

Abstract
The high genetic variability of RNA viruses is a significant factor limiting the discovery of effective biomarkers, the development of vaccines, and characterizations of the immune response during infection. Protein microarrays have been shown to be a powerful method in biomarker discovery and the identification of novel protein–protein interaction networks, suggesting that this technique could also be very useful in studies of infectious RNA viruses. However, to date, the amount of genetic material required to produce protein arrays, as well as the time- and labor-intensive procedures typically needed, have limited their more widespread application. Here, we introduce a method, protein microarray fabrication through gene synthesis (PAGES), for the rapid and efficient construction of protein microarrays particularly for RNA viruses. Using dengue virus as an example, we first identify consensus sequences from 3,604 different strains and then fabricate complete proteomic microarrays that are unique for each consensus sequence. To demonstrate their applicability, we show that these microarrays can differentiate sera from patients infected by dengue virus, related pathogens, or from uninfected patients. We anticipate that the microarray and expression library constructed in this study will find immediate use in further studies of dengue virus and that, more generally, PAGES will become a widely applied method in the clinical characterization of RNA viruses.
In the past decade, there have been a variety of viral infectious diseases that have significantly threatened global public health, from severe acute respiratory syndrome (SARS)1 that emerged in southern China in 2003 (1) to the recent outbreaks of Ebola (2) and Zika (3). In addition, many viruses have been a consistent global threat for many years, such as dengue virus that infects tens of millions of people worldwide each year, of which 500,000 develop hemorrhagic fever and 20,000 die (4).
To study these viruses, it is typically necessary to obtain their genetic material. However, due to the possibility of infection, the accessibility of this highly infectious genomic material is very limited. Moreover, owing to the fact that the genomes of most of these viruses are RNA-based and thus highly variable, it is difficult in practice to identify a single strain that could be representative of many of the variable strains of the same virus. As a result, there is a significant lack of effective tools that can be used to characterize the molecular details of these viruses for either the identification of diagnostic biomarkers, the development of broadly effective or strain-specific vaccines, or detailed characterization of on-going infections within individual patients at a systems-wide level.
Protein microarrays have become an established technology in a wide range of fields of biology and medicine owing to their ability to rapidly evaluate interactions with or between proteins in a high-throughput, low cost manner using only small sample volumes (5). The method is also relatively straightforward to perform such that the rate-limiting step in most protein microarray studies is the first one: its construction (5, 6). First, a sufficient amount of genetic material of the targeting species is required. All of the genes/predicted ORFs are then PCR amplified with primers containing proper restriction endonuclease sites/recombination sites (7), which can prove challenging for genes with high GC content (8, 9), and then cloned into expression vectors. This entire procedure is extremely labor- and time-consuming, generally requiring 3–4 years for a typical 4 Mb genome (6). In addition, due to codon usage bias in different species, it can also be challenging to obtain a sufficient amount of recombinant proteins from an exogenous host, such as Escherichia coli or yeast (10). Thus, while the construction of protein microarrays is challenging for many organisms, it has proven prohibitive for studies of species with highly variable genomes, such as RNA viruses, where the genetic material is obtained from the natural setting.
Promoted by advances in synthetic biology (11), the chemical synthesis of long DNA fragments is now a feasible and affordable means of obtaining genetic material. In comparison to traditional cloning technologies, cloning through de novo gene synthesis is advantageous since it is independent of the natural genetic material, is very flexible for designing specific elements/sites and codon optimization for better protein expression in a specific host, and requires much less time.
In this study, we have developed a technique, protein microarray fabrication through gene synthesis (PAGES), for the rapid and efficient construction of protein microarrays particularly suited for studies of RNA viruses. Focusing on the dengue virus, we first identified the consensus sequences of all serotypes from 3,604 strains. The usefulness of focusing on conserved residues within the serotypes is supported by the recent observation of broadly active antibodies isolated from patients infected with dengue virus (12) and HIV(13), suggesting that there are conserved immunogens present in these high-mutable pathogens. These consensus sequences are then de novo synthesized and inserted into expression vectors. After protein expression and purification, the dengue virus proteome microarray encompassing all serotypes is finally produced, all within one month. To our knowledge, this is first proteomic microarray of the complete population of serotypes of an RNA virus. We demonstrate the applicability of these microarrays to identify potential serum biomarkers and to monitor the humoral response of dengue virus infection, setting the stage for the wider application of PAGES in studies of RNA viruses.
MATERIALS AND METHODS
Serum Sample Collection
The cohort was comprised of 283 serum samples from 65 healthy volunteers, 159 PI (patients infected by dengue virus serotype I), 9 PII (patients infected by of dengue virus serotype II), 5 PIII (patients infected by dengue virus serotype III), 7 OP1 (patients diagnosed as viral hemorrhagic fever), 16 OP2 (patients infected by Scrub typus), and 13 OP3 (patients infected by epidemic encephalitis B). The characteristics of these sera are shown in Table I, Table II, and Fig. 7A. The healthy volunteers and patients had been given informed consent and approved by Center for Disease Control and Prevention of Guangdong. All the serum samples were collected during 2014–2015 by Center for Disease Control and Prevention of Guangdong. All the serum samples were collected according to a standard protocol. Briefly, 5 ml whole blood were collected to Improvacuter tube Company (Guangzhou, China) without anticoagulant. The whole blood was left at room temperature for 30 min and then centrifuged at 2,000 g for 10 min in a refrigerated centrifuge. At last, the serum was transferred to the Corning 2 ml vial and stored at −80 °C.
