

Abstract
Reliable quantitation of protein abundances in defined sets of cellular proteins is critical to numerous biological applications. Traditional immunodetection-based methods are limited by the quality and availability of specific antibodies, especially for site-specific post-translational modifications. Targeted proteomic methods, including the recently developed parallel reaction monitoring (PRM) mass spectrometry, have enabled accurate quantitative measurements of up to a few hundred specific target peptides. However, the degree of practical multiplexing in label-free PRM workflows remains a significant limitation for the technique. Here we present a strategy for significantly increasing multiplexing in label-free PRM that takes advantage of the superior separation characteristics and retention time stability of meter-scale monolithic silica-C18 column-based chromatography. We show the utility of the approach in quantifying kinase abundances downstream of previously developed active kinase enrichment methodology based on multidrug inhibitor beads. We examine kinase activation dynamics in response to three different MAP kinase inhibitors in colorectal carcinoma cells and demonstrate reliable quantitation of over 800 target peptides from over 150 kinases in a single label-free PRM run. The kinase activity profiles obtained from these analyses reveal compensatory activation of TGF-β family receptors as a response to MAPK blockade. The gains achieved using this label-free PRM multiplexing strategy will benefit a wide array of biological applications.
Kinases are involved in the regulation of most cellular processes and are implicated in numerous human disease states. Five hundred and eighteen human kinases are known, and between 200 and 400 are typically expressed in a given cell type (1–3). Quantitative measurement of kinase activity is of critical importance in numerous biological applications, including studies of cellular signaling, cancer biomarker discovery, and drug development. An ideal strategy for functional kinase analysis must combine an active state kinase enrichment component and a methodology to quantify their levels of activation, because kinase regulation is a major determinant of cellular function and disease phenotypes.
We and other groups have previously reported on the development and use of Multidrug Inhibitor Beads (MIB)1 strategy for active kinase enrichment (4–8). In this approach, kinase affinity enrichment scaffolds based on known kinase inhibitor structures targeted to conformationally active kinases are chemically coupled to a resin, which is used to capture broad classes of kinases from cell lysates (see recent review of this and related approaches by Daub (4)). When coupled to quantitative mass spectrometry-based proteomics, MIB enrichment can be used to determine kinase inhibitor specificity by comparing relative abundances of particular kinases enriched from drug treated and untreated samples or to study kinase activation dynamics as response to clinical therapy or various biological perturbations. For example, MIB experiments have demonstrated on-target suppression of kinase activity and activation of compensatory kinase signaling pathways driving resistance to therapy in triple negative breast cancer (5), drug-resistant leukemia (6), and ERBB2-positive breast cancer (7).
Despite significant kinase enrichment afforded by MIB, the eluates are still highly complex peptide mixtures in which reliable quantitative detection of low-abundance species remains challenging. To date, several workflows focused on this task have been described. Typically, around 110–125 kinases can be uniquely quantified by data dependent acquisition (DDA) LC-MS/MS in any single MIB enrichment with stringent exclusion of peptides shared by multiple kinases (6, 8). In addition, isobaric tags for relative and absolute quantitation (iTRAQ) (9), have been used downstream of MIB enrichment. However, this particular strategy suffers from non-uniform labeling efficiency and artificial compression of measured peptide ratios (10) and requires sample fractionation prior to MS analysis for adequate kinome coverage (5). Finally, selected reaction monitoring (SRM) is an established technique which allows quantitation of a selected set of desired target peptides with improved sensitivity of detection compared with DDA LC-MS/MS (11). For example, application of an SRM method downstream of ActiveX, a related total kinase enrichment strategy employing ADP and ATP analogs (12) allowed quantitation of 132 of 196 targeted kinases represented by 790 unique peptides (13). However, current SRM approaches require sufficient sample to carry out multiple runs of the same sample using conventional chromatography in order to overcome multiplexing limits and achieve broad kinase coverage. Given the pitfalls of the current approaches, development of an improved strategy for one-shot label-free MS quantitation in complex biological mixtures, including MIB-enriched samples, is necessary.
Parallel reaction monitoring (PRM) is a recently introduced targeted proteomic methodology that takes advantage of the unique capabilities of quadrupole-Orbitrap mass spectrometers (14, 15). PRM provides improved sensitivity, reproducibility, and quantitative dynamic range compared with the techniques described above. Like the older SRM methodology (11), PRM relies on scheduled precursor peptide selection by a quadrupole followed by analysis of fragment ions. However, unlike SRM where a few individual fragments are analyzed sequentially using low-resolution quadrupole-based measurement, PRM utilizes the Orbitrap to record full-range high mass resolution and high mass accuracy MS2 spectra. Thus, use of high resolution in selection of target precursor ions is coupled with high-accuracy MS2-level quantitation resulting in greatly improved specificity of detection. Additionally, evaluation of the PRM advantages for complex peptides mixtures has established that it achieves lower limits of detection and quantification than SRM (14, 15), making it particularly useful for analysis of unfractionated biological samples such as whole cell lysates.
Despite these inherent advantages of PRM, its multiplexing capacity, i.e. the ability to detect multiple targets simultaneously, remains a significant limitation of the technique. Detection of more than a few hundred targets, or roughly 100 proteins, in a single PRM run has been difficult to achieve in a reproducible and robust manner. Although a strategy for improved PRM multiplexing that takes advantage of real-time detection of synthetic isotope-labeled (SIL) peptide standards has been recently proposed (16), its utility is limited in applications where having a SIL standard for each target peptide is not practical, most notably in detection of peptides with post-translational modifications. Therefore, alternative strategies for improved PRM multiplexing that would allow a wider adoption of the methodology for large-scale quantitative targeted proteomic applications are needed.
Recently, remarkable gains in sensitivity and depth of peptide/protein detection have been achieved for DDA LC-MS/MS through the use of meter-scale monolithic silica-C18 silica capillary columns with ultra-long (up to 24-h) chromatographic gradients (17, 18). This approach is the current state of the art in chromatographic separation capabilities available for one-shot proteomics. Given the superb separation characteristics of the monolithic silica-C18 columns in long reverse phase chromatographic gradients (19, 20), we sought to exploit the performance characteristic of monolithic silica-C18 columns to greatly increase PRM multiplexing and evaluate the effectiveness of this strategy by specifically detecting activated kinases downstream of MIB enrichment (Fig. 1).
Fig. 1.

MIB-PRM workflow. Undigested protein lysate is passed through a gravity layered multidrug inhibitor beads column. Broad classes of activated kinases are preferentially enriched in the eluate. A tryptic digest of the eluate is loaded on a meter-scale monolithic silica-C18 column and resolved using a 6-h reverse phase chromatography gradient at a constant temperature of 40 °C. Online targeted detection of endogenous kinases by a highly multiplexed label-free PRM method provides high sensitivity and specificity of detection compatible with automated data processing.
EXPERIMENTAL PROCEDURES
On-line Liquid Chromatography
A prototype 200 cm fused silica monolithic silica-C18 column, 100 μm inner diameter, was used in all experiments. Columns with essentially identical characteristics are now available commercially from GL Sciences (Tokyo, Japan). Liquid chromatography was performed on a Waters NanoAcquity UPLC system (Waters, Milford, MA). The column was maintained at a constant temperature using Eppendorf CH30 column heater with TC50 temperature controller (Eppendorf, Hauppauge, NY). Reverse phase 6-h gradients for both DDA and PRM runs on the monolithic column were performed with a binary buffer system using 0.1% formic acid in water (buffer A) and 0.1% formic acid in acetonitrile (buffer B) as follows: load at 2% B at 600 nL/min for 20 min; gradient with 2–25% B over 288 min at 400 nL/min → 25–32% B over 36 min at 400 nL/min → 32–40% B over 18 min at 400 nL/min → 40–60% B for 18 min at 400 nL/min; wash at 60–80% B over 10 min at 400 nL/min; equilibrate at 2% B at 600 nL/min.
Six-hour gradients on the EasySpray 50 cm column (Thermo Fisher Scientific, Waltham, MA) were performed as above, except the flow rate was maintained at 200 nL/min to adjust for high column back pressure according to the manufacturer's recommendations.
