

Abstract
An organocatalytic method that achieves insertion of isatins into aryl difluoronitromethyl ketones under mild conditions is described. The reaction occurs in the presence of 20 mol% of DBU and with 100% atom economy. A series of isatin derived difluoronitromethyl substituted tertiary alcohol benzoates and naphthoates was prepared in 81–99% yield. The general synthetic usefulness of these 3-hydroxyoxindole derivatives is demonstrated with the selective reduction to fluorooximes.
Graphical abstract
Nitroalkanes are commonly used in Henry and Michael reactions,1 allylations,2 arylations,3 alkylations4 and for the construction of amide bonds.5 As a result of their general availability and high synthetic versatility, nitromethane and its homologues have become privileged building blocks for the formation of multifunctional target compounds and natural products. A remaining drawback of the nitroaldol reaction is that excessive amounts of the nitroalkane - the use of 5–10 equivalents is common - are typically required to achieve high yields.6 Although this might be still considered cost-effective when inexpensive nitromethane is used, it remains inefficient and produces considerable waste.
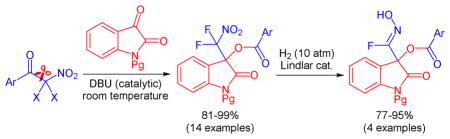
Following our longstanding interest in aldol reactions with nitroalkanes7 and fluorinated enolates,8 we recently introduced dihalonitronates to C-C bond formation with aldehydes using a LiBr/(i-Pr)2NEt pair as catalyst.9 Because of the high impact of fluorinated pharmaceuticals and agrochemicals the synthesis of new organofluorines and the development of methods to make them has received widespread attention.10 The steadily increasing structural diversity of fluorine-containing marketed pharmaceuticals and emerging drug candidates continues to provide a strong need for practical synthetic approaches. We now report an organocatalytic method that accomplishes the insertion of isatins into aryl difluoronitromethyl ketones under mild conditions based on a C-C bond cleavage-aldol reaction-acyl transfer sequence. The DBU catalyzed transformation occurs with stoichiometric amounts of the starting materials and it produces a 3-hydroxyoxindole scaffold, an important structural motif that occurs in a variety of biologically active compounds,11 in high yields and with 100% atom economy (Scheme 1). Moreover, we investigated the possibility to reduce the difluoronitromethyl motif and report for the first time the formation of fluorooximes.
Scheme 1.

Features of the DBU catalyzed C-C scission/nitronate addition/benzoyl transfer sequence.
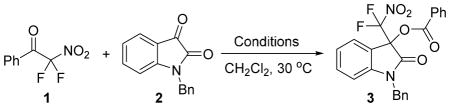
We began our search for a mild method that achieves the insertion of an isatin scaffold into an aryl difluoronitromethyl ketone through a bond scission-aldol reaction-acyl transfer sequence by screening a variety of nucleophiles and additives (Table 1). Using difluoronitroacetophenone, 1, and N-benzyl isatin, 2, as test compounds in stoichiometric amounts we were surprised that no reaction occurred in the presence of catalytic amounts of LiBr and (i-Pr)2NEt which is in stark contrast to our experience with aldehydes (entry 1). The propensity of fluorinated nucleophiles toward decomposition and uncontrolled carbene formation, in particular in the presence of a Lewis acid, has been identified as a general problem that often impedes fluoroalkylation chemistry.12 We therefore decided to probe metal-free conditions and found that the desired product 3 can be obtained when readily available N-nucleophiles were employed (a crystal structure of compound 3 is provided in the SI). The addition of one equivalent of (i-Pr)2NEt produced 3 in 63% yield after 48 hours (entries 2–4). To our delight, 3 was formed quantitatively in the presence of Et3N albeit a catalytic variant still gave unsatisfactory results (entries 5–7). At this point, we rationalized that the more nucleophilic 1,8-diazabicycloundec-7-ene (DBU) would facilitate the bond scission step with 1 and thus provide an entry to an effective catalytic process.13 Indeed, we found that the presence of 20 mol% of DBU yields quantitative formation of 3 within 24 hours in both dichloromethane and chloroform. Further reduction of the DBU loading, however, compromises the yield (entries 8–10). It is noteworthy that this protocol does not only eliminate the need for a Lewis acid but also the use of difluoronitromethane, which is volatile and difficult to handle, as the nitronate precursor.14
Table 1.
Optimization of the isatin insertion reaction.
| |||
|---|---|---|---|
| Entry | Additive (mol %) | t (h) | Yielda (%) |
| 1 | DIPEA (20)/LiBr (20) | 48 | 0 |
| 2 | 2,6-lutidine (40) | 48 | 0 |
| 3 | DMAP (50) | 48 | 25 |
| 4 | DIPEA (100) | 48 | 63 |
| 5 | Et3N (200) | 18 | 99 |
| 6 | Et3N (50) | 24 | 99 |
| 7 | Et3N (20) | 48 | 21 |
| 8 | DBU (20) | 24 | 99 |
| 9 | DBU (10) | 24 | 82 |
| 10b | DBU (20) | 24 | 99 |
Reaction conditions: A solution of 1 (0.18 mmol) and 2 (0.15 mmol) in the presence of the additive in 0.4 mL of CH2Cl2 was stirred at 30 °C.
Determined by 1H NMR analysis.
CHCl3 was used as solvent. DIPEA = N,N-diisopropylethylamine, DMAP = 4-dimethylaminopyridine, DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
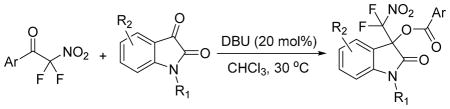
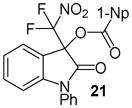
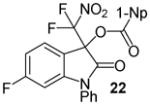
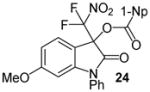
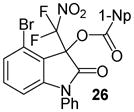
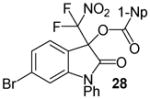
Having optimized the reaction conditions we continued with the evaluation of the substrate scope (Table 2). The DBU catalyzed insertion of isatins carrying various N-protecting groups into 1 produced 3, 5, and 7 in almost quantitative amounts (entries 1–3)15. We then examined N-phenylisatins with different aryl substituents R2. The reaction of 1 with 5-methyl-and 5-methoxyisatin, 8 and 12, or with the halogenated analogues 10 and 14 gave the cleavage-aldol reaction-acyl transfer products with more than 90% yield (entries 4–7). Employing N-phenyl 5-trifluoromethoxyisatin, 16, and the 5,6-difluoro derivative 18 in the reaction afforded 17 and 19 in 85% and 81% yield, respectively (entries 8 and 9). Finally, we introduced α-difluoronitroacetonaphthone, 20, as an alternative difluoronitronate precursor to the reaction sequence. The catalytic insertion of five different N-phenylisatins into 20 furnished 21, 22, 24, 26 and 28 in 91–99% yield (entries 10–14).