Table I. The characteristics of sera samples probed on dengue virus proteome microarray for screening sera biomarkers.
| Type | No. | Ages |
p* | Gender |
p | ||
|---|---|---|---|---|---|---|---|
| Mean | S.D. | Male | Female | ||||
| Healthy | 30 | 39.53 | 4.35 | 19 | 11 | ||
| PI | 98 | 34.95 | 17.14 | <0.05 | 52 | 46 | 0.921 |
| PII | 3 | 39.00 | 16.00 | ND | 3 | 0 | ND |
| PIII | 2 | 25.50 | 4.95 | ND | 0 | 2 | ND |
| OP1 | 2 | 34.50 | 4.95 | ND | 2 | 0 | ND |
| OP2 | 10 | 52.13 | 18.42 | ND | 6 | 4 | ND |
| OP3 | 8 | 31.25 | 11.85 | ND | 4 | 4 | ND |
* F-test.
Table II. The characteristics of sera samples used in ELISA for validation the candidate biomarkers.
| Type | No. | Ages |
p* | Gender |
p | ||
|---|---|---|---|---|---|---|---|
| Mean | S.D. | Male | Female | ||||
| Healthy | 35 | 33.86 | 17.95 | 8 | 27 | ||
| PI | 58 | 38.58 | 18.74 | 0.801 | 34 | 24 | 0.339 |
| PII | 6 | 34.33 | 13.22 | ND | 2 | 4 | ND |
| PIII | 3 | 33.00 | 16.46 | ND | 1 | 2 | ND |
| OP1 | 5 | 42.20 | 10.40 | ND | 4 | 1 | ND |
| OP2 | 6 | 57.80 | 8.66 | ND | 5 | 1 | ND |
| OP3 | 5 | 8.40 | 7.30 | ND | 2 | 3 | ND |
* F-test.
Fig. 7.

The dynamics of [IgG] specific against I-E and III-E. (A) Information of the three patients with sera drawn at different time points from day 2 to day 16. (B–D) The dynamics of [IgG] in serum that bound I-E and III-E for patient 1# (B), patient 2# (C), and patient 3# (D).
Chemical and Reagents
The polymer slide G was purchased from CapitalBio (Beijing, China). Human IgG, 3,3′,5,5′-tetramethylbenzidine and antiglutathione-S-transferase (GST) antibody produced in rabbit were provided by Sigma (Missouri). IgM was obtained from Thermo Scientific (Massachusetts). The secondary antibodies, CyTM5 affinipure goat anti-rabbit IgG(H+L) (abbreviated as G-A-R-Cy5), DyLightTM549 affinipure mouse anti-human IgG (M-A-H-IgG-549), DyLightTM649 affinipure donkey anti-human IgM (d-A-H-IgM-649), and peroxidase affinipure mouse anti-human IgG (M-A-H-IgG-HRP) were purchased from Jackson ImmunoResearch Inc. (Pennsylvania). IRDye 800C donkey anti-rabbit IgG(H+L) (d-A-R-800) was purchased from the LI-COR, Inc. (Nebraska).
Phylogenetic Analysis of Dengue Virus
The amino acid sequences of polyprotein of dengue virus were obtained from National Center for Biotechnology Information (NCBI, website http://www.ncbi.nlm.nih.gov/) after removed of the partial sequences. All the complete sequences were aligned by multiple sequence comparison by log-expectation (MUSCLE) (14) using a Linux system and analyzed by the FastTree 2 (15). The final results were visualized by the Archaeopteryx applet (16).
Consensus Sequences Generation
According to the serotypes of dengue virus, the aligned sequences were divided into four branches. All sequences in each part were realigned and then analyzed by function “seqconsensus” in Matlab R2014b (17). According to the dengue virus strains serotype I HAW (Hawaii), serotype II New Guinea C, serotype III H87, and serotype IV H241 (18), the consensus sequences of the polyproteins from each branch could be chopped into 11 ORFs.
Gene Synthesis and Transformants Construction
All the final consensus amino acid sequences were converted into DNA sequences. After being optimized by the OptimumGene algorithm (19), all genes were modified by adding elements for cloning, such as BamH I and EcoR I sites and stop codons. Subsequently, all the genes were synthesized by GenScript. (Nanjing, China). The synthesized genes were integrated into vector pGEX-4T-1 via the restriction endonuclease BamH I and EcoR I and transformed into competent cell E. coli BL21 strain to construct the transformants. All transformants were mixed with 50% glycerol and stored in −80 °C.
Protein Purification and Protein Microarray Fabrication
After optimization of induction conditions, all the transformants were induced by 0.5 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) at 16 °C overnight. The proteins were purified as previously described (6). Briefly, the GST-tagged proteins were purified by affinity-chromatography with glutathione agarose beads. The proteins were verified by Western blotting using an anti-GST antibody. All the purified proteins were spotted on polymer slides G with triplicate spots using a SmartArrayer 48 microarrayer from CapitalBio. In addition, some quantity controls were also included in the dengue virus proteome microarray, such as human IgG and IgM at different concentrations, GST, printing buffer, and land markers. The fabricated protein microarrays were stored at −80 °C.
Serum Profiling on Protein Microarray
Serum profiling on protein microarray was carried out as previously described (20). Briefly, the dengue virus proteome microarrays were blocked with 3% fat-free milk for 1h at the room temperature. After being washed with phosphate buffered saline containing 0.5% Tween-20 (PBST) for three times, 5 min each, the slides were incubated with 100X diluted sera for 1 h at room temperature, washed, and incubated with 1000X diluted secondary antibodies M-A-H-IgG-549 and d-A-H-IgM-649. Subsequently, the nonspecific binding was washed away. The slides were scanned by GenePix 4200A (Molecular Devices, Sunnyvale, CA). All the sera samples were repeated twice on the protein microarray. The microarray data were extracted and processed by GenePix Pro 6.0 (Molecular Devices). The heatmap was drawn by Heml (21).