DDA Mass Spectrometry
Q Exactive Plus instrument (Thermo Fisher Scientific) was used in Full-MS/ddMS2 mode with one survey scan (350–1500 m/z, r = 70,000 at 200 m/z, AGC target of 3e6), followed by up to 10 data-dependent HCD MS2 scans (AGC target of 5e4, max IT 120 ms, r = 17,500 at 200 m/z, isolation window 4.0 m/z, NCE 25%, 4% underfill ratio, and 10 s dynamic exclusion). Mouse bone marrow lysate sample was loaded at 100 ng per injection.
PRM Mass Spectrometry
Q Exactive Plus instrument (Thermo Fisher Scientific) was used in PRM mode with the following parameters: positive polarity, r = 17,500 at 200 m/z, AGC target 1e6, maximum IT 190 ms, MSX count 1, isolation window 3.0 m/z, NCE 35%. Mouse bone marrow lysate sample was loaded at 100 ng per injection unless otherwise specified. MIB pooled samples were loaded at 500 ng per injection. Experimental MIB enriched samples from HCT116 cells were loaded at estimated 200–500 ng per injection based on historical average yields determined in pooled samples using Peirce BCA protein assay kit (Thermo Fisher Scientific).
Mouse Bone Marrow Lysate Preparation and Tryptic Digest
Bone marrow cells were harvested by needle aspiration from femora and tibiae of wildtype C57BL/6 mice, snap-frozen in liquid nitrogen. Frozen cell pellets containing ∼1 × 10e8 cells were resuspended in 0.5 ml of TRIzol reagent (Thermo Fisher Scientific) and protein extraction was carried out according to the standard manufacturer's protocol. Precipitated protein pellets were washed with ethanol and vacuum-dried briefly. The pellets were reconstituted in 300 μl of 8 m urea, 0.2% Zwittergent 3–16 (Santa Cruz Biotechnology, Dallas, TX), and 50 mm Tris-HCl, pH 8.0. Twelve μl of 100 mm DTT were added and samples vortexed for 30 min at room temperature. The pellets were dissolved by microtip soniction (30 s on/30 s off on ice ×3). Eighteen microliters of 100 mm iodoacetamide was added, followed by water bath sonication for 30 min at room temperature. Two microliters of resuspended protein sample were quantified using Pierce BCA protein assay kit (Thermo Fisher Scientific) according to the standard manufacturer's protocol. Sample was diluted by adding 1.2 ml of 50 mm Tris-HCl, 1 mm CaCl2 (pH 8.0), and pH was adjusted to 8.0. Sequencing grade trypsin (Promega, Madison, WI) was added at a ratio of 1:50 (trypsin/protein), and the samples were incubated with gentle agitation at 37 °C overnight. Approximately 10 μl of formic acid per 1 ml of sample was added to adjust pH to ∼3.5. Digested peptides were desalted with Sep-Pak C18 cartridges (Waters) according to the manufacturer's protocol, dried to remove solvent under vacuum centrifugation, and stored at −80 °C for further use.
Cell Culture and Kinase Inhibitor Treatments
HCT116 cells obtained from the ATCC (Manassas, VA) (www.atcc.org) were cultured in DMEM supplemented with 10% fetal bovine serum. GDC-0973, GDC-0623, and SCH772984 were purchased from Selleckchem (Houston, TX) (www.selleckchem.com). For time course experiments, cells were grown in 6 cm dishes until near confluency, then treated for the indicated time and dose. For MIB experiments, cells were plated in 15-cm plates and grown to near confluency, then treated with drug or equivalent amounts of DMSO for 90 min. Cells were washed twice with phosphate buffered saline (PBS) then harvested by scrapping cells into PBS and centrifuging for 5 min at 300 × g and 4 °C yielding pellets of ∼0.5 ml, and the cell pellets were snap frozen.
Cell Lysate Preparation and Western Blotting
Cells were diluted in 3–5× pellet volume of lysis buffer (50 mm HEPES pH 7.5, 150 mm NaCl, 0.5% Trition X-100, 1 mm EDTA, 1 mm EGTA) with PhosStop phosphatase and cOmplete protease inhibitor cocktails (Roche, Basel, Switzerland). Cells were lysed on ice using two 3-s pulses in a water bath sonicator. Lysates were spin cleared, and protein concentration was checked by a BCA assay (Thermo Fisher Scientific). For immunobloting the following antibodies from Cell Signaling Technology (Danvers, MA) were used: MEK (4694), phospho-MEK (Ser217/221, 9154), ERK (9102), phospho-ERK (Thr202/Tyr204, 9106), RSK1 (8408), phospho-RSK (Ser380, 9341), Ret (14556), phospho-Ret (Tyr905, 3221), COX IV (4844), SMAD2 (5339), phospho-SMAD2 (Ser465/467, 3108), SMAD1 (6944), phospho SMAD1/5 (Ser463/465, 5753), and phospho-SMAD1/5/9 (Ser463/465/467, 13820).
MIB Kinase Enrichment and Tryptic Digestion
Compounds for Multiplex inhibitor beads were synthesized or commercially purchased and conjugated to resin as previously described (8). 10 mg of total protein per condition from lysed cell pellets was diluted to 10 ml final volume in high salt buffer (50 mm HEPES pH 7.5, 0.5% Triton X-100, 1 m NaCl, 1 mm EDTA, 1 mm EGTA) containing protease and phosphatase inhibitors (Roche). Lysates were precleared over unconjugated resin in a gravity column before passing onto the MIB gravity column. The MIB column was prepared by layering 50–100μl slurry of each individual inhibitor-conjugated bead in BioRad Poly-Prep Chromatography Columns with a 10 ml reservoir and 2 ml bed volume (BioRad, Hercules, CA, 731–1550). For binding and washing, a 26-guage needle was placed on the outlet of the MIB column to slow the flow rate. We estimate the flow rate to be around 0.3 ml/min. Lysate-bound columns were washed with 10 ml high salt buffer, 10 ml low salt lysis buffer (50 mm HEPES pH 7.5, 0.5% Triton X-100, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA) and last 1 ml of low salt buffer with 0.1% SDS. Samples were eluted off the MIB column with 2x incubations at 100 °C for 5 min with 0.5 ml of elution buffer (0.5% SDS, 1%BME in 0.1 m Tris-HCl ph 6.8). Samples were reduced with 5 mm DTT at 55°C for 30 min and alkylated at room temperature with 10 mm iodoacetimide for 45 min in the dark. Samples were spin concentrated to 100 μl using Amicon Ultra 10KDa centrifugal filters (Millipore, Billerica, MA). Proteins from the concentrated eluate were precipitated using a methanol-chloroform extraction, suspended in 75 μl of 50 mm ammonium bicarbonate and digested overnight with 3 μg sequencing grade modified trypsin (Promega, V5113). Samples were acidified to pH 3 with formic acid, desalted with C18 ZipTips (Millipore), speedvac evaporated to dryness and stored at −80°C until resuspension in 0.1% formic acid for MS analysis.
Data Analysis
1. Performance of Monolithic Silica-C18 Column by DDA MS
Raw data files of the mouse bone marrow lysate runs at 25, 40, 60, and 80 °C were converted to peak files using PAVA (21) and searched in Protein Prospector (22, 23) version 5.14.0 against UniprotKB mouse proteome database (24) downloaded on 09/18/13 (51,219 protein entries) and corresponding random concatenated decoy database with default “ESI-Q-high-res” parameters, including trypsin as the protease, up to one allowed missed cleavage site, Carbamidomethyl-C constant modification, default variable modifications (Acetyl (Protein N-term); Acetyl+Oxidation (Protein N-term M); Gln->pyro-Glu (N-term Q); Met-loss (Protein N-term M); Met-loss+Acetyl (Protein N-term M); Oxidation (M)), up to 3 modifications per peptide, and 20 ppm precursor mass and fragment mass accuracy. False discovery rate of <1% was used as the cutoff for peptide expectation values. The same high-specificity search parameters were used for both monolithic column and EasySpray column runs to minimize inclusion of false positive spectra in the spectral libraries. Spectral libraries of these runs in blib format were generated in Protein Prospector and imported into Skyline v. 3.5 (25). All identified peptides were added as targets in Skyline and raw data imported in MS1 filtering mode with the following settings: only scans within 1.5 min of MS/MS IDs imported, three isotope precursor peaks included, and Orbitrap mass analyzer with resolution of 70,000 at 200 m/z. Only peptides identified by the search engine in all runs were retained for further consideration. Peak widths were exported from Skyline as full width at half maximum (FWHM), and median peak widths as a function of the gradient time were calculated by binning peptides into RT windows of 30 min. Similarly, median peak capacity as a function of the gradient time was calculated as median peak widths within 30 min windows divided by the total gradient time, defined as the time between the earliest and the latest identified peptides. Retention time reproducibility was determined by evaluating RT standard deviations among triplicate runs of the bone marrow lysate sample at 40 °C.