Table 2.
Scope of the organocatalytic isatin insertion into aryl difluoronitromethyl ketones
| |||||
|---|---|---|---|---|---|
| Entry | Ar | Isatin R1/R2 | t (h) | Insertion product | Yield (%) |
| 1 | Ph (1) | Bn/H (2) | 24 |
|
99 |
| 2 | 1 | Me/H (4) | 24 |
|
99 |
| 3 | 1 | Ph/H (6) | 24 |
|
99 |
| 4 | 1 | Ph/5-Me (8) | 24 |
|
91 |
| 5 | 1 | Ph/6-F (10) | 24 |
|
91 |
| 6 | 1 | Ph/5-OMe (12) | 48 |
|
91 |
| 7 | 1 | Ph/5-Cl (14) | 48 |
|
97 |
| 8 | 1 | Ph/5,6-F2 (16) | 48 |
|
81 |
| 9 | 1 | Ph/5-OCF3 (18) | 48 |
|
85 |
| 10 | 1-Np (20) | Ph/H (6) | 24 |
|
96 |
| 11 | 20 | Ph/6-F (10) | 24 |
|
91 |
| 12 | 20 | Ph/6-OMe (23) | 48 |
|
99 |
| 13 | 20 | Ph/4-Br (25) | 24 |
|
99 |
| 14 | 20 | Ph/6-Br (27) | 24 |
|
99 |
General conditions: A solution of 1 or 20 (0.18 mmol), the isatin (0.15 mmol) and DBU (0.03 mmol) in 0.4 mL of CHCl3 was stirred for 24–48 hours.
The reaction mechanism was investigated by 19F NMR spectroscopic analysis (SI). We observed that addition of one equivalent of DBU into a solution of difluoronitroacetophenone, 1, in CDCl3 led to the formation of the difluoronitroacetophenone-DBU adduct A showing two diastereotopic fluorine NMR signals at −88.0 and −92.1 ppm that are upfield-shifted relative to the signal of 1 at −87.3 ppm. A constant ratio between 1 and A (1.2:1) was established within 5 minutes which indicates reversibility of this first step. Trapping of A with ethanol produced ethyl benzoate and led to complete conversion of both 1 and A to a mixture of difluoronitromethane and difluoronitronate within one hour according to 19F NMR analysis which further corroborates the proposed intermediate formation and reactivity of A.16 Finally, 19F NMR monitoring of the DBU catalyzed reaction showed that after addition of an isatin the intermediate A is first converted to B and subsequently to the benzoyl ester which is the final product of the organocatalytic isatin insertion reaction. The mechanistic studies are summarized in Scheme 2.
Scheme 2.

Mechanism of the organocatalytic isatin insertion.
The incorporation of a difluoronitromethyl group into the oxindole motif is unprecedented. This prompted us to investigate the possibility of mild reduction of the nitroalkyl moiety. For this purpose we first applied a reported procedure that uses trichlorosilane to reduce the nitro group in 3.17 We found that defluorination occurred and the nitrile 29 was isolated in low yields (Scheme 3). Because direct nitrile addition to isatins can be more practically accomplished by cyanobenzoylation we turned our attention to other methods.18 We were pleased to find that catalytic hydrogenation of 3 using the Lindlar catalyst in the presence of acetic acid and di-tert-butyl dicarbonate gave the (Z)-fluorooxime 30 in 95% yield and with excellent diastereoselectivity.19 We were able to grow a single crystal of 30 and X-ray analysis proved the Z-oxime structure (see SI). To the best of our knowledge, this is the first example of the synthesis of a fluorooxime. We believe that hydrogenation of the difluoronitromethyl moiety in 3 gives the corresponding N-hydroxy difluoromethylamine derivative which is expected to spontaneously eliminate HF to form 30.20 When we applied the same hydrogenation procedure to compounds 5, 13 and 21, we obtained the corresponding N-hydroxyimidoyl fluorides 31–33 in 77–90% yield. The fluooroximes were stored at 6 °C and showed no sign of degradation after 2 months.
Scheme 3.

Reduction of the difluoronitromethylated oxindoles 3, 5, 13 and 21.
In summary, we have introduced an organocatalytic method that achieves insertion of isatins into aryl difluoronitromethyl ketones. A series of isatin derived difluoronitromethyl substituted tertiary alcohol benzoates and naphthoates was prepared in excellent yields using 20 mol% of DBU as inexpensive, readily available catalyst. The irreversible cleavage-nitroaldol reaction-acyl transfer sequence occurs with 100% atom economy and at room temperature. A key feature of this protocol is that the difluoronitronate nucleophile, which is formed in situ under metal-free conditions, is used in stoichiometric amounts. This is in stark contrast to the vast majority of nitroaldol reactions that require the use of nitromethane or another nitroalkane in large excess. The synthetic utility of the reaction products was demonstrated by catalytic hydrogenation to unprecedented N-hydroxyimidoyl fluoride compounds.
Experimental Section
General information
All commercially available reagents and solvents were used without further purification unless otherwise noted. Chloroform was dried over activated 4 A molecular sieves prior to use as solvent. NMR spectra were obtained at 400 MHz (1H NMR), 100 MHz (13C NMR) and 376 MHz (19F NMR) in CDCl3. Chemical shifts are reported in ppm relative to tetramethylsilane. Difluoronitroacetophenone 1 and isatins 8, 10, 12, 14, 16, 18, 23, 25, 27 were synthesized as described in the literature.9,21 Reaction products were purified by column chromatography on silica using a flash purification system as described below.
