

Abstract
Proteolytic cascades are deeply involved in critical stages of cancer progression. During the course of peptide-wise analysis of shotgun proteomic data sets representative of colon adenocarcinoma (AC) and ulcerative colitis (UC), we detected a cancer-specific proteolytic fingerprint composed of a set of numerous protein fragments cleaved C-terminally to V, I, A, T, or C residues, significantly overrepresented in AC. A peptide set linked by a common VIATC cleavage consensus was the only prominent cancer-specific proteolytic fingerprint detected. This sequence consensus indicated neutrophil elastase as a source of the fingerprint. We also found that a large fraction of affected proteins are RNA processing proteins associated with the nuclear fraction and mostly cleaved within their functionally important RNA-binding domains. Thus, we detected a new class of cancer-specific peptides that are possible markers of tumor-infiltrating neutrophil activity, which often correlates with the clinical outcome. Data are available via ProteomeXchange with identifiers: PXD005274 (Data set 1) and PXD004249 (Data set 2). Our results indicate the value of peptide-wise analysis of large global proteomic analysis data sets as opposed to protein-wise analysis, in which outlier differential peptides are usually neglected.
Cancer-specific proteolytic activity is one of the highlights of cancer progression, and specific changes in the tumor degradosome may serve as a reservoir for potential diagnostic molecular features (1). For example, cancer-specific fragments of cytokeratins are among the most frequently tested protein-based cancer markers. Cytokeratin typing and quantitation for cancer studies, as well as diagnosis, prognosis, and therapy monitoring, have become routine in recent years (2–6). Moreover, keratins are cleaved in a cancer-dependent manner, and some of the keratin-based antibodies used in the context of cancer recognize such fragments (7). For instance, keratins 18 and 19 are known substrates for caspase degradation (8) which occurs in the intermediate stage of apoptosis (6), when well-controlled proteolysis of many components of the cytoplasmic and nuclear cytoskeleton orchestrates cellular breakdown. The resulting abundant keratin fragments are released into the circulation by tumor cells and can be detected there. Currently, several antibodies with epitopes linked to keratins and/or their fragments, such as tissue polypeptide antigen (TPA)1, tissue polypeptide-specific antigen, keratin 19 fragment (Cyfra21–1), and keratin 18 fragment (M30, M65), are widely used for cancer studies, and their clinical value is frequently assessed by numerous groups in different experimental settings. In the case of certain carcinomas, such as nonsmall cell lung cancer (6), oral cancer (7), bladder cancer (BC) (9), and colon cancer (10), serum assays derived from keratins 8, 18, and 19 are increasingly used to monitor tumor load and disease progression and for the early detection of recurrence and fast assessment of the efficacy of the response to therapy (11). In addition, tumors derived from epithelial cells are known to retain the cytokeratin expression pattern characteristic of the originating cell types, development, and differentiation stage (12), (13). Thus, the cytokeratin profile is indicative of a tumor's origin and generally retained, even in mobilized cells far from the primary source of malignancy.
However, knowledge regarding the proteolytic fragments populating the plasma/serum/urine/saliva of cancer patients is very limited. A plethora of commercially available antibodies, in general of not precisely defined affinities, are used to explore the potential of protein fragments as cancer markers. Such a “black-box” approach lacks a precise definition of targets and is carried out without selection of the best fragments. In addition, antibody-based approaches suffer from cross-reactivity with other targets, leading to numerous controversies, as for instance raised in the case of keratin-based markers (14–16). Although cross-reactivity decreases the specificity of existing tests, many currently available kits have reasonable levels of sensitivity and specificity. Assessment of the true sensitivity and specificity of cytokeratin fragment-based tests, excluding cross-reactivity phenomena, would require the application of more selective methods, such as methods based on mass spectrometry. To the best of our knowledge, these approaches have not yet been attempted.
In the present study, we reanalyzed the data sets collected during global proteomic studies of adenocarcinoma (AC) and ulcerative colitis, using data obtained previously by our group (17) and those available from the MS-data PRIDE repository (18), focusing on disease-specific proteolytic events. In addition to detecting peptides resulting from the caspase cleavage of keratins in AC, we identified other disease-specific proteolytic cleavages in a wider set of proteins. In some cases, the resulting peptides are an order of magnitude more abundant in cancer than in control noncancerous tissue, whereas the abundance ratio for a caspase-cleaved keratin 18 peptide is only 3. The cleavage pattern of peptides detected in cancer tissue revealed a strong preference for V, I, A, T, and C residues at the P1 cleavage site (VIATC consensus), making neutrophil elastase the most probable candidate for the protease responsible for the observed cancer-specific cleavage. In result, by reanalyzing the existing data sets we identified a new class of cancer-specific protein fragments that are potential markers of the presence and progression of cancer.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
In the presented study four distinct data sets have been processed, as summarized in Table I and Scheme 1, namely: Data set 1—iTRAQ differential proteomic data sets obtained during the course of global proteomic analyses of tubulovillous adenoma (TVA), tubular adenoma (TA), and AC tissue (17)—data deposited in PRIDE archive: PXD005274. Data set 2—the results of label free proteomic analysis of bladder cancer and colon cancer tissues-data deposited in PRIDE archive: PXD004249. Data set 3—data deposited in the proteomic data repository PRIDE, comprising 32 LC-MS raw files derived from 16 samples of colon cancer tissue and 16 healthy donor samples uploaded by the CPTAC consortium (PRIDE archive identifiers PXD002041 - PXD002050), selected at random from the larger available data set and Data set 4—data deposited in the proteomic data repository PRIDE, comprising 20 LC-MS raw files originating from the analysis of endoscopic mucosal colon biopsies from noninflamed tissue in 10 patients with UC and 10 controls (PRIDE archive identifier PXD001608). For each of the four data sets different approach to data analysis was used, as described below:
Table I. Four types of data sets analyzed in this work, along with source of data set, number of samples, quantitation method, and search engine used.
| Data set | Tissue type | Reanalysis? | Source | Num. of samples | Pooled? | Quantitation | Search engine |
|---|---|---|---|---|---|---|---|
| Data set 1 | Colon cancer | yes | Previously published data (17) | 3 AC | yes | iTRAQ | Mascot |
| PXD005274 | 3 TVA | ||||||
| 3 TA | |||||||
| 3 NC | |||||||
| Data set 2 | Colon cancer | Experimental data | 10 AC | ||||
| 10 NC | |||||||
| Bladder cancer | no | PXD004249 | 10 BC | no | label-free | Mascot | |
| 10 NC | |||||||
| Data set 3 | Colorectal cancer | yes | PRIDE (PXD002041–2050) | 16 AC | no | spectral count | MS-GF+ |
| 16 NC | |||||||
| Data set 4 | Ulcerative colitis | yes | PRIDE (PXD001608) | 10 UC | no | spectral count | MS-GF+ |
| 10 NC |
Scheme 1.

A flowchart explaining the data analysis approaches used for each of the Data sets 1–4.
Data Set 1—Reanalysis of iTRAQ Labeled Proteome of Tumor Tissues
Raw data sets of iTRAQ labeled proteomes of tubulovillous adenoma (TVA), tubular adenoma (TA), and AC tissue were obtained as described previously (17). The mass spectrometry proteomics data for Data set 1 have been deposited to the ProteomeXchange Consortium via the PRIDE (19) partner repository with the data set identifier PXD005274 and 10.6019/PXD005274. For the reanalysis a two-step procedure was applied. First, MS/MS spectra were extracted from raw files using Mascot Distiller (v 2.4.1, Matrix Science), followed by an initial Mascot (v. 2.4.1, Matrix Science) database search restricted to human sequences against the SwissProt database (release 10_2012, 20 232 entries actually searched). Search parameters were: enzyme specificity, trypsin; number of missed cleavage sites, 1; precursor tolerance window, 30 ppm; MS/MS tolerance, 0.2 Da; variable modification, methionine oxidation; fixed modification, cysteine carbamidomethylation, labeling method, iTRAQ 4-plex. Mascot search results were exported as *.dat files and subsequently internally calibrated using in house DatViewer software procedure described in supplementary Method S1 in ref (20). In short, the internal calibration procedure implemented in the DatViewer software used differences between observed and theoretical m/z values of precursor and fragment ions for a subset of high quality spectra selected from the peptide list obtained during an initial Mascot search to calculate systematic m/z measurement errors. Identifications with scores above both homology and identity thresholds in the initial search were used as an inclusion criterion for reference calibration list. A linear relationship between m/z value and measurement error was assumed and robust least squares method was used for fitting the calibration curve. The value of a systematic error calculated for a given m/z range was then subtracted from measured m/z values of all ions. Ion m/z tolerances were calculated separately for each sample based on standard deviation of m/z error of reference list peptides after calibration. Tolerance values were set for the next round of database search at 3 and 5 standard deviations for precursor and fragment ions, respectively. Calibrated spectra with sample-specific mass tolerance values were saved as mass-error corrected *.mgf files. Preliminary search results were discarded after this step and in a second step a new database search was performed on calibrated data, again using Mascot search engine, with nearly the same search parameters, except enzyme specificity which was set to “no enzyme specificity” and sample specific mass tolerance values. Also, in the second database search, target-decoy search strategy was applied to estimate the FDR of resulting identifications, and the quantitative analysis was carried out only using a set of peptides which fulfilled the criterion of FDR < 1%. Individual peptide abundance AC/NC ratios were normalized by their corresponding AC/NC protein ratios. Peptides and proteins identified are listed in Supporting Material supplemental Table S4.
Data Set 2—Quantitative Analysis of Colon Cancer and Bladder Cancer Tissue Samples Using Label-free Method
Patient Selection
Clinical tissue samples were collected at the Maria Sklodowska-Curie Memorial Cancer Center as described previously (21). The study protocol was approved by the Cancer Center Bioethics Committee, and all patients provided signed informed consent before inclusion. Samples of malignant tissue and adjacent healthy tissue were obtained after surgical resection. The tissue samples were immediately snap-frozen in liquid nitrogen and stored at −72 °C until use.