2. PRM Multiplexing in Mouse Bone Marrow Lysate
Spectral library generated in the DDA experiments was used as the reference library in Skyline. Target proteins were chosen based on the GO (26) category “Cellular Process.” Up to eight target peptides per protein were included by matching to the spectral library if the following criteria were met: RT standard deviation in the triplicate DDA runs is less than 2 min, peptide does not share identity with any other protein in the human SwissProt database (27), peptide length is between 9 and 26 residues, and no more than one miscleavage site is present. Up to 10 most intense y and b ions (with preference for y ions) were chosen as monitored transitions. A total of 1067 target peptides from 325 proteins were selected. Target RT for each peptide was the average RT from the triplicate DDA experiments. RT target windows were set at ± 8 min between minutes 0 and 150, ± 3.8 min between minutes 150 and 350 and −5/+6 min after minute 350. Larger windows at the tails of the gradient were used to accommodate greater degree of RT variation in those areas and were possible because of lower numbers of eluting precursors in these regions. Raw data files were imported into Skyline for automated peak detection using “targeted” MS/MS filtering mode with mass analyzer set to Orbitrap, r = 17,000 at 200 m/z. Total peak area (sum of areas of all transition peaks), library dot product, peaks found ratio, and MS2 fragment mass accuracy were calculated by Skyline and used to generate the plots in Fig. 3B–3F. Skyline document with PRM data collected in mouse bone marrow lysate is available on Panorama Web (28) (https://panoramaweb.org/labkey/Urisman_MIB-PRM.url).
Fig. 3.

Highly multiplexed label-free PRM is achieved with long gradient chromatography on meter-scale monolithic silica-C18 column. 1067 peptides from 325 mouse proteins belonging to GO category “Cellular Process” were included in a single PRM method utilizing a 6-h reverse phase chromatography gradient on the 200-cm monolithic silica-C18 column. Tryptic digest of the mouse bone marrow lysate was used to test the method. Data were analyzed in Skyline using automated peak detection. A, Scheduling diagram shows the expected maximum number of targets as a function of time along the gradient based on the overlap of target detection windows. B, Distributions of library dot product and peaks found ratio, two metrics of detection specificity calculated by Skyline, are shown as box plots (solid line = median, box = interquartile range, whiskers = 1.5 × interquartile distance, dots = individual observations outside whiskers). C, Histogram of MS2 fragment mass accuracy in parts per million (ppm) calculated by Skyline. D–F, Specificity of detection in relationship to protein abundance is estimated by library dot product (D), MS2 fragment mass accuracy (E), and peaks found ratio (F) as a function of peak area.
3. PRM Method for Kinase Detection
Approximately 150 MIB individual samples from prior unrelated diverse experiments in human cell lines were combined into four pools of 30–40 samples in each. The pools were separately analyzed using DDA runs on the Q Exactive instrument equipped with the monolithic silica-C18 column. Raw data files were converted to peak files using PAVA (21) and searched in Protein Prospector (22, 23) version 5.14.0 against human SwissProt database (27), downloaded on 07/29/2013 (39,457 entries), and corresponding random concatenated decoy database with default “ESI-Q-high-res” parameters. Protein Prospector search parameters were as above. FDR of <0.2% was used as the cutoff for peptide expectation values to ensure specificity. Spectral libraries of these runs in blib format were generated in Protein Prospector and imported into Skyline v. 3.5 (25). Raw data files have been uploaded to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the MassIVE partner repository (http://massive.ucsd.edu/ProteoSAFe/static/massive.jsp) with the data set identifiers PXD003934 and MSV000079644, respectively.
Target peptides matching a list of 509 human kinases (1) were selected from the discovery spectral library; only canonical splicing isoforms were included. All unique peptides, i.e. those that do not share identity with any other human protein in SwissProt database (downloaded on 07/29/2013; 39,457 entries), were initially selected. PTM-containing peptides were excluded. Given the high degree of conservation between kinases within individual families, some kinases (13%) could not be represented by more than one unique peptide. We also included 10 proteins (11 peptides) commonly detected in DDA analyses of human samples to serve as controls for nonspecific carry-over during MIB enrichment (supplemental Table S1). For each selected precursor, up to 10 most intense y and b ions (with preference for y ions) were chosen as monitored transitions. Initial scheduling method contained 944 target precursors from 182 proteins, was split into two injections with half of the targets in each, and used variable target detection windows as follows: ± 20 min between minutes 0 and 150, ± 10 min between minutes 150 and 350 and ±20 min after minute 350. The method was tested on two independent MIB enriched pools, each combining ∼30 individual MIB samples. Raw data were imported into Skyline, and positions of auto-detected peaks were checked manually, only 15 of which were clearly erroneous (background noise in the absence of visually identifiable peaks) and were excluded from the final method. The final single-injection method contained 925 precursor peptides from 182 kinases, and used the following target detection windows: ± 8 min between minutes 0 and 160, ± 3.5 min between minutes 150 and 350 and ±8 min after minute 310. The same two pooled MIB samples were used to test the single-injection method. For both two-injection and single-injection methods, precursors were considered successfully detected if Skyline auto-detected peaks satisfied the following criteria: library dot product >0. 5, peaks found ratio >0.5 (with at least 3 fragments detected), and fragment mass error <20 ppm.
4. MIB-PRM Experiments in HCT116 Cells
The single-injection kinase detection method was used without any modifications on the HCT116 experimental samples. Raw data were imported into Skyline v. 3.5, and auto-detected peaks in each sample were filtered according to the same specificity criteria as above (library dot product >0.5, peaks found ratio >0.5, and fragment mass error <20 ppm). Successfully detected peaks were used to normalize data by median centering log2 peak areas in each sample to match the global data set median. Because undetected peaks could mean either biologically relevant depletion in kinase binding or a technical failure of the method to detect the target precursors, the following pre-processing was applied to the undetected peaks. If in a given condition, a precursor was detected in one of two biological replicates, the undetected value was set to the mean of all non-zero observations for that precursor (i.e. lack of detection is likely because of a technical failure and thus its statistical significance is penalized). Log2 peak area of a precursor was set to 1 (a small number not equal 0 to allow calculation of ratios) only if it was undetected in both biological replicates of a given condition and only if no other precursor from that kinase was detected in that condition (i.e. lack of detection is likely because of a real biological kinase depletion). This pre-processing strategy is similar to that used for imputing missing values in MSStats (29), except it is more conservative in that it is not applied to all undetected peaks and results in fewer false positives. The number of precursors quantified versus targeted as well as percent imputed measurements for each kinase are listed in supplemental Table S1. The data were then converted to a matrix of log2 ratios of inhibitor conditions versus DMSO control and analyzed in MSStats version 2.6.0 using no further pre-processing. MSStats input file with PRM measurements listed by precursor and output file with estimated aggregate kinase abundances are available as supplementary Files S1 and S2, respectively; Skyline report with PRM measurements by transitions is available as Supplementary File S3 (see Supplementary Data). Based on the distributions of log2 ratios and corresponding p values calculated by MSStats for each inhibitor condition, a conservative cutoff of log2 ratio > 4 and p value < 0.05 was chosen to identify the most significant observations. Heat map diagram of kinases with statistically significant inhibition or activation was constructed by clustering all kinases with a statistically significant log2 ratio in at least one of the three inhibitor conditions. Complete linkage clustering using city block distance and image generation were performed in GENE-E software (http://www.broadinstitute.org/cancer/software/GENE-E/).
