

Abstract
Epigenetic changes, including CpG island hypermethylation, occur frequently in bladder cancer (BC) and may be exploited for BC detection and distinction between high-grade (HG) and low-grade (LG) disease. Genome-wide methylation analysis was performed using Agilent Human CpG Island Microarrays to determine epigenetic differences between LG and HG cases. Pathway enrichment analysis and functional annotation determined that the most frequently methylated pathways in HG BC were enriched for anterior/posterior pattern specification, embryonic skeletal system development, neuron fate commitment, DNA binding, and transcription factor activity. We identified 990 probes comprising a 32-gene panel that completely distinguished LG from HG based on methylation. Selected genes from this panel, EOMES, GP5, PAX6, TCF4, and ZSCAN12, were selected for quantitative polymerase chain reaction–based validation by MethyLight in an independent series (n = 84) of normal bladder samples and LG and HG cases. GP5 and ZSCAN12, two novel methylated genes in BC, were significantly hypermethylated in HG versus LG BC (P ≤ .03). We validated our data in a second independent cohort of LG and HG BC cases (n = 42) from The Cancer Genome Atlas (TCGA). Probes representing our 32-gene panel were significantly differentially methylated in LG versus HG tumors (P ≤ .04). These results indicate the ability to distinguish normal tissue from cancer, as well as LG from HG, based on methylation and reveal important pathways dysregulated in HG BC. Our findings were corroborated using publicly available data sets from TCGA. Ultimately, the creation of a methylation panel, including GP5 and ZSCAN12, able to distinguish between disease phenotypes will improve disease management and patient outcomes.
Introduction
Bladder cancer (BC), the fifth most common cancer in developed countries, is comprised of two distinct pathological entities: low-grade (LG) and high-grade (HG) BC [1], [2]. LG BC tumors seldom progress but often recur; thus, patients require long-term follow-up. Meanwhile, HG tumors are aggressive and have poor prognosis [3], [4]. BC is further defined by the extent of muscle invasiveness, presenting as either non–muscle invasive (NMI, comprising stages Tis, Ta, and T1) in ~80% of cases or muscle invasive (MI, comprising of stage T2 and above) in ~20% of cases [5]. NMIBC, also called superficial or papillary bladder cancer, consists of both LG and HG disease, whereas muscle invasiveness is found mostly in HG BC. The 5-year disease-specific survival rate of LG NMIBC is above 95%, whereas at the other end of the spectrum, HG MIBC has a poor prognosis, with a 5-year survival rate of 63% at stage T2 and with even lower rates for more advanced disease [6].
Currently, cystoscopy and urine cytology are the gold standard for BC diagnosis [7]. However, cystoscopy is costly, invasive in nature, and limited in its accuracy to predict the behavior of BC [8], [9]. Urine cytology, although noninvasive, has low sensitivity for detecting LG BC and is dependent on the pathologist's experience [10]. Therefore, there is a need for more sensitive biomarkers to improve the diagnostic accuracy of urine cytology and assist in distinguishing LG from HG BC. A number of urinary biomarker tests such as UroVysion and NMP22 BladderChek have been approved for use alongside cystoscopy [11], [12]. However, due to their inconsistent performance in terms of specificity and/or sensitivity, the markers proposed to date have not been widely adopted in routine clinical practice.
BC arises from the accumulation of not only genetic but also epigenetic changes. Among epigenetic mechanisms, DNA methylation is the best studied, and aberrant CpG island methylation has been shown to contribute to the development and progression of numerous cancer types including BC [13], [14]. DNA methylation-based biomarkers hold considerable promise to predict the biological behavior of various cancers including biologically and clinically distinct LG and HG BC [14], [15]. Therefore, identifying aberrant DNA methylation events that initiate and/or promote BC development can highlight biological markers for distinguishing LG from HG BC, improving diagnostic accuracy and treatment stratification.
In this study, we used an array-based approach to uncover genome-wide DNA methylation profiles of LG and HG BC, as well as to identify novel differentially methylated genes (DMGs) between the two types of tumors. We further evaluated the methylation patterns of five selected candidate genes in two independent cohorts of BC cases using two different strategies to assess their potential as biomarkers for HG BC.
Material and Methods
Patients and Pathology
For genome-wide CpG island methylation analysis, six fresh frozen urothelial carcinoma BC samples (three LG and three HG) obtained from transurethral resection were collected from the tissue bank at the University Health Network, Toronto, Ontario, Canada. Further stage or other clinicopathological data for these cases are unavailable. For gene-specific methylation analysis by a quantitative polymerase chain reaction (qPCR)–based methylation detection assay, MethyLight [16], a series of 80 (40 LG and 40 HG) formalin-fixed, paraffin-embedded (FFPE) urothelial carcinoma BC samples and 4 normal urothelial tissue samples were similarly collected. The clinicopathological characteristics of the cohort are listed in Table 1. All patients consented to the donation of removed tissue to the University Health Network tissue bank, and samples were obtained according to protocols approved by the Research Ethics Board from University Health Network, Toronto. The complete set of hematoxylin and eosin–stained slides from each transurethral resection or radical cystectomy case was collected and reviewed by an expert pathologist (T.H.V.D.K.) for stage and grade (WHO 2004 classification).
Table 1.
Clinicopathological Variables of LG and HG BC Patients of Cohort Used for MethyLight Gene-Specific Methylation Analysis
| Clinicopathological Variable | LG, N (%) | HG, N (%) |
|---|---|---|
| Gender | ||
| Male | 27 (67.5) | 27 (67.5) |
| Female | 13 (32.5) | 13 (32.5) |
| Age group | ||
| < 50 | 2 (5.0) | 4 (10.0) |
| 50-59 | 9 (22.5) | 8 (20.0) |
| 60-69 | 16 (40.0) | 15 (37.5) |
| 70-79 | 12 (30.0) | 12 (30.0) |
| 80-89 | 1 (2.5) | 1 (2.5) |
| Grade | ||
| LG | 40 (100.0) | 0 (0.0) |
| HG | 0 (0.0) | 40 (100.0) |
| Pathological stage | ||
| Ta | 21 (52.5) | 0 (0.0) |
| T1 | 18 (45.0) | 34 (85.0) |
| T2a | 0 (0.0) | 1 (2.5) |
| T2b | 1 (2.5) | 0 (0.0) |
| T3a | 0 (0.0) | 4 (10.0) |
| T3b | 0 (0.0) | 1 (2.5) |
| Surgery | ||
| Transurethral resection of bladder tumor | 16 (40.0) | 33 (82.5) |
| Radical cystectomy | 7 (17.5) | 7 (17.5) |
| No data | 17 (42.5) | 0 (0.0) |
| Recurrence | ||
| No recurrence | 4 (10.0) | 14 (35.0) |
| Recurrence | 12 (30.0) | 18 (45.0) |
| No data | 24 (60.0) | 8 (20.0) |
| FGFR3 mutation | ||
| Wild type | 10 (25.0) | 30 (75.0) |
| Mutation | 13 (32.5) | 9 (22.5) |
| No data | 17 (42.5) | 1 (2.5) |
| FGFR3 expression | ||
| Not expressed | 6 (15.0) | 18 (45.0) |
| Expressed | 17 (42.5) | 21 (52.5) |
| No data | 17 (42.5) | 1 (2.5) |
| P53 expression | ||
| Not expressed | 22 (55.0) | 18 (45.0) |
| Expressed | 1 (2.5) | 22 (55.0) |
| No data | 17 (42.5) | 0 (0.0) |
| P27 expression | ||
| Not expressed | 13 (32.5) | 21 (52.5) |
| Expressed | 10 (25.0) | 19 (47.5) |
| No data | 17 (42.5) | 0 (0.0) |
| EOMES median PMR (range) | 30.2 (0.0-100.0) | 41.8 (1.8-100.0) |
| GP5 median PMR (range) | 0.0 (0.0-53.7) | 1.6 (0.0-87.6) |
| PAX6 median PMR (range) | 19.0 (0.0-69.4) | 22.4 (0.0-100.0) |
| TCF4 median PMR (range) | 0.0 (0.0-37.5) | 0.2 (0.0-37.9) |
| ZSCAN12 median PMR (range) | 0.0 (0.0-73.56) | 10.9 (0.0-74.9) |
DNA Extraction and Bisulfite Modification
DNA was extracted from fresh frozen and FFPE tissues using QIAamp DNA Mini Kit (Qiagen, Mississauga, ON, Canada) according to the kit protocol for fresh frozen tissues and using a modified protocol which has previously been described for FFPE tissue [17]. DNA extraction was performed in areas containing a minimum of 70% tumor cells as assessed by histological examination. One hundred to 400 ng of DNA extracted from primary tumors and normal tissue was converted using the EZ DNA Methylation Gold Kit (Zymo Research, Orange, CA) according to the manufacturer's protocol and eluted to a final concentration of 10 ng/μl.