Synthesis of 1-difluoronitroacetonaphthone, 20
To a solution of 1-naphthoylnitromethane 22 (410.0 mg, 1.90 mmol) in anhydrous acetonitrile (15 mL) was added Selectfluor (1451.0 mg, 4.18 mmol) and anhydrous potassium phosphate (443.5 mg, 2.09 mmol). The reaction was stirred at room temperature for 2 days and monitored by F NMR. Upon completion, the mixture was poured onto saturated aqueous ammonium chloride solution (100 mL) and extracted with diethyl ether (3 × 15 mL). The combined organic layers were dried over MgSO4 and concentrated under reduced pressure. The residue was purified by chromatography using 2 – 8% hexanes/ethyl acetate as mobile phase to give 1-difluoronitroacetonaphthone (450.0 mg, 1.79 mmol) as a yellow oil in 94% yield. 1H NMR (400 MHz, CDCl3): δ 7.60 (dd, J = 8.2, 7.5 Hz, 1H), 7.65 (ddd, J = 8.2, 6.9, 1.2 Hz, 1H), 7.74 (ddd, J = 8.6, 6.9, 1.5 Hz, 1H), 7.96 (dd, J = 8.2, 1.8 Hz, 1H), 8.15 (ddd, J = 7.6, 3.6, 2.4 Hz, 1H), 8.21 (dd, J = 8.3, 1.1 Hz, 1H), 8.74 (dd, J = 8.7, 1.8 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 115.4 (t, J C-F = 302.4 Hz), 124.1, 125.0, 126.0, 127.6, 129.1, 130.0, 131.0, 131.2 (t, J C-F = 5.1 Hz), 134.0, 137.0, 180.6 (t, J C-F = 25.5 Hz). 19F NMR (376 MHz, CDCl3): δ −85.34 (s, 2F). Anal. Calcd. for C12H7F2NO3: C, 57.38; H, 2.81; N, 5.58. Found: C, 57.32; H, 2.91; N, 5.60.
General procedure for isatin insertion into aryl difluoronitromethyl ketones
To a solution of the isatin (0.15 mmol), difluoronitroacetophenone (36.2 mg, 0.18 mmol) or difluoronitroacetonaphthone (45.3 mg, 0.18 mmol) in anhydrous chloroform (0.4 mL) was added DBU (4.6 mg, 0.03 mmol) under nitrogen atmosphere. The reaction was stirred at room temperature for 24 – 48 hours. Upon completion, the mixture was dry-loaded on silica gel and purified by chromatography using 5 – 10% hexanes/ethyl acetate as mobile phase.
Hydrogenation procedure
A solution of 3 or 21 (0.16 mmol) and di-tert-butyl dicarbonate (52.4 mg, 0.24 mmol) in dichloromethane (2 mL) was transferred to a glass-lined hydrogenation apparatus containing the Lindlar catalyst (3 mol% Pd) and glacial acetic acid (50 μL, 0.87 mmol) in methanol (6 mL). The apparatus was charged with H2 (10 bar) and stirred at room temperature for 7 – 20 hours. The reaction mixture was then filtered through celite and concentrated under reduced pressure. The residue was dry-loaded on silica gel and purified by flash chromatography using 15 – 25% hexanes/ethyl acetate as mobile phase.
N-Benzyl 3-cyano-2-oxoindolin-3-yl benzoate, 29
A solution of 3 (70.1 mg, 0.16 mmol) and diisopropylethylamine (139.3 μL, 0.8 mmol) in dichloromethane (5 mL) was cooled to 0°C and then trichlorosilane (56.7 μL, 0.56 mmol) was added. The mixture was stirred for 8 hours and concentrated under reduced pressure. The crude product was purified by flash chromatography using 5 – 15% hexanes/ethyl acetate as mobile phase to give 22.3 mg of 29 (0.06 mmol, 38%) of a white solid, Mp 136.8 – 139.4 °C. 1H NMR (400 MHz, CDCl3): δ 4.94 (d, J = 15.7 Hz, 1H), 5.01 (d, J = 15.7 Hz, 1H), 6.75 (dd, J = 8.0, 0.9 Hz, 1H), 7.03 (dd, J = 7.6, 1.0 Hz, 1H), 7.24 – 7.43 (m, 7H), 7.45 (dd, J = 8.3, 7.6 Hz, 2H), 7.59 (dd, J = 8.0, 1.0 Hz, 1H), 8.13 (dd, J = 7.4, 1.0 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 44.0, 70.4, 109.5, 113.8, 123.1, 124.4, 125.6, 127.3, 127.7, 128.4, 128.8, 129.0, 130.1, 130.2, 133.5, 135.3, 143.7, 165.8, 172.3. Anal. Calcd. for C23H16N2O3: C, 74.99; H, 4.38; N, 7.60. Found: C, 75.31; H, 4.63; N, 7.49.
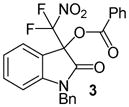
N-Benzyl 3-(difluoronitromethyl)-2-oxoindolin-3-yl benzoate, 3
N-Benzyl isatin (35.6 mg, 0.15 mmol) and difluoronitroacetophenone (36.2 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 24 hours. Flash purification gave 64.9 mg (0.15 mmol, 99%) of a white solid, Mp 127.3 – 129.8 °C. 1H NMR (400 MHz, CDCl3): δ 4.98 (d, J = 15.9 Hz, 1H), 5.12 (d, J = 15.9 Hz, 1H), 6.77 (d, J = 7.9 Hz, 1H), 7.09 (dd, J = 7.6, 7.6 Hz, 1H), 7.28 – 7.45 (m, 7H), 7.49 (dd, J = 8.2, 7.4 Hz, 2H), 7.64 (dd, J = 8.0, 1.0 Hz, 1H), 8.08 (dd, J = 7.1, 0.8 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 45.1, 77.7 (t, J C-F = 26.0 Hz), 110.7, 119.0, 120.3 (t, J C-F = 294.6 Hz), 123.7, 125.0, 127.4, 127.6, 128.1, 128.9, 129.1, 130.5, 132.7, 134.6, 134.6, 144.6, 163.0, 166.7. 19F NMR (376 MHz, CDCl3): δ −96.07 (d, J = 169.2 Hz, 1F), −95.62 (d, J = 169.9 Hz, 1F). Anal. Calcd. for C23H16F2N2O5: C, 63.02; H, 3.68; N, 6.39. Found: C, 62.91; H, 3.96; N, 6.39.
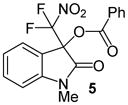
N-Methyl 3-(difluoronitromethyl)-2-oxoindolin-3-yl benzoate, 5
N-Methyl isatin (24.2 mg, 0.15 mmol) and difluoronitroacetophenone (36.2 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 24 hours. Flash purification gave 53.3 mg (0.15 mmol, 98%) of a white solid, Mp 167.1 – 169.2 °C. 1H NMR (400 MHz, CDCl3): δ 3.35 (s, 3H), 6.98 (d, J = 7.9 Hz, 1H), 7.12 (dd, J = 7.6, 7.6 Hz, 1H), 7.37 (d, J = 7.5 Hz, 1H), 7.43 – 7.54 (m, 3H), 7.63 (dd, J = 8.5, 1.1 Hz, 1H), 8.04 (dd, J = 7.4, 1.0 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 27.3, 77.5 (t, J C-F = 25.9 Hz), 109.6, 119.1, 120.2 (t, J C-F = 294.5 Hz), 123.7, 125.0, 127.6, 128.9, 130.4, 132.9, 134.5, 145.5, 163.1, 166.6. 19F NMR (376 MHz, CDCl3): δ −96.09 (d, J = 169.6 Hz, 1F), −95.62 (d, J = 169.6 Hz, 1F). Anal. Calcd. for C17H12F2N2O5: C, 56.36; H, 3.34; N, 7.73. Found: C, 56.47; H, 3.49; N, 7.53.
