in a glass Dounce. The homogenate was centrifuged for 10 min at 3,000 g at 4° C. Supernatants (including the pink salmon layer above the pellet) were collected in isolation buffer and then centrifuged for 10 min at 10,000 g at 4° C. The resulting pellets were finally re-suspended in ~1 ml isolation buffer and retained on ice until use (within 4 hours).
Determination of reduced glutathione (GSH) levels in mitochondria
Liver mitochondrial levels of reduced GSH were determined using a commercially kit (Cayman Chemical) following the manufacturer's instructions. The protein concentration of each sample was determined using a bicinchoninic acid assay (BCA; Thermo Scientific, Rockford, IL).
Determination of ATP production levels
ATP generation in LPS-treated liver homogenate was analyzed by a luciferase substrate assay (Invitrogen, Carlsbad, CA). Briefly, LPS-treated mice, in the presence or absence of LA, were sacrificed 4 hours after the LPS injection. The liver was homogenized, and the homogenate was sonicated in ATP Assay Buffer provided by the company. After centrifugation at 12,000 g for 5 minutes at 4° C, the supernatant was removed and used for ATP measurement in an Orion Microplate Luminometer (Berthold Detection Systems, Titertek Instruments, Inc., Huntsville, AL). For calculations the amount of ATP was normalized by protein concentration as determined using the BCA method.
Alpha-ketoglutarate dehydrogenase and complex III activities
KDH activity was determined by monitoring NAD+ reduction at 340 nm. Isolated liver mitochondria were suspended in medium containing 25 mM KH2PO4, 5 mM MgCl2, 0.5 mM EDTA, 0.1% (v/v) Triton X-100, 40 mM rotenone, 1.0 mM NAD+, 0.2 mM thiamin pyrophosphate, and 0.1 mM Coenzyme A, pH 7. The mitochondria were sonicated in a water bath (Denville Scientific, South Plainfield, NJ) at room temperature for 1 min. A reaction mixture containing 5 mM MgCl2, 40 mM rotenone, 0.1 mM Coenzyme A, 0.2 mM thymine pyrophosphate, and 1 mM NAD+ were added to the mitochondrial medium to reach a final concentration of 0.4 mg mitochondrial protein/ml. The reaction was initiated by adding 2.5 mM α-ketoglutarate [32-34].
Complex III (ubiquinol-ferricytochrome C oxidoreductase; EC 1.10.2.2) activity was measured by following an increase in absorbance at 550 nm due to the reduction of ferricytochrome C [35, 36]. The reaction mixture consisted of 25 mM KH2PO4, pH 7.4, 5 mM MgCl2, 2 mM KCN, 2.5 mg bovine serum albumin, 50 μM cytochrome C, and 4 mg rotenone. Mitochondrial protein (25 μg) was added and the reaction was initiated by the addition of 60 μM decylubiquinol; any increase in absorbance at 550 nm was monitored for 2 minutes. The assay was repeated, but with the inclusion of 5 μg antimycin A to the reaction mixture to determine antimycin A-insensitive activity. Antimycin A-sensitive complex III activity was determined using the initial rate as baseline. Enzyme activity was expressed as nmole of reduced cytochrome C/minute per mg of protein.
PGE2 and cytokine analyses
The levels of prostaglandins E2 (PGE2) and cortisone were measured by an enzyme immunoassay kit (Cayman Chemical), according to the manufacturer's instructions. Plasma levels of interleukin-6 (IL-6) and tumor necrosis factor (TNF-α) in plasma were measured using a LINCO multiplex mouse cytokine kit (Linco Research, Inc., St. Charles, MO) on a Luminex 100 system, according to the manufactures’ protocols (Luminex Corporation, Austin, TX).
Statistical analysis
All data are reported as means ± S.E.M. Values of p<0.05 were considered to be statistically significant (95% confidence limits). The effects of NO and LA were analyzed using ANOVA. Effects of fluctuation in body temperature were assessed by MANOVA by repeated measurements of each mouse's physiological conditions.