ELISA Validation
The procedures for ELISA validation was as previously described with slight modifications (20). In short, the purified proteins I-E and III-E were diluted to 2 μg/ml by 50 mm carbonate buffer (pH 9.6). The 96-well plates were sensitized with 50 μl proteins per well. After centrifugation for 2 min at 550 g for distributing proteins evenly, the 96-well plates were incubated at 4 °C overnight. Subsequently, the plates were emptied and blocked with 100 μl blocking buffer (5% skim milk in PBST) at room temperature. For 1 h, the plates were washed three times by PBST. The plates were incubated with 50 μl 5,000X diluted sera for 2 h at 37 °C. The plates were further washed with PBST for another three times. The plates were incubated with 50 μl 16,000X diluted secondary antibody M-A-H-IgG-HRP and washed again. A total of 50 μl 3,3′,5,5′-tetramethylbenzidine solution was added into each well and incubated for 20 min at 37 °C. The plates were incubated with 50 μl of 1 m H2SO4 for stopping the reaction. The results were recorded with a multichannel spectrophotometer at 450 nm. Three replicates were carried out. A typical third-order polynomial (cubic) nonlinear regression model was used for standard curve-fitting for the ELISA, which was further used to normalize the absorbance measurement data of samples.
Statistics Analysis
The fluorescent intensity of each spot in all the scanned slides were extracted with GenePix Pro 6.0 Software (Molecular Devices) and analyzed by limma package in R 3.1.3 (22). The differences of the clinical characteristics in each groups were evaluated by Fisher's test. The Vioplot package implemented in R 3.1.3 was used to generate violin plot (23).
RESULTS
Schematic Diagram of PAGES
An overview of the PAGES method is presented in Fig. 1. After identifying the amino acid sequences of all of the proteins encoded in the target genome or consensus target genome, the protein sequences were converted into nucleic acid sequences and further optimized. These sequences were then de novo synthesized and inserted into appropriate expression vectors for propagation in E. coli DH5α. The plasmids were next transferred into the final expression host to construct the expression library. Following induction of the transformants for protein expression, the proteins were affinity purified and spotted onto the appropriately modified glass substrate to produce the microarray.
Fig. 1.
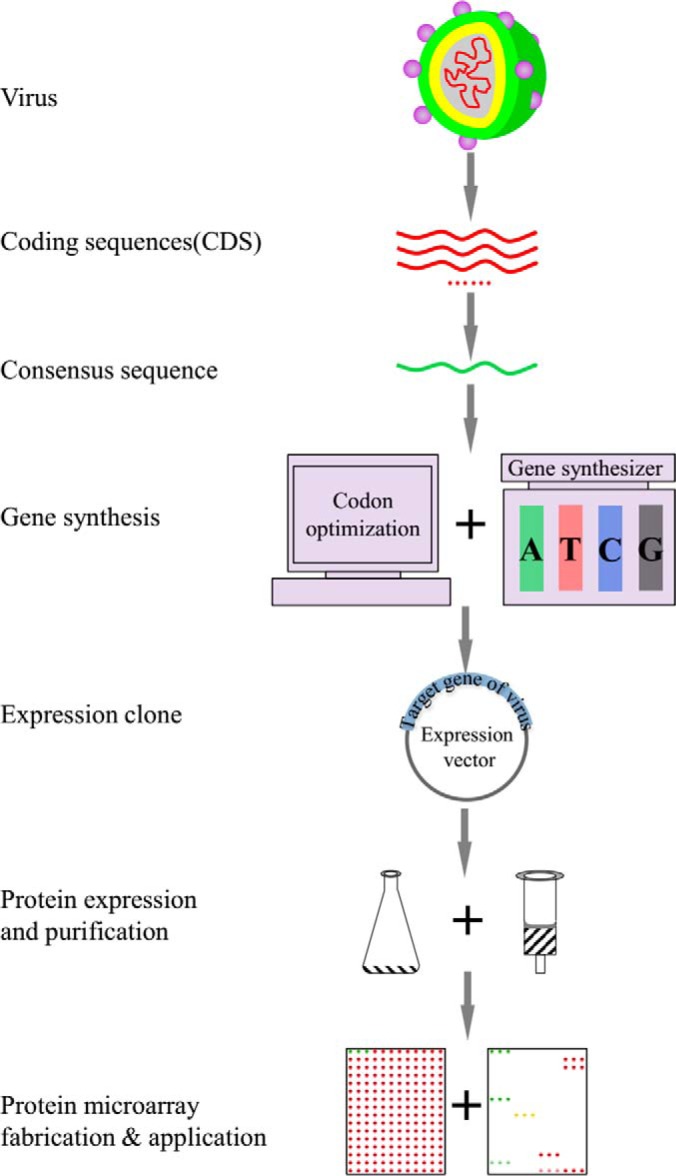
Schematic diagram of PAGES. The coding sequences of the virus of interest were retrieved from NCBI or other database. After the consensus sequences were generated, they were subjected for sequence optimization and gene synthesis. The proteins were then expressed in E. coli and affinity purified. The protein microarrays were fabricated and adopted for biology study and clinical research.
Generation of the Consensus Protein Sequences for All Serotypes of the Dengue Virus
To obtain polyprotein sequences of the dengue virus, candidate sequences in NCBI were identified by blasting the polyprotein sequences of prototypical strains of the dengue virus (GenBank accession number: ACF49259.1, AAC59275.1, AAA99437.1, and AAX48017.1). Following removal of partial or chimeric sequences, a set of 3,604 complete polyprotein sequences of the dengue virus were identified (Table S1). These sequences were then subjected to phylogenetic analysis and, as shown in Fig. 2, the well-established serotypes of the dengue virus were recapitulated (24). Additional details of the phylogenetic tree are shown in Fig. S1. In this way, the 3,604 polyprotein sequences could be separated into four groups, in which serotypes I, II, III, and IV contained 1,525, 1,104, 823, and 152 sequences, respectively.