FGFR3 Mutation Analysis
Mutation analysis of fibroblast growth factor receptor 3 (FGFR3) was performed using PCR-SNaPshot method as described previously [18]. Three regions (exon 7, 10, and 15) representing at least 99% of activating oncogenic FGFR3 mutations in BC were amplified by PCR. Excess primer and deoxynucleotides were removed, specific SNaPshot primers were annealed to the PCR products, and then the products were separated by capillary electrophoresis and analyzed in an automatic sequencer (Prism 3100 genetic analyzer).
Protein Expression Analysis
Protein expression of FGFR3, P53, and P27 was determined by immunohistochemistry. Positive and negative controls were included for each run and assessed by B.W.V.R. and T.H.V.D.K. Monoclonal antibodies were used against FGFR3 (FGFR3 B9, Santa Cruz, CA), P53 (clone DO-7), and P27 (clone 57) as described previously [19].
Human CpG Island Microarrays
Methylated DNA was analyzed using the differential methylation hybridization technique and was co-hybridized to Agilent Human CpG Island Microarrays at the University Health Network Microarray Centre according to the manufacturer's recommended protocol. Genomic DNA (100-200 ng) from bladder tumors was first digested with MseI. H-12/H-24 linker oligonucleotides were annealed together, creating overhangs that bind MseI-digested DNA. DNA was then ligated to the MseI cleaved ends using T4 DNA ligase and overnight ligation at 4°C. Ligated DNA was then sequentially digested first with HpaII followed by BstUI, both of which are methylation-sensitive enzymes. Amplification of intact fragments was then performed with ThermoPol Taq polymerase (New England Biolabs, Pickering, ON, Canada) with the following conditions: 72°C for 5 minutes, 95°C for 1 minute, 24 cycles of 95°C for 1 minute followed by 67°C for 2.5 minutes, and a final step of 72°C for 5 minutes. Following PCR, DNA was purified using the QIAquick PCR purification kit (Qiagen, Mississauga, ON, Canada) according to the manufacturer's instructions. Reference DNA, which was co-hybridized with tumor DNA to microarrays, was prepared in a similar fashion using lymphocyte DNA pooled from six healthy age-matched men.
Data Analysis
To identify the differentially methylated loci most robustly associated with HG disease, microarray data was analyzed using two distinct, commonly applied statistical approaches, Linear Models for Microarray Data (Limma) and local-pooled-error (LPE), suitable for analyzing small sample sizes. Probes were filtered to only include sequences with minimum two-fold methylation enrichment or depletion and P value ≤ .05 in both Limma and LPE methods. Only those gene loci that overlapped in significantly differentiating LG from HG tumors based on analysis by both Limma and LPE methods are reported.
Unsupervised hierarchical average linkage clustering with the most variant methylated probes was performed with the use of GenePattern Version 3.8.2 software (http://www.broad.mit.edu/cancer/software/genepattern/) [20].
Pathway Analysis
Pathway enrichment analyses and functional annotation were performed using the Genomic Regions Enrichment of Annotations Tool (GREAT) version 2.0.2 using the whole human genome as background [21]. Only significantly differentially methylated probes between LG and HG cases by Limma and LPE analyses were used for this analysis.
MethyLight
Methylation analysis was performed on tumor and normal tissue using the semiquantitative MethyLight assay, a TaqMan-based qPCR technique that assesses percent DNA methylation at a defined gene locus [16]. In brief, 10 ng of bisulfite-converted genomic DNA was amplified using locus-specific PCR primers flanking an oligonucleotide probe with a 5′ fluorescent reporter dye and a 3′ quencher dye. Primers and probe sequences used for eomesodermin (EOMES), glycoprotein V platelet (GP5), paired box 6 (PAX6), transcription factor 4 (TCF4), zinc finger and SCAN domain containing 12 (ZSCAN12), and the reference sequence ALU-C4 are shown in Supplementary Table 1. A percent methylation ratio (PMR) score was calculated for each gene locus by dividing the gene:ALU-C4 ratio of a sample by the gene:ALU-C4 ratio of commercially available fully methylated DNA (Millipore, Billerica, MA) and multiplying by 100. Samples were analyzed in duplicate on 96-well plates on an ABI 7500 RT-PCR thermocycler (Foster City, CA).
The Cancer Genome Atlas (TCGA) Data Set
Clinical data were downloaded from TCGA Data Portal for 412 bladder urothelial carcinoma cases in March 2016. All cases were muscle-invasive cancers. Of the 412 cases, 21 were LG. The LG cases consisted of 18 pathological stage T2 and 3 T3 cases. The HG cases consisted of eight T2, six T3, and seven T4 cases. All available LG cases were selected for analysis, of which 20 were male and 1 was female, with an average age of 58.7. From the remaining HG cases, 21 were randomly selected after matching based on age and sex to the LG cases with an average age of 60.3. For these 42 total selected cases, level 3 methylation data from Illumina Infinium HumanMethylation450 BeadChip arrays were downloaded from TCGA in March 2016. Methylation data were also downloaded for all 21 benign bladder tissue samples available. The benign tissue came from 11 males and 10 females with an average age of 69.2, all of which were from patients with HG BC.
Statistical Analysis
For each sample, following MethyLight analysis of EOMES, GP5, TCF4, PAX6, and ZSCAN12 genes, PMR values were obtained from averaging duplicate runs. Ten HG and two LG cases had both superficial and invasive tumor tissue available. No significant differences in methylation were observed between invasive and superficial tissue using the Wilcoxon test for related samples. Thus, the PMR values for both tissue types were averaged in subsequent statistical analysis. Comparison of median PMR values between LG, HG, and normal samples and other clinicopathological variables was performed using the Mann-Whitney U test or Kruskal-Wallis test, where appropriate. Receiver operating characteristic (ROC) curves were generated to obtain area under the curve (AUC) values. Mann-Whitney U test or Kruskal-Wallis test was utilized to compare benign tissue, LG, HG, and stage for select CpG probes from TCGA data. Multivariate analysis was not undertaken in order to prevent overfitting of data. For all described methods, two-sided P values ≤ .05 were considered significant. Statistics were performed using IBM SPSS Statistics 21 (Armonk, NY).
Results
Genome-Wide Methylation in LG and HG BC
We profiled the genome-wide DNA methylation status of three LG and three HG BCs using Human CpG Island Microarrays comprising 237,220 probes encompassing 27,800 CpG islands. Following genome-wide screening, we separated the analysis of our microarray data into LG versus HG BC tumors. Overall, there was a general increase in methylation in HG compared to LG tumors. Of 584 significantly differentially methylated probes, 568 had significantly increased methylation in HG compared to LG tumors (representing 232 genes, 139 unassigned intergenomic regions, and 2 microRNAs), whereas only 16 probes (representing 13 genes) had significantly decreased methylation. A clustering dendrogram and heat map of the top 50 hypermethylated and all of the hypomethylated probes are shown in Figure 1. Of the annotated significantly differentially methylated regions, 42% were located within gene promoters, 46% were intragenic, and 12% were intergenic. The top 32 genes, ranked by fold change, that were identified as significantly hyper- and hypomethylated in HG versus LG cancers are listed in Table 2.
Figure 1.