Proteome Extraction
Tissue samples were vigorously washed with cold phosphate buffered saline (PBS) containing protease (Roche, Basel, Switzerland) and phosphatase inhibitors (Sigma, St. Louis, MO) to remove residual blood and then centrifuged at 15,000 × g at 4 °C for 3 min. Proteins were extracted using the ProteoExtract® Subcellular Proteome Extraction Kit (Calbiochem, San Diego, CA, Cat. No. 539790) according to the manufacturer's protocol. The protein concentration was measured using the Bradford method. These fractions were stored as aliquots at −72 °C until use.
Protein Sample Preparation
Proteins were precipitated from equal amounts (by protein content) of sample from each tissue type using the ProteoExtract Protein Precipitation Kit (Calbiochem) according to the manufacturer's protocol. Protein pellets were resuspended in 200 μl of dissolution buffer (0.5 m triethylammonium bicarbonate with 0.1% SDS). To facilitate protein solubilization, samples were vortexed thoroughly and (optionally) treated with five pulses from a 500W Cole-Parmer ultrasonic homogenizer (amp. 24%, pulse 2 s, gap 2 s). The protein concentrations of the combined and subcellular samples were measured using the Bradford method. Aliquots of samples (100 μg) were stored at −72 °C.
LC-MS Settings
Peptides mixtures were analyzed by LC-MS (liquid chromatography coupled to tandem mass spectrometry) using Nano-Acquity (Waters, Milford, MA) LC system and Orbitrap Velos mass spectrometer (ThermoFisher Scientific, Waltham, MA). Prior to the analysis, proteins were subjected to standard “in-solution digestion” procedure during which proteins were reduced with 200 mm dithiothreitol (for 60 min at 60 °C), alkylated with 500 mm iodoacetamide (45 min in dark at room temperature) and digested overnight with trypsin (sequencing Grade Modified Trypsin - Promega, Madison, WI, V5111). Peptide mixture was applied to RP-18 precolumn (nanoACQUITY Symmetry® C18 - Waters 186003514) using water containing 0.1% TFA as mobile phase and then transferred to nano-HPLC RP-18 column (nanoACQUITY BEH C18 - Waters 186003545) using an acetonitrile gradient (5–35% AcN in 180 min) in the presence of 0.05% formic acid with the flowrate of 250 nl/min. Column outlet was directly coupled to the ion source of the spectrometer. A blank run ensuring lack of cross contamination from previous samples preceded each analysis.
Qualitative analyses (i.e. peptide and protein identification) were performed on pooled samples in data-dependent MS-to-MS/MS acquisition mode. Up to five MS/MS processes were allowed for each MS scan. To increase the number of peptide identifications, three LC-MS/MS runs were performed per pooled sample, each covering one of three ranges of m/z values: 300–600, 500–900, or 800–2000. This approach substantially improved the number of peptides. Quantitative analyses of individual samples were carried out in separate survey scan LC-MS runs with an m/z measurement range of 300–2000 using the same acetonitrile gradient as in the qualitative LC-MS/MS runs. The data-dependent MS-to-MS/MS switch was disabled, and the spectrometer resolution was set to 15,000.
Qualitative MS Data Processing and Database Search
Qualitative MS data processing and a database search were carried out as described for iTRAQ-labeled data (ref (17). and above), but with the following differences in search parameters: fixed modification, carbamidomethylation (C); variable modifications, oxidation (M). Only peptide spectrum matches with q-values ≤ 0.01 were considered to be confidently identified. Proteins identified by a subset of peptides from another protein were excluded from the analysis, and proteins matching the same set of peptides were clustered into single groups. Peptides and proteins identified are listed in Supporting Material supplemental Table S4. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (19) partner repository with the data set identifier PXD004249 and 10.6019/PXD004249.
Quantitative MS Data Processing
The list of peptides identified from the LC-MS/MS runs was overlaid onto two-dimensional maps generated from the LC-MS profile data for individual samples. A more detailed description of the feature extraction procedure is provided elsewhere (22). Briefly, the list of identified peptides was used to tag the corresponding peptide-related ion spectra on the basis of m/z value, the deviation from the predicted elution time, and the match between theoretical and observed isotopic envelopes. The relative abundance of each peptide ion was determined as the volume of the corresponding peak. To minimize the effects of nonbiological sources of variation, log-transformed peptide abundance was normalized by fitting a robust, locally weighted regression smoother (LOESS) between the individual samples and a median pseudosample. The parameters of the fit were established using a set of features exhibiting low variance in the nonnormalized data and then applied to the whole data set. For protein-wise analysis, the normalized peptide-level data were rounded up to relative protein abundance. The procedure involved rescaling the abundance of peptides originating from the same protein to a common level and computing their median value.
Statistical Analysis of Quantitative MS Results
The Mann-Whitney U test was used to select differentially expressed proteins. The resulting p values were corrected for multiple hypothesis testing using the Benjamini-Hochberg step-up procedure, which controls for the false discovery rate (23). Relative peptide abundances with adjusted p values ≤ 0.05 were considered significantly changed in at least one of the studied groups. All statistical analyses were performed using a combination of proprietary software running in the MATLAB environment (MathWorks; MStat, available at http://proteom.ibb.waw.pl) and in-house scripts for tasks such as identifying cleavage type from peptide and protein sequence or data format conversion.
Data Sets 3 and 4—Reanalysis of PRIDE Data Sets
For the analysis of PRIDE data, the extraction of MS/MS spectra was carried out, followed by an initial Mascot (v. 2.4.1, Matrix Science, London, UK) database search restricted to human sequences against the UniProt database (release 06_2015, 68 554 entries searched). Search parameters were: enzyme specificity, trypsin; number of missed cleavage sites, 1; precursor tolerance window, 30 ppm; MS/MS tolerance, 0.2 Da; variable modification, methionine oxidation; fixed modification, cysteine carbamidomethylation. Mascot search results were exported as *.dat files and internally calibrated using the same procedure as in the case of iTRAQ data, and saved as mass-error corrected *.mgf files with sample specific mass tolerance. The second, refined, database search was carried out using the MS-GF+ (v. 9979) (24) search engine against the Uniprot database restricted to human sequences. Search parameters were: mass tolerances calculated individually for each sample; enabled decoy search; no enzyme specificity; fixed modification, cysteine carbamidomethylation; variable modifications defined as methionine oxidation, N-terminal acetylation, and glutamine substitution by pyro-Glu. Following the extraction of peptides fulfilling the criterion of FDR< 1%, (peptides and proteins identified are listed in Supporting Material supplemental Table S4) the quantitative analysis was carried out using a “spectral count” approach (25).
RESULTS
Previously, we conducted a global proteomic analysis of iTRAQ labeled AC, TVA, and TA tissue in comparison to tumor-adjacent tissue (17). In this approach, pooled trypsin-digested iTRAQ-labeled samples were fractionated by isoelectrofocusing and then subjected to LC-MS (IEF-LC-MS approach)—Data set 1. Based on the stringent selection of iTRAQ-labeled peptides with q<0.01, we established a list of proteins significantly over- or under-represented in the AC proteome. The list includes for instance keratins 8 and 20, significantly downregulated in cancer, but not keratin 18. However, during the subsequent peptide-wise analysis of the same data sets, we noted the presence of a large subset of peptides regulated in a markedly different way than the other peptides originating from the same proteins. The commonality in these peptides was the presence of nontryptic cleavage sites. In Fig. 1 we present the AC/NC peptide-wise ratios (fold change, FC) calculated by the analysis of iTRAQ signals (tag 117 for AC, tag 114 for NC) for all peptides from keratin 8 and keratin 18 detected in three replicates of the experiment. Keratin 18 (Fig. 1A) tryptic peptides are characterized by an AC/NC ratio close to 1, in agreement with a lack of change in the K18 level detected during the protein-wise analysis. On the other hand, several outliers are present (colored circles), all resulting from nontryptic cleavage. Some of the outlier peptides were characterized by large FC values, some even exceeding 10, reproducibly in all three experiments. This finding indicates that these peptides are up to an order of magnitude more abundant in AC than NC samples. Caspases 3, 7, and 9 are known to cleave keratin 18 in a cancer-specific way, and caspase-cleaved keratin 18 fragments are established cancer biomarkers (26). Indeed, two keratin 18 peptides derived from caspase cleavage were found in our data set: Q225AQIASSGLTVEVD238 with FC>3 and N391LGDALD397 with FC = 1.5. These peptides are “semi-tryptic”, as the sample was subjected to tryptic cleavage preceding LC-MS and the caspase cleavage site is accompanied by a tryptic cleavage site at the other end of the peptide. However, stronger outliers with larger FC values represent other peptides cleaved at different, noncaspase sequence motifs. Cleaved sequences in these keratin 18 peptides include (P1-P1′ positions) Ser-Ser, Ser-Thr, Val-Thr, Thr-Gln, Gly-Asp, and Ile-Gln. The strongest overrepresented cleavage occurred at Val-Thr (FC>10), resulting in three detected peptides, one containing P1 residue (Val282) and two containing P1′ (Thr283) residue. All three of them are overrepresented in AC to varying degrees. Underrepresented cleavages were also found, with the most underrepresented peptide pair resulting from nontryptic cleavage at the Gly393-Asp site. The nontryptic cleavage sites localize to three regions of keratin 18: linker L12, the tail, and the N-terminal part of coil 2B, which are known to be unstructured in keratin filaments (27). Numerous nontryptic sites characterized by a large AC/NC ratio were also found in keratin 8 (Fig. 1B). The up-regulation of these peptides contrasts with the down-regulation of keratin 8 in AC versus NC samples, which is reflected by the median ratio of tryptic peptides of 0.5. Nontryptic cleavages with FC values as large as 10 occurred in five regions of the protein, including the unstructured head, linker L12, and an interface between linker L2 and coil 2B. Interestingly, the majority of nontryptic cleavages in both keratins occurred C-terminal to Val or Ile.