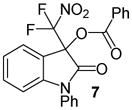
N-Phenyl 3-(difluoronitromethyl)-2-oxoindolin-3-yl benzoate, 7
N-Phenyl isatin (33.5 mg, 0.15 mmol) and difluoronitroacetophenone (36.2 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 24 hours. Flash purification gave 62.4 mg (0.15 mmol, 98%) of a white solid, Mp 109.2 – 112.1 °C. 1H NMR (400 MHz, CDCl3): δ 6.85 (d, J = 7.9 Hz, 1H), 7.14 (dd, J = 7.7, 7.7 Hz, 1H), 7.37 – 7.46 (m, 2H), 7.46 – 7.55 (m, 5H), 7.58 (dd, J = 8.3, 7.6 Hz, 2H), 7.64 (dd, J = 8.2, 1.2 Hz, 1H), 8.09 (dd, J = 7.6, 1.0 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 77.6 (t, J C-F = 25.5 Hz), 110.8, 118.7, 120.4 (t, J C-F = 294.5 Hz), 124.1, 125.2, 127.0, 127.7, 128.9, 129.2, 130.1, 130.5, 132.8, 133.7, 134.6, 146.0, 163.3, 166.0. 19F NMR (376 MHz, CDCl3): δ −95.94 (d, J = 169.1 Hz, 1F), −95.34 (d, J = 169.2 Hz, 1F). Anal. Calcd. for C22H14F2N2O5: C, 62.27; H, 3.33; N, 6.60. Found: C, 61.96; H, 3.43; N, 6.32.
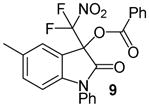
N-Phenyl 3-(difluoronitromethyl)-5-methyl-2-oxoindolin-3-yl benzoate, 9
N-Phenyl 5-methylisatin (35.6 mg, 0.15 mmol) and difluoronitroacetophenone (36.2 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 24 hours. Flash purification gave 61.2 mg (0.14 mmol, 93%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 2.32 (s, 3H), 6.75 (d, J = 8.1 Hz, 1H), 7.20 (d, J = 8.2 Hz, 1H), 7.23 (s, 1H), 7.43 – 7.52 (m, 5H), 7.57 (dd, J = 8.3, 6.5 Hz, 2H), 7.65 (dd, J = 6.4, 1.6 Hz, 1H), 8.09 (dd, J = 8.3, 1.4 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 21.1, 77.7 (t, J C-F = 24.9 Hz), 110.6, 118.6, 120.4 (t, J C-F = 295.4 Hz), 125.7, 126.9, 127.7, 128.9, 129.0, 130.0, 130.5, 133.2, 133.8, 134.0, 134.5, 143.6, 163.2, 165.9. 19F NMR (376 MHz, CDCl3): δ −95.93 (d, J = 168.9 Hz, 1F), −95.34 (d, J = 169.2 Hz, 1F). Anal. Calcd. for C23H16F2N2O5: C, 63.02; H, 3.68; N, 6.39. Found: C, 63.39; H, 4.01; N, 6.18.
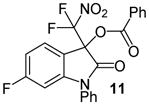
N-Phenyl 3-(difluoronitromethyl)-5-fluoro-2-oxoindolin-3-yl benzoate, 11
N-Phenyl 6-fluoroisatin (36.2 mg, 0.15 mmol) and difluoronitroacetophenone (36.2 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 24 hours. Flash purification gave 60.2 mg (0.14 mmol, 91%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 6.57 (dd, J = 8.8, 2.3 Hz, 1H), 6.81 (dd, J = 8.7, 2.3 Hz, 1H), 7.41 (dd, J = 8.4, 5.1 Hz, 1H), 7.46 – 7.54 (m, 5H), 7.59 (dd, J = 7.7, 6.5 Hz, 2H), 7.5 (dd, J = 6.3, 1.5 Hz, 1H), 8.07 (dd, J = 8.0, 1.4 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 77.8 (t, J C-F = 25.0 Hz), 99.8 (d, J C-F = 28.5 Hz), 110.8 (d, J C-F = 23.3 Hz), 114.1 (d, J C-F = 3.1 Hz), 120.2 (t, J C-F = 295.8 Hz), 126.9, 127.1 (d, J C-F = 10.5 Hz), 127.5, 128.9, 129.5, 130.3, 130.5, 133.2, 134.7, 148.0 (d, J C-F = 12.2 Hz), 163.3, 165.6 (d, J C-F = 252.5 Hz), 166.1. 19F NMR (376 MHz, CDCl3): δ −103.69 (ddd, J = 8.8, 8.7, 5.2 Hz, 1F), −95.89 (d, J = 169.5 Hz, 1F), −95.25 (d, J = 169.5 Hz, 1F). Anal. Calcd. for C22H13F3N2O5: C, 59.74; H, 2.96; N, 6.33. Found: C, 59.93; H, 3.22; N, 6.12.
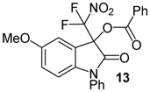
N-Phenyl 3-(difluoronitromethyl)-5-methoxy-2-oxoindolin-3-yl benzoate, 13
N-Phenyl 5-methoxyisatin (38.0 mg, 0.15 mmol) and difluoronitroacetophenone (36.2 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 48 hours. Flash purification gave 65.5 mg (0.14 mmol, 96%) of a yellow amorphous solid. 1H NMR (400 MHz, CDCl3): δ 3.77 (s, 3H), 6.79 (d, J = 8.7 Hz, 1H), 6.93 (dd, J = 8.7, 2.6 Hz, 1H), 7.01 (d, J = 2.5 Hz, 1H), 7.43 – 7.53 (m, 6H), 7.6 (dd, J = 8.7, 7.4 Hz, 2H), 7.65 (dd, J = 8.7, 1.3 Hz, 1H), 8.09 (dd, J = 7.3, 1.3 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 56.0, 77.8 (t, J C-F = 25.1 Hz), 111.5, 111.9, 117.4, 119.7, 120.3 (t, J C-F = 294.4 Hz), 126.8, 127.6, 128.9, 129.0, 130.0, 130.5, 133.9, 134.6, 139.2, 156.8, 163.2, 165.7. 19F NMR (376 MHz, CDCl3): δ −95.94 (d, J = 169.6 Hz, 1F), −95.38 (d, J = 169.2 Hz, 1F). Anal. Calcd. for C23H16F2N2O6: C, 60.80; H, 3.55; N, 6.17. Found: C, 60.69; H, 3.66; N, 6.08.
