

Abstract
Background:
Small-cell lung cancer (SCLC) represents one of the most aggressive forms of lung cancer. Despite the fair sensitivity of SCLC to chemotherapy and radiotherapy, the current standard treatment regimens have modest survival rates and are associated with potential life-threatening adverse events. Therefore, research into new optimised regimens that increase drug efficacy while respecting toxicity constraints is of primary importance.
Methods:
A PK/PD model for the combination of cisplatin and etoposide to treat extensive-stage SCLC patients was generated. The model takes into consideration both the efficacy of the drugs and their haematological toxicity. Using optimisation techniques, the model can be used to propose new regimens.
Results:
Three new regimens with varying timing for combining cisplatin and etoposide have been generated that respect haematological toxicity constraints and achieve better or similar tumour regression. The proposed regimens are: (1) Protocol OP1: etoposide 80 mg m−2 over 1 h D1, followed by a long infusion 12 h later (over 3 days) of 160 mg m−2 plus cisplatin 80 mg m−2 over 1 h D1, D1–D1 21 days; (2) Protocol OP2: etoposide 80 mg m−2 over 1 h D1, followed by a long infusion 12 h later (over 4 days) of 300 mg m−2 plus cisplatin 100 mg m−2 over 1 h D1, D1–D1 21 days; and (3) Protocol OP3: etoposide 40 mg m−2 over 1 h, followed by a long infusion 6 h later (3 days) of 105 mg m−2 plus cisplatin 50 mg m−2 over 1 h, D1–D1 14 days.
Conclusions:
Mathematical modelling can help optimise the design of new cisplatin plus etoposide regimens for managing extensive-stage SCLC patients.
Keywords: small cell lung cancer, cisplatin, etoposide, mathematical modelling, chemotherapy
Lung cancer is the leading cause of cancer death worldwide. Among the different lung cancer subtypes, small-cell lung cancer (SCLC) accounts for 15 to 20% of cases. The SCLC is characterised by an important metastatic-spread ability and a 60 to 80% response rate to chemotherapy±radiotherapy. Despite these high response rates, the median overall survival (OS) remains short (∼10 months) that is mainly because of distant relapses. To date, the most commonly used chemotherapy regimen is based on the combination of cisplatin (75 mg m−2 on day 1) or carboplatin (with a dose calculated using Calvert or Chatelut formulas) plus etoposide (120 mg m−2 on days 1 to 3) for 4 to 6 cycles (Evans et al, 1984; Porter et al, 1985; Calvert et al, 1989; Chatelut et al, 1995). Other regimens are less commonly used (Bunn et al, 1986; Fukuoka et al, 1991; Roth et al, 1992; Trillet-Lenoir et al, 1993; Sculier et al, 1998). The safety profile of these combinations is characterised by haematological (neutropenia and thrombocytopenia) and extra-haematological (nausea and vomiting, renal, peripheral neuropathy and alopecia) adverse events. Therefore, further research progress is needed.
Several studies have shown that changes in the doses and schedules of widely used regimens might lead to improved efficacy and tolerability (Gurney et al, 1991; van Warmerdam et al, 1997; Freyer et al, 2001; Moore et al, 2006). Could this be the case for SCLC regimens? As an example, the importance of scheduling etoposide administration was shown in a study by Slevin et al (1989). The authors compared two distinct protocols (etoposide 500 mg m−2 over 24 h at D1 vs 100 mg m−2 over 2 h from D1 to D5) for chemo-naive patients with stage IV SCLC. Surprisingly, the ORR increased from 10% to 90% for the two modalities. Although all patients were given the same total dose, and the average area under the curve (AUC) was comparable for all patients, the times during which the blood concentration of etoposide stayed above 1 mg l−1 were 46 and 94 h, respectively. Concomitantly, several studies suggested that intensive regimens could have better response and survival rates with an increase in the toxicity, making these regimens difficult to use in practice (Cohen et al, 1977; Tourani et al, 1993, 2000). Is there room for improvement of our currently used cisplatin plus etoposide regimen? Because of the multitude of possibilities, empirical approaches are impractical and impossible to test. As a result, mathematical modelling is needed.