Fig. 1.

(A) Adenocarcinoma (AC)/noncancer (NC) ratio of different peptides originating from keratin 18 and (B) keratin 8. Peptides are shown as black bars positioned at their corresponding location in sequence at the horizontal axis. Error bars are S.D. of three independent experiments. Lack of an error bar indicates that a peptide was detected in one of the three experiments. Nontryptic cleavages are marked by color circles, either N-terminal (red) or C-terminal (blue). Arrows indicate the positions of nontryptic cleavage of the highest AC/NC ratio in protein regions.
To explore this observation in a more systematic manner we listed all nontryptic cleavage sites in the whole data set of 17,965 peptides, corresponding to 4,254 proteins, and calculated their abundance AC/NC ratios, normalized by their corresponding AC/NC protein ratios. Next, we calculated the median AC/NC ratio for each type of amino acid at positions P4….P4′ of the cleavage site. Though no preference was detected for a particular amino acid type at positions P2, P1′, P2′ (Fig. 2) and also P4′, P3′, P3, and P4 (data not shown) Mann-Whitney U test indicated a significant overrepresentation (p value < 10−6) of peptides cleaved after Val, Ile, Ala, Thr, or Cys (P1 position) in cancer samples. Hereafter, this is referred to as the VIATC consensus. In contrast, peptides cleaved after Trp or Glu generally had a lower ratio in cancer. Of the 180 strongly (FC>4) up-regulated nontryptic peptides identified, 176 bear the VIATC consensus at the P1 cleavage site. A total of 456 VIATC peptides were found in the whole data set, so 38% of VIATC peptides are strongly up-regulated (FC>4) in AC. Exemplary fragmentation spectra of VIATC peptides are shown in supplemental Fig. S1.
Fig. 2.

Box-plots correlating the adenocarcinoma (AC)/noncancer (NC) ratio and nature of the cleavage site with P2′-P2 positions based on the analysis of 17 965 iTRAQ-labeled peptides. Boxes denote interquartile range whereas whiskers extend to the most extreme data point which is no more than 1.5 times the interquartile range from the box. Asterisks denote p value < 10−6. A significant cancer-dependent shift in the AC/NC ratio is prominent if the P1 position is Val, Ile, Thr, Ala, or Cys.
This analysis provides a strong indication that cleavage at a VIATC consensus in a large number of proteins is a much more frequent event in AC tissue than in normal tissue. We analyzed all cleavage sites within the MEROPS (28) and PMAP-CutDB (29) databases to find proteases characterized by the P1 VIATC consensus and lack of preferences at other positions. Our analysis indicated neutrophil elastase (NE), a serine protease (S01.131) known to cleave collagen and elastin, as the only protease to strictly fulfill these criteria. In the MEROPS database, the NE consensus for P4 - P4′ positions is defined as −/−/−/viat|−/−/−/−, indicating a nearly exact match to the VIATC consensus at P1 and agreement with a lack of preference at other positions. Among 488 NE cleavage sites collected in the MEROPS database, 392 (80%) match the VIATC consensus (P1: Ile (n = 142), Val (n = 129), Ala (n = 61), Thr (n = 56), Cys (n = 4)). Other proteases cleaving at a VIATC consensus have much broader specificities and could not explain the results shown in Fig. 2. We concluded that NE is the most probable candidate for the cancer-specific proteolytic activity detected in AC samples.
Next, we investigated proteins undergoing cancer-dependent cleavage at VIATC consensus sites in AC. From 456 VIATC peptides in the data set, we selected peptides with FC>4 that were detected in at least two of the three experiments. This narrowed down the set of peptides to 170 originating from 118 proteins (supplemental Table S1). Analysis of this protein set by STRING (30) (Fig. 3) revealed significant interconnections between a majority of these proteins. Cell component gene ontology analysis in STRING showed significant overrepresentation of proteins annotated to focal adhesion, the endoplasmic reticulum (ER) lumen, mitochondrion, and ribonucleoprotein complex. The group of ER lumen proteins contained four collagens, and a set of 10 heterogeneous nuclear ribonucleoproteins (hnRNPs) was prominent among the ribonucleoprotein complex proteins.
Fig. 3.
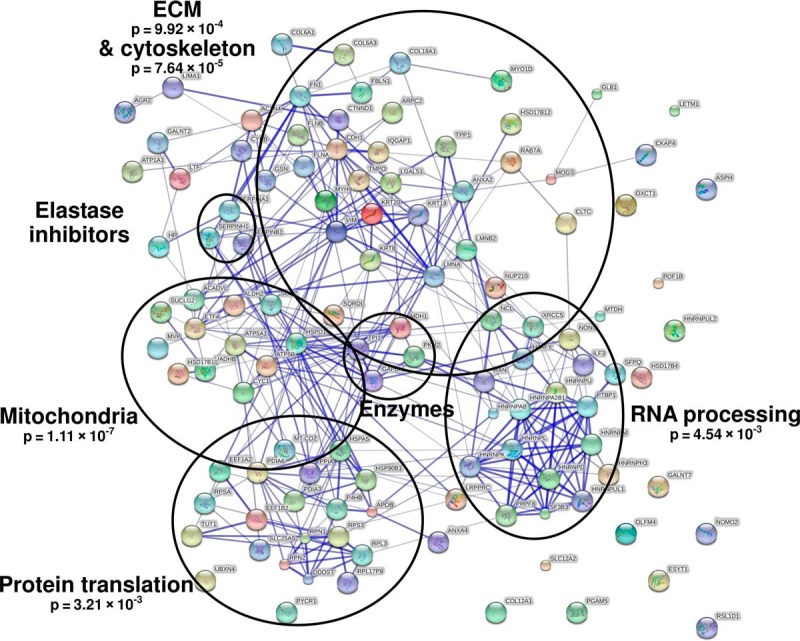
STRING analysis of proteins cleaved by neutrophil elastase in a cancer-specific manner. Adjusted p values calculated by STRING are provided for significantly overrepresented groups of proteins.
We also reanalyzed differential proteomic experiments of AC tissue fractionated into cytoskeleton, cytoplasmic, membrane + organelle, and nuclear fractions based on the total data set of 52,007 peptides (q<0.01) (17). The analysis of VIATC cut sites in fractionated AC samples (with total of 5170 peptides bearing a VIATC consensus) revealed much fewer number of such peptides in the cytoskeletal fraction than in the remaining fractions (Fig. 4A). In addition, we observed a prominent cancer-specific increase in the abundance of VIATC consensus peptides only in the nuclear fraction (average 2.5-fold increase in AC than NC control; Fig. 4B). In the three other fractions, including cytoskeleton, the increase in the abundance of VIATC peptides was much less pronounced or insignificant. In the cytoskeleton VIATC peptides are markedly less common than in other fractions, and cancer-dependent cleavage, as observed with keratins, is an exception rather than a rule. In contrast, in the nuclear fraction the abundance of a majority of the VIATC peptides was cancer-dependent; thus, in this case VIATC cleavage en gros may depend strongly on the presence of cancer. In the nuclear fraction, 628 VIATC peptides had an AC/NC ratio >4 (supplemental Table S1). These peptides belonged to 398 proteins (nucVIATC proteins) that could be classified into a few major groups, mainly representing proteins engaged in pre-mRNA processing, mRNA transport, and chromatin modifications, many of them were found in the unfractionated sample analysis (Fig. 3), including a group of nine hnRNPs, out of 16 major known hnRNPs. These proteins were accompanied in the nucVIATC set by 75 other proteins categorized as RNPs by the gene ontology algorithm (supplemental Table S1). Thus, a large fraction (75/398) of nucVIATC proteins are RNPs. In the nucVIATC set, a large number of proteins were also classified as ribosome assembly proteins, likely originating from preribosomal nuclear/nucleolar intermediates (31). We also noted the presence of numerous mitochondrial proteins, possibly originating from the contamination of nuclear protein preparations, indicating that the overrepresentation of RNPs in the nuclear set is even more pronounced. Of these 75 RNPs, 22 contain RRM RNA-binding domains. Interestingly, of the 35 VIATC cut sites observed in these 22 proteins, 28 (80%) are located within RRM domains and 7 outside these domains. RRM domains cover ∼30% of the amino acid sequences of these 22 proteins; therefore, in the case of random proteolysis, the expected cleavage frequency within RRM domains should not exceed 30%. This finding indicates a strong preference for VIATC cleavage within RRM domains. The rationale for selective cleavage of RRM domains is not straightforward. These domains may be less structured in solution than the remaining regions. Also, RRM domains are cleaved with strong preference for Val or Ile (26 cleavage sites) compared with Thr, Ala, or Cys (2 cleavage sites) (supplemental Table S2). Moreover, 21 of the 28 cut sites inside RRM domains were located within known secondary structure motifs (helices or sheets).
Fig. 4.

A, Numbers of total tryptic peptides and VIATC consensus peptides detected in four fractions: cytoskeletal, cytoplasmic, membrane + organelle, and nuclear. B, Box-plots of adenocarcinoma (AC)/noncancer (NC) ratios for VIATC consensus peptide abundance in four fractions on the logarithmic scale. Boxes denote interquartile range whereas whiskers extend to the most extreme data point which is no more than 1.5 times the interquartile range from the box. VIATC peptides were significantly more abundant in AC only for the nuclear fraction, indicating that the majority of VIATC nuclear peptides are overrepresented in colon cancer tissue.