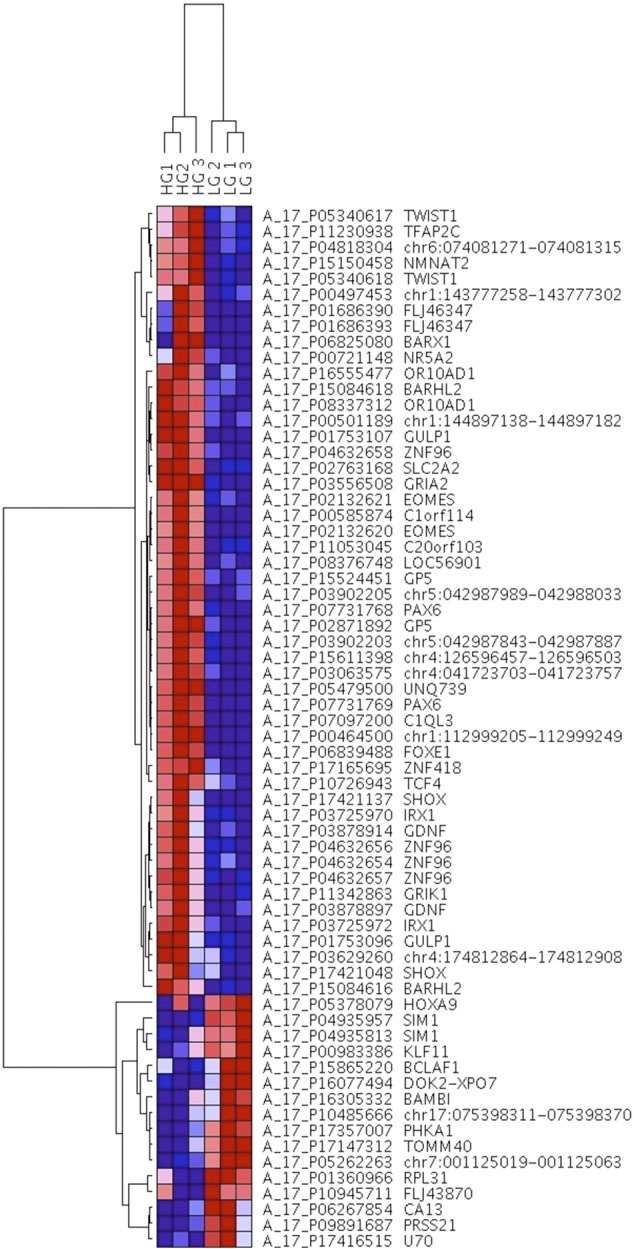
DNA methylation hierarchical clustering dendrogram and heat map of the top 50 hypermethylated probes and all 16 hypomethylated probes showing the greatest differential methylation across LG and HG samples, listed on the top. Gene probes and names are listed on the right. DNA methylation values are represented as colors, with red representing DNA hypermethylation and blue representing DNA hypomethylation.
Table 2.
List of the 19 Most Significantly Hypermethylated and 13 Hypomethylated Genes between LG and HG BC, Comprising a 32-Gene Panel
| Gene Name (Abbreviation) | Number of Probes | Fold Change |
P Value |
|
|---|---|---|---|---|
| LPE | Limma | |||
| Von Willebrand factor C domain containing 2 (UNQ739) | 1 | 66.7 | 3.51 × 10−6 | 2.16 × 10−5 |
| Uncharacterized LOC389064 (FLJ46347) | 2 | 41.3 | .0006 | .0332 |
| Eomesodermin (EOMES) | 3 | 36.5 | .0104 | .0016 |
| Glycoprotein V (platelet) (GP5) | 4 | 36.3 | .0108 | .0006 |
| Solute carrier family 2 (facilitated glucose transporter), member 2 (SLC2A2) | 2 | 36.1 | .0116 | .0002 |
| Glial cell–derived neurotrophic factor (GDNF) | 4 | 33.6 | .0033 | .0136 |
| Short stature homeobox (SHOX) | 3 | 32.6 | .0156 | .0238 |
| Olfactory receptor, family 10, subfamily AD, member 1 (OR10AD1) | 4 | 31.8 | .0013 | .0005 |
| Glutamate receptor, ionotropic, kainate 1 (GRIK1) | 1 | 29.4 | .0017 | .0035 |
| Zinc finger protein 418 (ZNF418) | 1 | 28.8 | 1.13 × 10−5 | .0006 |
| Forkhead box E1 (thyroid transcription factor 2) (FOXE1) | 1 | 28.3 | .0005 | .0001 |
| Zinc finger and SCAN domain containing 12 (ZSCAN12/ZNF96) | 6 | 26.8 | .0096 | .0062 |
| Homeobox D9 (HOXD9) | 1 | 24.4 | 7.63 × 10−6 | .0017 |
| Phosphatidylinositol-3,4,5-trisphosphate–dependent Rac exchange factor 1 (PREX1) | 1 | 23.4 | .0004 | .0311 |
| Engulfment adaptor PTB domain containing 1 (GULP1) | 6 | 23.1 | .0025 | .0069 |
| Myosin binding protein C, slow type (MYBPC1) | 1 | 23.0 | .0009 | .0071 |
| Motor neuron and pancreas homeobox 1 (HLXB9) | 1 | 22.3 | 4.02 × 10−5 | .0003 |
| Glutamate receptor, ionotropic, AMPA 2 (GRIA2) | 2 | 22.0 | .0002 | .0003 |
| Paired box 6 (PAX6) | 8 | 21.8 | .0063 | .004 |
| small nucleolar RNA, H/ACA box 70 (U70) | 1 | −8.9 | .033 | .011 |
| Protease, serine, 21 (PRSS21) | 1 | −7.8 | .043 | .008 |
| Carbonic anhydrase XIII (CA13) | 1 | −7.7 | .021 | .019 |
| Translocase of outer mitochondrial membrane 40 homolog (TOMM40) | 1 | −6.9 | .029 | .002 |
| Phosphorylase kinase, alpha 1 (PHKA1) | 1 | −6.9 | .047 | .004 |
| DOK2-XPO7 | 1 | −6.4 | .043 | .012 |
| Kruppel-like factor 11 (KLF11) | 1 | −6.3 | .018 | .018 |
| BCL2-associated transcription factor 1 (BCLAF1) | 1 | −6.2 | .027 | .024 |
| Single-minded homolog 1 (Drosophila) (SIM1) | 2 | −5.0 | .045 | .006 |
| FLJ43870 | 1 | −4.7 | .043 | .028 |
| BMP and activin membrane-bound inhibitor homolog (Xenopus laevis) (BAMBI) | 1 | −4.4 | .044 | .039 |
| Ribosomal protein L31 (RPL31) | 1 | −4.3 | .045 | .011 |
| Homeobox A9 (HOXA9) | 1 | −3.8 | .043 | .002 |
To distinguish potentially important patterns between LG and HG BC, DNA methylation profiles of the tumors were analyzed by unsupervised hierarchical clustering. Two robust DNA methylation clusters were identified: one encompassing all of the HG tumors and another containing all of the LG tumors, consisting of 32 genes (represented by 990 probes) with significant differential methylation. Thus, the 32 DMGs could separate LG from HG BC. Of the 32 DMGs, 19 showed hypermethylation and 13 hypomethylation in HG BC. As expected, some of the 32 DMGs were previously reported to be differentially methylated in BC including EOMES and PAX6 [22], [23], [24], [25], [26]. In addition, our analysis identified novel DMGs that have not been implicated in BC previously, such as GP5 and ZSCAN12.
Pathway Analysis of DMGs
To generate further insight into pathways targeted by the changes in DNA methylation between LG and HG BC, we used GREAT to determine if significantly differentially methylated regions were enriched for any functional processes [20]. The most frequently enriched biological processes as determined by gene ontology (GO) term analysis were anterior/posterior pattern specification (GO:0009952, GREAT Binom Raw P value = 5.8e-39), embryonic skeletal system development (GO:0048706, P value = 2.2e-28), and neuron fate commitment (GO:0048663, P value = 7.0e-24). Additionally, within the molecular function category, the most enriched GO terms were associated with transcriptional regulation including transcription regulatory region sequence-specific DNA binding (GO:0000976, GREAT Binom Raw P value = 6.4e-33), RNA polymerase II core promoter proximal region sequence-specific DNA binding transcription factor activity involved in positive regulation of transcription (GO:0001077, P value = 2.7e-20), and RNA polymerase II core promoter proximal region sequence-specific DNA binding transcription factor activity (GO:0000982, P value = 5.0e-20). Among these enriched DMG sets, we found numerous genes whose function in tumorigenesis is well documented in the literature, such as SMAD4, IHH, GLI3, WNT1, and WNT5a [27], [28], [29], [30], [31].