Results
α-Lipoic acid attenuates LPS-induced hypothermia mediated by decreasing excessive NO production during sepsis
Injection of LPS in C57BL/6 wild type (WT) mice induced a drop in body temperature from an initial 36.5 °C to a mean nadir of 28 °C during the 4 hours following LPS treatment (ΔT = − 8.5 °C) (Figure 1A). In contrast, a temperature drop in C57BL/6 mice lacking the inducible nitric oxide synthase (iNOS) was considerably attenuated (Figure 1A; ΔT = − 3.0 °C). Administering LA (40 mg/kg body weight) to WT mice 30 min prior to LPS injection protected them from hypothermia to a similar degree as untreated WT mice, but gave no further protection to the LPS-treated iNOS null mice (Figure 1). During the four hours of observation, the body temperature of the WT mice that were not treated with LPS remained unchanged (ΔT < 0.5 °C) and LA supplementation alone did not affect body temperature (data not shown). Supplementing with LA at 30 min post LPS injection was equally effective in preventing hypothermia as illustrated in Figure 1B. In addition, there was no significant attenuation of hypothermia in septic mice treated with ascorbate (Figure 1B).
Figure 1.


Time course of hypothermia in wild type (WT) and iNOS−/− mice induced by lipopolysaccharides (LPS). A). α-lipoic acid (LA) was given at 30 min prior to LPS injection. Animal number: WT+LA=17; WT-LA=14; iNOS−/−+LA=10; iNOS−/−-LA=10. B). LA or ascorbate were administered at 30 min after LPS injection. Data are expressed as means ± SEM. Animal number: WT+LA=12; WT-LA=13; iNOS−/−+LA=10; iNOS−/−-LA=10; and WT+ascrobate=4.
Next, we measured changes in total plasma nitrate/nitrite (NOx) after LPS injection since the circulating levels of NOx represent overall NOS activity. At 4 hours post-injection, NOx levels were increased 11-fold in WT mice (Figure 2). LA markedly attenuated LPS-induced NOx increase in WT mice but not in iNOS null mice (Figure 2). LPS injection decreased eNOS (endothelial nitric oxide synthase) gene expression by 5% and increased iNOS gene expression by 1700% in WT mouse liver. On the other hand, administration of LA did not significantly change the expression of eNOS and iNOS at 4 hours after LPS injection (P = 0.15 and 0.43, respectively; data not shown). Taken together, these data suggest that iNOS-derived NO is a major source of the NOx responsible for hypothermia during sepsis, and that the thermoregulatory effect of LA is mediated through NO.
Figure 2.

Total plasma nitrate/nitrite (NOx) immediately before, and 4 hours after LPS injection in septic mice. Values are the mean ± SEM. Statistical analysis are by one-way ANOVA.
Effect of LA on ATP production, mitochondrial functions and redox status
To investigate whether partial restoration of the body temperature is due to sustained energy metabolism, we measured ATP production in the liver. At 4 hours post LPS injection, liver ATP content in WT mice was reduced by 40% compared with un-treated mice, but addition of LA completely blocked this reduction (Figure 3A). ATP level in LPS-treated iNOS−/− mice did not significantly drop compared with non-treated control, and LA supplementation did not increase ATP production (Figure 3B).
Figure 3.

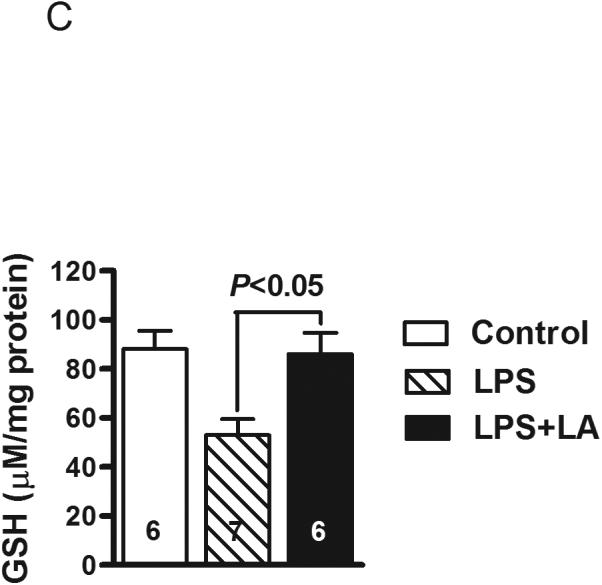
Effects of LA on energy production during sepsis. ATP content in the livers of the WT (A) and iNOS−/− mice (B) at 4 hours following LPS treatment. Control mice were injected with PBS. (C) Glutathione (GSH) levels in mitochondria isolated from liver of LPS-treated mice. All results are expressed as mean ± SEM. The numbers inside the bars indicate the number of animals used for each measurement.