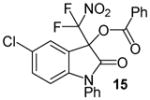
N-Phenyl 3-(difluoronitromethyl)-5-chloro-2-oxoindolin-3-yl benzoate, 15
N-Phenyl 5-chloroisatin (38.6 mg, 0.15 mmol) and difluoronitroacetophenone (36.2 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 48 hours. Flash purification gave 62.4 mg (0.14 mmol, 91%) of a pale yellow amorphous solid. 1H NMR (400 MHz, CDCl3): δ 6.80 (dd, J = 8.3, 2.8 Hz, 1H), 7.38 (dd, J = 8.5, 2.2 Hz, 1H), 7.44 (d, J = 2.3 Hz, 1H), 7.46 – 7.55 (m, 5H), 7.58 (dd, J = 7.8, 7.5 Hz, 2H), 7.66 (dd, J = 8.0, 1.7 Hz, 1H), 8.09 (dd, J = 7.6, 1.5 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 77.2 (t, J C-F = 24.9 Hz), 111.9, 120.0 (t, J C-F = 294.3 Hz), 120.2, 125.6, 126.9, 127.3, 129.0, 129.4, 129.5, 130.2, 130.6, 132.8, 133.3, 134.8, 144.6, 163.3, 165.6. 19F NMR (376 MHz, CDCl3): δ −95.64 (d, J = 170.3 Hz, 1F), −95.11 (d, J = 170.3 Hz, 1F). Anal. Calcd. for C22H13ClF2N2O5: C, 57.59; H, 2.86; N, 6.11. Found: C, 57.36; H, 3.21; N, 5.77.
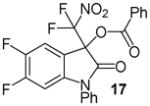
N-Phenyl 3-(difluoronitromethyl)-5,6-difluoro-2-oxoindolin-3-yl benzoate, 17
N-Phenyl 5,6-difluoroisatin (38.9 mg, 0.15 mmol) and difluoronitroacetophenone (36.2 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 48 hours. Flash purification gave 56.0 mg (0.12 mmol, 81%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 6.69 (dd, J = 9.8, 6.3 Hz, 1H), 7.32 (dd, J = 8.6, 7.5 Hz, 1H), 7.44 – 7.55 (m, 5H), 7.60 (dd, J = 8.3, 6.8 Hz, 2H), 7.66 (dd, J = 7.3, 1.3 Hz, 1H), 8.07 (dd, J = 8.2, 1.3 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 101.4 (d, J C-F = 23.9 Hz), 113.8 (dd, J C-F = 6.2, 4.5 Hz), 115.3 (d, J C-F = 21.2 Hz), 120.0 (t, J C-F = 295.9 Hz), 126.9, 127.2, 129.0, 129.7, 130.4, 130.6, 133.2, 134.9, 143.0 (dd, J C-F = 9.8, 2.8 Hz), 147.4 (dd, J C-F = 247.2, 13.6 Hz), 153.4 (dd, J C-F = 255.0, 13.7 Hz), 163.3, 165.7. 19F NMR (376 MHz, CDCl3): δ −142.23 (ddd, J = 19.9, 8.6, 6.1 Hz, 1F), −128.04 (ddd, J = 19.8, 9.8, 7.3 Hz, 1F), −95.79 (d, J = 170.3 Hz, 1F), −95.22 (d, J = 170.3 Hz, 1F). Anal. Calcd. for C22H12F4N2O5: C, 57.40; H, 2.63; N, 6.09. Found: C, 57.38; H, 2.78; N, 5.97.
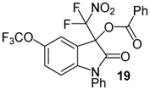
N-Phenyl 3-(difluoronitromethyl)-5-trifluoromethoxy-2-oxoindolin-3-yl benzoate, 19
N-Phenyl 5-trifluoromethoxyisatin (46.1 mg, 0.15 mmol) and difluoronitroacetophenone (36.2 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 48 hours. Flash purification gave 64.3 mg (0.13 mmol, 84%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 6.86 (d, J = 8.7 Hz, 1H), 7.29 (dd, J = 8.6, 2.8 Hz, 1H), 7.33 (d, J = 2.8 Hz, 1H), 7.47 – 7.53 (m, 5H), 7.59 (dd, J = 8.2, 7.7 Hz, 2H), 7.66 (dd, J = 7.9, 1.7 Hz, 1H), 8.08 (dd, J = 8.4, 1.3 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 77.79 (t, J C-F = 24.6 Hz), 111.60, 119.21, 119.97, 119.99 (t, J C-F = 294.9 Hz), 120.49 (q, J C-F = 257.0 Hz), 125.91, 126.92, 127.21, 129.00, 129.55, 130.28, 130.59, 133.28, 134.87, 144.58, 145.41 (q, J C-F = 1.9 Hz), 163.32, 165.74. 19F NMR (376 MHz, CDCl3): δ −95.82 (d, J = 169.9 Hz, 1F), −95.30 (d, J = 169.9 Hz, 1F), −58.52 (s, 3F). Anal. Calcd. for C23H13F5N2O6: C, 54.34; H, 2.58; N, 5.51. Found: C, 54.73; H, 2.93; N, 5.30.
N-Phenyl 3-(difluoronitromethyl)-2-oxoindolin-3-yl 1-naphthoate, 21
N-Phenyl isatin (33.5 mg, 0.15 mmol) and difluoronitroacetonaphthone (45.3 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 24 hours. Flash purification gave 68.7 mg (0.14 mmol, 97%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 6.89 (dd, J = 8.0, 1.4 Hz, 1H), 7.15 (dd, J = 7.7, 1.6 Hz, 1H), 7.42 (dd, J = 8.8, 7.6 Hz, 1H), 7.46 – 7.63 (m, 9H), 7.89 (dd, J = 8.0, 2.1 Hz, 1H), 8.12 (dd, J = 8.1, 1.7 Hz, 1H), 8.49 (dd, J = 7.2, 1.6 Hz, 1H), 8.81 (dd, J = 8.6, 2.3 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 77.7 (t, J C-F = 24.6 Hz), 110.8, 118.9, 120.5 (t, J C-F = 294.7 Hz), 123.6, 124.1, 124.8, 125.2, 125.5, 126.7, 127.1, 128.6, 128.9, 129.2, 130.1, 131.7, 132.3, 132.8, 133.7, 133.9, 135.5, 146.1, 163.6, 166.2. 19F NMR (376 MHz, CDCl3): δ −95.80 (d, J = 169.0 Hz, 1F), −95.06 (d, J = 169.0 Hz, 1F). Anal. Calcd. for C26H16F2N2O5: C, 65.82; H, 3.40; N, 5.90. Found: C, 65.86; H, 3.55; N, 5.82.