There are several approaches to model tumour regression while keeping drug toxicity effects below an acceptable level. In most of these approaches, however, it is difficult to efficiently control the toxicity constraints. Recently, in a convincing study, Meille and colleagues (Hénin et al, 2016; Meille et al, 2016) developed a mathematical model to optimise drug dosing regimens and redesign the dose intensification–dose escalation process with intensified cycles of combined anticancer drugs. As for the work by Meille and colleagues (Hénin et al, 2016; Meille et al, 2016), we propose a PK/PD mathematical model in this study for approaching the combination of cisplatin and etoposide to treat extensive-stage SCLC patients. The aim was to determine optimised temporal protocols that can minimise the tumour mass while avoiding harmful toxic side effects, namely severe neutropenia and thrombocytopenia.
Materials and methods
Mathematical model
The model assessed the combination of cisplatin and etoposide by evaluating both haematological toxicities (neutropenia and thrombocytopenia) and changes in the tumour volume. The model is mainly divided into three components (Meille et al, 2016). One is a PK component that describes the pharmacokinetics of cisplatin and etoposide (see Supplemental Material, section PK). This component consists of a two-compartment model for etoposide (Tranchand et al, 1999) and a three-compartment model for cisplatin (Monjanel-Mouterde et al, 2003). A PD-safety component consists of two systems of differential equations describing the changes in neutrophil and platelet counts and a PD-efficacy component describing the changes in the tumour volume (see Supplemental Material, sections Hematoxicity model and Modelling tumour growth). A fundamental problem in PK/PD modelling is the PK/PD link. To circumvent this difficulty, we used an adequate exposure model (labelled the ‘interface model' by the authors who proposed this concept) that can be considered a generalisation of both the AUC and the effect compartment model (Meille et al, 2008; see Supplementary Material sections Interface model and Interfaces).
To assess the validity of our model, simulation results were compared with the results of published clinical studies from the literature that assessed the cisplatin and etoposide combination for extensive SCLC patient management (Slevin et al, 1989; Ihde et al, 1994).
Toxicity constraints
The model was subject to three constraints in the risk of haematological adverse events. First, the model requires that the neutrophil and platelet counts permanently stay above an absolute threshold level (that is, w(t) ⩾wabs at any time t, with wabs=0.2 g l−1 and p(t) ⩾pabs at any time t and pabs=20 g l−1). Second, the model requires that the times (twrel and tprel) for which neutrophil and platelet counts are between defined threshold levels (wrel and prel) and the absolute threshold wabs, should not exceed predefined lengths of time (with wrel=1 g l−1, prel=50 g l−1, twrel=3 days and tprel=3 days). Third, the model requires haematological recovery at the time of the subsequent treatment cycle, with neutrophil and platelet counts that are above predefined threshold values (wrec=2 g l−1 and prec=150 g l−1). Erythropenia is also an important aspect. However, it rarely leads to treatment delay or interruption as it could be managed red blood transfusions and/or EPO-stimulating agents. Therefore, it would be preferable to not overload the model with additional equations and parameters by incorporating another toxicity constraint.
Optimisation of the cisplatin plus etoposide regimen
The optimisation procedure can be considered as finding a temporal distribution for the drug doses that achieve the best efficacy while respecting toxicity constraints for a particular patient (using the model). The problem can be described as finding a schedule 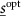 and distribution of doses
and distribution of doses 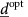 , such that the average value of the tumour size over an entire cycle
, such that the average value of the tumour size over an entire cycle 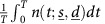 is minimal and the aforementioned toxicity constraints not violated. Our choice of minimising the integral of n rather than its value at the end of the cycle is based on the Goldie–Coldman hypothesis (Coldman and Goldie, 1983). This hypothesis suggests that the probability that a cancer would contain drug-resistant clones depends on the mutation rate and the size of the tumour. Therefore, we aimed at minimising the tumour size as much as possible during a cycle to avoid acquired resistance. This implies that we targeted the minimal size that can be reached in order to decrease the chances of developing resistant clones.