To evaluate whether administration of LA may compensate for possible loss of mitochondrial antioxidant capacity, we measured GSH levels in mitochondria. We observed that the liver mitochondrial GSH content was decreased by about 40% after LPS treatment (P<0.01) and LA-treated mice almost fully retained their original GSH levels (Figure 3C).
Complex III and KDH activities
Enzymatic assay of complex III in the mitochondria isolated from WT mouse liver demonstrates a 40% decrease in its activity at 4 hours post-injection of LPS, which is attenuated in the presence of LA (P<0.05) (Figure 4A). Likewise, KDH activity dropped by about 50% while LA partially prevented this reduction (P<0.01) (Figure 4C). We also found that KDH activity in liver mitochondria was not significantly altered in septic mice treated with ascorbate (Figure 4C). These results suggest that ascorbate supplementation does not completely share the same mechanisms as LA in terms of hypothermia treatment. In addition, both complex III and KDH activity in the mitochondria isolated from iNOS−/− mouse liver did not significantly decrease after injection of LPS and addition of LA did not show any obvious effect for the enzyme activities (Figures 4B and 4D).
Figure 4.


Activities of complex III and alpha-ketoglutarate dehydrogenase (KDH) in mitochondria isolated from liver of LPS-treated WT mice (A and C) and iNOS−/− mice (B and D) in the presence or absence of LA or ascorbate. (A and B) Complex III activity was expressed as nmole of reduced cytochrome C/minute per mg of protein. (C and D) KDH activity was determined by monitoring NAD+ reduction at 340 nm. The activities of two enzymes shown are the means ± SEM.
Examination of other possible regulatory factors for hypothermia
To explore other possible thermoregulatory mechanisms involving LA in addition to NO, we examined the effect of LA on PGE2 and cytokines. Our results showed that PGE2 levels in serum in all non-LPS treated mice were very low (with LA: 90 ± 12 pg/ml; without LA 85 ± 12 pg/ml). Serum PGE2 levels were increased at 2 hours (687 ± 72 pg/ml), continued to rise at 4 hours (1,115 ± 55 pg/ml) and reached a plateau at 24 hours post LPS challenge (Figure 5A). Treatment with LA did not significantly change serum PGE2 levels (At 2 hour: 654 ± 114 pg/ml; at 4 hours: 1,156 ± 65 pg/ml) (Figure 5A). Similarly, concentrations of serum TNF-α (Figure 5B) and IL-6 (Figure 5C) were markedly increased at 2 hours (TNF-α: 7.5 ± 0.98 ng/ml; IL-6: 25.4 ± 2.1 ng/ml) and 4 hours (TNF-α: 4.5 ± 0.32 ng/ml; IL-6: 20.4 ± 1.26 ng/ml) following LPS injection; but LA supplementation did not significantly affect their levels (TNF-α: 5.6 ± 1.1 ng/mg; IL-6: 26.2 ± 1.8 ng/ml at 2 hours; TNF-α: 3.9 ± 1.42 ng/ml; IL-6: 22.4 ± 2.1) at 4 hours; i.e. thermoregulation via LA neither directly affects levels of PGE2 nor pro-inflammatory cytokines.
Figure 5.

Serum concentrations of (A) prostaglandin E2 (PGE2), (B) tumor necrosis factor alpha (TNF-α), and (C) interleukin-6 (IL-6) at 2 hours and 4 hours after intraperitoneal injection of LPS in mice supplemented with LA compared to controls. Error bars represent SEM.