N-Phenyl 3-(difluoronitromethyl)-6-fluoro-2-oxoindolin-3-yl 1-naphthoate, 22
N-Phenyl 6-fluoroisatin (36.2 mg, 0.15 mmol) and difluoronitroacetonaphthone (45.3 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 24 hours. Flash purification gave 70.4 mg (0.14 mmol, 95%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 6.61 (dd, J = 8.8, 2.3 Hz, 1H), 6.83 (ddd, J = 8.7, 8.7, 2.3 Hz, 1H), 7.45 (dd, J = 8.4, 5.1 Hz, 1H), 7.49 – 7.64 (m, 8H), 7.89 (dd, J = 8.4, 1.5 Hz, 1H), 8.13 (dd, J = 8.2, 1.6 Hz, 1H), 8.47 (dd, J = 7.4, 1.3 Hz, 1H), 8.80 (dd, J = 8.4, 1.2 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 77.6 (t, J C-F = 24.6 Hz), 99.8 (d, J C-F = 28.5 Hz), 110.8 (d, J C-F = 23.3 Hz), 114.3 (d, J C-F = 3.3 Hz), 120.4 (t, J C-F = 294.8 Hz), 123.4, 124.8, 125.4, 126.8, 126.9, 127.0 (d, J C-F = 10.2 Hz), 128.7, 128.9, 129.5, 130.2, 131.7, 132.4, 133.3, 133.9, 135.6, 148.0 (d, J C-F = 12.2 Hz),163.6, 165.6 (d, J C-F = 252.3 Hz) 166.3. 19F NMR (376 MHz, CDCl3): δ −103.84 (ddd, J = 8.8, 8.7, 5.1 Hz, 1F), −95.75 (d, J = 169.3 Hz, 1F), −94.98 (d, J = 169.3 Hz, 1F). Anal. Calcd. for C26H15F3N2O5: C, 63.42; H, 3.07; N, 5.69. Found: C, 63.50; H, 3.28; N, 5.63.
N-Phenyl 3-(difluoronitromethyl)-6-methoxy-2-oxoindolin-3-yl 1-naphthoate, 24
N-Phenyl 6-methoxyisatin (38.0 mg, 0.15 mmol) and difluoronitroacetonaphthone (45.3 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 48 hours. Flash purification gave 73.4 mg (0.15 mmol, 97%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 3.77 (s, 3H), 6.40 (d, J = 2.3 Hz, 1H), 6.62 (dd, J = 8.5, 2.3 Hz, 1H), 7.38 (dd, J = 8.4, 1.0 Hz, 1H), 7.47 – 7.63 (m, 8H), 7.89 (dd, J = 8.1, 1.5 Hz, 1H), 8.11 (dd, J = 8.2, 2.0 Hz, 1H), 8.47 (dd, J = 7.4, 1.3 Hz, 1H), 8.82 (dd, J = 8.8, 1.3 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 55.8, 77.7 (t, J C-F = 24.6 Hz), 98.3, 108.5, 110.3, 120.6 (t, J C-F = 296.0 Hz), 123.9, 124.8, 125.6, 126.4, 126.7, 127.1, 128.6, 128.9, 129.2, 130.1, 131.7, 132.2, 133.7, 133.9, 135.4, 147.6, 163.5, 163.7, 166.7. 19F NMR (376 MHz, CDCl3): δ −95.82 (d, J = 168.4 Hz, 1F), −95.01 (d, J = 168.4 Hz, 1F). Anal. Calcd. for C27H18F2N2O6: C, 64.29; H, 3.60; N, 5.55. Found: C, 64.30; H, 3.61; N, 5.52.
N-Phenyl 3-(difluoronitromethyl)-4-bromo-2-oxoindolin-3-yl 1-naphthoate, 26
N-Phenyl 4-bromoisatin (45.3 mg, 0.15 mmol) and difluoronitroacetonaphthone (45.3 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 24 hours. Flash purification gave 74.3 mg (0.14 mmol, 90%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 6.80 (dd, J = 7.7, 1.1 Hz, 1H), 7.20 – 7.32 (m, 2H), 7.48 – 7.65 (m, 8H), 7.91 (dd, J = 8.0, 2.0 Hz, 1H), 8.14 (dd, J = 8.2, 1.6 Hz, 1H), 8.62 (dd, J = 7.4, 1.3 Hz, 1H), 8.84 (dd, J = 8.7, 1.3 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 79.98 (dd, J C-F = 26.0, 22.9 Hz), 109.66, 118.26, 119.97, 120.26 (dd, J C-F = 301.2, 295.0 Hz), 122.81, 124.69, 125.40, 126.57, 127.04, 128.51, 128.65, 128.76, 129.41, 130.08, 131.67, 132.65, 133.16, 133.18, 133.81, 135.43, 147.53, 163.40, 165.79. 19F NMR (376 MHz, CDCl3): δ −91.99 (d, J = 163.1 Hz, 1F), −90.97 (d, J = 163.0 Hz, 1F). Anal. Calcd. for C26H15BrF2N2O5: C, 56.44; H, 2.73; N, 5.06. Found: C, 56.55; H, 3.05; N, 4.95.
N-Phenyl 3-(difluoronitromethyl)-6-bromo-2-oxoindolin-3-yl 1-naphthoate, 28
N-Phenyl 6-bromoisatin (45.3 mg, 0.15 mmol) and difluoronitroacetonaphthone (45.3 mg, 0.18 mmol) were employed in the general procedure described above and the reaction was stirred for 24 hours. Flash purification gave 77.8 mg (0.14 mmol, 94%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 7.01 (d, J = 1.9 Hz, 1H), 7.27 – 7.35 (m, 2H), 7.49 – 7.65 (m, 8H), 7.90 (dd, J = 7.9, 1.2 Hz, 1H), 8.13 (dd, J = 8.3, 1.4 Hz, 1H), 8.46 (dd, J = 7.6, 1.2 Hz, 1H), 8.79 (dd, J = 8.6, 1.5 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 77.8 (t, J C-F = 24.6 Hz), 114.3, 117.2, 120.2 (t, J C-F = 294.7 Hz), 123.1, 123.3, 124.8, 125.4, 126.4, 126.8, 127.0, 127.1, 128.8, 129.0, 129.6, 130.3, 131.7, 132.4, 133.2, 133.9, 135.7, 147.2, 163.6, 166.0. 19F NMR (376 MHz, CDCl3): δ −95.68 (d, J = 169.4 Hz, 1F), −94.97 (d, J = 169.5 Hz, 1F). Anal. Calcd. for C26H15BrF2N2O5: C, 56.44; H, 2.73; N, 5.06. Found: C, 56.52; H, 3.10; N, 4.85.