is minimal and the aforementioned toxicity constraints not violated. Our choice of minimising the integral of n rather than its value at the end of the cycle is based on the Goldie–Coldman hypothesis (Coldman and Goldie, 1983). This hypothesis suggests that the probability that a cancer would contain drug-resistant clones depends on the mutation rate and the size of the tumour. Therefore, we aimed at minimising the tumour size as much as possible during a cycle to avoid acquired resistance. This implies that we targeted the minimal size that can be reached in order to decrease the chances of developing resistant clones.
Results
Model validation
The model includes a large number of parameters that are usually individual dependent and thus vary from one patient to another, especially the pharmacokinetic parameters of cisplatin and etoposide. Because not all necessary clinical data from stage IV SCLC patients were available, a standard pharmacokinetic population fit approach could not be performed. To circumvent this issue, parameters were adjusted such that our simulations fit with the clinical results in the literature. Therefore, we searched for a set of parameters following a log-normal distribution that achieved, for a standard treatment, tumour regression and haematologic profiles that were similar to the results reported in the literature. As an example, we first tested the validity of the model using the results from Slevin et al (1989). We assessed the outcomes for 1000 ‘virtual' patients (each patient has a distinct set of parameters drawn from a log-normal distribution with the same mean and s.d. values for all patients) over 6 cycles to mimic a clinical study. The results reported in Table 1 showed a good correlation between the model and the reported clinical data. Then, we tested the model validity using the results from Ihde et al (1994). Using the model, grade 4 neutropenia over 5 days (Figure 1) was observed that is in line with the study results. Therefore, the model confirmed severe haematological toxicity using this regimen. Furthermore, the comparison of the response rate outcomes between our model and the results reported by Ihde et al (1994) were in agreement (Table 2). Additional results given in Supplementary Tables 4–7 showed a good correlation between the model and the reported clinical data for both the etoposide/cisplatin (EP) and intensified EP regimens.
Table 1. Comparison of the overall response rate between the model and clinical results after 6 cycles of protocols proposed by Slevin et al (1989).
| Protocol | Slevin et al (1989) | Model | 95% CI |
|---|---|---|---|
| 500 mg m−2 D1 | 10% | 8% | (11–36%) |
| 100 mg m−2 D1–D5 | 90% | 91% | (74–95%) |
Abbreviations: CI=confidence interval; D, day.
Figure 1.

Absolute neutrophil count (ANC) profile using the high dose protocol by Ihde et al (1994).
Table 2. Comparison of the response rate between the model and clinical results after 4 cycles of the EP protocol (Ihde et al, 1994).
| Response | Ihde et al (1994) | Model | 95% CI |
|---|---|---|---|
| Complete | 22% | 25% | (11–36%) |
| Partial+complete | 83% | 85% | (74–95%) |
Abbreviations: CI=confidence interval; EP=etoposide/cisplatin.
Improved standard regimen: OP1 regimen (focussing on response)
This optimised regimen should have the same characteristics of the EP protocol, that is: (1) a maximal total dose for etoposide equal to 240 mg m−2 and (2) a maximal total dose for cisplatin equal to 80 mg m−2 per cycle. The model suggested a new infusion schedule for etoposide, with a short infusion of 80 mg m−2 followed by a long infusion of 160 mg m−2 12 h later over 3 days. Cisplatin (80 mg m−2) was infused for 1 h after the first etoposide infusion (Table 3). The model suggests that the timing of cisplatin infusion does not affect the optimised regimen and that cisplatin can be administered at any time during day 1. Compared with the standard EP protocol, the OP1 regimen yielded better tumour regression, with a response rate of 50% instead of 30% after one treatment cycle (Figure 2). Simulations for 1000 patients led to a response rate of 98% for the OP1 vs 85% for the EP after 4 treatment cycles. Moreover, the OP1 regimen was less toxic than the standard EP regimen as assessed by the ANC counts (data not shown).