Selection of Candidate Biomarker Genes
We next selected 5 promising genes from the 32 DMGs for further analysis based on statistical significance from microarray results, number of significant probes, biological function of the gene, involvement in BC, and novelty. EOMES and PAX6 were found to be significantly hypermethylated by our microarray profiling, with fold change values of 36.5 and 21.8 in HG versus LG, respectively. GP5 and ZSCAN12 are novel methylated genes identified in our study whose methylation has not previously been established in BC. GP5 exhibited hypermethylation 36.3-fold higher in HG versus LG in our array profiling, and ZSCAN12 methylation was 26.8-fold higher in HG versus LG. TCF4, although not in the 32-gene panel, showed 21.3-fold hypermethylation in HG versus LG tumors and was in the top 100 significantly DMGs.
Gene-Specific Methylation Analysis
To verify the quantitative promoter methylation patterns of five DMGs identified from our microarray findings, we extended our analysis to an independent cohort of normal urothelial tissue samples (n = 4) plus 80 LG and HG BCs (n = 40 for each group) using MethyLight assay. Median PMR values for all tumor cases were 31.8% for EOMES, 0% for GP5, 20.2% for PAX6, 0.1% for TCF4, and 3.1% for ZSCAN12. We next compared median PMR values of each gene of interest between normal tissue, LG, and HG cases (Table 3). Figure 2 shows the distribution of relative methylation values for each gene of interest. None of the normal cases were highly methylated for any of the five genes, and median PMR values increased with grade. This association was statistically significant for the methylation of EOMES, GP5, and ZSCAN12 (P = .001, .008, and .04, respectively). Comparing LG and HG, median tumor DNA methylation was significantly higher in HG for ZSCAN12 (0% vs 10.9%, P = .03) and GP5 (0% vs 1.6%, P = .006), whereas EOMES methylation trended towards significance (30.2% vs 41.8%, P = .05). TCF4 and PAX6 did not show significant differences between normal and tumor DNA or LG versus HG.
Table 3.
Associations between Methylation of Five Genes and Clinicopathological Variables in BC Patients
| Clinicopathological Variable | EOMES PMR | GP5 PMR | PAX6 PMR | TCF4 PMR | ZSCAN12 PMR |
|---|---|---|---|---|---|
| Normal vs cancer | |||||
| Normal | 0.0 | 0.0 | 3.9 | 0.0 | 0.9 |
| Cancer | 31.8 | 0.0 | 20.2 | 0.1 | 3.1 |
| P value | 1.2 × 10−4 | .22 | .06 | .19 | .29 |
| Normal vs LG vs HG | |||||
| Normal | 0.0 | 0.0 | 3.9 | 0.0 | 0.9 |
| LG | 30.2 | 0.0 | 19.0 | 0.01 | 0.0 |
| HG | 41.8 | 1.6 | 22.4 | 0.2 | 10.9 |
| P value | .001 | .008 | .11 | .32 | .04 |
| LG vs HG | |||||
| LG | 30.2 | 0.0 | 19.0 | 0.01 | 0.0 |
| HG | 41.8 | 1.6 | 22.4 | 0.2 | 10.9 |
| P value | .05 | .006 | .34 | .53 | .03 |
| Normal vs LG | |||||
| Normal | 0.0 | 0.0 | 3.9 | 0.0 | 0.9 |
| LG | 30.2 | 0.0 | 19.0 | 0.01 | 0.0 |
| P value | .002 | .43 | .07 | .23 | .66 |
| Normal vs HG | |||||
| Normal | 0.0 | 0.0 | 3.9 | 0.0 | 0.9 |
| HG | 41.8 | 1.6 | 22.4 | 0.2 | 10.9 |
| P value | 1.8 × 10−5 | .11 | .06 | .19 | .10 |
| Stage | |||||
| Ta | 9.7 | 0.0 | 9.3 | 0.01 | 0.0 |
| T1 | 46.1 | 0.0 | 26.7 | 0.2 | 10.9 |
| T2a | 31.0 | 16.4 | 16.0 | 0.0 | 0.0 |
| T2b | 15.8 | 5.8 | 7.4 | 4.6 | 0.8 |
| T3a | 18.4 | 1.7 | 3.4 | 0.6 | 3.4 |
| T3b | 55.7 | 62.7 | 1.0 | 0.0 | 12.4 |
| P value | .047 | .51 | 4.6 × 10−4 | .03 | .001 |
| Recurrence | |||||
| No recurrence | 43.5 | 0.0 | 22.8 | 0.04 | 0.3 |
| Recurrence | 46.1 | 2.5 | 30.7 | 0.7 | 11.7 |
| P value | .80 | .17 | .17 | .64 | .23 |
| P53 expression | |||||
| Not expressed | 21.6 | 0.0 | 12.9 | 0.01 | 0.3 |
| Expressed | 41.4 | 0.0 | 26.3 | 0.01 | 1.1 |
| P value | .09 | .02 | .31 | .35 | .39 |
| P27 expression | |||||
| Not expressed | 31.0 | 0.0 | 23.9 | 0.0 | 4.6 |
| Expressed | 22.8 | 0.0 | 12.9 | 0.02 | 0.0 |
| P value | .91 | .001 | .80 | .88 | .36 |
| FGFR3 mutation | |||||
| Wild-type | 28.5 | 0.0 | 16.6 | 0.0 | 2.1 |
| Mutated | 36.4 | 0.0 | 23.9 | 0.2 | 0.0 |
| P value | .09 | .11 | .81 | .37 | .005 |
| FGFR3 expression | |||||
| Not expressed | 30.0 | 0.0 | 14.6 | 0.02 | 0.5 |
| Expressed | 27.9 | 0.0 | 23.8 | 0.01 | 0.3 |
| P value | .30 | .002 | .29 | .34 | .005 |
Median PMR and Kruskal-Wallis or Mann-Whitney U test P values are shown.
Figure 2.
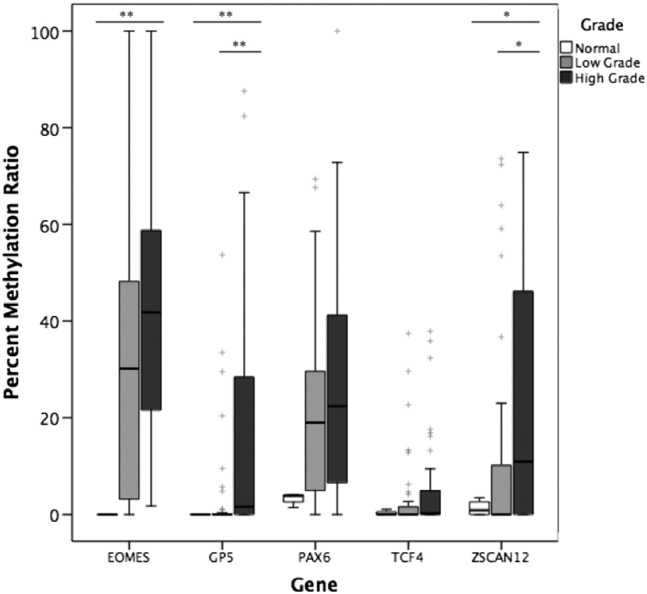
Methylation in normal urothelial tissue, LG BC, and HG BC tissue in EOMES, GP5, PAX6, TCF4, and ZSCAN12 genes measured by MethyLight. Mann-Whitney U test was used to compare methylation between normal urothelial tissue (n = 4), LG (n = 40), and HG (n = 40). Kruskal-Wallis test was used to compare methylation between LG and HG. *P < .05, **P < .01. + represents outliers.
Comparisons among different pathological tumor stages were performed for each DMG. Methylation differed significantly among stages for four of the five candidate genes: EOMES, PAX6, TCF4, and ZSCAN12 (P = .047, 4.6 × 10−4, .03, and .001, respectively). There were no significant differences among patients that recurred versus those that did not recur.