Skyline document with PRM data collected in MIB samples, pool injections and HCT116 experiments, is available on Panorama Web (https://panoramaweb.org/labkey/Urisman_MIB-PRM.url).
RESULTS
Monolithic Silica-C18 Column Performance in 6-h DDA Gradients
As the first step toward developing a highly multiplexed PRM pipeline, we examined the performance of a 200 cm monolithic silica-C18 column in a variety of conditions by considering three factors: estimating its peak capacity (PC), peak width, retention time (RT) reproducibility, and sensitivity of detection (Fig. 2). PC is defined as the number of elution peaks that can be separated from baseline over the total duration of a given gradient. RT refers to the elapsed time between sample injection and detection of a given peak.
Fig. 2.

Performance characteristics of a meter-scale monolithic silica-C18 column desirable for targeted proteomic applications. Tryptic digest of mouse bone marrow lysate was separated on the 200-cm monolithic silica-C18 column at 25, 40, 60, and 80 °C and analyzed by LC-MS/MS. A, Peak width as a function of gradient time is calculated as a running median of full width at half maximum. B, Peak capacity as a function of gradient time is calculated as the running median of peak width divided by gradient duration. C, Number of peptides and proteins identified at each temperature. D, Triplicate runs of the mouse bone marrow lysate sample were performed on the 200 cm monolithic silica-C18 column (at 40 °C) and 50 cm C18 silica particle-packed column (at 45 °C). Histogram of RT standard deviations for peptides identified in all replicates is plotted.
Temperature is an important determinant of separation efficiency in reverse phase chromatography and ion spray performance, where even small changes in temperature can introduce significant variation in RTs of individual analytes (30, 31). Thus, we evaluated the performance of the monolithic column at different temperatures. For this purpose, a tryptic digest of mouse bone marrow lysate was used in triplicate DDA runs employing 6-h HPLC gradients while maintaining the column at 25 °C, 40 °C, 60 °C, or 80 °C. In general, peak widths (Fig. 2A) and PCs (Fig. 2B) observed were similar to the results previously reported for the meter-scale monolithic silica-C18 columns (19). Only minor changes in peak width and PC were observed as a function of temperature. However, the number of identified proteins and peptides was clearly superior at 40 °C (3384 proteins/21,263 peptides), with more than doubling of identified peptides compared with 25 °C (2262 proteins/9537 peptides). Therefore, we chose 40 °C as the standard monolithic column temperature for the remainder of this study.
We next examined RT reproducibility, a prerequisite for high multiplexing, on the monolithic silica-C18 column as compared with a conventional C18 silica particle-packed column. The same biological sample was analyzed in triplicate on the monolithic column at 40 °C and on a widely used 50 cm Thermo EasySpray C18 silica particle-packed column maintained at 45 °C (previously identified as the optimal temperature, data not shown) using the same gradient (Fig. 2D). Results obtained using the monolithic column established significantly lower RT variability. RT standard deviations under 3 min were documented for over 95% of all identified peptides, while on the packed column, close to 50% of peptides had RT standard deviations above 3 min (t test p < 1e-15). Consistent with prior reports (19), ∼25% more proteins and peptides were identified on the monolithic silica-C18 column compared with the C18 silica particle-packed column when using loading amounts of <1 μg (supplemental Fig. S1). Both improved sensitivity, as indicated by the increased depth of peptide detection, and lower RT variability afforded by the monolithic silica-C18 column are very desirable characteristics for developing highly multiplexed targeted proteomic applications. In particular, based on the observed low RT variability characteristic of the monolithic column, we deduced that target detection windows of ±4 min would permit the detection of well over 95% of target peptides in a PRM assay.
PRM Multiplexing with Monolithic Silica-C18 Columns
Based on our experience and consistent with prior reports (14, 15), optimal PRM sensitivity in complex biological samples on currently available instruments such as Q Exactive Plus (Thermo) requires Orbitrap injection times between 100 and 300 ms. Assuming a cycle time of 6 s, where cycle time is the time delay between adjacent MS2 scans of the same target peptide, is required for five measurements over a 30 s elution peak and injection time of 120 ms, no more than 50 targets can be scheduled at any given time (supplemental Fig. S2). Furthermore, based on the typical distributions of overlapping target detection windows in the long gradient runs of complex peptide digests, we estimated that use of target windows of ±4 min (see above) would be adequate to permit scheduling of around 1000 target peptides in a single run.
To test this possibility, we used a subset of peptides identified in the DDA runs of the mouse bone marrow lysate sample to schedule a highly multiplexed PRM assay (Fig. 3). In order to minimize bias for high abundance targets, all detected proteins belonging to Gene Ontology (GO) (26) category “cellular process” were included, independent of the observed intensity in the DDA runs. A total of 1067 target peptides from 325 proteins were selected and scheduled for detection by a single PRM method with variable length detection windows ranging from ±3.8 min in the most complex part of the gradient (150–350 min) up to ±8 min in the less complex tail portions (<150 min and >350 min) (Fig. 3A). PRM was performed on the same mouse sample and using the same LC gradient as in the DDA runs. Spectra were analyzed with Skyline software, which allows automated detection of extracted MS2 chromatographic peaks (25). Three metrics available in Skyline to monitor specificity of target detection (32) were used to assess the quality of all peaks auto-detected by the software. The distributions of library dot product, a measure of similarity between the observed MS2 spectra and those in the reference DDA library, showed that over 90% of peptides had a dot product > 0.5, where a perfect match equals 1. The distribution of peaks found ratio, the fraction of MS2 component peaks detected of those monitored by PRM, showed that over 95% of peptides had a peaks-found ratio > 0.6 (Fig. 3B). The high median library dot product and peaks found ratios support a high overall specificity of detection. Similarly, fragment ion mass error distribution demonstrated that more than 90% of all detected peaks were within ±20 ppm of expected (Fig. 3C). Based on these results, we chose a combination of the following three parameters as the threshold for classifying a given target peptide as successfully detected: library dot product > 0.5, peak found ratio > 0.5, and mass error < ±20 ppm.
Employing these strict specificity criteria, a total of 856 target peptides (80%) from 293 proteins (90%) were successfully detected. Considering only these successfully detected peptides and using integrated peak areas as a rough estimate of their abundance, we found no significant correlation between abundance and library dot product (Fig. 3D) or mass error (Fig. 3E). However, a trend for lower peak found ratio with decreasing abundance was observed (Fig. 3F), consistent with loss of lower-intensity peaks in the MS2 spectra recorded at the limits of detection. Additionally, we saw no apparent relationship between abundance and the number of peptides per protein detected among those scheduled (supplemental Fig. S3). A comparison of relative intensities of the MS1 precursor ion intensities obtained in the DDA runs to the measured by PRM total MS2 fragment ion intensities demonstrated that the MS1-level and MS2-level quantitation were highly correlated (supplemental Fig. S4). Collectively, these results indicate that PRM multiplexing approaching 1000 target peptides per single 8-h (6-h gradient) run on a 200-cm monolithic silica-C18 column is appropriate for routine quantitative PRM detection of label-free endogenous peptides in complex biological samples, providing high sensitivity and specificity of detection.
Monolithic Silica-C18 Column-Coupled PRM for High-coverage Kinase Detection After MIB Enrichment
Quantitative detection of a broad span of kinase activity is needed for comprehensive studies of cellular signaling mechanisms as well as in cancer biomarker discovery and drug development applications. To achieve high coverage of kinases in a quantitative fashion, we developed and validated a highly multiplexed PRM method for detecting human kinases following MIB kinase enrichment. Target precursors were chosen from DDA spectral libraries representing ∼150 diverse MIB samples, analyzed in pools of 30 to 40 samples each using a 6-h gradient on the monolithic silica-C18 column. Initial scheduling runs used two injections per sample with ∼465 targets per injection and target windows of ±10 min. In these runs, detection of up to 79% of target precursors was achieved in several independent pools of ∼30 unrelated MIB samples each (supplemental Fig. S5A). A single-injection method was then created by reducing target windows to ±3.5 min, which successfully detected >99% of targets (up to 738 peptides from 152 kinases in a single sample) previously detected by the 2-injection method (supplemental Figs. S5B, S5C). The final method employed a 6-h gradient on the 200-cm monolithic column and targeted 929 unique precursors (peptides of specific charge) from 182 human kinases. Having validated the efficiency of the MIB-PRM platform for detecting a broad set of kinases, we next sought to use the single injection MIB-PRM method to profile kinase regulation in a biologically relevant context.