Table 3. Summary of the optimised protocols proposed by the model.
|
Etoposide |
Cisplatin |
|||
|---|---|---|---|---|
| Protocol | Time | Dose | Time | Dose |
| OP1 | 0–1 h | 80 mg m−2 | 1–2 h | 80 mg m−2 |
| 12–84 h | 160 mg m−2 | |||
| OP2 (intensified) | 0–1 h | 80 mg m−2 | 1–2 h | 80 mg m−2 |
| 12–108 h | 300 mg m−2 | |||
| OP3 (14-day cycle) | 0–1 h | 40 mg m−2 | 1–2 h | 50 mg m−2 |
| 6–78 h | 105 mg m−2 | |||
Figure 2.

Comparison of tumour growth. Dashed line indicates standard protocol and solid line indicates optimised protocol OP1.
Improved high dose regimen: OP2 regimen
The model demonstrated that the intensified EP protocol (etoposide 80 mg m−2 J1–J5 and cisplatin 27 mg m−2 J1–J5) did not fit with the predefined safety constraints. Indeed, the model showed that (for 2 cycles) the intensified EP protocol produced grade 3 neutropenia during 5 days that broke the predefined limit of 3 days for this toxicity. The model proposed an optimised regimen called the OP2 regimen (etoposide 80 mg m−2 over 1 h on day 1 followed 1 h later by cisplatin 100 mg m−2 over 1 h and then followed 12 h later by a prolonged infusion of 4 days of etoposide at a dose of 300 mg m−2, Table 3). The OP2 regimen yielded a better tumour response than the high EP regimen (Figure 3) while respecting the haematological predefined constraints (grade 3 neutropenia for <3 days).
Figure 3.

Comparison of tumour growth. Dashed line indicates high EP protocol and solid line indicates optimised protocol OP2.
Intensified EP regimen: OP3 regimen
The model was set up to explore the possibility of an intensified EP regimen by reducing the cycle length (2 weeks instead of 3 weeks). The model assessed a regimen consisting of 3 cycles of 14 days compared with 2 cycles of the standard EP protocol. Using our model, optimisation led to a regimen called the OP3 regimen (Table 3). After 2 treatment cycles, the OP3 regimen led to a response rate of 80% compared with 60% for the standard EP protocol without violating the toxicity constraints (Figure 4).
Figure 4.

Comparison of tumour growth. Dashed line indicates standard protocol and solid line indicates intensified protocol OP3.
Discussion
The SCLC patients still present with several unmet needs that are partially linked to the limited long-term efficacy and adverse event rate of the currently available standard cisplatin plus etoposide regimen. Intensifying strategies that increase the standard dose (Cohen et al, 1977; Tourani et al, 2000) or administer additional drugs (Pujol et al, 2001) yield slightly better OS, but they are more toxic. Modified schedules are more efficacious in some cases and are much worse in others (Slevin et al, 1989) without a rational understanding of the reason for this enormous difference. Improving the toxicity to efficacy balance of the etoposide plus cisplatin regimen is not empirically simple, as demonstrated by the relative failure of all newly proposed regimens.
The results reported here propose three newly optimised regimens based on mathematical modelling of the cisplatin plus etoposide combination. The mathematical modelling takes advantage of the many combinations that could be simultaneously tested in silico to propose regimens that provide the highest response rates while respecting the haematologic toxic constraints. The OP1 regimen involved adjustment of the temporal schedule of the standard regimen. The OP2 and OP3 regimens aimed to intensify the standard regimen by increasing the dose or reducing the cycle length. The model enabled the proposal of an optimised solution for all these situations with regimens that improve the expected response rate while respecting the constraints of the risk of haematological toxicity.