Of seven sites localized outside RRM domains, five were located in Asp/Glu rich regions: two in the helical leucine zipper oligomerization domain spanning positions 194–216 of hnRNP C1/C2 (32) critical for RNA-binding activity, and three in the C-terminal flanking smart00582 nuclear pre-mRNA regulation domain spanning positions 689–746 of U2 snRNP-associated SURP motif-containing protein. Another cleavage outside RRM domains affects the flanking region of the PWI RNA/DNA binding domain of RNA-binding protein 25. The PWI domain regulates Bcl-x-pre-mRNA alternative splicing and its flanking domain serves as a binding partner; thus, it participates in directing Bcl to either its pro-apoptotic Bcl-xS or anti-apoptotic Bcl-xL form, with profound consequences on cell fate (33). Therefore, the observed proteolytic activity is not an indiscriminate cleavage of cellular debris, but was executed on proteins in their native state and overwhelmingly affected functionally important domains.
Next, we investigated whether cancer-specific VIATC cleavage peptides allow differentiation of TVA and TA samples (labeled with iTRAQ 115 and 116, respectively) from NC and/or AC samples (17). The abundance of VIATC-cleaved peptides in TVA and TA samples compared with AC is shown in Fig. 5A. The average TVA/NC and TA/NC ratios for VIATC did not shift from 1 (TVA) or were much smaller than the AC/NC ratio (TA). Generally, cleavage at VIATC in TVA and TA seemed to be disease-independent, in contrast to AC. Principal component analysis based on all VIATC peptides (Fig. 5B) revealed that TVA and TA states were not well separated from NC controls in the dimension of the first component. However, TVA and TA were well separated from NC controls in the dimension of the second component. The analysis of a set of peptides leading to this separation revealed consistently that, for example, keratin 19 peptide ASDGLAGNEK (supplemental Fig. S2) was less abundant in TA and TVA than NC or AC samples. Many more nontryptic peptides with large FC values, differentiating TVA and TA from NC controls, were found in a set of other (nonVIATC) nontryptic peptides (supplemental Table S3). An overall down-regulation of other nontryptic cleavages in TVA and TA (Fig. 5A) was noted. This set is exemplified by cleavage at Gly-Asp in keratin 18, already mentioned in the analysis of K18 peptides at (Fig. 1) as most downrepresented in AC. As shown in supplemental Fig. S3, the peptides LLEDGEDFNLG- and -DALDSSNSMQTIQK resulting from cleavage at this site were much less abundant not only in AC, but also TVA and TA compared with NC controls. Interestingly, the Gly-Asp site in keratin 18 is known to be cleaved by meprin beta protease (34). Meprin beta is expected to be co-expressed with meprin alpha in the small intestine, but not the colon (35). However, we detected both proteins in colon tissue, and both were found to be much more abundant in normal tissue than in TVA, TA, or AC (17). Meprin alpha (Q16819) and beta (Q16820) are interdependent proteases involved in tumor progression in colon cancer (36) and are expected to be up-regulated in tumors, in contrast to the observed down-regulation of their putative cleavage products in the present study. Such interesting cases require further studies, as no commonality was found to link this set of peptides. However, when all nontryptic peptides were taken into account, principal component analysis (Fig. 6) revealed a significant separation of TA and TVA samples from NC samples along the first component axis.
Fig. 5.
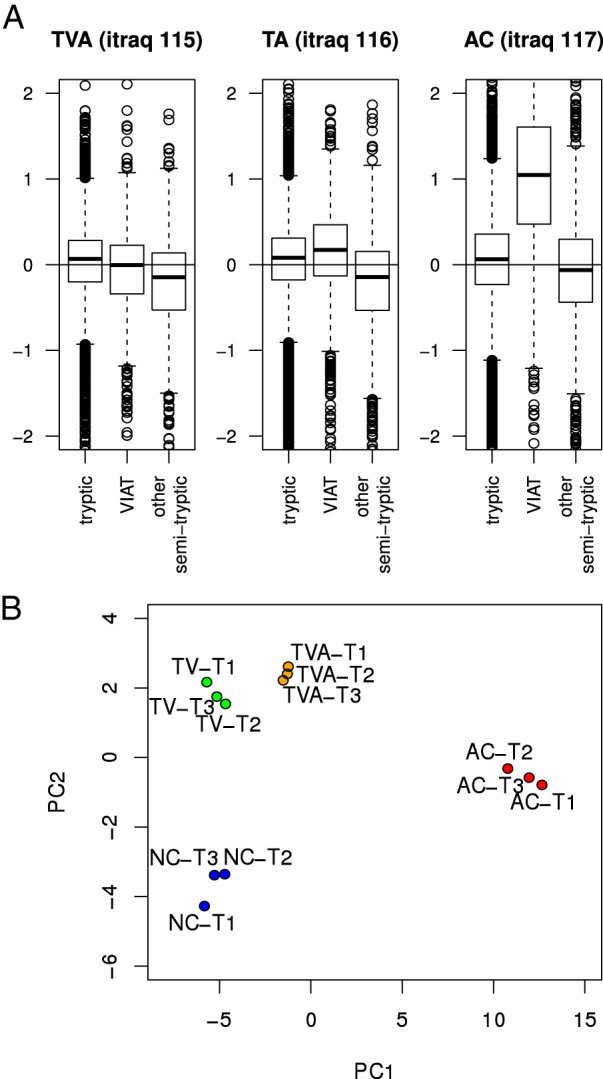
A, Box-plots correlating FC values for VIATC consensus cleaved peptides and all other peptides when tubulo-villous adenoma (TVA - iTRAQ115), tubular adenoma (TA - iTRAQ116), and adenocarcinoma (AC - iTRAQ117) samples were compared with normal tissue (NC - iTRAQ114). Boxes denote interquartile range whereas whiskers extend to the most extreme data point which is no more than 1.5 times the interquartile range from the box. Only in AC an increased abundance of VIATC peptides is observed, as compared with NC. B, Principal component analysis based on a set of VIATC consensus cleaved peptides shows a good separation of NC, TVA, TA, and AC groups. The first component separated AC from NC best, whereas the second component separated TVA/TA from NC.
Fig. 6.
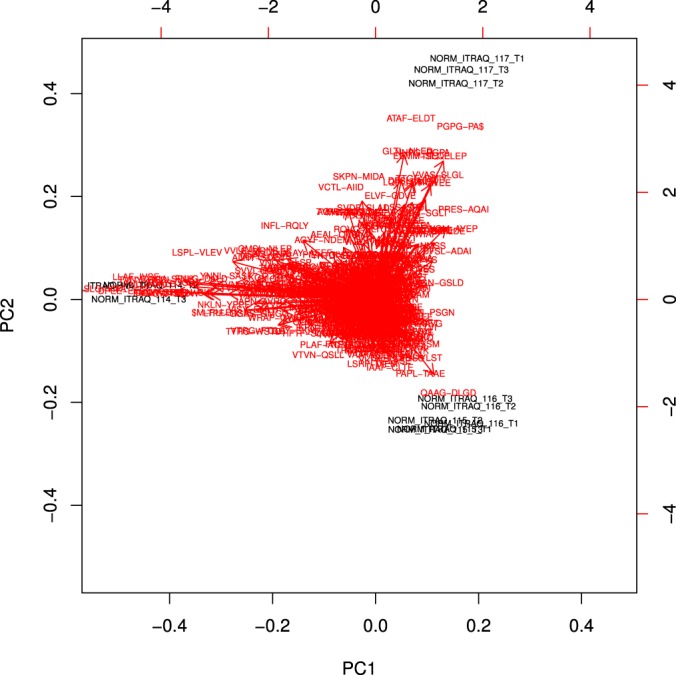
Principal component analysis of AC - iTRAQ117, TVA - iTRAQ115, TA - iTRAQ116, and NC - iTRAQ114 in triplicate based on the abundance of nontryptic peptides.
The results of Data set 1 presented above are based on analyses of pooled samples obtained by mixing tissue samples collected from different patients (17). Pooling allowed for in-depth analysis and access to quantitative information on low-abundance peptides/proteins at the cost of a decreased number of replicate experiments. Therefore, we analyzed a set of 40 independently collected tumor tissue/control samples using a label-free quantitation single pass LC-MS/MS approach. In the new set, 10 samples originated from a colon tumor (i.e. AC) and 10 from control nonmalignant tissue samples (NCA), as well as 10 from BC samples and 10 from control bladder tissue (NCB) samples. Each sample was analyzed during a single 3-hour LC-MS run, allowing the samples to be individually processed without pooling. However, this led to a much smaller number of only the most abundant peptides/proteins (Data set 2 peptides/proteins, listed in supplemental Table S4) that could be quantified, as the information was extracted from a single LC-MS run and not from a series of LC-MS/MS runs of IEF sample fractions, which was the case for the pooling experiment. Nevertheless, a label-free approach was necessary to verify whether the significance of AC versus NC differentiation using VIATC peptide can be retained in a larger sample set. Using the same approach, BC samples were compared with nontransformed control tissue samples. We identified 126 VIATC consensus nontryptic peptides in the AC/NCA data set and 153 peptides in the BC/NCB data set. The results are illustrated for 15 select peptides in Fig. 7, showing significantly different levels in tumor samples than in controls for both AC and BC. Similar to the pooled experiments, peptides with a cancer-specific VIATC consensus were found in tumor samples. As the majority of peptides with differential expression detected in the IEF-LC-MS experiment originated from the nuclear fraction, the number of VIATC differential peptides detected in LC-MS was relatively small. More sensitive, targeted approaches are necessary to detect peptides originating from low abundance proteins, bearing a more distinct cancer-specific signature. The number of VIATC peptides differentiating BC from NCB samples was 53 (31 up-regulated and 22 downregulated) thus, a large fraction (approximately one-third) of all VIATC peptides are differential for BC, i.e. either significantly overrepresented or underrepresented. Interestingly, several peptides were shared in the AC and BC differential sets (Fig. 7), with a common direction of change. As such, these results reflect general changes in cancer tissue that are not dependent on cancer type. However, other peptides were differential only in AC or BC. In general, LC-MS experiments on nonpooled sample series confirmed the presence of cancer-specific VIATC consensus protein fragments in both AC and BC tumor samples. To obtain analytical access to a panel of peptides originating from low abundance proteins, which could provide the most clinically relevant information, approaches with higher sensitivity than simple one-pass LC-MS are required.