Fig. 2.

The workflow of constructing expression library for dengue virus.
The consensus sequence for each serotype was then determined (Table S2). Comparing these sequences to those of prototypical strains of the four serotypes (namely, serotype I HAW (Hawaii), serotype II New Guinea C, serotype III H87, and serotype IV H241) enabled the identification of the 11 individual protein sequences encoded therein (Table S3).
Purification of the Dengue Proteins from Codon Optimized and De Novo Synthesized Genes
To construct expression clones, the consensus protein sequences (Table S3) were converted into DNA sequences, which were then optimized for expression in E. coli (Table S4). In particular, the sequences were optimized after considering factors such as the frequency of codon usage in E. coli, GC content, mRNA secondary structures, potential splicing sites, and repetitive motifs. The 44 genes were then de novo synthesized and cloned into the vector pGEX-4T-1, and transformed into the E. coli strain BL21 to produce the final expression library (Fig. 3A).
Fig. 3.

Dengue virus proteins of all the four serotypes were successfully purified using a high-throughput way. (A) The synthesized genes were integrated into the expression vector pGEX-4T-1. (B) Expected molecular weight (MW) of elected proteins. (C) The proteins were expressed and purified in a high-throughput format. The E. coli expression clones were stored in a 96-well plate. The clones were inoculated to a 12-channel plate for culture and protein expression. The cells were harvested, and the proteins were affinity purified in a 96-deep well plate. The purified proteins were stored at −80 °C prior to microarray printing. (D) All the purified proteins were validated by Western blotting. The red asterisk indicated the target proteins.
We examined a wide range of conditions for optimal protein expression, using transformants that correspond to I-2k (29 kDa), I-E (81 kDa), I-NS4B (53 kDa), and I-NS5 (130 kDa) to represent a wide range of protein sizes and biochemical characteristics (Fig. 3B). After several rounds of testing, the best conditions for protein expression were determined to be 0.5 mm IPTG, 16°C, with overnight induction (data not shown). Under these conditions, all 44 proteins were expressed and purified with a single high-throughput procedure (Fig. 3C) as previously described (6). The purified proteins were then verified by Western blotting and silver straining. All the proteins were successfully purified, of which eight proteins were shown single band purity, 23 proteins with dominant bands, and remaining proteins with low purity. When proteins were purified at large scale for constructing protein microarray, usually, ∼85% purity for most of the proteins could be obtained, and the purity could be further improved by optimize the expression and purification. In Fig. 3D, the proteins were checked by an anti-GST antibody. Given the standard protein purification procedure and high specificity of the anti-GST antibody, we speculated that the bands other than the expected band might be due to the partial degradation of the target proteins while still retaining the GST tag during protein expression and purification. To support our speculation, indeed, most of the “nonspecific” bands were smaller than the proteins of expected size. In a word, these results clearly demonstrated that the abundance and purity of most of the 44 proteins were suitable for protein microarray fabrication (Fig. 3D and Fig. S2).
The Dengue Virus Proteome Microarray Is of High Quality
To fabricate the proteome microarray, each of the purified proteins was spotted in triplicate using a microarrayer (Fig. 4A). In addition, two location markers of fluorescent-labeled proteins, namely Alexa-549-labeled IgG and Alexa-649-labeled IgM, were included to facilitate data analysis. Serially diluted IgG and IgM from human sera were also included to provide for the possibility of quantitative analysis (as described below) (Fig. 4A).
Fig. 4.

Characteristics of the dengue virus proteome microarray. (A) The layout of the proteome microarray. (B) The microarray was incubated with an anti-GST antibody followed with secondary antibody G-A-R-Cy5. (C) Histogram analysis of fluorescence intensity of all the purified proteins according to the signal of secondary antibody G-A-R-Cy5. (D) The microarray was incubated with a secondary antibody M-A-H-IgG-549. (E) The microarray was incubated with a secondary antibody d-A-H-IgM-649. (F) The correlation between the concentration of the serially diluted IgG (left) and IgM (right) on the microarray and corresponding fluorescence intensities.
The quality of the microarray was first evaluated using rabbit anti-GST antibodies and G-A-R-Cy5 secondary antibodies, which showed that all 44 purified dengue proteins were successfully immobilized on the microarray (Fig. 4B). Quantitative analysis of the fluorescence intensity demonstrated that the distribution of the foreground and background intensities exhibited typical bell-shaped curves that were well separated (Fig. 4C). The information of the dengue virus proteome microarray was deposited on the Protein Microarray Database (www.proteinmicroarray.cn) with number PMDA 173.
Since one of the major applications of this proteome microarray was serum profiling, we also examined whether fluorescently labeled secondary antibodies, such as M-A-H-IgG-549 and d-A-H-IgM-649, bound directly to dengue proteins, which would produce false positive signals. As shown in Figs. 4D and 4E, there were no nonspecific bindings of d-A-H-IgM-649 or d-A-H-IgG-549, except, with the latter, to protein C and NS5 of some serotypes. Further, this experiment showed that there was no cross-reactivity between d-A-H-IgM-649 and IgG or M-A-H-IgG-549 and IgM.
We also examined whether the microarray could be applied for quantitative analyses by measuring the signals at the serially diluted IgG and IgM spots following incubation with M-A-H-IgG-549 and d-A-H-IgM-649. As shown in Fig. 4F, these bindings were indeed concentration dependent and strongly correlated with the concentrations of the immobilized proteins.