Two recent studies showed the benefits of using mathematical modelling to optimise dosing regimens. The first dealt with optimising metronomic oral vinorelbine in metastatic NSCLC and malignant pleural mesothelioma (Barbolosi et al, 2014; Bocci and Kerbel, 2016; Elharrar et al, 2016). A mathematical PK/PD model suggested an alternative weekly D1, D2 and D4 schedule with respective doses of 60, 30 and 60 mg that could lead to better safety and efficacy profiles. Currently, 12 patients are enrolled in a phase 1a study under the proposed optimised regimen, and the analysis has shown promising results. The second study focussed on redesigning the dose escalation process using densified cycles of combined docetaxel plus epiribucin anticancer therapy in metastatic breast cancer patients (Hénin et al, 2016; Meille et al, 2016). Using optimisation techniques, a mathematical model was developed to compute the total drug distribution for each escalation dose level and each drug in the combination. This enabled minimisation of the average tumour mass for each cycle while respecting predefined toxicity constraints.
One of the main drawbacks of the approach proposed in this study is the lack of clinical data that can weaken the model reliability. To circumvent this difficulty, we assessed the model validity by comparing its results with clinical study results taken from the literature. Our model results correlated well with the literature and suggest that it can be consolidated and used in clinical studies. Another issue with the modelling approach is its only indirect prediction of the OS. Indeed, the model cannot take into account the OS rate as a formal output of the optimisation procedure. This is inherent to the modelling structure as the input variables consist of pharmacokinetic and pharmacodynamic parameters. Therefore, the model can only address the relationship of these input parameters to output variables, such as haematological profiles and tumour volume regression, according to the RECIST. However, minimising toxicities while improving response rates might have the potential to produce better OS rates than standard protocols. This was proven in previous studies (Hénin et al, 2016; Meille et al, 2016) where the good management of toxicity constraints was associated to a doubling of the median overall survival. Nonetheless, this remains to be prospectively tested in a properly designed randomised study
The ultimate goal of this work is to establish a prospective phase I study in which patients are enrolled and treated according to the optimised regimens proposed in this study. During the first cycle, data samples measuring neutrophil and platelet counts, as well as drug concentrations, are collected for each patient and integrated into the model to recalibrate the parameters. Having data samples for each patient would allow for the use of Bayesian analysis to individualise treatments and propose patient-specific optimised regimens.
Footnotes
Supplementary Information accompanies this paper on British Journal of Cancer website (http://www.nature.com/bjc)
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
The authors declare no conflict of interest.
Supplementary Material
References
- Barbolosi D, Ciccolini J, Meille C, Elharrar X, Faivre C, Lacarelle B, André N, Barlesi F (2014) Metronomics chemotherapy: time for computational decision support. Cancer Chemother Pharmacol 74: 647–652. [DOI] [PubMed] [Google Scholar]
- Bocci G, Kerbel RS (2016) Pharmacokinetics of metronomic chemotherapy: a neglected but crucial aspect. Nat Rev Clin Oncol 13(11): 659–673. [DOI] [PubMed] [Google Scholar]