MIB-PRM Identifies Activation of TGF-β Signaling as a Compensatory Response to MAPK Signaling Blockade
The RAS/RAF/MEK/ERK MAP kinase (MAPK) signaling pathway is an important regulator of cell proliferation and survival (33). Activating driver mutations in the pathway are present in over 20% of all human malignancies, including BRAF mutations in ∼60% of melanomas and RAS mutations in ∼30% of lung and over 50% of colorectal and pancreatic adenocarcinomas (34, 35). Thus, inhibition of MAPK signaling is of significant therapeutic interest (36, 37). Interestingly, recent studies have shown that different classes of inhibitors of MEK and ERK have varying degrees of effectiveness depending on the oncogenic genotype (38, 39). GDC-0973, or cobimetinib, has demonstrated effectiveness in BRAF V600E melanomas by inhibiting MEK, but it also stabilizes MEK in its active conformation. In contrast, GDC-0623 also inhibits MEK but preferentially stabilizes its inactive conformation. As GDC-0973 and GDC-0623 are both allosteric inhibitors of MEK, they are potentially sensitive to signaling bypass mechanisms that occur within complexes containing inhibited MEK, with complex composition and activity often determined by genotype. The stabilization of an inactive conformation of MEK by GDC-0623 blocks cell growth and disrupts this feedback mechanism effectively in KRAS-mutant cell lines, whereas GDC-0973 is ineffective in KRAS-mutant cell line models (38). Inhibition of ERK, which has no direct interaction with either RAS or RAF, with SCH772984 shows broad effectiveness in BRAF, NRAS, and KRAS mutant cell lines, and MEK-inhibitor resistant cells (39). Given the distinct mechanisms of target inhibition by GDC-0973, GDC-062,3 and SCH772984, we hypothesized that an evaluation of the kinome response to these inhibitors by MIB-PRM, in addition to the known patterns of MEK and ERK activation, would likely highlight unique signatures of kinase response to feedback regulation and crosstalk with other signaling pathways.
We first validated this approach by Western blotting for known kinase regulation phenotypes in response to the three inhibitors in HCT116 KRAS mutant colorectal cancer cells (Fig. 4). As previously reported (38), GDC-0973 maintained MEK phosphorylation, while inhibiting phosphorylation of downstream kinases ERK and RSK. In contrast, GDC-0623 stabilized inactive dephosphorylated form of MEK, while similarly inhibiting phosphorylation of ERK and RSK. SCH772984 eliminated ERK phosphorylation, but feedback activation of MEK was observed (38, 40). We next used the MIB-PRM approach to identify differences in kinome activation in response to the three types of MAPK pathway inhibitors in HCT116 cells.
Fig. 4.
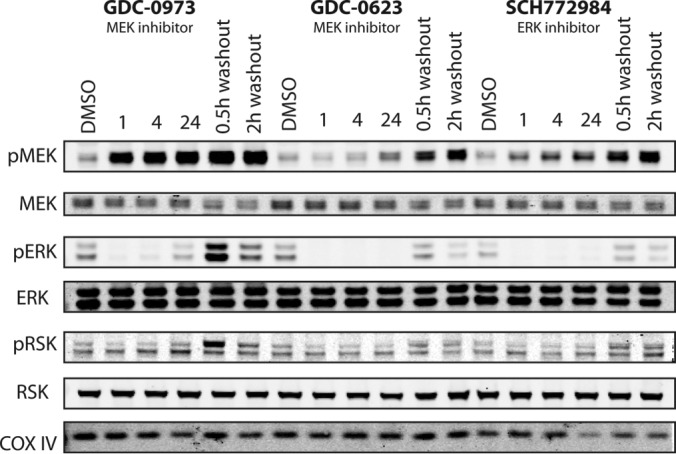
Three types of selective inhibitors of MAPK signaling produce expected differential kinase inhibition and activation responses in HCT116 colorectal cancer cells. HCT116 cells were treated with 250 nm of GDC-0973, GDC-0623, SCH772984 or DMSO for 1, 4, or 24 h. In the washout samples, cells were drug treated for 24 h, then changed into fresh media and harvested after 0.5 or 2 h. Lysates were used for Western blots of total and phosphorylated MEK, ERK, and RSK; blotting for COX IV was used as the loading control.
The validated single-injection PRM method was used to quantify kinase abundance in the eight experimental samples from two biological replicates of 4 experimental conditions. To ensure high specificity, targets were considered successfully detected using the same criteria as those used in the mouse digest highly multiplexed PRM validation experiments with a library dot product > 0.5, peak found ratio > 0.5, and mass error < ±20 ppm. No additional individual fragment ion filters were used (such as threshold signal-to-noise ratio or coefficient of variance in repeat experiments) based on the observation that total areas (i.e. sum of the peak areas of all detected fragments) and sum area of only the top three most intense fragments with highest signal-to-noise separation are highly correlated (r >0.97) (supplemental Fig. S6). Overall, 814 target precursors, 88% of the 929 scheduled, from 154 kinases were quantified (supplemental Table S1 and supplemental Fig. S7). A lower than 100% detection rate is expected in MIB experiments, given the preferential enrichment of activated kinases and cell-specific variation in expression of individual kinases. Comparison of PRM measurements in biological replicates revealed variable condition-dependent heterogeneity, with correlation coefficients ranging between 0.47 and 0.86 (supplemental Fig. S8). This heterogeneity was attributed mainly to biological differences between the samples, given the almost negligible in comparison differences between technical repeats (supplemental Fig. S5C). MSStats software (29) was used to normalize the data and assign statistical significance to the observed differences among the three drug conditions and DMSO control (supplemental Fig. S9). Plots of individual peptide ratios in the two biological replicates for all quantified kinases are provided in supplementary File S4, and plots of aggregated by condition median ratios for each quantified kinase, with corresponding variance and significance estimated by MSStats, are provided in supplementary File S5.
As expected, the PRM results demonstrated a highly significant reduction in detectable MAPK1 (ERK2) and MAPK3 (ERK1) following SCH772984 treatment compared with DMSO control (Fig. 5A), consistent with inhibition of the two ERK proteins by the compound. Furthermore, increased abundance of MAP2K1 (MEK1) was observed in both GDC-0973 and SCH772984 treatments (Fig. 5C), consistent with the concurrent increase in MEK phosphorylation measured by Western blot in these two conditions (Fig. 4) and in agreement with prior evidence that both inhibitors induce compensatory activation of MEK1 (38–40). Consistent enrichment of MAP2K1 (MEK1) and MAP2K2 (MEK2) remains an area for further optimization of MIB proteomics and is likely the root cause of the less than statistically significant quantitation of these kinases.
Fig. 5.

Highly multiplexed PRM method downstream of MIB kinase enrichment quantifies kinome response to three types of MAPK signaling inhibitors in HCT116 colorectal cancer cells. HCT116 cells were treated with 250 nm GDC-0973, GDC-0623, SCH772984, or DMSO for 90 min. The lysates were collected and passed over MIB columns. The eluates enriched for activated kinases were digested with trypsin and analyzed by a single PRM method utilizing the long monolithic silica-C18 column and targeting 929 precursor peptides from 182 kinases. A total of 814 precursors from 154 kinases were quantified using strict specificity criteria. A, Abundance of MAPK1 (ERK2), MAPK3 (ERK1) and MAP2K1 (MEK1) in MIB eluates measured by PRM are plotted as log2 ratios to DMSO control. B, All kinases with statistically significant inhibition or activation relative to DMSO control were ordered by hierarchical clustering of the corresponding log2 ratios. C, HCT116 cells were treated with 250 nm of GDC-0973, GDC-0623, SCH772984, or DMSO for 90 min, or with TGF-β at 50 pg/ml for 30 min, followed by Western blotting.