Fig. 7.

Nonpooled sample series analysis of 10 adenocarcinoma (AC) samples and 10 bladder cancer (BC) samples. Box-plots representing peptide abundance for AC versus control samples (NCA) and BC versus control tissue (NCB) for selected VIATC consensus peptides. Boxes denote interquartile range whereas whiskers extend to the most extreme data point which is no more than 1.5 times the interquartile range from the box. Asterisks denote * - q<0.05, ** - q<0.01.
To verify whether our findings can be confirmed in an analysis of data sets obtained in other laboratories, we searched for the disease-specific VIATC consensus proteolytic fingerprint in data sets deposited in PRIDE (Data sets 3 and 4). We analyzed 52 LC-MS raw files obtained from the analysis of colon cancer samples (16 AC versus 16 controls—Data set 3) and noncancer samples originating from an analysis of mucosal colon biopsies taken by endoscopy from noninflamed UC patients (10 UC versus 10 controls—Data set 4). For quantitation we used the “spectral count” approach, comparing the fraction of VIATC cleaved peptide queries in a given protein in disease and control groups. We detected 757 proteins characterized by the presence of 10 or more queries assigned to VIATC consensus cleavages in the AC or control sample set. For each of these proteins, we calculated the fraction of queries identifying VIATC peptides compared with the total number of queries assigned to this protein (qVIATC/qTotal). Next, we extracted a list of 250 proteins for which qVIATC/qTotal was at least 2-fold larger in AC than in controls (supplemental Table S5A). The number of proteins with the VIATC fraction underrepresented in AC by more than 2-fold was much smaller (Fig. 8A, red marks). A similar comparison was obtained for the UC versus control sample set, identifying 166 proteins with qVIATC/qTotal >2. Thus, the overrepresentation of VIATC cleavages in UC was also substantial. However, as Fig. 8A shows, for proteins from the UC list the fraction of VIATC cleavages was much smaller on average than for the AC list, indicating markedly less frequent VIATC cleavage in UC than AC. A Venn diagram comparing the protein identities from the UC (Data set 4), AC (Data set 3), and previous iTRAQ AC analyses of Data set 1 (Fig. 8B) shows the number of proteins common and unique for each condition studied. Sixty-one of the 250 proteins identified in the PRIDE database were also identified in the iTRAQ experiment as being preferentially cleaved at the VIATC consensus in cancer. Forty-nine of these proteins were not found among those preferentially cleaved in UC samples (supplemental Table S5B). Therefore, the proteome may be affected by VIATC cleaving protease in a disease-specific manner.
Fig. 8.
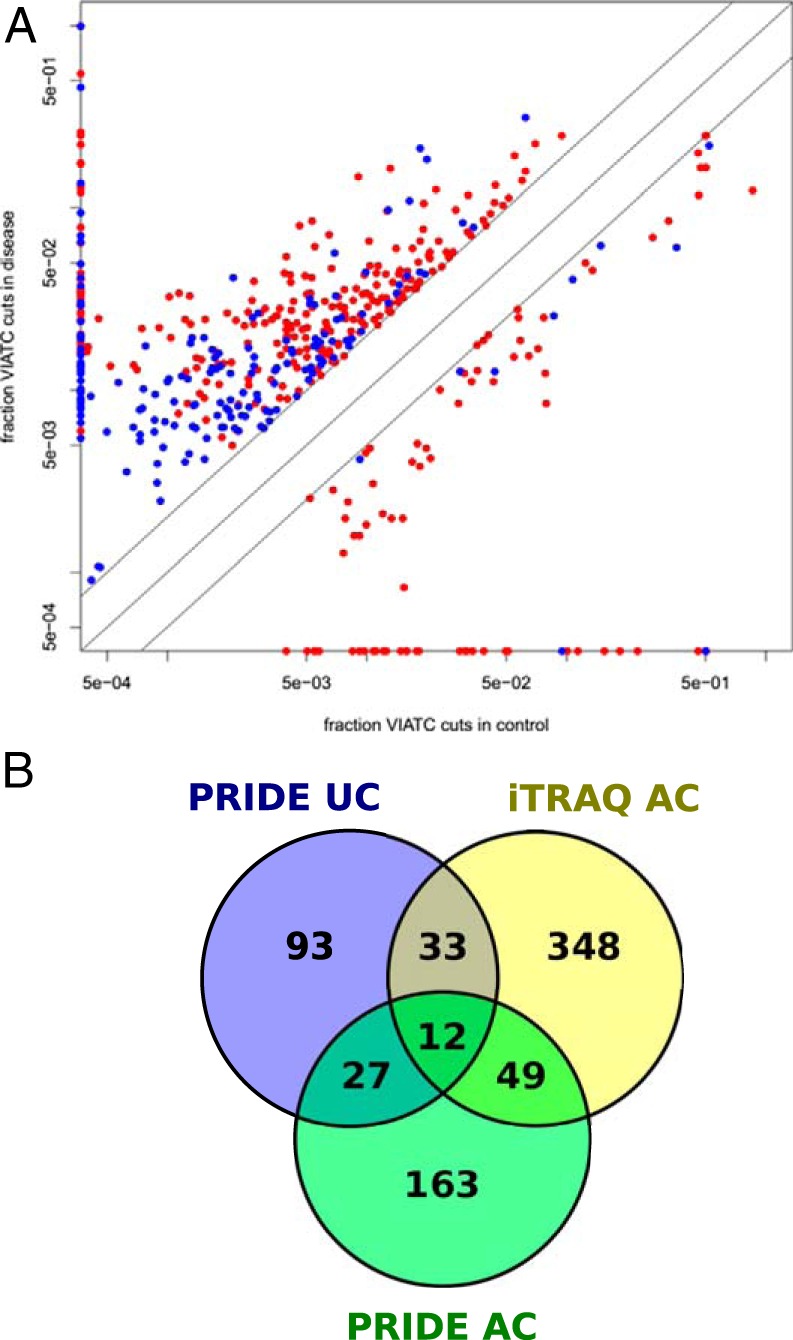
A, Fraction of queries assigned to VIATC peptides (qVIATC/qTotal) for a subset of PRIDE database proteins in disease (vertical axis) and control samples (horizontal axis) for adenocarcinoma (AC; red) and ulcerative colitis (UC; blue). Data for proteins characterized by the presence of more than 10 VIATC queries are shown. For clarity, data from proteins with a qVIATC/qTotal between 0.5 and 2 are not shown. Proteins marked on the vertical axis had no VIATC queries in control samples, whereas proteins marked on the horizontal axis had no VIATC queries in disease samples. Note the higher number of proteins with qVIATC/qTotal >2 (placed above the diagonal) than qVIATC/qTotal <0.5 (placed below the diagonal) in disease samples, both AC and UC. Also note the significantly lower average fraction of VIATC cleavages in UC than AC. B, Venn diagram illustrating number of common and unique proteins containing significantly overrepresented VIATC-cleaved peptides in iTRAQ adenocarcinoma, PRIDE adenocarcinoma and PRIDE ulcerative colitis data set. Note 49 proteins commonly identified in iTRAQ and PRIDE adenocarcinoma data set and absent in ulcerative colitis data set.
DISCUSSION
In the course of peptide-wise proteomic analyses of AC samples, we detected abundant cancer-specific proteolytic events that strictly fulfill the cleavage consensus VIATC characteristic for neutrophil elastase. We have shown that cancer-specific elastase cleavage mostly affects the nuclear fraction of AC tumor tissue, with multiple RNPs affected and many cleavage sites localized within functionally important RRM RNA-binding domains. Differential peptide set linked by a common VIATC consensus was the strongest proteolytic fingerprint marking AC. In contrast, VIATC cleavage was not disease-dependent in TVA and TA samples. Other cleavage events, such as cleavage of the Asp-Gly site in keratin 18, can be detected much more frequently in noncancerous tissue than in TVA, TA, or AC. In addition, multiple differentially proteolytic peptides with the VIATC consensus were also detected in BC samples. Furthermore, we confirmed our findings by analyzing cancer and UC data from the publicly accessible PRIDE database.
NE, or elastase-2, is a serine protease (37) that accumulates in azurophil granules in the cytoplasm of neutrophils. Unlike elastase-1, NE is characterized by a well-defined narrow cleavage consensus. Neutrophils are a nearly exclusive source of NE and produce large amounts of this protease (38). Neutrophils, predominantly circulation leukocytes, are traditionally thought to be terminally differentiated effector cells because of their role during the acute phase of inflammation and host defense against pathogens. NE is able to cleave numerous targets, including elastin, collagens, and Omp bacterial proteins. In the traditional view of the role of neutrophils, the presence of the NE proteolytic signature inside tumors may seem to be unexpected. Protein Atlas contains IHC data for neutrophil elastase, however staining is negative for several CRC samples tested there. The reason could be that IHC tissue arrays used in Protein Atlas are not representative to spot elastase staining which seems to be focal and associated with neutrophil extracellular traps (NETs) presence. In contrast to Protein Atlas Arelaki et al. (39) using same antibody as the one utilized in Protein Atlas (sc-55549) have clearly shown neutrophil elastase IHC staining in CRC samples, and importantly, elastase presence overlapped with NETs distribution.