- Bunn PA, Greco FA, Einhorn L (1986) Cyclophosphamide, doxorubicin, and etoposide as first-line therapy in the treatment of small-cell lung cancer. Semin Oncol 13: 45–53. [PubMed] [Google Scholar]
- Calvert AH, Newell DR, Gumbrell LA, O'Reilly S, Burnell M, Boxall FE, Siddik ZH, Judson IR, Gore ME, Wiltshaw E (1989) Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol 7: 1748–1756. [DOI] [PubMed] [Google Scholar]
- Chatelut E, Canal P, Brunner V, Chevreau C, Pujol A, Boneu A, Roché H, Houin G, Bugat R (1995) Prediction of carboplatin clearance from standard morphological and biological patient characteristics. J Natl Cancer Inst 87: 573–580. [DOI] [PubMed] [Google Scholar]
- Cohen MH, Creaven PJ, Fossieck BE, Broder LE, Selawry OS, Johnston AV, Williams CL, Minna JD (1977) Intensive chemotherapy of small cell bronchogenic carcinoma. Cancer Treat Rep 61: 349–354. [PubMed] [Google Scholar]
- Coldman AJ, Goldie JH (1983) A model for the resistance of tumor cells to cancer chemotherapeutic agents. Math Biosci 65: 291–307. [Google Scholar]
- Elharrar X, Barbolosi D, Ciccolini J, Meille C, Faivre C, Lacarelle B, André N, Barlesi F (2016) A phase Ia/Ib clinical trial of metronomic chemotherapy based on a mathematical model of oral vinorelbine in metastatic non-small cell lung cancer and malignant pleural mesothelioma: rationale and study protocol. BMC Cancer 16: 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans WK, Feld R, Osoba D, Shepherd FA, Dill J, Deboer G (1984) VP-16 alone and in combination with cisplatin in previously treated patients with small cell lung cancer. Cancer 53: 1461–1466. [DOI] [PubMed] [Google Scholar]
- Freyer G, Ligneau B, Tranchand B, Ardiet C, Souquet PJ, Court-Fortune I, Riou R, Rebattu P, Morignat E, Boissel JP, Trillet-Lenoir V, Girard P (2001) The prognostic value of etoposide area under the curve (AUC) at first chemotherapy cycle in small cell lung cancer patients: a multicenter study of the groupe Lyon-Saint-Etienne d'Oncologie Thoracique (GLOT). Lung Cancer 31: 247–256. [DOI] [PubMed] [Google Scholar]
- Fukuoka M, Furuse K, Saijo N, Nishiwaki Y, Ikegami H, Tamura T, Shimoyama M, Suemasu K (1991) Randomized trial of cyclophosphamide, doxorubicin, and vincristine versus cisplatin and etoposide versus alternation of these regimens in small-cell lung cancer. J Natl Cancer Inst 83: 855–861. [DOI] [PubMed] [Google Scholar]
- Gurney H, de Campos ES, Dodwell D, Kamthan A, Thatcher N (1991) Ifosfamide and mitomycin in combination for the treatment of patients with progressive advanced non-small cell lung cancer. Eur J Cancer 27: 565–568. [DOI] [PubMed] [Google Scholar]
- Hénin E, Meille C, Barbolosi D, You B, Guitton J, Iliadis A, Freyer G (2016) Revisiting dosing regimen using PK/PD modeling: the MODEL1 phase I/II trial of docetaxel plus epirubicin in metastatic breast cancer patients. Breast Cancer Res Treat 156: 331–341. [DOI] [PubMed] [Google Scholar]
- Ihde DC, Mulshine JL, Kramer BS, Steinberg SM, Linnoila RI, Gazdar AF, Edison M, Phelps RM, Lesar M, Phares JC (1994) Prospective randomized comparison of high-dose and standard-dose etoposide and cisplatin chemotherapy in patients with extensive-stage small-cell lung cancer. J Clin Oncol 12: 2022–2034. [DOI] [PubMed] [Google Scholar]
- Meille C, Barbolosi D, Ciccolini J, Freyer G, Iliadis A (2016) Revisiting dosing regimen using pharmacokinetic/pharmacodynamic mathematical modeling: densification and intensification of combination cancer therapy. Clin Pharmacokinet 55(8): 1015–1025. [DOI] [PubMed] [Google Scholar]
- Meille C, Iliadis A, Barbolosi D, Frances N, Freyer G (2008) An interface model for dosage adjustment connects hematotoxicity to pharmacokinetics. J Pharmacokinet Pharmacodyn 35: 619–633. [DOI] [PubMed] [Google Scholar]