A total of 23 kinases demonstrated significant changes of abundance in MIB eluates as measured by PRM in at least one of the three inhibitor treatments relative to the DMSO control and using a conservative threshold of significance set at log2 fold change of >4 and MSStats estimated p value of <0.05 (Fig. 5B and supplemental Fig. S10). Overall, although negative changes (loss of kinase detection) produced by the three inhibitors were almost entirely non-overlapping, 5 of 11 kinases that showed significant positive changes (gain of detection) were shared by all three inhibitors. This suggests, that although negative changes reflect largely direct inhibition of target and off-target kinases, positive changes are more likely to represent shared compensatory effects in response to inhibition of MAPK signaling. The data further suggest very high specificity of SCH772984 with only on-target inhibition of MAPK1 and MAPK3 detected, as well as higher specificity of GDC-0623 compared with GDC-0973 based on the higher number of off-target inhibited kinases detected for the latter.
Among the kinases with shared activation in response to the three inhibitors of MAPK signaling in the HCT116 cells were TGF-β superfamily receptors TGF-β receptor 1 (TGFBR1), activin receptor type 1B (ACTR1B), and bone morphogenic protein receptor type 1A (BMPR1A). These closely related receptors are activated upon binding of TGF-β and TGF-β-like ligands and are involved in diverse cellular processes such as cell migration and differentiation in response to paracrine signals (41). All three receptors function through downstream activation of SMAD2/3 and/or SMAD1/5 pathways, depending on cell type-specific context (42, 43). In our experiments, the apparent activation of all three receptors (TGFBR1, ACTR1B, and BMPR1A) was observed by PRM in response to GDC-0623 and SCH772984 and was accompanied by a concurrent increase in phosphorylation of SMAD2 (Fig. 5C). In contrast, GDC-0973 treatment resulted in activation of TGFBR1 and ACTR1B but not BMPR1A, and no changes in SMAD2 phosphorylation were observed. Phosphorylation of SMAD1, SMAD5, and SMAD9 was not affected by any of the inhibitors (Fig. 5C). Previously, increased expression of TGF-β was shown to be a predictor of recurrence and metastasis in colorectal cancer (CRC) (44). It is also known that activation of TGF-β signaling leads to SMAD-independent activation of MAPK signaling in multiple systems (45, 46). In this light, our findings suggest that activation of TGF-β signaling is a compensatory mechanism that in addition to downstream activation of SMAD2 may include a SMAD-independent effect aimed at restoring MAPK signaling in the context of MAPK signaling blockade in these KRAS-dependent cells.
DISCUSSION
We have developed a strategy for highly multiplexed PRM relying on ultra-long monolithic silica-C18 column based liquid chromatography and demonstrated its utility by quantifying the activity of a broad segment of the human kinome in vivo in response to kinase inhibitor therapy. By coupling MIB with highly-multiplexed PRM, we eliminate the need for isobaric tagging reagents and off-line sample fractionation prior to LC-MS/MS, while maintaining broad coverage and highly specific, reproducible and sensitive quantitation.
Higher kinome detection coverage of up to 260 kinases by DDA LC-MS/MS has been reported in proof-of-principle experiments on pooled lysates comprised of diverse cell lines using kinobead enrichment approach (47), a strategy similar to MIB that is not activity-dependent. However, consistent and specific quantitation at such high kinome coverage has not been demonstrated in practical biological applications employing a single cell type. With the ability to specifically target kinases for quantitation across a large dynamic range, MIB-PRM has the advantage of higher sensitivity, reproducibility, and quantitative accuracy in realistic biological applications.
A recently published report by Ruprecht et al. calls into question the reliability of MIB enrichment as a functional readout of kinase activity (48). In this work, the authors compare the state of kinase activation inferred from phosphorylation changes of the downstream substrate sites measured by phosphoproteomics to the patterns of kinase enrichment following MIB. We agree with the authors that MIB enrichment is not as comprehensive or sensitive as phosphoproteomics in detecting kinase activity changes. However, based on our experience and consistent with the authors' findings, the changes in kinase regulation that are detected by MIB are typically in good agreement with those observed by phosphoproteomics. Therefore, based on our studies, MIB strategy remains a biologically relevant assay of kinase activity, albeit less sensitive than phosphoproteomics. The MIB platform can also be preferable in applications where phosphoproteomics are not feasible, e.g. the workflow used by Ruprecht et al. required metabolic isotopic protein labeling by SILAC (49) and extensive offline fractionation of the digested lysates prior to LC-MS/MS analysis.
We used the MIB-PRM approach to study the effects of MAPK signaling inhibition by two MEK inhibitors, GDC-0973 and GDC-0623, the former being less potent in mutant KRAS driven cells, and an ERK inhibitor, SCH772984, in KRAS-mutant HCT116 colorectal cancer cells and found that all three inhibitors produced activation of TGF-β family of receptors. Although GDC-0623 and SCH772984 led to activation of TGFBR1, ACTR1B, and BMPR1A with concurrent activation of SMAD2; GDC-0973 treatment resulted in activation of only TGFBR1 and ACTR1B (not BMPR1A), and no activation of SMAD2 was observed. Given prior evidence of SMAD-independent MAPK activation downstream of TGF-β signaling (45, 46), we hypothesize that the observed activation of TGF-β signaling might be a compensatory mechanism either partially or primarily aimed at SMAD-independent MAPK signaling activation upstream of RAS to counteract MAPK blockade produced by the inhibitors. These results provide further support for the argument that combination therapy with MAPK and TGF-β inhibitors may have potential benefits in the treatment of KRAS-driven cancers (50, 51).
In this report, we demonstrate the utility of a meter-scale monolithic silica-C18 column in targeted proteomic applications. Long monolithic silica-C18 columns have been previously shown to be highly effective in achieving unprecedented chromatographic separation of peptides in complex biological samples in long (>6 h) gradients, enabling near-proteome-scale protein detection in one-shot DDA proteomics applications (18). Here, we used the superior chromatographic characteristics of the long monolithic silica-C18 columns, specifically high peak capacity and low RT variability, to address one of the principal challenges of PRM - its limited multiplexing capacity.
Furthermore, we demonstrate the feasibility of targeting ∼1000 endogenous target precursors with expected detection of over 85% in a single 8-h PRM run in routine applications not employing SIL standard peptides. Previously, Burgess et al., used an in-house 30 cm C18 silica particle-packed column with a 3-h reverse phase gradient to test an 800-plex SRM assay to detect 400 heavy and 262 counterpart light SIL peptides (52). A total of 257 peptide pairs (514 total precursors) were detected in that effort, effectively defining the practical limits of SRM multiplexing. More recently, Gallien et al. addressed the limits of PRM multiplexing using a novel approach in which detection of endogenous target peptides is aided by real-time detection of spiked in SIL standard peptides, which once detected trigger on the fly adjustments to Orbitrap™ injection time, scan resolution, and target window duration to maximize sensitivity and minimize overlap between multiple windows (16). This so-called standard-triggered PRM used 600 SIL standard peptides and was capable of detecting up to 300 corresponding unlabeled endogenous peptides in serum samples run on a conventional 15 cm C18 silica particle-packed column with a 66-min reverse phase HPLC gradient. Although, this approach is perfectly suited for clinical and other analytical applications for which synthesis of SIL peptides is warranted, many biological applications in targeted proteomics would not be feasible if SIL standards were required for each desired target.
Not using SIL standards in PRM applications puts particular emphasis on specificity of detection and reliability of automated peak detection. We have demonstrated that high specificity can be achieved via use of high-resolution discovery spectral libraries for accurate RT scheduling followed by application of high-stringency criteria that simultaneously ensure similarity with reference spectra, acceptable fraction of MS2 peaks detected, and high mass accuracy during routine automated analysis in Skyline (25). Furthermore, the general PRM workflow and data analysis strategy illustrated above do not require any custom software or hardware beyond a PRM-enabled instrument, making it straight-forward to implement in most proteomic labs.