Discussion
In this study, we found that treatment with LA significantly attenuates LPS-induced hypothermia in septic mice. Thermoregulation by LA is likely associated with its ability to protect mitochondria by markedly decreasing NO production and increasing mitochondrial antioxidant capacity. Our data shows that LA treatment significantly decreased LPS-induced accumulation of the major nitric oxide end products (NO3− and NO2−) without significantly affecting eNOS and iNOS gene expression, suggesting that decrease of NO by LA is primarily through inhibiting NO biosynthesis. Excessive NO produced during sepsis is expected to be involved in impairment of mitochondrial enzymes. It is well-known that excessive NO interacts with the mitochondrial respiratory chain by inhibiting cytochrome c oxidase (complex IV) and NADH-ubiquinone oxidoreductase (complex I) [37-39]. We find that activities of two other enzymes, KDH and complex III, are also decreased by excessive NO in LPS-treated WT mice and the activities are partially restored by LA supplementation. Our study presents new RNS targets and extends the knowledge about the protective capabilities of LA. KDH is in a key regulatory step of the Krebs cycle [40], which determines the capacity of mitochondrial metabolism [41], and could play a vital role in the ROS-associated bioenergetic deficit [42]. Complex III is a central component of all primary energy transduction systems. It functions in maintaining the proton gradient, which provides about 30% of energy production [43]. Therefore, it is possible that the decrease in catalytic activities of the two enzymes contributes to decreased ATP production, leading to hypothermia during sepsis, since mitochondria generate more than 90-95% of ATP in the body [44]. This assumption is supported by the observation in iNOS−/− mice in which body temperature does not significantly drop upon injection of LPS. In addition, ATP production, enzymatic activities of KDH and complex III in iNOS−/− mice neither significantly decrease after LPS injection, nor respond to LA supplementation. These data strongly suggest that excessive NO induced by LPS injection plays a critical role in hypothermia formation and a link may exist between NO levels and amount of ATP synthesis. On the other hand, since GSH is predominantly an intra-mitochondrial antioxidant [12] that protects mitochondrial complexes from NO-induced damage [45], a declined GSH level weakens mitochondrial antioxidant defense and deteriorates mitochondrial function. It has been observed that GSH decreases when NO production increases [38]. Our result showed that the decreased mitochondrial GSH levels were partially restored with LA treatment. Since both GSH and LA are thiol antioxidants, treatment with LA may spare consumption of GSH [46], and reduce formation of protein disulfide bridges [47]. In addition, LA can raise GSH levels by reducing cystine to cysteine, a GSH precursor [48]. However, it is not certain whether this reaction can take place under the oxidative stress conditions. The increased mitochondrial GSH levels by LA may provide protection from NO-induced damage to mitochondrial components related to energy production like KDH, and consequently attenuate hypothermia. It is known that KDH is sensitive to ROS/RNS such as hydrogen peroxide, peroxynitrite and nitric oxide [49, 50]. Thus, it is not surprising at the outcome that enzyme activity decreases during sepsis.
These data are also consistent with previous observations from our and other investigators. For instance, human endothelial cells exposed to serum from patients with sepsis have shown lower rates of mitochondrial respiration and significantly reduced ATP production than cells treated with serum from healthy volunteers [51, 52]. Likewise, in vitro studies showed mitochondrial complex inhibition by RNS with decreased ATP synthesis [52, 53]. Additionally, mitochondrial damage develops into energetic failure during sepsis, leading to cell death and organ failure [54, 55]. Moreover, sepsis-associated oxidative stress can be effectively quenched by LA treatment [18, 56]. Our previous data showed that LA in hepatocytes decreased noticeably in LPS treated mice [18]. At this point it is not clear whether decreasing LA levels affect the availability of the cofactor for normal KDH function. The fact that KDH activity does not change with LA supplementation in LPS-treated iNOS−/− mice indicates that KDH activity under these conditions is not dependent on the cofactor. Earlier reports have shown that LA administration improved the activities of KDH by improving the co-factor availability [57, 58]; however, these observations were made in aged animals, and they cannot be applied to studies in animals as young as those used in our studies.
Evidence shows that LA and ascorbate are powerful antioxidants that catalyze denitrosylation [59, 60]. However, ascorbate fails to prevent hypothermia. Thermoregulatory properties of LA may be related to its unique structure and physical/chemical characteristics. We believe that the presence of a thiol group in LA could result in better protection of mitochondria [61].
Since hypothermia may be a reversible process, we extended the time of observation for body temperature from 4 hours to 24 hours to probe possible thermo-regulative mechanisms of LA. We found that low body temperature persisted for 24 hours post LPS injection and that the highest level of plasma NO occurred at 24 hours in septic mice. The thermoregulatory effect of LA weakened over time and body temperature increased <1°C 6-8 hours after LPS injection, suggesting that extended exposure to NO could lead to irreversible inhibition of mitochondrial function, which is difficult to attenuate with a one-time treatment of LA. Repeated treatment or a higher dose may be required to overcome this situation.
Highlights.
α-lipoic acid (LA) significantly attenuates hypothermia.
LA restores enzymatic activities inhibited by excessive nitric oxide.
Our results should provide a new strategy for effective treatment of sepsis.
Acknowledgments
This project was supported by HL42630 and HL087946 (N.M.), by UNC University Research Council Grants, and in part by DK056350 to the UNC-CH Nutrition Obesity Research Center (X.Yi.).
The authors thank Dr. Rosaline Coleman for her insightful suggestions.