Next, we examined associations between several key prognostic molecular alterations in BC (FGFR3 mutations; expression of FGFR3, P53, and P27) and methylation patterns of the five DMGs. FGFR3 mutation and expression are associated with LG BC and NMIBC, P53 expression is associated with LG BC, and P27 expression is associated with recurrence and progression [19]. Of the five genes assessed, GP5 methylation was significantly differentially methylated between tumors with or without P53 expression (P = .02), P27 expression (P = .001), and FGFR3 expression (P = .002). Higher ZSCAN12 methylation was significantly associated with wild-type FGFR3 (P = .005) and inversely associated with its expression (P = .005). The methylation of the remaining DMGs was not significantly different when stratified according to FGFR3 mutations or FGFR3, P53, and P27 expression. Similar results were seen for the above gene-specific methylation analysis when MIBC cases were removed and only NMIBC cases were analyzed (Supplementary Table 2).
We also generated ROC curves to determine the ability of each individual gene marker to discriminate between LG and HG (Figure 3). GP5 and ZSCAN12 had significant P values (P = .02 and P = .03, respectively). The AUC for GP5 was 0.656 and for ZSCAN12 was 0.652. The combination of GP5 and ZSCAN12 methylation together achieved the highest AUC of any single gene or other genes combined, P = .006 and AUC = 0.679. EOMES was borderline significant, with P = .051 and AUC = 0.630. PAX6 and TCF4 were not significant.
Figure 3.

ROC curves for discrimination of HG BC. ROC curves and AUC values were generated for 5 genes to compare methylation in 40 LG and 40 HG cases. The genes assessed were (A) EOMES, (B) GP5, (C) PAX6, (D) TCF4, (E) ZSCAN12, and (F) combination of GP5 and ZSCAN12.
Gene-Specific Validation in TCGA Data Set
To further validate our findings in an independent cohort, we utilized publicly available methylation data from TCGA bladder urothelial carcinoma cases (TCGA Research Network: http://cancergenome.nih.gov/). Methylation between 21 LG and 21 HG BC cases was compared for each Illumina 450K probe located within or in proximity to the 32 genes of our panel.
A total of 865 CpG probes on the Illumina 450K array, incorporating associated upstream and downstream regulatory regions including CpG islands, shores, and shelves, represented the genes of our panel. The loci FLJ46347, FLJ43870, and SHOX from our 32-gene panel were not represented on the 450K arrays. One hundred thirty-five of 865 (15.6%) probes were significantly differentially methylated between LG and HG. Every gene from our panel except four (SLCA2, ZNF418, GRIA2, CA13) had at least one significant probe, including probes from all five selected candidate genes we validated by MethyLight. The most significant differentially methylated probe between LG and HG cases is listed in Table 4 for EOMES, GP5, PAX6, TCF4, and ZSCAN12. Figure 4 shows the distribution of relative methylation values for each gene of interest. All five genes were significantly hypermethylated in HG compared to LG BC (P < .05). Methylation at the most significantly differentially methylated probe between LG and HG was not significantly different among different stages (comparison of stages 2-4).
Table 4.
Associations between Methylation of a Five-Gene Panel and Grade in BC Patients from TCGA
| Gene | EOMES | GP5 | PAX6 | TCF4 | ZSCAN12 |
|---|---|---|---|---|---|
| Probe ID | cg21473142 | cg18780769 | cg24701575 | cg23482397 | cg20271532 |
| Benign vs cancer | |||||
| Benign | 0.261 | 0.137 | 0.137 | 0.022 | 0.072 |
| Cancer | 0.606 | 0.129 | 0.281 | 0.028 | 0.098 |
| P value | .003 | .94 | .02 | 1.9 × 10−4 | .31 |
| Benign vs LG vs HG | |||||
| Benign | 0.261 | 0.137 | 0.137 | 0.022 | 0.072 |
| LG | 0.314 | 0.092 | 0.156 | 0.026 | 0.056 |
| HG | 0.753 | 0.232 | 0.465 | 0.046 | 0.225 |
| P value | 2.7 × 10−5 | 2.9 × 10−4 | 1.3 × 10−4 | 1.7 × 10−4 | .02 |
| LG vs HG | |||||
| LG | 0.314 | 0.092 | 0.156 | 0.046 | 0.225 |
| HG | 0.753 | 0.232 | 0.565 | 0.026 | 0.056 |
| P value | .003 | .001 | .001 | .04 | .02 |
| Benign vs LG | |||||
| Benign | 0.261 | 0.137 | 0.137 | 0.022 | 0.072 |
| LG | 0.314 | 0.092 | 0.156 | 0.026 | 0.056 |
| P value | .77 | .009 | .97 | .01 | .46 |
| Benign vs HG | |||||
| Benign | 0.246 | 0.137 | 0.137 | 0.022 | 0.072 |
| HG | 0.753 | 0.232 | 0.565 | 0.046 | 0.225 |
| P value | 2.3 × 10−6 | .01 | 5.5 × 10−5 | 9.3 × 10−5 | .01 |
| Stage | |||||
| T2 | 0.321 | 0.125 | 0.226 | 0.027 | 0.079 |
| T3 | 0.671 | 0.098 | 0.280 | 0.046 | 0.070 |
| T4 | 0.732 | 0.195 | 0.347 | 0.027 | 0.429 |
| P value | .12 | .39 | .77 | .59 | .44 |
The most significant probe within each gene for the comparison of LG to HG was used for each association. Median beta values and Mann-Whitney U test or Kruskal-Wallis test P values are shown.
Figure 4.
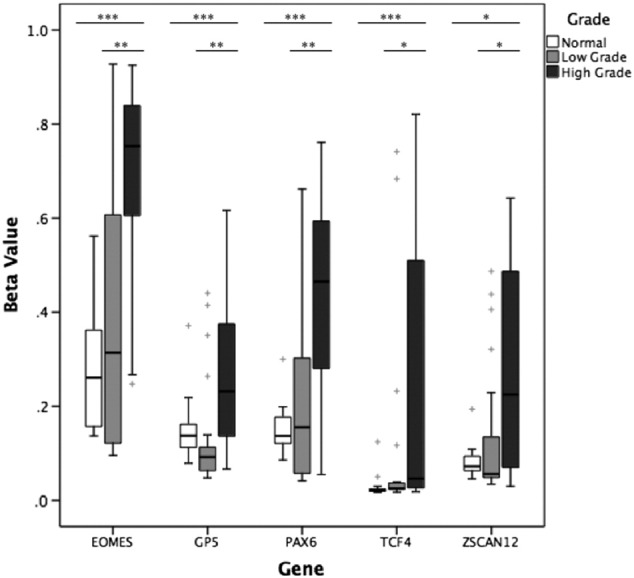
Methylation in benign urothelial tissue, LG BC, and HG BC tissue in EOMES, GP5, PAX6, TCF4, and ZSCAN12 genes measured by Illumina 450K array through TCGA. Mann-Whitney U test was used to compare methylation between benign urothelial tissue (n = 21), LG MIBC (n = 21), and HG MIBC (n = 21). Kruskal-Wallis test was used to compare methylation between LG and HG. *P < .05, **P < .01, and ***P < .001. + represents outliers.
Methylation from 21 benign bladder tissue samples from TCGA BC patients was also assessed in comparison to cancer cases. Of 865 probes within 29 genes of our panel, 525 (60.7%) were significantly differentially methylated between benign and LG, 593 (68.6%) were significantly differentially methylated between benign and HG, and 652 (75.4%) were significantly differentially methylated between benign and all cancer cases (LG + HG), P < .05. Every gene from our panel represented by CpG probes on the 450K array had at least three significant probes for each comparison. Comparison between benign and LG, HG, or cancer of any grade is indicated in Table 4 at the CpG probe that was most significant for the comparison of LG versus HG as described earlier. EOMES, PAX6, and TCF4 were significantly hypermethylated in cancer versus benign (P < .05). When stratified by tumor grade, TCF4 was significantly hypermethylated in LG compared to benign tissue (P = .01), whereas all five genes were significantly hypermethylated in HG (P < .05).
Discussion
In this study, we used a combination of different strategies to identify and characterize methylation biomarkers that will discriminate between LG versus HG tumors. We used human CpG island microarrays to identify DMGs in LG versus HG BC. We have shown that there are considerable methylation changes in BC with a significant overall shift to increased localized methylation profile within CpG islands in higher-grade tumors. Furthermore, we were able to identify a DNA methylation signature of 32 genes that can completely discriminate LG from HG BC. Of these, significant tumor-specific DNA hypermethylation of EOMES, GP5, and ZSCAN12 in HG BC was confirmed in our validation cohort and TCGA cohort.