It should be acknowledged that workflows incorporating very long chromatographic gradients, such as the 6-h gradient used here, present a significant demand on instrument time and are not practical in all PRM applications. The proposed workflow is chiefly intended to overcome the scan time limitations of the existing Quadrupole-Orbitrap instruments in order to achieve the highest possible multiplexing capacity in PRM runs. Coupled with the higher sensitivity of detection stemming from the superior separation achieved in the long gradients on the monolithic silica-C18 column, the described workflow is particularly beneficial in the most demanding applications characterized by high sample complexity, low starting protein amounts prohibiting multiple injections, and a desire for the highest possible target multiplexing.
The demonstrated high degree of PRM multiplexing without SIL standards, combined with high detection specificity achieved by routine automated data processing and the inherent highly quantitative nature of the methodology, opens unprecedented possibilities for biological applications. In particular, we foresee the strategy applied to large scale screens of protein function in hundreds of proteins simultaneously. In addition to the MIB platform described above, other kinase enrichment modalities would benefit from downstream PRM detection (4). Other attractive targets include functional classes of proteins that can be enriched via biochemical or biophysical methods, including transcription factors and other nucleic acid binding proteins, proteins restricted to specific subcellular compartments, or specialized extracellular structures. In addition, the approach is perfectly suited for studies of signaling regulation by post-translational modifications (PTM) in large signaling networks. For example, phosphorylation states at many hundreds of sites in hundreds of proteins can be quantified by highly multiplexed PRM following phosphopeptide enrichment. The approach is also well suited for “one-shot targeted proteomics” applications that require flexible on-demand detection of hundreds of precursors relevant to a specific biological question in minute samples such as fluorescence sorted subpopulations of specialized cells, difficult-to-scale organoid cultures, or amount-limited clinical materials.
Supplementary Material
Footnotes
Author contributions: A.U., Y.I., H.H., and A.L.B. developed highly multiplexed PRM methodology using ultra-long gradients on monolithic silica-C18 columns. R.S.L., J.D.G., and K.M.S. developed MIB methodology for activated kinase enrichment. A.U., R.S.L., and J.D.G. designed, conducted, and analyzed the experiments. J.T.W. and R.S.L. performed MSStats analysis. A.U., R.S.L., and J.D.G. wrote the manuscript. K.M.S., Y.I., and A.L.B. provided input on experimental design and contributed comments on the manuscript.
* This work was supported by the National Institute of General Medical Sciences grant 8P41GM103481 (AlB), National Institutes of Health shared instrumentation program grant 1S10OD016229-01 (ALB), JST-AMED SENTAN program (YI), Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (ALB and AU), and the Howard Hughes Medical Institute (KMS). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- MIB
- multidrug inhibitor beads
- AGC
- automatic gain control
- CRC
- colorectal cancer
- DDA
- data dependent acquisition
- iTRAQ
- isobaric tags for relative and absolute quantitation
- HCD
- high-energy collisional dissociation
- ESI
- electrospray ionization
- FWHM
- full width at half maximum
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- MSX
- multiplexed MS/MS
- NCE
- normalized collision energy
- PC
- peak capacity
- PRM
- parallel reaction monitoring
- PTM
- post-translational modification
- RT
- retention time
- SRM
- selected reaction monitoring
- SIL
- synthetic isotope-labeled
- SILAC
- stable isotope labeling by amino acids in culture
- UPLC
- ultra performance liquid chromatography.
REFERENCES
- 1. Manning G., Whyte D. B., Martinez R., Hunter T., and Sudarsanam S. (2002) The protein kinase complement of the human genome. Science 298, 1912–1934 [DOI] [PubMed] [Google Scholar]
- 2. Blume-Jensen P., and Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365 [DOI] [PubMed] [Google Scholar]
- 3. Uhlen M., Oksvold P., Fagerberg L., Lundberg E., Jonasson K., Forsberg M., Zwahlen M., Kampf C., Wester K., Hober S., Wernerus H., Bjorling L., and Ponten F. (2010) Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 28, 1248–1250 [DOI] [PubMed] [Google Scholar]
- 4. Daub H. (2015) Quantitative proteomics of kinase inhibitor targets and mechanisms. ACS Chem. Biol. 10, 201–212 [DOI] [PubMed] [Google Scholar]
- 5. Duncan J. S., Whittle M. C., Nakamura K., Abell A. N., Midland A. A., Zawistowski J. S., Johnson N. L., Granger D. A., Jordan N. V., Darr D. B., Usary J., Kuan P. F., Smalley D. M., Major B., He X., Hoadley K. A., Zhou B., Sharpless N. E., Perou C. M., Kim W. Y., Gomez S. M., Chen X., Jin J., Frye S. V., Earp H. S., Graves L. M., and Johnson G. L. (2012) Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cooper M. J., Cox N. J., Zimmerman E. I., Dewar B. J., Duncan J. S., Whittle M. C., Nguyen T. A., Jones L. S., Ghose Roy S., Smalley D. M., Kuan P. F., Richards K. L., Christopherson R. I., Jin J., Frye S. V., Johnson G. L., Baldwin A. S., and Graves L. M. (2013) Application of multiplexed kinase inhibitor beads to study kinome adaptations in drug-resistant leukemia. PLoS ONE 8, e66755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stuhlmiller T. J., Miller S. M., Zawistowski J. S., Nakamura K., Beltran A. S., Duncan J. S., Angus S. P., Collins K. A., Granger D. A., Reuther R. A., Graves L. M., Gomez S. M., Kuan P. F., Parker J. S., Chen X., Sciaky N., Carey L. A., Earp H. S., Jin J., and Johnson G. L. (2015) Inhibition of lapatinib-induced kinome reprogramming in ERBB2-positive breast cancer by targeting BET family bromodomains. Cell Rep. 11, 390–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sos M. L., Levin R. S., Gordan J. D., Oses-Prieto J. A., Webber J. T., Salt M., Hann B., Burlingame A. L., McCormick F., Bandyopadhyay S., and Shokat K. M. (2014) Oncogene mimicry as a mechanism of primary resistance to BRAF inhibitors. Cell Rep. 8, 1037–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wiese S., Reidegeld K. A., Meyer H. E., and Warscheid B. (2007) Protein labeling by iTRAQ: a new tool for quantitative mass spectrometry in proteome research. Proteomics 7, 340–350 [DOI] [PubMed] [Google Scholar]
- 10. Ow S. Y., Salim M., Noirel J., Evans C., Rehman I., and Wright P. C. (2009) iTRAQ underestimation in simple and complex mixtures: “the good, the bad and the ugly”. J. Proteome Res. 8, 5347–5355 [DOI] [PubMed] [Google Scholar]
- 11. Lange V., Picotti P., Domon B., and Aebersold R. (2008) Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Patricelli M. P., Nomanbhoy T. K., Wu J., Brown H., Zhou D., Zhang J., Jagannathan S., Aban A., Okerberg E., Herring C., Nordin B., Weissig H., Yang Q., Lee J. D., Gray N. S., and Kozarich J. W. (2011) In situ kinase profiling reveals functionally relevant properties of native kinases. Chem. Biol. 18, 699–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Worboys J. D., Sinclair J., Yuan Y., and Jorgensen C. (2014) Systematic evaluation of quantotypic peptides for targeted analysis of the human kinome. Nat. Methods 11, 1041–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Peterson A. C., Russell J. D., Bailey D. J., Westphall M. S., and Coon J. J. (2012) Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11, 1475–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gallien S., Duriez E., Crone C., Kellmann M., Moehring T., and Domon B. (2012) Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol. Cell. Proteomics 11, 1709–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gallien S., Kim S. Y., and Domon B. (2015) Large-scale targeted proteomics using internal standard triggered-parallel reaction monitoring. Mol. Cell. Proteomics 14, 1630–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Miyamoto K., Hara T., Kobayashi H., Morisaka H., Tokuda D., Horie K., Koduki K., Makino S., Nunez O., Yang C., Kawabe T., Ikegami T., Takubo H., Ishihama Y., and Tanaka N. (2008) High-efficiency liquid chromatographic separation utilizing long monolithic silica capillary columns. Anal. Chem. 80, 8741–8750 [DOI] [PubMed] [Google Scholar]