The manuscript has been reviewed by the US Environmental Protection Agency, NHEERL and approved for publication. The authors would like to thank Drs. P.R. Kodavanti and J. Royland for their invaluable suggestions. Approval does not signify that the contents reflect the views of the US EPA, nor that the mention of trade names or commercial products could constitute endorsement or recommendation for use.
Abbreviations
- iNOS
inducible Nitric Oxide Synthase
- eNOS
endothelial Nitric Oxide Synthase
- IL-6
interleukin-6
- KDH
α-ketoglutarate dehydrogenase complex
- LA
α-lipoic acid
- LPS
lipopolysaccharide
- NO
nitric oxide
- GSH
glutathione
- TNF-α
tumor necrosis factor alpha
- WT
wild type
- ROS
Reactive Oxygen Species
- RNS
Reactive Nitrogen Species
Footnotes
Author Disclosure Statement
The authors declare that no competing financial interests exist.
References
- 1.Feihl F, Waeber B, Liaudet L. Is nitric oxide overproduction the target of choice for the management of septic shock? Pharmacol Ther. 2001;91(3):179–213. doi: 10.1016/s0163-7258(01)00155-3. [DOI] [PubMed] [Google Scholar]
- 2.Angus DC, Wax RS. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–10. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM, Sibbald WJ. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101(6):1644–55. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 4.Kurz A, Sessler DI, Lenhardt R. Perioperative normothermia to reduce the incidence of surgical-wound infection and shorten hospitalization. Study of Wound Infection and Temperature Group. N Engl J Med. 1996;334(19):1209–15. doi: 10.1056/NEJM199605093341901. [DOI] [PubMed] [Google Scholar]
- 5.Marik PE, Zaloga GP. Hypothermia and cytokines in septic shock. Norasept II Study Investigators. North American study of the safety and efficacy of murine monoclonal antibody to tumor necrosis factor for the treatment of septic shock. Intensive Care Med. 2000;26(6):716–21. doi: 10.1007/s001340051237. [DOI] [PubMed] [Google Scholar]
- 6.Peres Bota D, Lopes Ferreira F, Mélot C, Vincent JL. Body temperature alterations in the critically ill. Intensive Care Med. 2004;30(5):811–6. doi: 10.1007/s00134-004-2166-z. [DOI] [PubMed] [Google Scholar]
- 7.Remick DG, Ward PA. Evaluation of endotoxin models for the study of sepsis. Shock. 2005;24(Suppl 1):7–11. doi: 10.1097/01.shk.0000191384.34066.85. [DOI] [PubMed] [Google Scholar]
- 8.Reid MB, Durham WJ. Generation of reactive oxygen and nitrogen species in contracting skeletal muscle: potential impact on aging. Ann N Y Acad Sci. 2002;959:108–16. doi: 10.1111/j.1749-6632.2002.tb02087.x. [DOI] [PubMed] [Google Scholar]
- 9.Rocha M, Herance R, Rovira S, Hernández-Mijares A, Victor VM. Mitochondrial dysfunction and antioxidant therapy in sepsis. Infect Disord Drug Targets. 2012;12(2):161–78. doi: 10.2174/187152612800100189. [DOI] [PubMed] [Google Scholar]
- 10.Lawler JM, Song W. Specificity of antioxidant enzyme inhibition in skeletal muscle to reactive nitrogen species donors. Biochem Biophys Res Commun. 2002;294(5):1093–100. doi: 10.1016/S0006-291X(02)00602-2. [DOI] [PubMed] [Google Scholar]
- 11.Luiking YC, Engelen MP, Deutz NE. Regulation of nitric oxide production in health and disease. Curr Opin Clin Nutr Metab Care. 2010;13(1):97–104. doi: 10.1097/MCO.0b013e328332f99d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brealey D, Singer M. Mitochondrial Dysfunction in Sepsis. Curr Infect Dis Rep. 2003;5(5):365–371. doi: 10.1007/s11908-003-0015-9. [DOI] [PubMed] [Google Scholar]
- 13.Huttemann M, Lee I, Pecinova A, Pecina P, Przyklenk K, Doan JW. Regulation of oxidative phosphorylation, the mitochondrial membrane potential, and their role in human disease. J Bioenerg Biomembr. 2008;40(5):445–56. doi: 10.1007/s10863-008-9169-3. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J, Bang ML, Gokhin DS, Lu Y, Cui L, Li X, Gu Y, Dalton ND, Scimia MC, Peterson KL, Lieber RL, Chen J. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am J Physiol Cell Physiol. 2005;288(4):C840–9. doi: 10.1152/ajpcell.00325.2004. [DOI] [PubMed] [Google Scholar]