The presence of NE in tumor tissues has been studied for some time (40), (41), also in the context of breast cancer (42). Its potential prognostic value was also assessed (43–45). A role of protease-inhibitor imbalance in cancer progression has been suggested (46), and NE has been proposed as a new therapeutic target (47–50). For many cancer types, increased NE presence has been correlated with cancer stage, grade, and survival. Currently, the discussion focuses on the prognostic relevance of tumor-associated neutrophils (51) and their anti-tumor and pro-tumor activities (52–53), which are most likely connected to the neutrophil maturation pathway (54). Similar to macrophages, neutrophils infiltrate tumors, but their role in the modification of cancer progression has just begun to emerge (47, 55). These cells are a major source of stromal proteases in tumors, and elastase secreted by neutrophils has been shown to be taken up into lung and breast cancer cells (56) via an endocytic, clathrin-mediated pathway (42, 57). Neutrophil infiltration was found to be gradually reduced from the tumor mass to the distal margin (39). However, the presence of neutrophils in tumors is not sufficient to explain the observation that a large set of nuclear proteins is also affected. Apoptotic tumor cells may provide nuclear proteins for elastase cleavage, or NE proteolytic fragments of nuclear proteins may be neutrophil proteins degraded during NET-osis (58), a neutrophil-specific process of controlled cell death during which extruded fibrillary networks composed of nuclear components and granule proteins, the so called neutrophil extracellular traps, are formed (59). Thus, during the course of NET-osis, the nuclear envelope disintegrates, granting access to its nuclear proteins. NET-osis has been detected in tumors (60), and a causative link between NET formation and cancer-associated thrombosis, one of the major causes of death in cancer patients, has been established (61). Moreover, proteomic analysis of NET-associated substrate profiles indicates a predominant contribution by NE (62). Irrelevant if their source are tumor cells or neutrophils elastase-cleaved peptides mark the tumor presence.
Many other noncaspase proteases have emerged as active players in apoptotic proteolytic cascades (1, 63). These proteases can target proteins that are not cleaved by caspases (64) or the same proteins. Many proteins found in the present study to be cleaved at VIATC consensus are also known to be cleaved by caspases (e.g. keratin 18, nucleolin, vimentin, lamin B2, annexins, alpha-actinin, gelsolin, hnRNPs, ribosomal proteins, elongation factors; Table II in (65), Table I in (66)). 111 proteins containing VIATC peptides characterized by a large AC/NC ratio in our study are also present on the list of caspase targets compiled recently (see supplemental Tables S2 and S4 in (67)). Thus, ca. 25% of the proteins found in our study are known caspase targets. Notably, a large fraction of proteins modified in apoptosis contain RNA-binding motifs, including RRM domains (66, 68, 69), similar to the overrepresentation of RNPs in the VIATC protein set with a preference for cleavage at RRM domains.
Currently, the most widespread protein fragments used for cancer studies originate from caspase cleavage of keratins. TPA is a protein antigen that was identified in 1957 by raising antibodies against pooled tumors. TPA has been identified as consisting of keratins 8, 18, and 19 and/or their fragments (70). TPA reactivity is derived from specific epitopes of human cytokeratin 18, mainly fragment 284–396 (71). In keratin 18 the so-called M30 epitope comes from a >20 kDa keratin fragment resulting from two caspase cleavages at sequence consensus sites V235EVD and D394ALD (7). The target sites for the keratin 19-derived Cyfra 21–1 monoclonal antibodies BM 19.21 and KS 19.1 lie within amino acids 346–367 and amino acids 311–335, respectively. These mAbs detect a keratin 19 fragment most likely resulting from cleavage at caspase site S233VEVD (72) in the helix 2B region of the rod domain of keratin 19. The Cyfra 21–1 assay has found broad clinical application, allowing the monitoring of treatment and the response to therapy, and has proven to be particularly useful in the case of squamous cell carcinomas of the lung (6). However, some authors have found that these antibodies detect intact keratin 19 instead of the Cyfra 21–1 fragment (73). A more detailed comparison of fragment-specific and total keratin 18 antibodies has been carried out in the context of lung cancer (74).
NE has not previously been mentioned in the context of noncaspase proteases involved in mediating/promoting cell death; nevertheless, its secondary role cannot be excluded as the VIATC P1 cleavage was the strongest cancer-specific proteolytic fingerprint that could be detected in cancer tissues. Proteolytic cleavage is not only a simple degradation event, but also an important signaling event (75). Thus, we can reasonably expect that proteolytic fingerprints reflect important processes, including cancer development, and can be a source of biomarker panels. Neutrophil-specific elastase cleavage may be of value, as the presence of tumor-infiltrating neutrophils often correlates with clinical outcome.
A 2002 paper claiming an efficient SELDI-based ovarian cancer biomarker serum panel offered high hopes for efficient biomarkers (76). However, further work dampened the initial optimism, providing knowledge on multiple factors that mask disease-related changes in the proteome of biofluids (77, 78) and the successes are currently very limited (79). To overcome these problems, more targeted approaches coupled with new high-confidence MS methods are necessary. Jimenez et al. (80) summarized this well: “We anticipate that in the near future, these novel mass spectrometry-based in-depth approaches will uncover many novel, specific CRC marker candidates in clinical tissues and that their targeted validation with multi-reaction monitoring MS will speed up development of noninvasive tests in feces and serum/plasma.”
In conclusion, during the course of global analyses of cancer versus control tissue samples, we detected a very strong cancer-specific proteolytic fingerprint of nontryptic cleavages. Proteomic experiments on both colon and bladder tumor tissue indicated the presence of numerous proteolytic protein fragments, whose abundance strongly correlated with the presence of cancer. We noted a significant number of differential peptides characterized by VIATC consensus cleavage sites. As outliers in the protein-wise analysis, these peptides are usually ignored during routine global differential proteomics, but they may provide important information on disease-related proteolytic events. Currently, only a few protein fragments are the object of more detailed tests exploring their diagnostic/prognostic potential, but our work shows that the spectrum of possible cancer-specific peptide markers may be much broader. Similar to caspase-derived keratin fragments, elastase- and/or meprin-derived peptides can be released into the circulation during cellular breakdown and be detected by sensitive enough methods. The newly detected fragments, bearing the strict fingerprint of elastase cleavage specificity, in our opinion most likely originate from the activity of neutrophil elastase infiltrating tumors, which is a not-unexpected well known phenomenon. The substrate proteins may originate from the tumor cells, and/or neutrophil cells undergoing netosis. Although we do not speculate what is the major source of substrates we identify, we conclude that their presence is strongly correlated with tumor presence, providing a source of potential disease markers. We are aware that the identification of a marker is merely a small initial step toward diagnosis, early detection, prognosis or personalization of the treatment and the ultimate tests should be carried out on body fluids, preferably plasma/serum samples. However, we believe that the direct detection of highly diluted peptide fragments in plasma/serum, without prior enrichment, is not possible with the present state of the art LC-MS systems. We also believe that enrichment strategies when worked out and optimized will enable such detection and quantification of low abundance fragments, as was already shown by numerous examples (81, 82).
Supplementary Material
Acknowledgments
We thank Krzysztof Drabikowski for valuable suggestions and Piotr Zarebski for data handling and storage.
Footnotes
Author contributions: J.D., J.K., M.M., J. Ostrowski, and M.D. designed research; M.K., J.D., J.K., and A.P. performed research; M.K., J.D., J.K., M.M., and M.D. analyzed data; J.D., M.M., J. Ostrowski, and M.D. wrote the paper; J. Olędzki mass spectrometry measurements.
* We acknowledge the financial support from CEPT (POIG.02.02.00-14-024/08-00) and Nano-Fun (POIG.02.02.00-00-025/09) programs as well as the support from National Science Centre (NCN) within project MAESTRO (UMO-2014/14/A/NZ1/00306).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- TPA
- tissue polypeptide antigen
- AC
- adenocarcinoma
- BC
- bladder cancer
- FC
- fold change
- hnRNPs
- heterogeneous nuclear ribonucleoproteins
- NC
- noncancer
- NCA
- nonmalignant tissue samples
- NCB
- control bladder tissue
- NE
- neutrophil elastase
- TA
- tubular adenoma
- TPS
- Tissue polypeptide-specific antigen
- TVA
- tubulovillous adenoma
- UC
- ulcerative colitis.
