

Summary
Our previous studies revealed that the interaction of fibrin with the very low density lipoprotein receptor (VLDLR) promotes transendothelial migration of leukocytes and thereby inflammation, and localized the fibrin-binding site to CR-domains 2–4 of this receptor. In the present study, we tested interaction of three anti-VLDLR monoclonal antibodies, mAb 1H10, 1H5, and 5F3, with recombinant fragments of VLDLR containing various combinations of its CR-domains and found that the epitopes for mAb 1H10 and mAb 1H5 overlap with the fibrin-binding site of VLDLR. Based on these findings, we hypothesized that mAb 1H10 and mAb 1H5 should inhibit fibrin-VLDLR interaction and modulate leukocyte transmigration. To test this hypothesis, we first demonstrated that these monoclonal antibodies both have high affinity to the fibrin-binding fragments of the VLDL receptor and efficiently inhibit interaction between the VLDLR-binding fragment of fibrin and the fibrin-binding fragments of VLDLR. Next, in the in vitro experiments using leukocyte transendothelial migration assay we found that both monoclonal antibodies efficiently inhibit leukocyte transmigration induced by fibrin mimetic NDSK-II. Finally, in vivo experiments using mouse model of peritonitis revealed that mAb 1H10 and mAb 1H5 both significantly reduce infiltration of leukocytes into the peritoneum. Furthermore, our experiments using mouse model of myocardial ischemia-reperfusion injury revealed that both monoclonal antibodies significantly reduce myocardial injury induced by ischemia-reperfusion. Thus, the results obtained indicate that monoclonal antibodies 1H10 and 1H5 are novel specific inhibitors of fibrin-VLDLR-dependent leukocyte transmigration pathway. They may represent potential therapeutics for treatment of fibrin-dependent inflammation including myocardial ischemia-reperfusion injury.
Keywords: Fibrinogen/fibrin, VLDL receptor, leukocyte transmigration, monoclonal antibodies
Introduction
Fibrinogen is the major plasma protein involved in hemostasis and other important physiological and pathological processes. Activation of the blood coagulation cascade upon vascular injury results in generation of thrombin, which converts soluble fibrinogen into an insoluble polymeric fibrin. Fibrin polymer serves as the basis for blood clots that seal the injured vasculature to prevent blood loss and as a provisional matrix that participates in subsequent wound healing process (1), which includes inflammation, angiogenesis, tissue formation, etc. Numerous data indicate the involvement of fibrin(ogen) in inflammation. It was proposed earlier that fibrinogen or fibrin degradation products promote transendothelial migration of leukocytes and thereby inflammation through their interaction with endothelial receptors ICAM-1 or VE-cadherin, respectively (2, 3). Our recent study revealed that fibrin promotes leukocyte transmigration through its interaction with another endothelial cell receptor, the very low density lipoprotein (VLDL) receptor (4).
Fibrinogen is a chemical dimer consisting of two identical subunits, each of which includes three non-identical polypeptide chains, Aα, Bβ, and γ (5, 6). The central region of the fibrinogen molecule is formed by N-terminal portions of all 6 chains linked together by 11 disulfide bonds, and is often called the N-terminal disulfide knot (NDSK) (7). Upon conversion of fibrinogen into fibrin, thrombin removes from this region, namely from the N-terminal portions of the Aα and Bβ chains, fibrinopeptides A and B (FpA and FpB), respectively (6). Digestion of fibrinogen with CNBr produces NDSK fragment, which corresponds to the central region of the fibrinogen molecule. Subsequent treatment of NDSK with thrombin results in NDSK-II fragment corresponding to the central region of fibrin. NDSK-II retains some binding sites of fibrin and is often used as a simple fibrin mimetic in functional studies. Specifically, it was shown that this fragment interacts with endothelial VE-cadherin (8) and this interaction promotes angiogenesis (9) and inflammation (3). Our previous study (10) localized the VE-cadherin-binding site to a pair of fibrin βN-domains formed by the β chain residues 15–64 that are present in the NDSK-II fragment. We also demonstrated that the recombinant (β15–66)2 fragment, mimicking the dimeric arrangement of these domains in fibrin, interacts with VE-cadherin with practically the same affinity as fibrin (10). Furthermore, our recent study (4) revealed that fibrin interacts with the VLDL receptor through its βN-domains and (β15–66)2 corresponding to these domains has practically the same affinity to VLDLR as fibrin. Thus, the (β15–66)2 fragment retains functional properties of fibrin βN-domains.