- 15.Alvarez S, Evelson PA. Nitric oxide and oxygen metabolism in inflammatory conditions: sepsis and exposition to polluted ambients. Front Biosci. 2007;12:964–74. doi: 10.2741/2117. [DOI] [PubMed] [Google Scholar]
- 16.Packer L, Witt EH, Tritschler HJ. alpha-Lipoic acid as a biological antioxidant. Free Radic Biol Med. 1995;19(2):227–50. doi: 10.1016/0891-5849(95)00017-r. [DOI] [PubMed] [Google Scholar]
- 17.Bast A, Haenen GR. Lipoic acid: a multifunctional antioxidant. Biofactors. 2003;17(1-4):207–13. doi: 10.1002/biof.5520170120. [DOI] [PubMed] [Google Scholar]
- 18.Yi X, Kim K, Yuan W, Xu L, Kim HS, Homeister JW, Key NS, Maeda N. Mice with heterozygous deficiency of lipoic acid synthase have an increased sensitivity to lipopolysaccharide-induced tissue injury. J Leukoc Biol. 2009;85(1):146–53. doi: 10.1189/jlb.0308161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reed LJ. From lipoic acid to multi-enzyme complexes. Protein Sci. 1998;7(1):220–4. doi: 10.1002/pro.5560070125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tirosh O, Shilo S, Aronis A, Sen CK. Redox regulation of mitochondrial permeability transition: effects of uncoupler, lipoic acid and its positively charged analog LA-plus and selenium. Biofactors. 2003;17(1-4):297–306. doi: 10.1002/biof.5520170129. [DOI] [PubMed] [Google Scholar]
- 21.Yi X, Maeda N. Endogenous production of lipoic acid is essential for mouse development. Mol Cell Biol. 2005;25(18):8387–92. doi: 10.1128/MCB.25.18.8387-8392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blanque R, Meakin C, Millet S, Gardner CR. Hypothermia as an indicator of the acute effects of lipopolysaccharides: comparison with serum levels of IL1 beta, IL6 and TNF alpha. Gen Pharmacol. 1996;27(6):973–7. doi: 10.1016/0306-3623(95)02141-8. [DOI] [PubMed] [Google Scholar]
- 23.Akarsu ES, Mamuk S. Escherichia coli lipopolysaccharides produce serotype-specific hypothermic response in biotelemetered rats. Am J Physiol Regul Integr Comp Physiol. 2007;292(5):R1846–50. doi: 10.1152/ajpregu.00786.2006. [DOI] [PubMed] [Google Scholar]
- 24.Can C, Demirci B, Uysal A, Akçay YD, Koşay S. Contradictory effects of chlorpromazine on endothelial cells in a rat model of endotoxic shock in association with its actions on serum TNF-alpha levels and antioxidant enzyme activities. Pharmacol Res. 2003;48(3):223–30. doi: 10.1016/s1043-6618(03)00093-8. [DOI] [PubMed] [Google Scholar]
- 25.Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, Choyke PL, Pooput C, Kirk KL, Buettner GR, Levine M. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A. 2007;104(21):8749–54. doi: 10.1073/pnas.0702854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melhem MF, Craven PA, Derubertis FR. Effects of dietary supplementation of alpha-lipoic acid on early glomerular injury in diabetes mellitus. J Am Soc Nephrol. 2001;12(1):124–33. doi: 10.1681/ASN.V121124. [DOI] [PubMed] [Google Scholar]
- 27.Min AK, Kim MK, Kim HS, Seo HY, Lee KU, Kim JG, Park KG, Lee IK. Alpha-lipoic acid attenuates methionine choline deficient diet-induced steatohepatitis in C57BL/6 mice. Life Sci. 2012;90(5-6):200–5. doi: 10.1016/j.lfs.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 28.Thirunavukkarasu V, Anitha Nandhini AT, Anuradha CV. Lipoic acid attenuates hypertension and improves insulin sensitivity, kallikrein activity and nitrite levels in high fructose-fed rats. J Comp Physiol B. 2004;174(8):587–92. doi: 10.1007/s00360-004-0447-z. [DOI] [PubMed] [Google Scholar]