REFERENCES
- 1. Mason S. D., and Joyce J. A. (2011) Proteolytic networks in cancer. Trends Cell Biol. 21, 228–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Karantza V. (2011) Keratins in health and cancer: more than mere epithelial cell markers. Oncogene 30, 127–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chu P. G., Lau S. K., and Weiss L. M. (2009) Keratin expression in endocrine organs and their neoplasms. Endocr. Pathol. 20, 1–10 [DOI] [PubMed] [Google Scholar]
- 4. Chu P. G., and Weiss L. M. (2002) Keratin expression in human tissues and neoplasms. Histopathology 40, 403–439 [DOI] [PubMed] [Google Scholar]
- 5. Moll R., Divo M., and Langbein L. (2008) The human keratins: biology and pathology. Histochem. Cell Biol. 129, 705–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barak V., Goike H., Panaretakis K. W., and Einarsson R. (2004) Clinical utility of cytokeratins as tumor markers. Clin. Biochem. 37, 529–540 [DOI] [PubMed] [Google Scholar]
- 7. Sawant S. S., Zingde S. M., and Vaidya M. M. (2008) Cytokeratin fragments in the serum: Their utility for the management of oral cancer. Oral Oncol. 44, 722–732 [DOI] [PubMed] [Google Scholar]
- 8. Schutte B., Henfling M., Kölgen W., Bouman M., Meex S., Leers M. P., Nap M., Björklund V., Björklund P., Björklund B., Lane E. B., Omary M. B., Jörnvall H., and Ramaekers F. C. (2004) Keratin 8/18 breakdown and reorganization during apoptosis. Exp. Cell Res. 297, 11–26 [DOI] [PubMed] [Google Scholar]
- 9. Washino S., Hirai M., Matsuzaki A., and Kobayashi Y. (2011) Clinical usefulness of CEA, CA19–9, and CYFRA 21–1 as tumor markers for urothelial bladder carcinoma. Urol. Int. 87, 420–428 [DOI] [PubMed] [Google Scholar]
- 10. Holdenrieder S., Stieber P., Liska V., Treska V., Topolcan O., Dreslerova J., Matejka V. M., Finek J., and Holubec L. (2012) Cytokeratin serum biomarkers in patients with colorectal cancer. Anticancer Res. 32, 1971–1976 [PubMed] [Google Scholar]
- 11. Linder S. (2007) Cytokeratin markers come of age. Tumour Biol. 28, 189–195 [DOI] [PubMed] [Google Scholar]
- 12. Moll R., Franke W. W., Schiller D. L., Geiger B., and Krepler R. (1982) The catalog of human cytokeratins: patterns of expression in normal epithelia, tumors and cultured cells. Cell 31, 11–24 [DOI] [PubMed] [Google Scholar]
- 13. Herrmann H., and Aebi U. (2004) Intermediate filaments: molecular structure, assembly mechanism, and integration into functionally distinct intracellular Scaffolds. Annu. Rev. Biochem. 73, 749–789 [DOI] [PubMed] [Google Scholar]
- 14. Fulzele A., Malgundkar S. A., Govekar R. B., D'Cruz A. K., Chaturvedi P., Patil A., Kane S. V., and Zingde S. M. (2012) Keratins in oral cancer: necessity of mass spectrometry for validation of antibody based identifications. J. Proteomics 75, 2404–2416 [DOI] [PubMed] [Google Scholar]
- 15. Yao C. C., Tseng S. W., and Han C. P. (2010) Monoclonal antibody CAM5.2 can detect the cytokeratin 8, not cytokeratin 18. Comment on: “Occult disseminated tumor cells in lymph nodes of patients with gastric carcinoma. A critical appraisal of assessment and relevance. Langenbecks Arch Surg. 2009 Jan; 394(1): 105–113.” Langenbeck's Arch. Surgery 395, 291–292 [DOI] [PubMed] [Google Scholar]
- 16. Leei C. K., Lin W. L., and Han C. P. (2010) Monoclonal antibody Cam 5.2 targeted mainly CK8, but not CK18–Comment on: “Chromophobe renal cell carcinoma with liposarcomatous dedifferentiation–report of a unique case. Int J Clin Exp Pathol. 2010 May 5; 3(5):534–40.” Int. J. Clin. Exp. Pathol. 3, 742. [PMC free article] [PubMed] [Google Scholar]
- 17. Mikula M., Rubel T., Karczmarski J., Goryca K., Dadlez M., and Ostrowski J. (2011) Integrating proteomic and transcriptomic high-throughput surveys for search of new biomarkers of colon tumors. Funct. Integr. Genomics 11, 215–224 [DOI] [PubMed] [Google Scholar]
- 18. Slebos R. J., Wang X., Wang X., Zhang B., Tabb D. L., and Liebler D. C. (2015) Proteomic analysis of colon and rectal carcinoma using standard and customized databases. Sci. Data 2, 150022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vizcaíno J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q.-W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mikula M., Gaj P., Dzwonek K., Rubel T., Karczmarski J., Paziewska A., Dzwonek A., Bragoszewski P., Dadlez M., and Ostrowski J. (2010) Comprehensive analysis of the palindromic motif TCTCGCGAGA: a regulatory element of the HNRNPK promoter. DNA Res. 17, 245–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Skrzypczak M., Goryca K., Rubel T., Paziewska A., Mikula M., Jarosz D., Pachlewski J., Oledzki J., and Ostrowski J. (2010) Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One 5, e13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bakun M., Karczmarski J., Poznanski J., Rubel T., Rozga M., Malinowska A., Sands D., Hennig E., Oledzki J., Ostrowski J., and Dadlez M. (2009) An integrated LC-ESI-MS platform for quantitation of serum peptide ladders. Application for colon carcinoma study. Proteomics Clin Appl 3, 932–946 [DOI] [PubMed] [Google Scholar]
- 23. Benjamini Y., and Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. JSTOR 57, 289–300 [Google Scholar]
- 24. Kim S., and Pevzner P. A. (2014) MS-GF+ makes progress towards a universal database search tool for proteomics. Nat Commun 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nahnsen S., Bielow C., Reinert K., and Kohlbacher O. (2013) Tools for Label-free Peptide Quantification. Mol. Cell. Proteomics 12, 549–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Simpson K. L., Cawthorne C., Zhou C., Hodgkinson C. L., Walker M. J., Trapani F., Kadirvel M., Brown G., Dawson M. J., MacFarlane M., Williams K. J., Whetton A. D., and Dive C. (2013) A caspase-3 “death-switch” in colorectal cancer cells for induced and synchronous tumor apoptosis in vitro and in vivo facilitates the development of minimally invasive cell death biomarkers. Cell Death and Disease 4, e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Premchandar A., Kupniewska A., Tarnowski K., Mücke N., Mauermann M., Kaus-Drobek M., Edelman A., Herrmann H., and Dadlez M. (2015) Analysis of distinct molecular assembly complexes of keratin K8 and K18 by hydrogen-deuterium exchange. J. Struct. Biol. 192, 426–440 [DOI] [PubMed] [Google Scholar]
- 28. Rawlings N. D., Barrett A. J., and Bateman A. (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 40, D343–D350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Igarashi Y., Heureux E., Doctor K. S., Talwar P., Gramatikova S., Gramatikoff K., Zhang Y., Blinov M., Ibragimova S. S., Boyd S., Ratnikov B., Cieplak P., Godzik A., Smith J. W., Osterman A. L., and Eroshkin A. M. (2009) PMAP: databases for analyzing proteolytic events and pathways. Nucleic Acids Res. 37, D611–D618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., and von Mering C. (2011) The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kressler D., Hurt E., and Bassler J. (2010) Driving ribosome assembly. Biochim. Biophys. Acta 1803, 673–683 [DOI] [PubMed] [Google Scholar]
- 32. Whitson S. R., LeStourgeon W. M., and Krezel A. M. (2005) Solution structure of the symmetric coiled coil tetramer formed by the oligomerization domain of hnRNP C: implications for biological function. J. Mol. Biol. 350, 319–337 [DOI] [PubMed] [Google Scholar]
- 33. Gong D., Yang F., Li F., Qian D., Wu M., Shao Z., Wu M., Wu J., and Shi Y. (2013) Crystal structure and functional characterization of the human RBM25 PWI domain and its flanking basic region. Biochem. J. 450, 85–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Becker-Pauly C., Barré O., Schilling O., Auf dem Keller U., Ohler A., Broder C., Schütte A., Kappelhoff R., Stöcker W., and Overall C. M. (2011) Proteomic analyses reveal an acidic prime side specificity for the astacin metalloprotease family reflected by physiological substrates. Mol. Cell. Proteomics 10, M111.009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lottaz D., Hahn D., Müller S., Müller C., and Sterchi E. E. (1999) Secretion of human meprin from intestinal epithelial cells depends on differential expression of the alpha and beta subunits. Eur. J. Biochem. 259, 496–504 [DOI] [PubMed] [Google Scholar]
- 36. Lottaz D., Maurer C. A., Noël A., Blacher S., Huguenin M., Nievergelt A., Niggli V., Kern A., Müller S., Seibold F., Friess H., Becker-Pauly C., Stöcker W., and Sterchi E. E. (2011) Enhanced Activity of Meprin-α, a Pro-Migratory and Pro-Angiogenic Protease, in Colorectal Cancer. PLoS ONE 6, e26450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sinha S., Watorek W., Karr S., Giles J., Bode W., and Travis J. (1987) Primary structure of human neutrophil elastase. Proc. Natl. Acad. Sci. U.S.A. 84, 2228–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee W. L., and Downey G. P. (2001) Leukocyte elastase: physiological functions and role in acute lung injury. Am. J. Respir. Crit. Care Med. 164, 896–904 [DOI] [PubMed] [Google Scholar]
- 39. Arelaki S., Arampatzioglou A., Kambas K., Papagoras C., Miltiades P., Angelidou I., Mitsios A., Kotsianidis I., Skendros P., Sivridis E., Maroulakou I., Giatromanolaki A., and Ritis K. (2016) Gradient Infiltration of Neutrophil Extracellular Traps in Colon Cancer and Evidence for Their Involvement in Tumour Growth. PLOS ONE 11, e0154484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Powell D. R., and Huttenlocher A. (2016) Neutrophils in the Tumor Microenvironment. Trends Immunol. 37, 41–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Coffelt S. B., Wellenstein M. D., and de Visser K. E. (2016) Neutrophils in cancer: neutral no more. Nat. Rev. Cancer 16, 431–446 [DOI] [PubMed] [Google Scholar]