The VLDL receptor (VLDLR) is a member of the low density lipoprotein (LDL) receptor family. It functions as a peripheral lipoprotein receptor involved in the delivery of triglyceride-rich lipoproteins to peripheral tissue (11, 12) and also plays an important role in reelin signaling (13, 14), angiogenesis and tumor growth (15), and fibrin-dependent inflammation (4). VLDLR consists of one polypeptide chain that forms the extracellular portion, the transmembrane domain, and the cytoplasmic domain (16, 17). The extracellular portion, which includes 8 complement-type repeats (CR-domains), and EGF-like, β-propeller, and the O-linked sugar domains, has been expressed in the insect expression system (18). The ligand-binding region of VLDLR including all 8 CR-domains has been expressed in the bacterial expression system and used in functional studies, as well as an antigen for preparation of anti-VLDLR monoclonal antibodies (18). Three such antibodies, 1H10, 1H5, and 5F3, have been prepared and partially characterized (18).
We have recently prepared a number of recombinant fragments corresponding to various regions of the extracellular portions of the VLDL receptor and, using them, localized the fibrin-binding site to CR-domains 2–4 of this receptor (19). The major aims of the present study were to further localize the epitopes for the above mentioned anti-VLDLR monoclonal antibodies and test the effects of these antibodies on the interaction of fibrin with the VLDL receptor and fibrin-VLDLR-dependent transendothelial migration of leukocytes in vitro and in vivo.
Materials and methods
Proteins, antibodies, and reagents
NDSK-II fragment was prepared by digestion of human fibrinogen (Enzyme Research Laboratories) with CNBr followed by cleavage of its fibrinopeptides with thrombin-agarose as described earlier (8, 20). Human receptor-associated protein (RAP) was expressed in E. coli and purified as described (21). Anti-human VLDLR monoclonal antibodies (mAb) 1H5, 1H10, and 5F3 (18) were purified from hybridoma supernatants by affinity chromatography on Protein A-Sepharose (Sigma-Aldrich). All three monoclonal antibodies cross-reacted with mouse VLDLR as revealed by immunostaining of mouse heart tissue sections. Anti-VLDLR mAb E8 and 6A6 and anti-β-tubulin mAb G-8 were obtained from Santa Cruz Biotechnology. Purified mouse IgG1, κ isotype control antibody, was from Biolegend. Goat secondary anti-mouse antibodies conjugated with HRP and HRP substrate SureBlue TMB were from KPL. The anti-His(C-term) antibody (anti-His tag mAb) conjugated with HRP was from Invitrogen. Calcein AM fluorescent dye, phorbol 12-myristate 13-acetate (PMA), and N-formyl-Met-Leu-Phe (fMLP) were obtained from BD Biosciences, Promega, and Sigma-Aldrich, respectively.
Mice
C57BL/6J mice aged 8–12 weeks were from The Jackson Laboratory. All mice were housed in a pathogen-free facility, and all procedures were performed with approval of the University of Maryland Institutional Animal Care and Use Committee.