- 29.Yi X, Nickeleit V, James LR, Maeda N. alpha-Lipoic acid protects diabetic apolipoprotein E-deficient mice from nephropathy. J Diabetes Complications. 2011;25(3):193–201. doi: 10.1016/j.jdiacomp.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yi X, Xu L, Kim K, Kim HS, Maeda N. Genetic reduction of lipoic acid synthase expression modestly increases atherosclerosis in male, but not in female, apolipoprotein E-deficient mice. Atherosclerosis. 2010;211(2):424–30. doi: 10.1016/j.atherosclerosis.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2(2):287–95. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 32.Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem. 2001;276(26):23357–61. doi: 10.1074/jbc.M100320200. [DOI] [PubMed] [Google Scholar]
- 33.Kaplan P, Tatarkova Z, Engler I, Calkovska A, Mokra D, Drgova A, Kovalska M, Lehotsky J, Dobrota D. Effects of long-term oxygen treatment on alpha-ketoglutarate dehydrogenase activity and oxidative modifications in mitochondria of the guinea pig heart. Eur J Med Res. 2009;14(Suppl 4):116–20. doi: 10.1186/2047-783X-14-S4-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pekovich SR, Martin PR, Singleton CK. Thiamine pyrophosphate-requiring enzymes are altered during pyrithiamine-induced thiamine deficiency in cultured human lymphoblasts. J Nutr. 1996;126(7):1791–8. doi: 10.1093/jn/126.7.1791. [DOI] [PubMed] [Google Scholar]
- 35.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 36.Kwong LK, Sohal RS. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch Biochem Biophys. 2000;373(1):16–22. doi: 10.1006/abbi.1999.1495. [DOI] [PubMed] [Google Scholar]
- 37.Brown GC, Borutaite V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc Res. 2007;75(2):283–90. doi: 10.1016/j.cardiores.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 38.Brealey D, Brand M, Hargreaves I, Heales S, Land J, Smolenski R, Davies NA, Cooper CE, Singer M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet. 2002;360(9328):219–23. doi: 10.1016/S0140-6736(02)09459-X. [DOI] [PubMed] [Google Scholar]
- 39.Brown GC, Borutaite V. Nitric oxide, cytochrome c and mitochondria. Biochem Soc Symp. 1999;66:17–25. doi: 10.1042/bss0660017. [DOI] [PubMed] [Google Scholar]
- 40.Cooney GJ, Taegtmeyer H, Newsholme EA. Tricarboxylic acid cycle flux and enzyme activities in the isolated working rat heart. Biochem J. 1981;200(3):701–3. doi: 10.1042/bj2000701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bunik VI, Fernie AR. Metabolic control exerted by the 2-oxoglutarate dehydrogenase reaction: a cross-kingdom comparison of the crossroad between energy production and nitrogen assimilation. Biochem J. 2009;422(3):405–21. doi: 10.1042/BJ20090722. [DOI] [PubMed] [Google Scholar]
- 42.Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: a target and generator of oxidative stress. Philos Trans R Soc Lond B Biol Sci. 2005;360(1464):2335–45. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crofts AR. Proton-coupled electron transfer at the Qo-site of the bc1 complex controls the rate of ubihydroquinone oxidation. Biochim Biophys Acta. 2004;1655(1-3):77–92. doi: 10.1016/j.bbabio.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 44.Acuna-Castroviejo D, Martín M, Macías M, Escames G, León J, Khaldy H, Reiter RJ. Melatonin, mitochondria, and cellular bioenergetics. J Pineal Res. 2001;30(2):65–74. doi: 10.1034/j.1600-079x.2001.300201.x. [DOI] [PubMed] [Google Scholar]
- 45.Bolanos JP, Heales SJ, Peuchen S, Barker JE, Land JM, Clark JB. Nitric oxide-mediated mitochondrial damage: a potential neuroprotective role for glutathione. Free Radic Biol Med. 1996;21(7):995–1001. doi: 10.1016/s0891-5849(96)00240-7. [DOI] [PubMed] [Google Scholar]
- 46.Meister A. In: Glutathione, Chemical, Biochemical and Medical Aspect, Part A. Dolphin D, Poulson K, Aavrannovic O, editors. Wiley-Interscience Publication; New York: 1989. [Google Scholar]