- 42. Mittendorf E. A., Alatrash G., Qiao N., Wu Y., Sukhumalchandra P., St John L. S., Philips A. V., Xiao H., Zhang M., Ruisaard K., Clise-Dwyer K., Lu S., and Molldrem J. J. (2012) Breast cancer cell uptake of the inflammatory mediator neutrophil elastase triggers an anticancer adaptive immune response. Cancer Res. 72, 3153–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Akizuki M., Fukutomi T., Takasugi M., Takahashi S., Sato T., Harao M., Mizumoto T., and Yamashita J. (2007) Prognostic significance of immunoreactive neutrophil elastase in human breast cancer: long-term follow-up results in 313 patients. Neoplasia 9, 260–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Foekens J. A., Ries C., Look M. P., Gippner-Steppert C., Klijn J. G., and Jochum M. (2003) The prognostic value of polymorphonuclear leukocyte elastase in patients with primary breast cancer. Cancer Res. 63, 337–341 [PubMed] [Google Scholar]
- 45. Yamashita J., Ogawa M., and Shirakusa T. (1995) Free-form neutrophil elastase is an independent marker predicting recurrence in primary breast cancer. J. Leukoc. Biol. 57, 375–378 [DOI] [PubMed] [Google Scholar]
- 46. Yang P., Bamlet W. R., Sun Z., Ebbert J. O., Aubry M. C., Krowka M. J., Taylor W. R., Marks R. S., Deschamps C., Swensen S. J., Wieben E. D., Cunningham J. M., Melton L. J., and de Andrade M. (2005) Alpha1-antitrypsin and neutrophil elastase imbalance and lung cancer risk. Chest 128, 445–452 [DOI] [PubMed] [Google Scholar]
- 47. Gregory A. D., and Houghton A. M. (2011) Tumor-associated neutrophils: new targets for cancer therapy. Cancer Res. 71, 2411–2416 [DOI] [PubMed] [Google Scholar]
- 48. Moroy G., Alix A. J., Sapi J., Hornebeck W., and Bourguet E. (2012) Neutrophil elastase as a target in lung cancer. Anticancer Agents Med. Chem. 12, 565–579 [DOI] [PubMed] [Google Scholar]
- 49. Hunt K. K., Wingate H., Yokota T., Liu Y., Mills G. B., Zhang F., Fang B., Su C. H., Zhang M., Yi M., and Keyomarsi K. (2013) Elafin, an inhibitor of elastase, is a prognostic indicator in breast cancer. Breast Cancer Res. 15, R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu Y., Zhang J., Han J., Pan X., Cao Y., Guo H., Pan Y., An Y., and Li X. (2012) Curcumin inhibits tumor proliferation induced by neutrophil elastase through the upregulation of α1-antitrypsin in lung cancer. Mol. Oncol. 6, 405–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Donskov F. (2013) Immunomonitoring and prognostic relevance of neutrophils in clinical trials. Semin. Cancer Biol. 23, 200–207 [DOI] [PubMed] [Google Scholar]
- 52. Galdiero M. R., Bonavita E., Barajon I., Garlanda C., Mantovani A., and Jaillon S. (2013) Tumor associated macrophages and neutrophils in cancer. Immunobiology 218, 1402–1410 [DOI] [PubMed] [Google Scholar]
- 53. Lungarella G., Cavarra E., Lucattelli M., and Martorana P. A. (2008) The dual role of neutrophil elastase in lung destruction and repair. Int. J. Biochem. Cell Biol. 40, 1287–1296 [DOI] [PubMed] [Google Scholar]
- 54. Fridlender Z. G., Sun J., Kim S., Kapoor V., Cheng G., Ling L., Worthen G. S., and Albelda S. M. (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 16, 183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Fridlender Z. G., and Albelda S. M. (2012) Tumor-associated neutrophils: friend or foe? Carcinogenesis 33, 949–955 [DOI] [PubMed] [Google Scholar]
- 56. Houghton A. M., Rzymkiewicz D. M., Ji H., Gregory A. D., Egea E. E., Metz H. E., Stolz D. B., Land S. R., Marconcini L. A., Kliment C. R., Jenkins K. M., Beaulieu K. A., Mouded M., Frank S. J., Wong K. K., and Shapiro S. D. (2010) Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med. 16, 219–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gregory A. D., Hale P., Perlmutter D. H., and Houghton A. M. (2012) Clathrin pit-mediated endocytosis of neutrophil elastase and cathepsin G by cancer cells. J. Biol. Chem. 287, 35341–35350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fuchs T. A., Abed U., Goosmann C., Hurwitz R., Schulze I., Wahn V., Weinrauch Y., Brinkmann V., and Zychlinsky A. (2007) Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 176, 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jaillon S., Galdiero M. R., Del Prete D., Cassatella M. A., Garlanda C., and Mantovani A. (2013) Neutrophils in innate and adaptive immunity. Semin. Immunopathol. 35, 377–394 [DOI] [PubMed] [Google Scholar]
- 60. Berger-Achituv S., Brinkmann V., Abed U. A., Kühn L. I., Ben-Ezra J., Elhasid R., and Zychlinsky A. (2013) A proposed role for neutrophil extracellular traps in cancer immunoediting. Front. Immunol. 4, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Demers M., Krause D. S., Schatzberg D., Martinod K., Voorhees J. R., Fuchs T. A., Scadden D. T., and Wagner D. D. (2012) Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. U.S.A. 109, 13076–13081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. O'Donoghue A. J., Jin Y., Knudsen G. M., Perera N. C., Jenne D. E., Murphy J. E., Craik C. S., and Hermiston T. W. (2013) Global substrate profiling of proteases in human neutrophil extracellular traps reveals consensus motif predominantly contributed by elastase. PLoS ONE 8, e75141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Moffitt K. L., Martin S. L., and Walker B. (2007) The emerging role of serine proteases in apoptosis. Biochem. Soc. Trans. 35, 559–560 [DOI] [PubMed] [Google Scholar]
- 64. Johnson D. E. (2000) Noncaspase proteases in apoptosis. Leukemia 14, 1695–1703 [DOI] [PubMed] [Google Scholar]
- 65. Thiede B., and Rudel T. (2004) Proteome analysis of apoptotic cells. Mass Spectrom. Rev. 23, 333–349 [DOI] [PubMed] [Google Scholar]
- 66. Thiede B., Treumann A., Kretschmer A., Söhlke J., and Rudel T. (2005) Shotgun proteome analysis of protein cleavage in apoptotic cells. Proteomics 5, 2123–2130 [DOI] [PubMed] [Google Scholar]
- 67. Stoehr G., Schaab C., Graumann J., and Mann M. (2013) A SILAC-based approach identifies substrates of caspase-dependent cleavage upon TRAIL-induced apoptosis. Mol. Cell. Proteomics 12, 1436–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schmidt F., Hustoft H. K., Strozynski M., Dimmler C., Rudel T., and Thiede B. (2007) Quantitative proteome analysis of cisplatin-induced apoptotic Jurkat T cells by stable isotope labeling with amino acids in cell culture, SDS-PAGE, and LC-MALDI-TOF/TOF MS. Electrophoresis 28, 4359–4368 [DOI] [PubMed] [Google Scholar]
- 69. Thiede B., Dimmler C., Siejak F., and Rudel T. (2001) Predominant identification of RNA-binding proteins in Fas-induced apoptosis by proteome analysis. J. Biol. Chem. 276, 26044–26050 [DOI] [PubMed] [Google Scholar]
- 70. Weber K., Osborn M., Moll R., Wiklund B., and Lüning B. (1984) Tissue polypeptide antigen (TPA) is related to the non-epidermal keratins 8, 18 and 19 typical of simple and non-squamous epithelia: re-evaluation of a human tumor marker. EMBO J. 3, 2707–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rydlander L., Ziegler E., Bergman T., Schoberl E., Steiner G., Bergman A. C., Zetterberg A., Marberger M., Bjorklund P., Skern T., Einarsson R., and Jornvall H. (1996) Molecular characterization of a tissue-polypeptide-specific-antigen epitope and its relationship to human cytokeratin 18. Eur. J. Biochem. 241, 309–314 [DOI] [PubMed] [Google Scholar]
- 72. Dohmoto K., Hojo S., Fujita J., Yang Y., Ueda Y., Bandoh S., Yamaji Y., Ohtsuki Y., Dobashi N., Ishida T., and Takahara J. (2001) The role of caspase 3 in producing cytokeratin 19 fragment (CYFRA21–1) in human lung cancer cell lines. Int. J. Cancer 91, 468–473 [DOI] [PubMed] [Google Scholar]
- 73. Alix-Panabières C., Vendrell J. P., Slijper M., Pellé O., Barbotte E., Mercier G., Jacot W., Fabbro M., and Pantel K. (2009) Full-length cytokeratin-19 is released by human tumor cells: a potential role in metastatic progression of breast cancer. Breast Cancer Res. 11, R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. De Petris L., Brandén E., Herrmann R., Sanchez B. C., Koyi H., Linderholm B., Lewensohn R., Linder S., and Lehtiö J. (2011) Diagnostic and prognostic role of plasma levels of two forms of cytokeratin 18 in patients with non-small-cell lung cancer. Eur. J. Cancer 47, 131–137 [DOI] [PubMed] [Google Scholar]
- 75. Turk B., Turk D., and Turk V. (2012) Protease signalling: the cutting edge. EMBO J. 31, 1630–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Petricoin E. F., Ardekani A. M., Hitt B. A., Levine P. J., Fusaro V. A., Steinberg S. M., Mills G. B., Simone C., Fishman D. A., Kohn E. C., and Liotta L. A. (2002) Use of proteomic patterns in serum to identify ovarian cancer. Lancet 359, 572–577 [DOI] [PubMed] [Google Scholar]
- 77. Pietrowska M., and Widłak P. (2012) MALDI-MS-based profiling of serum proteome: detection of changes related to progression of cancer and response to anticancer treatment. Int. J. Proteomics 2012, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Karczmarski J., Rubel T., Mikula M., Wolski J., Rutkowski A., Zagorowicz E., Dadlez M., and Ostrowski J. (2013) Pre-analytical-related variability influencing serum peptide profiles demonstrated in a mass spectrometry-based search for colorectal and prostate cancer biomarkers. Acta Biochim. Pol. 60, 417–425 [PubMed] [Google Scholar]
- 79. Leung F., Diamandis E. P., and Kulasingam V. (2012) From bench to bedside: discovery of ovarian cancer biomarkers using high-throughput technologies in the past decade. Biomark. Med. 6, 613–625 [DOI] [PubMed] [Google Scholar]
- 80. Jimenez C. R., Knol J. C., Meijer G. A., and Fijneman R. J. (2010) Proteomics of colorectal cancer: overview of discovery studies and identification of commonly identified cancer-associated proteins and candidate CRC serum markers. J. Proteomics 73, 1873–1895 [DOI] [PubMed] [Google Scholar]
- 81. Razavi M., Leigh Anderson N., Pope M. E., Yip R., and Pearson T. W. (2016) High precision quantification of human plasma proteins using the automated SISCAPA Immuno-MS workflow. Nat. Biotechnol. 33, 494–502 [DOI] [PubMed] [Google Scholar]
- 82. De Marchi T., Kuhn E., Dekker L. J., Stingl C., Braakman R. B., Opdam M., Linn S. C., Sweep F. C., Span P. N., Luider T. M., Foekens J. A., Martens J. W., Carr S. A., and Umar A. (2016) Targeted MS assay predicting tamoxifen resistance in estrogen-receptor-positive breast cancer tissues and sera. J. Proteome Res. 15, 1230–1242 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.