- Monjanel-Mouterde S, Ciccolini J, Bagarry D, Zonta-David M, Duffaud F, Favre R, Durand A (2003) Population pharmacokinetics of cisplatin after 120-h infusion: application to routine adaptive control with feedback. J Clin Pharm Ther 28: 109–116. [DOI] [PubMed] [Google Scholar]
- Moore AM, Einhorn LH, Estes D, Govindan R, Axelson J, Vinson J, Breen TE, Yu M, Hanna NH (2006) Gefitinib in patients with chemo-sensitive and chemo-refractory relapsed small cell cancers: a Hoosier Oncology Group phase II trial. Lung Cancer 52: 93–97. [DOI] [PubMed] [Google Scholar]
- Porter LL, Johnson DH, Hainsworth JD, Hande KR, Greco FA (1985) Cisplatin and etoposide combination chemotherapy for refractory small cell carcinoma of the lung. Cancer Treat Rep 69: 479–481. [PubMed] [Google Scholar]
- Pujol JL, Daurès JP, Rivière A, Quoix E, Westeel V, Quantin X, Breton JL, Lemarié E, Poudenx M, Milleron B, Moro D, Debieuvre D, Le Chevalier T (2001) Etoposide plus cisplatin with or without the combination of 4'-epidoxorubicin plus cyclophosphamide in treatment of extensive small-cell lung cancer: a French Federation of Cancer Institutes multicenter phase III randomized study. J Natl Cancer Inst 93: 300–308. [DOI] [PubMed] [Google Scholar]
- Roth BJ, Johnson DH, Einhorn LH, Schacter LP, Cherng NC, Cohen HJ, Crawford J, Randolph JA, Goodlow JL, Broun GO (1992) Randomized study of cyclophosphamide, doxorubicin, and vincristine versus etoposide and cisplatin versus alternation of these two regimens in extensive small-cell lung cancer: a phase III trial of the Southeastern Cancer Study Group. J Clin Oncol 10: 282–291. [DOI] [PubMed] [Google Scholar]
- Sculier JP, Berghmans T, Castaigne C, Luce S, Sotiriou C, Vermylen P, Paesmans M (1998) Maintenance chemotherapy for small cell lung cancer: a critical review of the literature. Lung Cancer 19: 141–151. [DOI] [PubMed] [Google Scholar]
- Slevin ML, Clark PI, Joel SP, Malik S, Osborne RJ, Gregory WM, Lowe DG, Reznek RH, Wrigley PF (1989) A randomized trial to evaluate the effect of schedule on the activity of etoposide in small-cell lung cancer. J Clin Oncol 7: 1333–1340. [DOI] [PubMed] [Google Scholar]
- Tourani J, Levy R, Coscas Y, Even P, Andrieu J (1993) Limited small-cell lung-carcinoma - high complete response rate and survival with short intensive chemotherapy and extensive irradiation - results of a pilot-study. Int J Oncol 3: 347–353. [DOI] [PubMed] [Google Scholar]
- Tourani JM, Jaillon-Abraham C, Coscas Y, Dabouis G, Andrieu JM (2000) Feasibility and preliminary results of intensive chemotherapy and extensive irradiation in selected patients with limited small-cell lung carcinoma—results of three consecutive phase II programs. Acta Oncol 39: 501–508. [DOI] [PubMed] [Google Scholar]
- Tranchand B, Amsellem C, Chatelut E, Freyer G, Iliadis A, Ligneau B, Trillet-Lenoir V, Canal P, Lochon I, Ardiet CJ (1999) A limited-sampling strategy for estimation of etoposide pharmacokinetics in cancer patients. Cancer Chemother Pharmacol 43: 316–322. [DOI] [PubMed] [Google Scholar]
- Trillet-Lenoir V, Mornex F, Chauvin F, Fournel P, Voloch A, Perol M, Laennec E, Piperno D, Boyer J, Ardiet JM (1993) Limited disease small cell lung cancer: alternating combination of doxorubicin, etoposide, ifosfamide and hyperfractionated radiotherapy. Final results of a multicentric pilot study for the Groupe Lyonnias d'Oncologie Thoracique (GLOT). Lung Cancer Amst Neth 10: 35–45. [DOI] [PubMed] [Google Scholar]
- van Warmerdam LJ, Huizing MT, Giaccone G, Postmus PE, ten Bokkel Huinink WW, van Zandwijk N, Koolen MG, Helmerhorst TJ, van der Vijgh WJ, Veenhof CH, Beijnen JH (1997) Clinical pharmacology of carboplatin administered in combination with paclitaxel. Semin Oncol 24(1 Suppl 2): S2–97-S2-104. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.