Preparation of recombinant fragments
The recombinant (β15–66)2 fragment was prepared as described earlier (10, 20). The soluble form of human VLDLR that contains its entire extracellular portion (sVLDLR) was prepared with the Drosophila Expression System as previously described (18, 19). Recombinant fragments of VLDLR containing various combinations of its CR-domains, VLDLR(1–8), VLDLR(1–4), VLDLR(5–8), VLDLR(1–2), VLDLR(2–3), VLDLR(2–4), and VLDLR(3–4), were expressed in E. coli, purified, and refolded as described earlier (19). Two additional fragments, VLDLR(5–6) and VLDLR(7–8) containing CR-domains 5–6 and 7–8 (a.a. residues 164–248 and 249–328, respectively), both with His tag, were expressed in E. coli strain BL21(DE2)pLysS using a pET-20b expression vector. The cDNA fragments encoding these regions were produced by PCR using following primers in which the restrictase-recognition sequences are underlined: 5′-GATCGCCAACATATGCCAACCTGTGGCGCCCATG-3′ (forward) and 5′-GCTGCTCGAGTCAGTGGTGGTGGTGGTGGTGAGAGGGACAGTTGACCTCATC-3′ (reverse) for VLDLR(5–6), and 5′-GATCGCCAACATATGCGAACTTGCCGACCTGAC-3′ (forward) and 5′-GCTGCTCGAGTCAGTGGTGGTGGTGGTGGTGACACTCTTTCAGGGGCTCATC-3′ (reverse) for VLDLR(7–8). The full-length cDNA encoding human VLDLR was used as a template. The PCR products were sub-cloned into the pET20b expression vector using NdeI and XhoI restriction sites and then transformed into DH5α E. coli host cells (Invitrogen). For preparation of VLDLR(5–6) and VLDLR(7–8), the BL21/pLysS E. coli host cells were transformed with the resulting plasmids and both fragments were produced, purified, and refolded following the procedures described earlier (19). Concentrations of the newly expressed VLDLR fragments were determined spectrophotometrically using extinction coefficients (E280,1%) estimated from fragments’ sequences by the ProtParam online tool (http://www.expasy.ch/tools/protparam.html); their molecular masses were also estimated using this tool. The following molecular masses and E280,1% values were obtained: VLDLR(5–6), 10.0 kDa and 11.7; VLDLR(7–8), 9.8 kDa and 6.4.
Solid-phase binding assay
To map epitopes for the anti-VLDLR(1–8) mAb 1H10, 1H5, and 5F3, wells of Immulon 2HB microtiter plates were coated overnight at 4°C with various VLDLR fragments, each at 1 μg/mL in 0.1M Na2CO3, pH 9.5 (coating buffer). The wells were then blocked with Blocker BSA in TBS (Thermo Scientific) for 1 hour at room temperature. Following washing with Tris-buffered saline (TBS) containing 0.05% Tween 20 and 1 mM CaCl2 (binding buffer), the anti-VLDLR(1–8) mAbs, each at 1 μg/mL in the binding buffer, were added to the wells and incubated for 1 hour at 37°C. Bound mAbs were detected by reaction with the HRP-conjugated goat anti-mouse antibodies (1 hour at 37°C) as previously described (19). To estimate equilibrium dissociation constants (Kd), the wells were coated overnight at 4°C with VLDLR(1–8) at 1 μg/mL in the coating buffer, blocked as above, and the anti-VLDLR mAbs at indicated concentrations were added to the wells and incubated for 1 hour at 37°C. Bound mAbs were detected as described above, and Kd values were determined as described previously (19). To test the inhibitory effect of mAb 1H10, 1H5, and 5F3, the wells were coated with (β15–66)2 at 2 μg/mL in the coating buffer overnight at 4°C and blocked as above, VLDLR(1–8) or sVLDLR at 10 nM in the binding buffer was pre-incubated with increasing concentrations of mAbs for 30 min at 37°C, and 100 μL aliquots of the mixture were added to the wells and incubated for 1 hour at 37°C. Bound VLDLR(1–8) or sVLDLR was detected by the reaction with the anti-His tag mAb as previously described (19).
Immunoblotting
For evaluation of VLDLR levels in human umbilical vein endothelial cells (HUVECs), the cells were cultured in the medium containing either 2 or 10% fetal bovine serum (FBS) for 48 hours. The cells were lysed in sample buffer (Invitrogen), the lysates were subjected to SDS-PAGE, and the proteins were transferred to nitrocellulose membranes. VLDLR or β-tubulin were detected by immunoblotting with a mixture of anti-VLDLR mAb E8 and 6A6, or anti-β-tubulin mAb G-8, respectively, followed by secondary anti-mouse HRP-conjugated IgG. The blots were developed with SuperSignal West Pico chemiluminescent substrate (Pierce).