Identification of Potential Serum Biomarkers of Virus Infection Using the Dengue Virus Proteome Microarray
To test the applicability of the proteome microarray for diagnostic applications, the sera of patients infected with dengue virus (PI, PII, and PIII) were probed on the microarray. Given that the clinical symptoms of patients infected by dengue virus were similar to those of other viral infections (25, 26), we also examined sera from patients diagnosed with viral hemorrhagic fever (OP1) or infected with Scrub typus (OP2) and epidemic encephalitis B (OP3). Sera from healthy people were also included as a control. The status of all sera was confirmed by a variety of assays, such as virus isolation, real-time PCR, or ELISA-based serological detection (data not shown).
After data extraction and deposition to the Protein Microarray Database (dataset No. PMDE228), the signal intensities were converted into concentrations of IgG and IgM according to the regression formulas obtained from each subarray and plotted as heatmaps for [IgG] (Fig. 5A) and [IgM] (Fig. 5B). Because protein C and NS5 of some serotypes bound to the secondary antibody M-A-H-IgG-549 directly, the [IgG] signals associated with these proteins were not included in this analysis.
Fig. 5.

Probing serum on the dengue virus proteome microarray. The microarray signal intensities were transformed to the serum antibody concentration according to the standard curve in each block. Heatmaps were generated based on the data for [IgG] (A) and [IgM] (B) OP1, OP2, OP3 indicated the sera that were collected from patients diagnosed as viral hemorrhagic fever or infected by Scrub typus and pidemic encephalitis B, respectively. (C) Candidate proteins with p value < 0.05 and fold change > 1.
Overall, the [IgG] and [IgM] signals against dengue proteins were higher in sera from all patients compared with those from the healthy group. Further, [IgG] and [IgM] were both higher in the PI group than in the OP1, OP2, and OP3 groups. As expected, IgG and IgM were found to bind to some dengue proteins of all serotypes, possibly because of the high sequence similarities. Generally, [IgG] and [IgM] against proteins of serotype I and III were higher than those of serotype II and IV, and this difference was more significant in [IgM]. We also note that the overall [IgG] signal was significantly higher than [IgM], which may be owing to the later immune response of IgG than IgM and the faster dynamics of IgM than IgG upon viral infection (27) since the sera were collected several days after the onset of symptoms.
After further statistical analysis, candidate proteins (Fig. 5C) were identified that exhibited significant differences of [IgG] between the healthy and PI groups. We found that the top two candidate proteins were I-E and III-E, which was consistent with other studies (27, 28). No significant differences were observed in [IgG] against II-E and IV-E between the PI group and healthy group, indicating that serological testing against specific proteins could differentiate dengue serotypes at least to some extent.
Validation of the Immune Response against I-E and III-E upon Dengue Virus Infection by ELISA
Following the same protocol and criteria of the samples used in the discovery stage (Table I), a new set of samples was collected for validation with ELISA (Table II) using purified proteins I-E and III-E (Fig. 6A). Phylogenetic analysis indicates I-E and III-E were more closely clustered among the four serotypes (Fig. 6B). A violin plot generated for both I-E and III-E revealed significant differences for the tail of the violin (Fig. 6C), which was typical of a small set of samples. These results indicated that protein E could be further developed as a biomarker for a subset of strains.
Fig. 6.

Validation of proteins I-E and III-E as potential markers by ELISA. (A) Purified protein I-E (left) and III-E (right) were assessed by silver staining. (B) The phylogenetic relationship of the four protein E. (C) ELISA was performed by using an independent set of samples against protein I-E (left) and III-E (right).
Detection of Dynamic IgG Responses for I-E and III-E after Dengue Virus Infection
To further demonstrate the applicability of this microarray, sera collected at different time points from the same patient after dengue virus infection were probed on the microarray to monitor the dynamics of the [IgG] response. Three sets of sera were collected from three patients after the onset of the symptoms (Fig. 7A). To reduce the experimental variability, sera from the same patient were probed side-by-side on individual subarrays of the same microarray. Time-dependent [IgG] responses against I-E and III-E were clearly observed in all three patients (Figs. 7B–7D). Moreover, the overall trend of the [IgG] temporal response was similar among the three patients: namely, a rapid increase after infection up to day 8 to 14, followed by a decrease, consistent with the general viral immune response (29). Differences were also observed for [IgG] among the three patients, indicating that this microarray could also be useful for personalized medicine applications. Finally, we note that the immune-response to I-E and III-E were similar for each patient, which could be owing to the high homology between these two proteins (Fig. 6B).
DISCUSSION
We have developed PAGES, a new strategy that combines consensus sequence identification, gene synthesis and high-throughput protein purification for the rapid and efficient construction of protein microarrays. We have demonstrated the effectiveness of this method using dengue virus as an example, fabricating the first proteomic microarray covering all four serotypes of this virus in one month. We have then demonstrated its usefulness with serum profiling, finding that protein I-E and III-E show potential as biomarkers of dengue virus infection for at least a subset of samples. In addition, we have found that, while there are similar general trends of the immune-response to dengue virus infection among different patients, clear differences are also apparent.