Using Agilent Human CpG Island Microarrays, we found 245 genes that significantly differed in methylation in HG compared to LG tumors. Consistent with previous genome-wide studies of DNA methylation, we observed an overall increase in DNA methylation in HG BC. Many of the hypermethylated genes we identified in our study have been previously reported in BC, including CDKN2A and TWIST1 [14]. Among our 32-gene panel, we selected PAX6 and EOMES for independent validation because their methylation has previously been investigated in tumor as well as urine samples of BC patients, with EOMES methylation being associated with recurrence and higher grade [22], [23], [25], [26]. Our study also identified novel hypermethylated loci for potential biomarkers such as GP5, TCF4, and ZSCAN12.
A number of other studies have investigated DNA methylation changes in BC with regards to grade, detection, prognosis, progression, recurrence, survival, and stage [14], [32]. Several methylation markers previously shown to be associated with grade in bladder cancer are CD99, CDH13, RARB, RASSF1A, SFRP5, and TMEFF2 [33], [34], [35], [36]. A number of other markers of BC grade have also been shown specifically in NMIBC including ACTL5B, ATP5G2, BCL2, BRCA1, CIDEA, DAPK, FRZB, HOXB2, ITPKB, IRX1, KRT13, MGMT, MLL3, RASSF1A, RUNX3, TERT, TET2, TRPA1, VAX2, and VHL [37], [38], [39], [40], [41], [42], [43]. Of these markers, several have been implicated across multiple studies, including BRCA1, CDH13, RASSF1A, and TMEFF2, whereas other novel markers have emerged from only single studies thus far [24], [33], [35], [37], [39], [44], [45], [46]. This is in line with our own results, in which several of the DMGs from our panel had previously been implicated in BC, such as EOMES and PAX6, whereas others were unique, including GP5 and ZSCAN12.
In our study, we included several MIBC cases in our analysis, and we were able to detect significant differences in methylation between LG and HG at GP5 and ZSCAN12. We also showed these significant differences in analysis excluding MIBC cases (Supplementary Table 2). Furthermore, methylation differences were apparent between LG and HG cases from the TCGA cohort despite the fact that all cases were MIBC. Thus, the methylation changes we observed are likely specific to high-grade cancers. Discrepancies between our results and previous findings may be due to a number of differences between our approach and those previously utilized. These include different positions of CpGs covered, methods of data analysis, array platforms used, and populations, as well as contributions from the tumor microenvironment and tumor heterogeneity. Wherever possible, we designed our MethyLight primers and probes to overlap some of the CpGs in the significant Agilent array probes. A potential limitation of our study is that we interrogated a limited number of LG and HG cases for genome-wide methylation and therefore could not detect differential methylation of these genes in BC.
Our analysis has shown that a signature of 32 DMGs represented by 990 probes can completely discriminate LG from HG BC. Among these, certain pathways of clear relevance to cancer, such as transcription factors within development and differentiation pathways, were enriched to a larger extent in HG BC. This suggests a functional role for DNA methylation in regulating the genes in these pathways, as multiple DNA methylation events at different levels may occur to assure that essential cancer-preventing pathways are downregulated or silenced in HG bladder tumors. DNA demethylating agents, currently approved for the treatment of lymphomas and myelomas, may potentially reverse these methylation patterns in BC or other solid tumors. Treatment of BC cell lines with demethylating agents inhibits their proliferation, migration, and invasion and has also been shown to sensitize cells resistant to cisplatin-based chemotherapy [47], [48].
The results of this study verify previous reports of EOMES hypermethylation in BC tissue and its association with HG disease [25], [26]. Additionally, EOMES hypermethylation has been detected in urine of BC patients and is associated with recurrence as well as HG BC. GP5 and ZSCAN12 methylation has not previously been reported in the literature. GP5 is part of the Ib-V-IX system of surface glycoproteins that constitute the receptor for von Willebrand factor, whereas ZSCAN12 is a transcription factor. The role of these two novel genes identified in our study in BC tumorigenesis is currently unknown and warrants further investigation. In addition, the role of EOMES, a transcription factor required for development of mesoderm and the central nervous system, in BC also requires further research. Despite previous studies showing significant methylation of the transcription factor PAX6 in BC, we did not find there to be any association between PAX6 methylation and tumor grade upon MethyLight analysis in 40 LG and 40 HG tumors. We also did not find any association between transcription factor TCF4 methylation and cancer, tumor grade, or other molecular characteristics of our BC cases when measured in a larger population. This may be due to differences in assay type as well as interpatient tumor heterogeneity, as we started with a smaller cohort of six samples. Whether methylation plays a role in the downregulation of expression of these genes in BC or is solely a marker of tumor grade remains to be determined. For our 40 LG and 40 HG BC cases, we do not have progression or survival data and have incomplete recurrence data; thus, these markers require further investigation to determine their association with specific disease outcomes.
Harnessing TCGA methylation data, we examined the same five genes in a second cohort. We found that all five genes validated by MethyLight analysis had at least four CpG probes per gene that were significantly differentially methylated on the 450K array for the comparison of LG versus HG. Thus, the independent TCGA array data validated our original Agilent array findings. A comparison of benign bladder tissue and HG cases was also significant for each of the five genes tested, indicating that these markers may have clinical utility for diagnosis as well as discernment of grade in BC. Despite our promising results, we recognize that this is not a comprehensive analysis of the TCGA data set and further exploration of this large data set should be performed. Also, although the TCGA data set provided a well-characterized independent cohort of cases, only 21 cases were LG and all were MIBC. Thus, our gene panel warrants further investigation of methylation markers in large, well-characterized patient cohorts representing all grades equally to determine their utility as prognostic and predictive biomarkers for aggressive BC. Additionally, the TCGA probes did not cover the same CpG sites as our Agilent arrays or MethyLight primers. Thus, we provided results for the most significant probes located within each of the five genes, regardless of their location.
The current grading system for BC is limited in its reproducibility among pathologists, which can impact patient management decisions [5], [49]. Methylation-based testing could complement pathological grading to allow for more accurate prediction of disease aggressiveness at the time of BC diagnosis, which will greatly improve clinical decision-making and impact on patient management and quality of life [7]. DNA methylation-based biomarkers have a great potential for clinical utility because some DNA methylation changes may represent early events and it is a stable mark that can be easily determined in archived material as well as samples collected in a noninvasive manner such as urine. Assessment of methylation levels in urine of BC patients could lead to the development of a noninvasive screen or a diagnostic aid for use in conjunction with traditional cystoscopy and/or urine cytology for BC. Urinalysis utilizing methylation markers could also potentially be applied to other aspects of BC, for example, monitoring response to Bacillus Calmette-Guérin therapy. Furthermore, DNA methylation can be analyzed with great sensitivity and specificity using an increasing number of high-throughput methods.
Conclusions
Our findings indicate significant differences in global methylation patterns between LG and HG BC, with HG tumors exhibiting a general increase in methylation. Among these, we identified a 32-gene methylation signature that can completely separate LG from HG BC. In addition, we validated hypermethylation marks in GP5, EOMES, and ZSCAN12 genes that should be further investigated as markers of BC detection in screening and differentiation of more aggressive disease.
The following are the supplementary data related to this article.
Primer and Probe Sequences Used in the MethyLight Assay for ALU-C4, EOMES, GP5, TCF4, PAX6, and ZSCAN12 Genes
Associations between Methylation of Five Genes and Clinicopathological Variables in Non–Muscle Invasive Bladder Cancer Patients (Median Beta Values and Kruskal-Wallis or Mann-Whitney U Test P Values Are Shown)
Funding
This work was supported by Ontario Graduate Scholarship (E.O.M.), Ontario Student Opportunity Trust Fund (E.O.M.), University of Toronto (A.J.S.), Sinai Health System (A.J.S.), and Canadian Institutes of Health Research Interdisciplinary Health Research Team Program (A.J.S.). The funding sources had no involvement in the study design, collection, analysis and interpretation of data, or in writing the report.
Footnotes
This work was supported by Ontario Graduate Scholarship (E.O.M.), Ontario Student Opportunity Trust Fund (E.O.M.), University of Toronto (A.J.S.), Sinai Health System (A.J.S.), and Canadian Institutes of Health Research Interdisciplinary Health Research Team Program (A.J.S.). The funding sources had no involvement in the study design, collection, analysis and interpretation of data, or in writing the report.