N-Benzyl (Z)-3-(fluoro(hydroxyimino)methyl)-2-oxoindolin-3-yl benzoate, 30
N-Benzyl 3-(difluoronitromethyl)-2-oxoindolin-3-yl benzoate (70.1 mg, 0.16 mmol) was employed in the catalytic hydrogenation procedure described above and the reaction was stirred for 7 hours. Flash purification gave 61.7 mg (0.15 mmol, 95%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 4.99 (d, J = 15.9 Hz, 1H), 5.10 (d, J = 15.9 Hz, 1H), 6.74 (d, J = 7.9 Hz, 1H), 7.05 (dd, J = 7.6, 1.0 Hz, 1H), 7.27 (dd, J = 7.8, 1.2 Hz, 2H), 7.31 – 7.38 (m, 2H), 7.39 – 7.49 (m, 5H), 7.62 (dd, J = 5.6, 1.5 Hz, 1H), 8.08 (dd, J = 8.3, 1.3 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): δ 44.8, 77.7, 110.4, 123.3, 123.4, 124.5, 127.3, 127.9, 128.4, 128.7, 129.0, 130.4, 131.3, 134.1, 135.1, 143.4, 148.43 (d, J C-F = 342.2 Hz), 163.9, 169.13 (d, J C-F = 3.2 Hz). 19F NMR (376 MHz, CDCl3): δ −79.21 (s, 1F). Anal. Calcd. for C23H17FN2O4: C, 68.31; H, 4.24; N, 6.93. Found: C, 68.11; H, 4.46; N, 6.89.
N-Methyl (Z)-3-(fluoro(hydroxyimino)methyl)-2-oxoindolin-3-yl benzoate, 31
N-Methyl-3-(difluoronitromethyl)-2-oxoindolin-3-yl benzoate (43.5 mg, 0.12 mmol) was employed in the catalytic hydrogenation procedure described above and the reaction mixture was stirred for 8 hours. Flash purification gave 35.3 mg (0.11 mmol, 90%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 3.33 (s, 3H), 6.95 (d, J = 7.9 Hz, 1H), 7.10 (dd, J = 8.0, 7.6 Hz, 1H), 7.37 – 7.50 (m, 4H), 7.59 (dd, J = 7.7, 1.5 Hz, 1H), 8.03 (dd, J = 7.5, 1.2 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): 27.2, 77.8, 109.3, 123.2, 123.5, 124.7, 128.4, 128.7, 130.4, 131.5, 134.1, 144.4, 148.30 (d, J C-F = 342.4 Hz), 164.0, 169.1 (d, J C-F = 3.0 Hz). 19F NMR (376 MHz, CDCl3): δ −79.2 (s, 1F). Anal. Calcd. for C17H13FN2O4: C, 62.20; H, 3.99; N, 8.53. Found: C, 62.37; H, 3.68; N, 8.32.
N-Phenyl (Z)-3-(fluoro(hydroxyimino)methyl)-5-methoxy-2-oxoindolin-3-yl benzoate, 32
N-Phenyl 3-(difluoronitromethyl)-5-methoxy-2-oxoindolin-3-yl benzoate (44.0 mg, 0.1 mmol) was employed in the catalytic hydrogenation procedure described above and the mixture was stirred for 8 hours. Flash purification gave 33.3 mg (0.08 mmol, 80%) of a white amorphous solid.1H NMR (400 MHz, CDCl3): δ 3.77 (s, 3H), 6.78 (d, J = 8.7 Hz, 1H), 6.87 (dd, J = 8.7, 2.6 Hz, 1H), 7.09 (d, J = 2.7 Hz, 1H), 7.46 (dd, J = 8.3, 7.6 Hz, 2H), 7.49 – 7.55 (m, 5H), 7.60 (dd, J = 8.4, 2.0 Hz, 1H), 8.09 (dd, J = 7.5, 1.9 Hz, 2H). 13C{1H} NMR (100 MHz, CDCl3): 56.0, 77.9, 111.2, 111.4, 116.2, 124.0, 126.7, 126.9, 128.4, 128.7, 129.9, 130.4, 134.1, 134.4, 138.2, 148.3 (d, J C-F = 342.6 Hz), 156.7, 164.1, 168.3 (d, J C-F = 2.7 Hz). 19F NMR (376 MHz, CDCl3): δ −79.4 (s, 1F). Anal. Calcd. for C23H17FN2O5: C, 65.71; H, 4.08; N, 6.66. Found: C, 65.75; H, 3.69; N, 6.41.
N-Phenyl (Z)-3-(fluoro(hydroxyimino)methyl)-2-oxoindolin-3-yl 1-naphthoate, 33
N-Phenyl 3-(difluoronitromethyl)-2-oxoindolin-3-yl 1-naphthoate (75.9 mg, 0.16 mmol) was employed in the catalytic hydrogenation procedure described above and the reaction was stirred for 7 hours. Flash purification gave 54.3 mg (0.12 mmol, 77%) of a white amorphous solid. 1H NMR (400 MHz, CDCl3): δ 6.88 (d, J = 8.0 Hz, 1H), 7.14 (dd, J = 7.6, 1.0 Hz, 1H), 7.36 (dd, J = 7.8, 1.3 Hz, 1H), 7.46 (m, 1H), 7.49 – 7.62 (m, 8H), 7.87 (dd, J = 8.0, 2.1 Hz, 1H), 8.08 (dd, J = 8.3, 1.9 Hz, 1H), 8.43 (dd, J = 7.3, 1.3 Hz, 1H), 8.83 (d, J = 8.6, 1.2 Hz, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 77.8, 110.5, 123.2, 123.8, 124.6, 124.8, 125.8, 126.6, 127.1, 128.4, 128.8, 128.9, 129.9, 131.3, 131.6, 131.8, 133.9, 134.2, 134.8, 144.9, 148.50 (d, J C-F = 342.2 Hz), 164.7, 168.6 (d, J C-F = 2.9 Hz). 19F NMR (376 MHz, CDCl3): δ −78.8 (s, 1F). Anal. Calcd. for C26H17FN2O4: C, 70.90; H, 3.89; N, 6.36. Found: C, 70.70; H, 4.09; N, 6.36.