Cell culture and treatments
HUVECs (Lonza, Cat. #C2519AS) were cultured in EBM-2 basal medium supplemented with EGM SingleQuot Kit (Lonza), which contained 2% FBS, according to the manufacturer’s instruction. HUVECs were also cultured in the same medium with 10% FBS. The HL-60 human promyelocytic cell line (ATCC) was cultured and differentiated to a neutrophil-like lineage as described earlier (20, 22). All cell cultures were maintained at 37°C in 5% CO2.
Leukocyte transendothelial migration assay
Transendothelial migration experiments were performed with 24-well plates containing 8-μm pore size PET membrane inserts (BD Biosciences) as described earlier (4, 20). Briefly, HUVECs were grown to confluence on the insert membrane and serum-starved for 2 hours before experiments. Calcein AM-labeled differentiated HL-60 cells were stimulated with PMA. Stimulated HL-60 cells in IMDM containing 1.5 μM NDSK-II without or with increasing concentrations of mAbs or 500 nM IgG1 (control) were added on top of the HUVEC monolayer. The inserts were placed into the wells containing chemoattractant fMLP, transmigration proceeded for 4 hours, and HL-60 cells migrated to the bottom wells were quantified with fluorescence plate reader.
Mouse model of peritonitis
Mice (6–8 per group) received an intravenous injection (via the tail vein) of 100 μg mAb 1H10 or 1H5, both in 200 μL Phosphate-buffered saline (PBS), 10 minutes prior to intraperitoneal injection of 3.85% Bacto Fluid thioglycollate (1 mL per mouse). Mice in control groups received an intravenous injection of the same volume and amount of IgG1 in PBS. Four hours after the injections, each group of mice was euthanized, peritoneal cells were lavaged by injecting intraperitoneally with 3 mL ice-cold PBS, and total lavage fluid was withdrawn. Total cell number in lavage fluid was determined using a hemocytometer and the percentage of neutrophils (~90%) was determined by cytospin, as previously described (23).
Mouse model of myocardial ischemia-reperfusion injury
Anesthesia in C57BL/6J mice was induced with 3–4% isoflurane and then maintained at 2% isoflurane. An ocular lubricant paralube was applied to the animal eyes to prevent corneal desiccation. With orotracheal intubation with a 20G catheter (0.9-mm outside diameter), mouse was ventilated mechanically (Harvard Apparatus, Model 687), supplemented with oxygen at a rate of 105 breaths/min and with a tidal volume of 10–15 mL/kg body weight. After a front mid-cervical incision, the left jugular vein was cannulated with a saline-filled PE 10 tube. A left thoracotomy was then made between the 3rd and 4th rib. The left coronary artery at the level of the atrial appendage was ligated with an 8–0 silk suture against a piece of 1–0 silk suture. Ischemia was maintained for 30 min after which both sutures were removed to initiate blood flow into the ischemic myocardium (reperfusion). 100 μg in 50 μL PBS of each mAb or IgG1 (control) was bolus injected via the jugular vein catheter into the mice twice, 1 min prior and 30 min after reperfusion. After 2-hours reperfusion, mice were euthanized and the hearts were retrieved. To identify infarcted areas, the hearts were perfused with 1% triphenyltetrazolium in PBS for 15 min, cut into 2 mm-thick sections using a standard heart Matrix (Roboz), and the sections were additionally incubated in 1% triphenyltetrazolium for 15 min at 37°C. The size of infarcted areas, which appears in pale color, was estimated using ImageJ program (NIH).
Statistical analysis
Statistical analysis was done using Student’s t-test with a P value of less than 0.05 being considered significant. All statistical analyses were performed in SigmaPlot 13.0 software (Systat Software).
Results
Epitope mapping
To localize epitopes for the anti-VLDLR monoclonal antibodies, 1H5, 1H10, and 5F3, prepared earlier (18) by immunizing VLDLR-deficient mice with the VLDLR(1–8) fragment containing eight CR-domains of VLDLR, we used a number of previously described recombinant VLDLR fragments including CR-domains 1–4, 5–8, 1–2, 2–3, 3–4, 2–4 (19). In addition, we prepared two new fragments, VLDLR(5–6) and VLDLR(7–8), containing CR-domains 5–6 and 7–8 that were also used for epitope mapping.