Contributor Information
Ekaterina Olkhov-Mitsel, Email: olkhov@lunenfeld.ca.
Andrea J. Savio, Email: savio@lunenfeld.ca.
Ken J. Kron, Email: kenjkron@gmail.com.
Vaijayanti V. Pethe, Email: pethev@aol.com.
Thomas Hermanns, Email: thomas.hermanns@usz.ch.
Neil E. Fleshner, Email: neil.fleshner@uhn.ca.
Bas W. van Rhijn, Email: basvanrhijn@hotmail.com.
Theodorus H. van der Kwast, Email: theodorus.vanderkwast@uhn.ca.
Alexandre R. Zlotta, Email: azlotta@mtsinai.on.ca.
Bharati Bapat, Email: bapat@lunenfeld.ca.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Epstein JI, Amin MB, Reuter VR, Mostofi FK. The World Health Organization/International Society of Urological Pathology consensus classification of urothelial (transitional cell) neoplasms of the urinary bladder. Bladder Consensus Conference Committee. Am J Surg Pathol. 1998;22:1435–1448. doi: 10.1097/00000478-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Hernández V, Llorente C, de la Peña E, Pérez-Fernández E, Guijarro A, Sola I. Long-term oncological outcomes of an active surveillance program in recurrent low grade Ta bladder cancer. Urol Oncol. 2016;34:e19–e23. doi: 10.1016/j.urolonc.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 4.Jacobs BL, Lee CT, Montie JE. Bladder cancer in 2010: how far have we come? CA Cancer J Clin. 2010;60:244–272. doi: 10.3322/caac.20077. [DOI] [PubMed] [Google Scholar]
- 5.van Rjihn BWG, van Leenders GJLH, Ooms BCM, Kirkels WJ, Zlotta AR, Boevé ER, Jöbsis AC, van der Kwast TH. The pathologist's mean grade is constant and individualizes the prognostic value of bladder cancer grading. Eur Urol. 2010;57:1052–1057. doi: 10.1016/j.eururo.2009.09.022. [DOI] [PubMed] [Google Scholar]
- 6.Trenta P, Calabrò F, Cerbone L, Sternberg CN. Chemotherapy for muscle-invasive bladder cancer. Curr Treat Options Oncol. 2016;17:6. doi: 10.1007/s11864-015-0376-y. [DOI] [PubMed] [Google Scholar]
- 7.Babjuk M, Burger M, Zigeuner R, Shariat SF, van Rhijn BW, Compérat E, Sylvester RJ, Kaasinen E, Böhle A, Palou Redorta J. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder: update 2013. Eur Urol. 2013;64:639–653. doi: 10.1016/j.eururo.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Konety BR. Molecular markers in bladder cancer: a critical appraisal. Urol Oncol. 2006;24:326–337. doi: 10.1016/j.urolonc.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 9.Tilki D, Burger M, Dalbagni G, Grossman HB, Hakenberg OW, Palou J, Reich O, Rouprêt M, Shariat SF, Zlotta AR. Urine markers for detection and surveillance of non-muscle-invasive bladder cancer. Eur Urol. 2011;60:484–492. doi: 10.1016/j.eururo.2011.05.053. [DOI] [PubMed] [Google Scholar]
- 10.Planz B, Jochims E, Deix T, Caspers HP, Jakse G, Boecking A. The role of urinary cytology for detection of bladder cancer. Eur J Surg Oncol. 2005;31:304–308. doi: 10.1016/j.ejso.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Seideman C, Canter D, Kim P, Cordon B, Weizer A, Oliva I, Rao J, Inman BA, Posch M, Herr H. Multicenter evaluation of the role of UroVysion FISH assay in surveillance of patients with bladder cancer: does FISH positivity anticipate recurrence? World J Urol. 2015;33:1309–1313. doi: 10.1007/s00345-014-1452-9. [DOI] [PubMed] [Google Scholar]
- 12.Önal B, Han Ü, Yilmaz S, Köybasioglu F, Altug U. The use of urinary nuclear matrix protein 22 (NMP22) as a diagnostic adjunct to urine cytology for monitoring of recurrent bladder cancer — institutional experience and review. Diagn Cytopathol. 2015;43:307–314. doi: 10.1002/dc.23239. [DOI] [PubMed] [Google Scholar]
- 13.Weisenberger D. Characterizing DNA methylation alterations from The Cancer Genome Atlas. J Clin Invest. 2014;124:17–23. doi: 10.1172/JCI69740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kandimalla R, van Tilborg AA, Zwarthoff EC. DNA methylation-based biomarkers in bladder cancer. Nat Rev Urol. 2013;10:327–335. doi: 10.1038/nrurol.2013.89. [DOI] [PubMed] [Google Scholar]
- 15.Maruyama R, Toyooka S, Toyooka KO, Harada K, Virmani AK, Zöchbauer-Müller S, Farinas AJ, Vakar-Lopez F, Minna JD, Sagalowsky A. Aberrant promoter methylation profile of bladder cancer and its relationship to clinicopathological features. Cancer Res. 2001;61:8659–8663. [PubMed] [Google Scholar]
- 16.Eads CA, Danenberg KD, Kawakami K, Saltz LB, Blake C, Shibata D, Danenberg PV, Laird PW. MethyLight: a high-throughput assay to measure DNA methylation. Nucleic Acids Res. 2000;28:E32. doi: 10.1093/nar/28.8.e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kron K, Pethe V, Briollais L, Sadikovic B, Ozcelik H, Sunderji A, Venkateswaran V, Pinthus J, Fleshner N, van der Kwast T. Discovery of novel hypermethylated genes in prostate cancer using genomic CpG island microarrays. PLoS One. 2009;4:e4830. doi: 10.1371/journal.pone.0004830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pouessel D, Neuzillet Y, Mertens LS, van der Heijden MS, de Jong J, Sanders J, Peters D, Leroy K, Manceau A, Maille P. Tumor heterogeneity of fibroblast growth factor receptor 3 (FGFR3) mutations in invasive bladder cancer: implications for peri-operative anti-FGFR3 treatment. Ann Oncol. 2016;27:1311–1316. doi: 10.1093/annonc/mdw170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Rhijn BW, Liu L, Vis AN, Bostrom PJ, Zuiverloon TCM, Fleshner NE, van der Aa MNM, Alkhateeb SS, Bangma CH, Jewett MAS. Prognostic value of molecular markers, sub-stage and European Organisation for the Research and Treatment of Cancer risk scores in primary T1 bladder cancer. BJU Int. 2012;110:1169–1176. doi: 10.1111/j.1464-410X.2012.10996.x. [DOI] [PubMed] [Google Scholar]
- 20.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 21.McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Agundez M, Grau L, Palou J, Algaba F, Villavicencio H, Sanchez-Carbayo M. Evaluation of the methylation status of tumor suppressor genes for predicting bacillus Calmette-Guerin response in patients with T1G3 high-risk bladder tumors. Eur Urol. 2011;60:131–140. doi: 10.1016/j.eururo.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 23.Hellwinkel OJ, Kedia M, Isbarn H, Budäus L, Friedrich MG. Methylation of the TPEF- and PAX6-promoters is increased in early bladder cancer and in normal mucosa adjacent to pTa tumors. BJU Int. 2008;101:753–757. doi: 10.1111/j.1464-410X.2007.07322.x. [DOI] [PubMed] [Google Scholar]
- 24.Sacristan R, Gonzalez C, Fernández-Gómez JM, Fresno F, Escaf S, Sánchez-Carbayo M. Molecular classification of non–muscle-invasive bladder cancer (pTa low-grade, pT1 low-grade, and pT1 high-grade subgroups) using methylation of tumor-suppressor genes. J Mol Diagn. 2014;16:564–572. doi: 10.1016/j.jmoldx.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 25.Reinert T, Borre M, Christiansen A, Hermann GG, Ørntoft TF, Dyrskjøt L. Diagnosis of bladder cancer recurrence based on urinary levels of EOMES, HOXA9, POU4F2, TWIST1, VIM, and ZNF154 hypermethylation. PLoS One. 2012;7:e46297. doi: 10.1371/journal.pone.0046297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reinert T, Modin C, Castano FM, Lamy P, Wojdacz TK, Hansen LL, Wiuf C, Borre M, Byrskjøt L, Orntoft TF. Comprehensive genome methylation analysis in bladder cancer: identification and validation of novel methylated genes and application of these as urinary tumor markers. Clin Cancer Res. 2011;17:5582–5592. doi: 10.1158/1078-0432.CCR-10-2659. [DOI] [PubMed] [Google Scholar]