In comparison to the traditional techniques used to construct protein microarrays, there are several advantages of PAGES (Fig. 8). First, PAGES is completely independent of natural genomic materials. Thus, it is possible for an investigator to construct any protein microarray desired, even when the accessibility of natural genomic materials is limited or prohibited, such as with viruses of high infectivity and high pathogenicity. Second, PAGES is suitable for fabricating protein microarrays for species with highly variable genomes, taking advantage of the usefulness of consensus sequences as accurate representatives for large populations of serotypes. Third, the expression clones are rationally designed. Because the genes are synthesized de novo, the sequences could be easily adjusted for any purpose, such as the addition/removal of specific elements or sites, and codon optimization. This step affords a high degree of flexibility to the PAGES method. Finally, PAGES is much less time intensive. Overall, the steps from the initial identification of the consensus sequences to their de novo synthesis could be easily accomplished in two weeks, enabling the complete fabrication of the dengue virus proteome microarray in one month. By contrast, construction of the proteome from a similarly sized genome using traditional method can require up to one year.
Fig. 8.

The comparison the PAGES and the traditional strategy for protein microarray construction.
Nonetheless, there is also some ways by which PAGES can be improved. First, the cost is slightly higher than traditional strategies (Fig. 8), primarily owing to expenses incurred for gene synthesis. Further, current technologies for artificial gene synthesis is still not well suited for synthesis of long genes, for example, longer than 5 kb. However, we anticipate that costs will be sharply reduced as improvements of this technology are made in the near future, resulting from its growing demand in synthetic biology. Second, sequence information must be known so as to enable gene synthesis. Still, with the incredible power of next-generation DNA sequencing technology, it should always be possible to obtain a rich collection of sequence information immediately after the outbreak of infectious diseases (30, 31). Third, there are greater demands on computation and bioinformatics required in PAGES. From sequence retrieving to consensus sequence generation and codon optimization to sequencing tailing, each of these steps is presently performed manually. Still, all of the tools are publically available, and so they could be easily integrated, simplifying the procedures in the near future.
We note though that our microarray is not the first protein microarray of the dengue virus. In particular, Fernandez et al. (18) constructed a protein microarray using a traditional strategy in which “prototypical” strains of the dengue virus were selected for its fabrication. However, this microarray is limited to only the proteomes of the strains included, regardless of how representative these sequences are for the serotypes more generally. Further, this microarray might not be suitable for studying current dengue virus epidemics that exhibit highly variable genomes. Finally, the lack of some specific proteins in this microarray, such as protein 2k of all serotypes and protein NS5 of serotype I and IV, may compromise its broad effectiveness. By comparison, our dengue virus proteome microarray could represent many, if not all, dengue viruses. In addition, our microarray contains all of the dengue virus proteins, including the NS5 proteins from serotype I and IV of dengue virus, which may be owing to the codon optimization that facilitated protein expression.
By probing our microarray with sera from dengue-infected patients and healthy people, we find that proteins I-E and III-E could serve as the most potential biomarkers (Fig. 5C). These findings are consistent with previous studies (27, 28). Further, the candidate protein I-E and III-E are validated by a new set of sera and can be used as biomarkers for a subset of patients (Fig. 6C). Moreover, examining the serum collected from the same patient at different time points during infection demonstrated that [IgG] against I-E and III-E are dynamic, with trends consistent with the traditional humoral response to viral infection (29). However, a big difference among the patients was also observed (Fig. 7). Therefore, the candidate protein I-E and III-E can be applicable in clinical detection if the patients are characterized in more detail and divided into more precise subgroups. We note that our sera were collected during the 2014 dengue outbreak in Guangzhou, China, where serotype I is predominant (32). As a result, only a few sera of other serotypes were collected, which limited our ability to identify serotype-specific biomarkers. Clearly, though, access to a greater range of patient sera is expected to yield a wider range of these specific biomarkers.
While we focused on consensus sequences, genes of a predominant epidemic dengue virus strain could also be synthesized, subjected to protein purification, and added to this microarray, as needed. Besides sera profiling and immune-response monitoring, the dengue virus proteome microarray could also be applied for many other studies of the dengue virus, such as host–pathogen interactions and vaccine development. Because PAGES is general, it could easily be adopted for the rapid construction of protein microarrays for other viruses, either in advance or right after an outbreak of a specific disease, thus providing a powerful option for investigating these diseases. PAGES could also be applied to construct a “pan-virus” protein microarray, covering many of the important viruses, from SARS to Zika, whether or not the virus is RNA based. Due to its flexibility, PAGES could also be used for many other applications, such as constructing microarrays of proteins containing a series of mutations, truncations, or protein domains.
To conclude, viral outbreaks are a significant concern for global health. Rapidly obtaining diagnostic profiles, applying effective vaccines, and accurately monitoring therapy efficacy are all serious, generally unmet present challenges. We anticipate that PAGES will develop into a highly effective methodology to specifically address these concerns.
Supplementary Material
Footnotes
Author contributions: S.T. designed the research; H.Q., H.Z., S.G., Y.L., N.W., and Y.S. performed the research; L.L. and D.W. contributed new reagents or analytic tools; H.Q. and J.W. analyzed data; and H.Q., D.M.C., and S.T. wrote the paper.
* This study was funded in part by National Natural Science Foundation of China Grants (31600672, 31370813, and 31670831), the National Key Research and Development Program of China (2016YFA0500601), and China Postdoctoral Science Foundation (2016M591667).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- SARS
- severe acute respiratory syndrome
- PCR
- polymerase chain reaction
- ORF
- open reading frame
- ELISA
- enzyme linked immunosorbent assay
- GST
- glutathione S-transferase
- PMD
- protein microarray database.