In ELISA, when the three mAbs were incubated with all above mentioned VLDLR fragments immobilized on microtiter plates, mAb 5F3 bound to VLDLR(1–8), VLDLR(5–8), and VLDLR(7–8); no reasonable binding was observed with the other fragments (Figure 1A). These results indicate that the epitope for mAb 5F3 is located in CR-domains 7–8. In contrast, mAb 1H10 did not interact with VLDLR(7–8) while its interaction with the remaining fragments except VLDLR(1–2) was obvious, indicating that the epitope for this antibody is located in CR-domains 3–6. The third monoclonal antibody, 1H5, interacted with all fragments except VLDLR(3–4) and VLDLR(7–8), indicating that the epitope for this mAb is located in CR-domains 1–2 and 5–6. However, the fact that mAb 1H5 bound equally well to both VLDLR(1–2) and VLDLR(2–3) suggests that the first CR-domain may not be involved in mAb-binding. Similar results were obtained when all above mentioned VLDLR fragments were incubated with immobilized mAb 1H5, 1H10, and 5F3 (not shown). Location of the epitopes for all three mAbs is presented in Figure 1B. It should be noted that the affinity of these mAbs to VLDLR(1–8) was very high. The Kd values for the interaction of mAb 1H10, 1H5, and 5F3 with the VLDLR(1–8) fragment determined by ELISA were found to be 0.49 ± 0.11, 0.31 ± 0.06, and 0.12 ± 0.02 nM, respectively (Figure 2).
Figure 1. Localization of the epitopes for the anti-VLDLR monoclonal antibodies used in the present study.
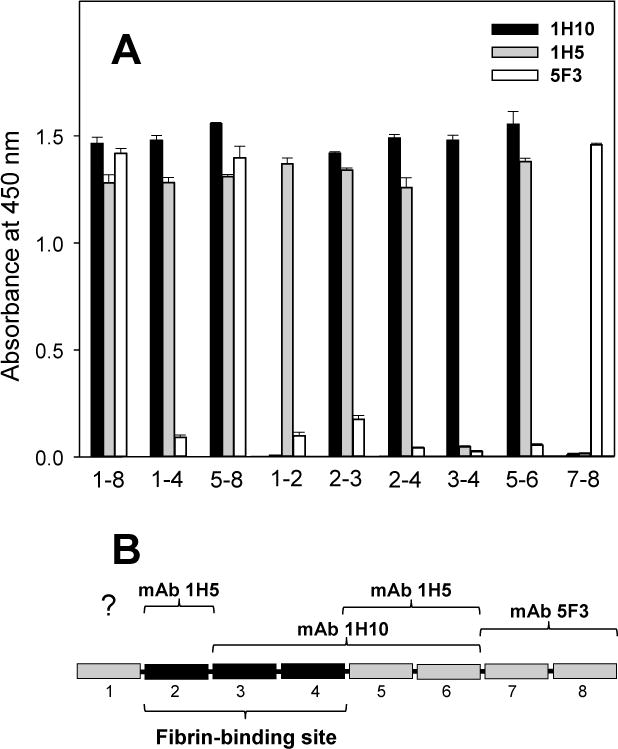
(A) ELISA-detected interaction between the anti-VLDLR monoclonal antibodies, 1H10 (black bars), 1H5 (grey bars), and 5F3 (empty bars), and various VLDLR fragments. The three monoclonal antibodies, each at 1 μg/mL, were incubated with microtiter wells coated with the recombinant VLDLR(1–8), VLDLR(1–4), VLDLR(5–8), VLDLR(1–2), VLDLR(2–3), VLDLR(2–4), VLDLR(3–4), VLDLR(5–6), and VLDLR(7–8) fragments, and the bound mAbs were detected with the goat anti-mouse secondary antibodies as described in Materials and methods. The corresponding VLDLR fragments are indicated by numbers (1–8, 1–4, 5–8, etc.). The bars are representative of at least 2 independent experiments; error bars represent the standard deviation of triplicate determinations. (B) Schematic representation of the ligand-binding region of the VLDL receptor including 8 CR-domains; the previously localized fibrin-binding CR-domains 2–4 (19) are presented by black bars; the epitopes localized for each of the three mAb are indicated.