Supplementary Material
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (GM106260).
Footnotes
Supporting Information Available. NMR spectra and crystallographic details. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Ono N. The Nitro Group in Organic Synthesis. Wiley; New York: 2001. [Google Scholar]
- 2.a) Tsuji J, Yamada T, Minami L, Yuhara M, Nisar M, Shimizu I. J Org Chem. 1987;52:2988–2995. [Google Scholar]; (b) Rieck H, Helmchen G. Angew Chem, Int Ed Engl. 1996;34:2687–2689. [Google Scholar]; (c) Trost BM, Surivet J-P. Angew Chem, Int Ed. 2000;39:3122–3124. doi: 10.1002/1521-3773(20000901)39:17<3122::aid-anie3122>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]; (d) Maki K, Kanai M, Shibasaki M. Tetrahedron. 2007;63:4250–4257. [Google Scholar]
- 3.Vogl EM, Buchwald SL. J Org Chem. 2000;67:106–111. doi: 10.1021/jo010953v. [DOI] [PubMed] [Google Scholar]
- 4.a) Seebach D, Lehr F. Angew Chem, Int Ed Engl. 1976;15:505–506. [Google Scholar]; b) Katritzky AR, Kashmiri MA, De Ville GZ, Patel RC. J Am Chem Soc. 1983;105:90–96. [Google Scholar]; c) Gildner PG, Gietter AAS, Cui D, Watson DA. J Am Chem Soc. 2012;134:9942–9945. doi: 10.1021/ja304561c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Shen B, Makley DM, Johnston JN. Nature. 2010;465:1027–1032. doi: 10.1038/nature09125. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schwieter KE, Shen B, Shackleford JP, Leighty MW, Johnston JN. Org Lett. 2014;16:4714–4717. doi: 10.1021/ol502089v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Schwieter KE, Johnston JN. Chem Sci. 2015;6:2590–2595. doi: 10.1039/c5sc00064e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stoichiometric nitroaldol reactions: Majhi A, Kadam ST, Kim SS. Bull Korean Chem Soc. 2009;30:1767–1770.Soengas RG, Silva AMS. Synlett. 2012;23:873–876.Rokhum L, Bez G. Can J Chem. 2013;91:300–306.
- 7.a) Liu S, Wolf C. Org Lett. 2008;10:1831–1834. doi: 10.1021/ol800442s. [DOI] [PubMed] [Google Scholar]; b) Yearick Spangler K, Wolf C. Org Lett. 2009;11:4724–4727. doi: 10.1021/ol9018612. [DOI] [PubMed] [Google Scholar]; c) Xu H, Wolf C. Chem Commun. 2010:8026–8028. doi: 10.1039/c0cc02378g. [DOI] [PubMed] [Google Scholar]; d) Xu H, Wolf C. Angew Chem Int Ed. 2011;50:12249–12252. doi: 10.1002/anie.201105778. [DOI] [PubMed] [Google Scholar]
- 8.a) Zhang P, Wolf C. J Org Chem. 2012;77:8840–8844. doi: 10.1021/jo3017583. [DOI] [PubMed] [Google Scholar]; b) Zhang P, Wolf C. Angew Chem Int Ed. 2013;52:7869–7873. doi: 10.1002/anie.201303551. [DOI] [PubMed] [Google Scholar]; c) Balaraman K, Moskowitz M, Liu Y, Wolf C. Synthesis. 2016;48:2376–2384. doi: 10.1055/s-0035-1561433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding R, Wolf C. Chem Commun. 2016;52:3576–3579. doi: 10.1039/c5cc09753c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Yang X, Wu T, Phipps RJ, Toste FD. Chem Rev. 2015;115:826–870. doi: 10.1021/cr500277b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mei H, Acena JL, Soloshonok VA, Roeschenthaler GV, Han J. Eur J Org Chem. 2015:6401–6412. [Google Scholar]; c) Wang J, Sanchez-Rosello M, Acena JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 11.a) Ren Q, Huang J, Wang L, Li W, Liu H, Jiang X, Wang J. ACS Catal. 2012;2:2622–2625. [Google Scholar]; b) Duan Z, Han J, Qian P, Zhang Z, Wang Y, Pan Y. Org Biomol Chem. 2013;11:6456–6459. doi: 10.1039/c3ob41460d. [DOI] [PubMed] [Google Scholar]; c) Saidalimu I, Fang X, He XP, Liang J, Yang X, Wu F. Angew Chem Int Ed. 2013;52:5566–5570. doi: 10.1002/anie.201301443. [DOI] [PubMed] [Google Scholar]; d) Prathima PS, Rajesh P, Rao JV, Kailash US, Sridhar B, Rao MM. Eur J Med Chem. 2014;84:155–159. doi: 10.1016/j.ejmech.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Ni C, Hu J. Chem Soc Rev. 2016;45:5441–5454. doi: 10.1039/c6cs00351f. [DOI] [PubMed] [Google Scholar]
- 13.Kaljurand I, Kuett A, Soovaeli L, Rodima T, Maeemets V, Leito I, Koppel IA. J Org Chem. 2005;70:1019–1028. doi: 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- 14.a) Bissell ER. J Org Chem. 1963;28:1717–1720. [Google Scholar]; b) Butler P, Golding BT, Laval G, Loghmani-Khouzani H, Ranjbar-Karimib R, Sadeghib MM. Tetrahedron. 2007;63:11160–11166. [Google Scholar]
- 15.The reaction also occurs with unprotected isatin, however, the insertion product was isolated in only 60% yield.
- 16.Ethanol and 1 do not react in the absence of DBU.
- 17.Orlandi M, Tosi F, Bonsignore M, Benaglia M. Org Lett. 2015;17:3941–3943. doi: 10.1021/acs.orglett.5b01698. [DOI] [PubMed] [Google Scholar]
- 18.Esmaeili AA, Ghalandarabad SA, Zangouei M. Tetrahedron Lett. 2012;42:5605–5607. [Google Scholar]
- 19.The (E)-diastereomer was not detected by NMR analysis.
- 20.The –NH-C(R)F-moiety is unstable: Annedi SC, Li W, Samson S, Kotra LP. J Org Chem. 2003;68:1043–1049. doi: 10.1021/jo026310c.
- 21.Chan DMT, Monaco KL, Wang RP, Winters MP. Tetrahedron Lett. 1998;39:2933–2936. [Google Scholar]
- 22.Katritzky AR, Abdel-Fattah AAA, Gromova AV, Witek R, Steel PJ. J Org Chem. 2005;70:9211–9214. doi: 10.1021/jo051231x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.