REFERENCES
- 1. Peiris J. S., Guan Y., and Yuen K. Y. (2004) Severe acute respiratory syndrome. Nat. Med. 10, S88–S97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gire S. K., Goba A., Andersen K. G., Sealfon R. S., Park D. J., Kanneh L., Jalloh S., Momoh M., Fullah M., Dudas G., Wohl S., Moses L. M., Yozwiak N. L., Winnicki S., Matranga C. B., Malboeuf C. M., Qu J., Gladden A. D., Schaffner S. F., Yang X., Jiang P. P., Nekoui M., Colubri A., Coomber M. R., Fonnie M., Moigboi A., Gbakie M., Kamara F. K., Tucker V., Konuwa E., Saffa S., Sellu J., Jalloh A. A., Kovoma A., Koninga J., Mustapha I., Kargbo K., Foday M., Yillah M., Kanneh F., Robert W., Massally J. L., Chapman S. B., Bochicchio J., Murphy C., Nusbaum C., Young S., Birren B. W., Grant D. S., Scheiffelin J. S., Lander E. S., Happi C., Gevao S. M., Gnirke A., Rambaut A., Garry R. F., Khan S. H., and Sabeti P. C. (2014) Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345, 1369–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cohen J. (2016) The race for a Zika vaccine is on. Science 351, 543–544 [DOI] [PubMed] [Google Scholar]
- 4. Normile D. (2014) Tropical diseases. Dengue vaccine trial poses public health quandary. Science 345, 367–368 [DOI] [PubMed] [Google Scholar]
- 5. Zhu H., Bilgin M., Bangham R., Hall D., Casamayor A., Bertone P., Lan N., Jansen R., Bidlingmaier S., Houfek T., Mitchell T., Miller P., Dean R. A., Gerstein M., and Snyder M. (2001) Global analysis of protein activities using proteome chips. Science 293, 2101–2105 [DOI] [PubMed] [Google Scholar]
- 6. Deng J., Bi L., Zhou L., Guo S. J., Fleming J., Jiang H. W., Zhou Y., Gu J., Zhong Q., Wang Z. X., Liu Z., Deng R. P., Gao J., Chen T., Li W., Wang J. F., Wang X., Li H., Ge F., Zhu G., Zhang H. N., Gu J., Wu F. L., Zhang Z., Wang D., Hang H., Li Y., Cheng L., He X., Tao S. C., and Zhang X. E. (2014) Mycobacterium tuberculosis proteome microarray for global studies of protein function and immunogenicity. Cell Rep. 9, 2317–2329 [DOI] [PubMed] [Google Scholar]
- 7. Reboul J., Vaglio P., Rual J. F., Lamesch P., Martinez M., Armstrong C. M., Li S., Jacotot L., Bertin N., Janky R., Moore T., Hudson J. R. Jr, Hartley J. L., Brasch M. A., Vandenhaute J., Boulton S., Endress G. A., Jenna S., Chevet E., Papasotiropoulos V., Tolias P. P., Ptacek J., Snyder M., Huang R., Chance M. R., Lee H., Doucette-Stamm L., Hill D. E., and Vidal M. (2003) C. elegans ORFeome version 1.1: experimental verification of the genome annotation and resource for proteome-scale protein expression. Nat. Genet. 34, 35–41 [DOI] [PubMed] [Google Scholar]
- 8. Bachmann H. S., Siffert W., and Frey U. H. (2003) Successful amplification of extremely GC-rich promoter regions using a novel “slowdown PCR” technique. Pharmacogenetics 13, 759–766 [DOI] [PubMed] [Google Scholar]
- 9. Kumar A and Kaur J. (2014) Primer based approach for PCR amplification of high GC content gene: Mycobacterium gene as a model. Mol. Biol. Int. 10.1155/2014/937308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cartegni L., Chew S. L., and Krainer A. R. (2002) Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 3, 285–298 [DOI] [PubMed] [Google Scholar]
- 11. Gibson D. G., Benders G. A., Andrews-Pfannkoch C., Denisova E. A., Baden-Tillson H., Zaveri J., Stockwell T. B., Brownley A., Thomas D. W., Algire M. A., Merryman C., Young L., Noskov V. N., Glass J. I., Venter J. C., Hutchison C. A., and Smith H. O. 3rd. (2008) Complete chemical synthesis, assembly, and cloning of a Mycoplasma genitalium genome. Science 319, 1215–1220 [DOI] [PubMed] [Google Scholar]
- 12. Rouvinski A., Guardado-Calvo P., Barba-Spaeth G., Duquerroy S., Vaney M. C., Kikuti C. M., Navarro Sanchez M. E., Dejnirattisai W., Wongwiwat W., Haouz A., Girard-Blanc C., Petres S., Shepard W. E., Desprès P., Arenzana-Seisdedos F., Dussart P., Mongkolsapaya J., Screaton G. R., and Rey F. A. (2015) Recognition determinants of broadly neutralizing human antibodies against dengue viruses. Nature 520, 109–113 [DOI] [PubMed] [Google Scholar]
- 13. Jardine J. G., Ota T., Sok D., Pauthner M., Kulp D. W., Kalyuzhniy O., Skog P. D., Thinnes T. C., Bhullar D., Briney B., Menis S., Jones M., Kubitz M., Spencer S., Adachi Y., Burton D. R., Schief W. R., and Nemazee D. (2015) Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 349, 156–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Edgar R. C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Price M. N., Dehal P. S., and Arkin A. P. (2010) FastTree 2—Approximately maximum-likelihood trees for large alignments. PloS One 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Han MV and Zmasek C. M. (2009) phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinformatics 10, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. O'Donnell C. R., Wang H., and Dunbar W. B. (2013) Error analysis of idealized nanopore sequencing. Electrophoresis 34, 2137–2144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fernandez S., Cisney E. D., Tikhonov A. P., Schweitzer B., Putnak R. J., Simmons M., and Ulrich R. G. (2011) Antibody recognition of the dengue virus proteome and implications for development of vaccines. Clin. Vaccine Immunol. 18, 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chutrakul C., Jeennor S., Panchanawaporn S., Cheawchanlertfa P., Suttiwattanakul S., Veerana M., and Laoteng K. (2016) Metabolic engineering of long chain-polyunsaturated fatty acid biosynthetic pathway in oleaginous fungus for dihomo-gamma linolenic acid production. J. Biotechnol. 218, 85–93 [DOI] [PubMed] [Google Scholar]