Figure 2. ELISA-detected interaction between the anti-VLDLR monoclonal antibodies and the ligand-binding fragment of VLDLR.
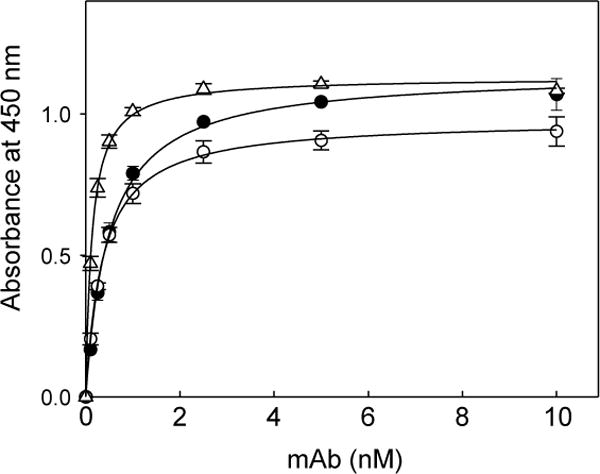
Increasing concentrations of the anti-VLDLR monoclonal antibodies 1H10 (filled circles), 1H5 (empty circles), and 5F3 (empty triangles) were incubated with microtiter wells coated with the recombinant VLDLR(1–8) fragment, and the bound antibodies were detected with goat anti-mouse secondary antibodies as described in Materials and methods. The data are representative of 3 independent experiments; error bars represent the standard deviation of triplicate determinations.
Inhibitory effect of anti-VLDLR mAbs on the interaction of VLDLR with fibrin
We previously localized the VLDLR-binding site to the βN-domains of fibrin (4) and the complementary fibrin-binding site to the CR-domains 2–4 of VLDLR (19). Since the epitopes for mAb 1H10 and 1H5 include CR-domains 3–4 and 2, respectively, we hypothesized that these two mAbs should inhibit interaction of the VLDL receptor with fibrin. To test this hypothesis, we examined the effect of all three mAbs on the interaction of the VLDLR(1–8) fragment with the (β15–66)2 fragment representing the VLDLR-binding βN-domains of fibrin.
In ELISA experiments, when VLDLR(1–8) was incubated with increasing concentrations of each of the three antibodies and then added to immobilized (β15–66)2, mAb 1H10 and 1H5 both inhibited binding of VLDLR(1–8) to (β15–66)2 in a concentration-dependent manner while mAb 5F3 exhibited little effect (Figure 3). The inhibition was practically complete at about a 2-fold molar excess of mAb 1H10 and 1H5 over VLDLR(1–8) while only about 20% inhibition was achieved even with a 10-fold molar excess of mAb 5F3. Similar results were obtained when sVLDLR representing the extracellular portion of the VLDLR receptor (sVLDLR) was used instead of VLDLR(1–8) (not shown). These experiments clearly indicate that mAb 1H10 and 1H5 are efficient inhibitors of fibrin-VLDL receptor interaction while the inhibitory potency of mAb 5F3 is very low.
Figure 3. Inhibitory effect of the anti-VLDLR monoclonal antibodies on the interaction between fibrin- and VLDLR-derived fragments detected by ELISA.