- 27.Yang G, Yang X. Smad4-mediated TGF-beta signaling in tumorigenesis. Int J Biol Sci. 2010;6:1–8. doi: 10.7150/ijbs.6.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riobo NA. Cholesterol and its derivatives in Sonic Hedgehog signaling and cancer. Curr Opin Pharmacol. 2012;12:736–741. doi: 10.1016/j.coph.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fei DL, Sanchez-Mejias A, Wang Z, Flaveny C, Long J, Singh S, Rodriguez-Blanco J, Tokhunts R, Giambelli C, Briegel KJ. Hedgehog signaling regulates bladder cancer growth and tumorigenicity. Cancer Res. 2012;72:4449–4458. doi: 10.1158/0008-5472.CAN-11-4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majid S, Saini S, Dahiya R. Wnt signaling pathways in urological cancers: past decades and still growing. Mol Cancer. 2012;11:7. doi: 10.1186/1476-4598-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Y. Wnt/Planar cell polarity signaling: a new paradigm for cancer therapy. Mol Cancer Ther. 2009;8:2103–2109. doi: 10.1158/1535-7163.MCT-09-0282. [DOI] [PubMed] [Google Scholar]
- 32.Schulz WA, Goering W. DNA methylation in urothelial carcinoma. Epigenomics. 2016;8:1415–1428. doi: 10.2217/epi-2016-0064. [DOI] [PubMed] [Google Scholar]
- 33.Costa VL, Henrique R, Danielsen SA, Duarte-Pereira S, Eknaes M, Skotheim RI, Rodrigues A, Magalhães JS, Oliveira J, Lothe RA. Three epigenetic biomarkers, GDF15, TMEFF2, and VIM, accurately predict bladder cancer from DNA-based analyses of urine samples. Clin Cancer Res. 2010;16:5842–5851. doi: 10.1158/1078-0432.CCR-10-1312. [DOI] [PubMed] [Google Scholar]
- 34.Lin YL, Liu XQ, Li WP, Sun G, Zhang CT. Promoter methylation of H-cadherin is a potential biomarker in patients with bladder transitional cell carcinoma. Int Urol Nephrol. 2012;44:111–117. doi: 10.1007/s11255-011-9961-6. [DOI] [PubMed] [Google Scholar]
- 35.Serizawa RR, Ralfkiaer U, Steven K, Lam GW, Schmiedel S, Schüz J, Hansen AB, Horn T, Guldberg P. Integrated genetic and epigenetic analysis of bladder cancer reveals an additive diagnostic value of FGFR3 mutations and hypermethylation events. Int J Cancer. 2011;129:78–87. doi: 10.1002/ijc.25651. [DOI] [PubMed] [Google Scholar]
- 36.Xuan Y, Kim S, Lin Z. Protein expression and gene promoter hypermethylation of CD99 in transitional cell carcinoma of urinary bladder. J Cancer Res Clin Oncol. 2011;137:49–54. doi: 10.1007/s00432-010-0858-z. [DOI] [PubMed] [Google Scholar]
- 37.Cabello MJ, Grau L, Franco N, Orenes E, Alvarez M, Blanca A, Heredero O, Palacios A, Urrutia M, Fernández JM. Multiplexed methylation profiles of tumor suppressor genes in bladder cancer. J Mol Diagn. 2011;13:29–40. doi: 10.1016/j.jmoldx.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ibragimova I, Dulaimi E, Slifker MJ, Chen DY, Uzzo RG, Cairns P. A global profile of gene promoter methylation in treatment-naïve urothelial cancer. Epigenetics. 2014;9:760–773. doi: 10.4161/epi.28078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim JS, Chae Y, Ha YS, Kim IY, Byun SS, Yun SJ, Kim WJ. Ras association domain family 1A: a promising prognostic marker in recurrent nonmuscle invasive bladder cancer. Clin Genitourin Cancer. 2012;10:114–120. doi: 10.1016/j.clgc.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 40.Kitchen MO, Bryan RT, Emes RD, Glossop JR, Luscombe C, Cheng KK, Zeegers MP, James ND, Devall AJ, Mein CA. Quantiative genome-wide methylation analysis of high-grade non–muscle invasive bladder cancer. Epigenetics. 2016;11:237–246. doi: 10.1080/15592294.2016.1154246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marsit CJ, Houseman EA, Christensen BC, Gagne L, Wrensch MR, Nelson HH, Wiemels J, Zheng S, Wiencke JK, Andrew AS. Identification of methylated genes associated with aggressive bladder cancer. PLoS One. 2010;5:e12334. doi: 10.1371/journal.pone.0012334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vinci S, Giannarini G, Selli C, Kuncova J, Villari D, Valent F, Orlando C. Quantitative methylation analysis of BCL2, hTERT, and DAPK promoters in urine sediment for the detection of non-muscle-invasive urothelial carcinoma of the bladder: a prospective, two-center validation study. Urol Oncol. 2011;29:150–156. doi: 10.1016/j.urolonc.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Yan C, Kim YW, Ha YS, Kim IY, Kim YJ, Yun SJ, Moon SK, Bae SC, Kim WJ. RUNX3 methylation as a predictor for disease progression in patients with non–muscle-invasive bladder cancer. J Surg Oncol. 2012;105:425–430. doi: 10.1002/jso.22087. [DOI] [PubMed] [Google Scholar]
- 44.Lin HH, Ke HL, Huang SP, Wu WJ, Chen YK, Chang LL. Increase sensitivity in detecting superficial, low grade bladder cancer by combination analysis of hypermethylation of E-cadherin, p16, p14, RASSF1A genes in urine. Urol Oncol. 2010;28:597–602. doi: 10.1016/j.urolonc.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 45.Monteiro-Reis S, Leça L, Almeida M, Antunes L, Monteiro P, Dias PC, Morais A, Oliveira J, Henrique R, Jerónimo C. Accurate detection of upper tract urothelial carcinoma in tissue and urine by means of quantitative GDF15, TMEFF2 and VIM promoter methylation. Eur J Cancer. 2014;50:226–233. doi: 10.1016/j.ejca.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 46.Xiong G, Liu J, Tang Q, Fan Y, Fang D, Yang K, Xie F, Zhang M, Zhang L, Liu L. Prognostic and predictive value of epigenetic biomarkers and clinical factors in upper tract urothelial carcinoma. Epigenomics. 2015;7:733–744. doi: 10.2217/epi.15.34. [DOI] [PubMed] [Google Scholar]
- 47.Zhang H, Qi F, Cao Y, Zu X, Chen M, Li Z, Qi L. 5-aza-2′-deoxycytidine enhances maspin expression and inhibits proliferation, migration, and invasion of the bladder cancer T24 cell line. Cancer Biother Radiopharm. 2013;28:343–350. doi: 10.1089/cbr.2012.1303. [DOI] [PubMed] [Google Scholar]
- 48.Xylinas E, Hassler MR, Zhuang D, Krzywinski M, Erdem Z, Robinson BD, Elemento O, Clozel T, Shariat SF. An epigenomic approach to improving response to neoadjuvant cisplatin chemotherapy in bladder cancer. Biomolecules. 2016;6:37. doi: 10.3390/biom6030037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humphrey PA, Moch H, Cubilla AL, Ulbright TM, Reuter VE. The 2016 WHO classification of tumours of the urinary system and male genital organs—part B: prostate and bladder tumours. Eur Urol. 2016;70:106–119. doi: 10.1016/j.eururo.2016.02.028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primer and Probe Sequences Used in the MethyLight Assay for ALU-C4, EOMES, GP5, TCF4, PAX6, and ZSCAN12 Genes
Associations between Methylation of Five Genes and Clinicopathological Variables in Non–Muscle Invasive Bladder Cancer Patients (Median Beta Values and Kruskal-Wallis or Mann-Whitney U Test P Values Are Shown)