- 18. Yamana R., Iwasaki M., Wakabayashi M., Nakagawa M., Yamanaka S., and Ishihama Y. (2013) Rapid and deep profiling of human induced pluripotent stem cell proteome by one-shot NanoLC-MS/MS analysis with meter-scale monolithic silica columns. J. Proteome Res. 12, 214–221 [DOI] [PubMed] [Google Scholar]
- 19. Horie K., Sato Y., Kimura T., Nakamura T., Ishihama Y., Oda Y., Ikegami T., and Tanaka N. (2012) Estimation and optimization of the peak capacity of one-dimensional gradient high performance liquid chromatography using a long monolithic silica capillary column. J. Chromatogr. A 1228, 283–291 [DOI] [PubMed] [Google Scholar]
- 20. Iwasaki M., Miwa S., Ikegami T., Tomita M., Tanaka N., and Ishihama Y. (2010) One-dimensional capillary liquid chromatographic separation coupled with tandem mass spectrometry unveils the Escherichia coli proteome on a microarray scale. Anal. Chem. 82, 2616–2620 [DOI] [PubMed] [Google Scholar]
- 21. Guan S., Price J. C., Prusiner S. B., Ghaemmaghami S., and Burlingame A. L. (2011) A data processing pipeline for mammalian proteome dynamics studies using stable isotope metabolic labeling. Mol. Cell. Proteomics 10, M111.010728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baker P. R., and Chalkley R. J. (2014) MS-viewer: a web-based spectral viewer for proteomics results. Mol. Cell. Proteomics 13, 1392–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chalkley R. J., Baker P. R., Huang L., Hansen K. C., Allen N. P., Rexach M., and Burlingame A. L. (2005) Comprehensive analysis of a multidimensional liquid chromatography mass spectrometry data set acquired on a quadrupole selecting, quadrupole collision cell, time-of-flight mass spectrometer: II. New developments in Protein Prospector allow for reliable and comprehensive automatic analysis of large data sets. Mol. Cell. Proteomics 4, 1194–1204 [DOI] [PubMed] [Google Scholar]
- 24. UniProt C0nsortium (2015) UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., and Sherlock G. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bairoch A., and Apweiler R. (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 28, 45–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sharma V., Eckels J., Taylor G. K., Shulman N. J., Stergachis A. B., Joyner S. A., Yan P., Whiteaker J. R., Halusa G. N., Schilling B., Gibson B. W., Colangelo C. M., Paulovich A. G., Carr S. A., Jaffe J. D., MacCoss M. J., and MacLean B. (2014) Panorama: a targeted proteomics knowledge base. J. Proteome Res. 13, 4205–4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi M., Chang C. Y., Clough T., Broudy D., Killeen T., MacLean B., and Vitek O. (2014) MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526 [DOI] [PubMed] [Google Scholar]
- 30. Wolcott R. G., Dolan J. W., Snyder L. R., Bakalyar S. R., Arnold M. A., and Nichols J. A. (2000) Control of column temperature in reversed-phase liquid chromatography. J. Chromatogr. A 869, 211–230 [DOI] [PubMed] [Google Scholar]
- 31. Hancock W. S., Chloupek R. C., Kirkland J. J., and Snyder L. R. (1994) Temperature as a variable in reversed-phase high-performance liquid chromatographic separations of peptide and protein samples. I. Optimizing the separation of a growth hormone tryptic digest. J. Chromatogr. A 686, 31–43 [DOI] [PubMed] [Google Scholar]
- 32. Stergachis A. B., Maclean B., Lee K., Stamatoyannopoulos J. A., and MacCoss M. J. (2011) Rapid empirical discovery of optimal peptides for targeted proteomics. Nat. Methods 8, 1041–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boutros T., Chevet E., and Metrakos P. (2008) Mitogen-activated protein (MAP) kinase/MAP kinase phosphatase regulation: roles in cell growth, death, and cancer. Pharmacol. Rev. 60, 261–310 [DOI] [PubMed] [Google Scholar]
- 34. Dhillon A. S., Hagan S., Rath O., and Kolch W. (2007) MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290 [DOI] [PubMed] [Google Scholar]
- 35. Schubbert S., Shannon K., and Bollag G. (2007) Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7, 295–308 [DOI] [PubMed] [Google Scholar]
- 36. Downward J. (2003) Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3, 11–22 [DOI] [PubMed] [Google Scholar]
- 37. Roberts P. J., and Der C. J. (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310 [DOI] [PubMed] [Google Scholar]
- 38. Hatzivassiliou G., Haling J. R., Chen H., Song K., Price S., Heald R., Hewitt J. F., Zak M., Peck A., Orr C., Merchant M., Hoeflich K. P., Chan J., Luoh S. M., Anderson D. J., Ludlam M. J., Wiesmann C., Ultsch M., Friedman L. S., Malek S., and Belvin M. (2013) Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature 501, 232–236 [DOI] [PubMed] [Google Scholar]
- 39. Morris E. J., Jha S., Restaino C. R., Dayananth P., Zhu H., Cooper A., Carr D., Deng Y., Jin W., Black S., Long B., Liu J., Dinunzio E., Windsor W., Zhang R., Zhao S., Angagaw M. H., Pinheiro E. M., Desai J., Xiao L., Shipps G., Hruza A., Wang J., Kelly J., Paliwal S., Gao X., Babu B. S., Zhu L., Daublain P., Zhang L., Lutterbach B. A., Pelletier M. R., Philippar U., Siliphaivanh P., Witter D., Kirschmeier P., Bishop W. R., Hicklin D., Gilliland D. G., Jayaraman L., Zawel L., Fawell S., and Samatar A. A. (2013) Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discovery 3, 742–750 [DOI] [PubMed] [Google Scholar]
- 40. Carlino M. S., Todd J. R., Gowrishankar K., Mijatov B., Pupo G. M., Fung C., Snoyman S., Hersey P., Long G. V., Kefford R. F., and Rizos H. (2014) Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol. Oncol. 8, 544–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heldin C. H., Miyazono K., and ten Dijke P. (1997) TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471 [DOI] [PubMed] [Google Scholar]
- 42. Goumans M. J., Valdimarsdottir G., Itoh S., Rosendahl A., Sideras P., and ten Dijke P. (2002) Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 21, 1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. James D., Levine A. J., Besser D., and Hemmati-Brivanlou A. (2005) TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 132, 1273–1282 [DOI] [PubMed] [Google Scholar]
- 44. Calon A., Espinet E., Palomo-Ponce S., Tauriello D. V., Iglesias M., Cespedes M. V., Sevillano M., Nadal C., Jung P., Zhang X. H., Byrom D., Riera A., Rossell D., Mangues R., Massague J., Sancho E., and Batlle E. (2012) Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 22, 571–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Derynck R., and Zhang Y. E. (2003) Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584 [DOI] [PubMed] [Google Scholar]
- 46. Javelaud D., and Mauviel A. (2005) Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene 24, 5742–5750 [DOI] [PubMed] [Google Scholar]
- 47. Medard G., Pachl F., Ruprecht B., Klaeger S., Heinzlmeir S., Helm D., Qiao H., Ku X., Wilhelm M., Kuehne T., Wu Z., Dittmann A., Hopf C., Kramer K., and Kuster B. (2015) Optimized chemical proteomics assay for kinase inhibitor profiling. J. Proteome Res. 14, 1574–1586 [DOI] [PubMed] [Google Scholar]
- 48. Ruprecht B., Zecha J., Heinzlmeir S., Medard G., Lemeer S., and Kuster B. (2015) Evaluation of kinase activity profiling using chemical proteomics. ACS Chem. Biol. 10, 2743–2752 [DOI] [PubMed] [Google Scholar]
- 49. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., and Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 50. Miyata N., Azuma T., Hozawa S., Higuchi H., Yokoyama A., Kabashima A., Igarashi T., Saeki K., and Hibi T. (2012) Transforming growth factor beta and Ras/MEK/ERK signaling regulate the expression level of a novel tumor suppressor Lefty. Pancreas 41, 745–752 [DOI] [PubMed] [Google Scholar]
- 51. Nagaraj N. S., and Datta P. K. (2010) Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin. Investig. Drugs 19, 77–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Burgess M. W., Keshishian H., Mani D. R., Gillette M. A., and Carr S. A. (2014) Simplified and efficient quantification of low-abundance proteins at very high multiplex via targeted mass spectrometry. Mol. Cell. Proteomics 13, 1137–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.