- 20. Yang L., Wang J., Li J., Zhang H., Guo S., Yan M., Zhu Z., Lan B., Ding Y., Xu M., Li W., Gu X., Qi C., Zhu H., Shao Z., Liu B., and Tao S. C. (2016) Identification of serum biomarkers for gastric cancer diagnosis using a human proteome microarray. Mol. Cell. Proteomics 15, 614–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Deng W., Wang Y., Liu Z., Cheng H., and Xue Y. (2014) HemI: A toolkit for illustrating heatmaps. PloS One 9, e111988, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kauffmann A., and Huber W. (2010) Microarray data quality control improves the detection of differentially expressed genes. Genomics 95, 138–142 [DOI] [PubMed] [Google Scholar]
- 23. Hintze J. L., and Nelson R. D. (1998) Violin plots: A box plot-density trace synergism. Am. Stat. 52, 181–184 [Google Scholar]
- 24. Costa R. L., Voloch C. M., and Schrago C. G. (2012) Comparative evolutionary epidemiology of dengue virus serotypes. Infect. Genet. Evol. 12, 309–314 [DOI] [PubMed] [Google Scholar]
- 25. Watt G., Jongsakul K., Chouriyagune C., and Paris R. (2003) Differentiating dengue virus infection from scrub typhus in Thai adults with fever. Am. J. Trop. Med. Hyg. 68, 536–538 [DOI] [PubMed] [Google Scholar]
- 26. Li J., Gao N., Fan D., Chen H., Sheng Z., Fu S., Liang G., and An J. (2016) Cross-protection induced by Japanese encephalitis vaccines against different genotypes of dengue viruses in mice. Sci. Rep. 6, 19953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peeling R. W., Artsob H., Luis Pelegrino J., Buchy P., Cardoso M. J., Devi S., Enria D. A., Jeremy F., Gubler D. J., Guzman M. C., Halstead S. B., Hunsperger E., Kliks S., Margolis H. S., Nathanson C. M., Vinh Chau N., Rizzo N., Vázquez S., and Yoksan S. (2010) Evaluation of diagnostic tests: Dengue. Nat. Rev. Microbiol. 8, S30–S38 [DOI] [PubMed] [Google Scholar]
- 28. Guzman M. G., Halstead S. B., Artsob H., Buchy P., Farrar J., Gubler D. J., Hunsperger E., Kroeger A., Margolis H. S., Martínez E., Nathan M. B., Pelegrino J. L., Simmons C., Yoksan S., and Peeling R. W. (2010) Dengue: A continuing global threat. Nat. Rev. Microbiol. 8, S7–S16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bernardo L., Izquierdo A., Prado I., Rosario D., Alvarez M., Santana E., Castro J., Martínez R., Rodriguez R., Morier L., Guillén G., and Guzman M. G. (2008) Primary and secondary infections of Macaca fascicularis monkeys with Asian and American genotypes of dengue virus 2. Clin. Vaccine Immunol. 15, 439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quick J., Loman N. J., Duraffour S., Simpson J. T., Severi E., Cowley L., Bore J. A., Koundouno R., Dudas G., Mikhail A., Ouédraogo N., Afrough B., Bah A., Baum J. H., Becker-Ziaja B., Boettcher J. P., Cabeza-Cabrerizo M., Camino-Sánchez Á., Carter L. L., Doerrbecker J., Enkirch T., García-Dorival I., Hetzelt N., Hinzmann J., Holm T., Kafetzopoulou L. E., Koropogui M., Kosgey A., Kuisma E., Logue C. H., Mazzarelli A., Meisel S., Mertens M., Michel J., Ngabo D., Nitzsche K., Pallasch E., Patrono L. V., Portmann J., Repits J. G., Rickett N. Y., Sachse A., Singethan K., Vitoriano I., Yemanaberhan R. L., Zekeng E. G., Racine T., Bello A., Sall A. A., Faye O., Faye O., Magassouba N., Williams C. V., Amburgey V., Winona L., Davis E., Gerlach J., Washington F., Monteil V., Jourdain M., Bererd M., Camara A., Somlare H., Camara A., Gerard M., Bado G., Baillet B., Delaune D., Nebie K. Y., Diarra A., Savane Y., Pallawo R. B., Gutierrez G. J., Milhano N., Roger I., Williams C. J., Yattara F., Lewandowski K., Taylor J., Rachwal P., Turner D. J., Pollakis G., Hiscox J. A., Matthews D. A., O'Shea M. K., Johnston A. M., Wilson D., Hutley E., Smit E., Di Caro A., Wölfel R., Stoecker K., Fleischmann E., Gabriel M., Weller S. A., Koivogui L., Diallo B., Keïta S., Rambaut A., Formenty P., Günther S., and Carroll M. W. (2016) Real-time, portable genome sequencing for Ebola surveillance. Nature 530, 228–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Enfissi A., Codrington J., Roosblad J., Kazanji M., and Rousset D. (2016) Zika virus genome from the Americas. Lancet 387, 227–228 [DOI] [PubMed] [Google Scholar]
- 32. Lin Y. P., Luo Y., Chen Y., Lamers M. M., Zhou Q., Yang X. H., Sanyal S., Mok C. K., and Liu Z. M. (2016) Clinical and epidemiological features of the 2014 large-scale dengue outbreak in Guangzhou city, China. BMC Infect. Dis. 16, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.