- 47.Slepneva IA, Sergeeva SV, Khramtsov VV. Reversible inhibition of NADPH-cytochrome P450 reductase by alpha-lipoic acid. Biochem Biophys Res Commun. 1995;214(3):1246–53. doi: 10.1006/bbrc.1995.2420. [DOI] [PubMed] [Google Scholar]
- 48.Han D, Handelman G, Marcocci L, Sen CK, Roy S, Kobuchi H, Tritschler HJ, Flohé L, Packer L. Lipoic acid increases de novo synthesis of cellular glutathione by improving cystine utilization. Biofactors. 1997;6(3):321–38. doi: 10.1002/biof.5520060303. [DOI] [PubMed] [Google Scholar]
- 49.Chinopoulos C, Adam-Vizi V. Depolarization of in situ mitochondria by hydrogen peroxide in nerve terminals. Ann N Y Acad Sci. 1999;893:269–72. doi: 10.1111/j.1749-6632.1999.tb07834.x. [DOI] [PubMed] [Google Scholar]
- 50.Park YS, Suzuki K, Taniguchi N, Gutteridge JM. Glutathione peroxidase-like activity of caeruloplasmin as an important lung antioxidant. FEBS Lett. 1999;458(2):133–6. doi: 10.1016/s0014-5793(99)01142-4. [DOI] [PubMed] [Google Scholar]
- 51.Boulos M, Astiz ME, Barua RS, Osman M. Impaired mitochondrial function induced by serum from septic shock patients is attenuated by inhibition of nitric oxide synthase and poly(ADP-ribose) synthase. Crit Care Med. 2003;31(2):353–8. doi: 10.1097/01.CCM.0000050074.82486.B2. [DOI] [PubMed] [Google Scholar]
- 52.Belikova I, Lukaszewicz AC, Faivre V, Damoisel C, Singer M, Payen D. Oxygen consumption of human peripheral blood mononuclear cells in severe human sepsis. Crit Care Med. 2007;35(12):2702–8. doi: 10.1097/01.ccm.0000295593.25106.c4. [DOI] [PubMed] [Google Scholar]
- 53.Brookes PS, Bolanos JP, Heales SJ. The assumption that nitric oxide inhibits mitochondrial ATP synthesis is correct. FEBS Lett. 1999;446(2-3):261–3. doi: 10.1016/s0014-5793(99)00217-3. [DOI] [PubMed] [Google Scholar]
- 54.Carre JE, Orban JC, Re L, Felsmann K, Iffert W, Bauer M, Suliman HB, Piantadosi CA, Mayhew TM, Breen P, Stotz M, Singer M. Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am J Respir Crit Care Med. 2010;182(6):745–51. doi: 10.1164/rccm.201003-0326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crouser ED. Mitochondrial dysfunction in septic shock and multiple organ dysfunction syndrome. Mitochondrion. 2004;4(5-6):729–41. doi: 10.1016/j.mito.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 56.Vanasco V, Cimolai MC, Evelson P, Alvarez S. The oxidative stress and the mitochondrial dysfunction caused by endotoxemia are prevented by alpha-lipoic acid. Free Radic Res. 2008;42(9):815–23. doi: 10.1080/10715760802438709. [DOI] [PubMed] [Google Scholar]
- 57.Arivazhagan P, Ramanathan K, Panneerselvam C. Effect of DL-alpha-lipoic acid on mitochondrial enzymes in aged rats. Chem Biol Interact. 2001;138(2):189–98. doi: 10.1016/s0009-2797(01)00268-x. [DOI] [PubMed] [Google Scholar]
- 58.Savitha S, Sivarajan K, Haripriya D, Kokilavani V, Panneerselvam C. Efficacy of levo carnitine and alpha lipoic acid in ameliorating the decline in mitochondrial enzymes during aging. Clin Nutr. 2005;24(5):794–800. doi: 10.1016/j.clnu.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 59.Stoyanovsky DA, Tyurina YY, Tyurin VA, Anand D, Mandavia DN, Gius D, Ivanova J, Pitt B, Billiar TR, Kagan VE. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein S-nitrosothiols. J Am Chem Soc. 2005;127(45):15815–23. doi: 10.1021/ja0529135. [DOI] [PubMed] [Google Scholar]
- 60.May JM. How does ascorbic acid prevent endothelial dysfunction? Free Radic Biol Med. 2000;28(9):1421–9. doi: 10.1016/s0891-5849(00)00269-0. [DOI] [PubMed] [Google Scholar]
- 61.Korotchkina LG, Yang H, Tirosh O, Packer L, Patel MS. Protection by thiols of the mitochondrial complexes from 4-hydroxy-2-nonenal. Free Radic Biol Med. 2001;30(9):992–9. doi: 10.1016/s0891-5849(01)00491-9. [DOI] [PubMed] [Google Scholar]