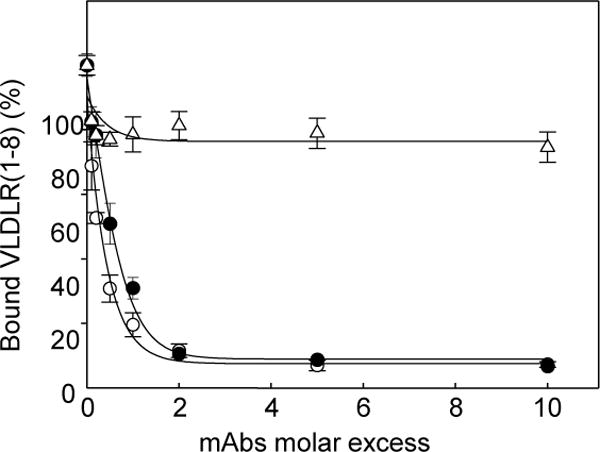
Increasing concentrations of the anti-VLDLR monoclonal antibodies 1H10 (filled circles), 1H5 (empty circles), and 5F3 (empty triangles) were preincubated with the VLDLR(1–8) fragment, the mixtures were added to microtiter wells coated with the fibrin (β15–66)2 fragment, and bound VLDLR(1–8) was detected with the anti-His tag monoclonal antibody, as described in Materials and methods. The data are expressed as a percentage of control binding in the absence of the anti-VLDLR mAbs and are representative of 3 independent experiments; error bars represent the standard deviation of triplicate determinations.
Inhibitory effect of the mAb 1H10 and 1H5 on leukocyte transmigration in vitro
Since mAb 1H5 and 1H10 both efficiently inhibited interaction between the (β15–66)2 fragment and VLDLR(1–8) or sVLDLR, we hypothesized that these mAbs should also inhibit previously discovered fibrin-VLDLR-dependent transendothelial migration of leukocytes and thereby inflammation. To test this hypothesis, we studied the effect of mAb 1H10 and 1H5 on leukocyte transmigration in vitro using a leukocyte transendothelial migration assay. For this assay, HL-60 cells were differentiated into neutrophil-like cells using a procedure described by Hauert et al. (24), who demonstrated that such cells constitute a valid model system for the analysis of human neutrophil migration. Transmigration of the differentiated cells across a confluent HUVEC monolayer was stimulated by fibrin mimetic NDSK-II, which was shown to be a potent stimulator of leukocyte transmigration (3, 4, 20).
In our previous studies (4, 20), the transmigration experiments were performed with HUVECs grown in the medium containing 2% FBS. In the present study, we compared leukocyte transmigration across HUVEC monolayers grown in the medium containing either 2% or 10% FBS and found that NDSK-II much better stimulates leukocyte transmigration when HUVECs are grown in 10% FBS (Figure 4). The fact that NDSK-II-stimulated transmigration was almost completely inhibited by RAP, a well-known antagonist of ligand binding of VLDLR and other LDL receptor family members, further confirmed that this process is VLDLR-dependent. Additional immunoblot experiments revealed that expression of VLDLR on HUVECs grown in 10% FBS medium was about 3-fold higher than that on HUVECs grown in 2% BFS medium while expression of tubulin (control) was practically the same in both media (Figure 4, inset). Such increased expression of VLDLR may explain the observed superior stimulating effect of NDSK-II on the VLDLR-dependent pathway of leukocyte transmigration. Thus, all subsequent transmigration experiments were performed with HUVECs grown in 10% FBS-containing media.
Figure 4. Effect of serum concentration on VLDLR expression levels in HUVECs and NDSK-II-induced migration of neutrophils through HUVEC monolayer.
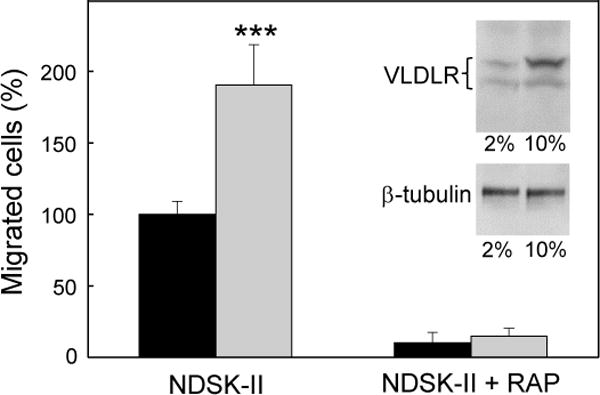
HUVECs were cultured in the medium containing 2% (black bars) or 10% (grey bars) FBS for 48 hours before transmigration assays. HUVECs were grown to confluency on gelatin-coated cell culture inserts. Calcein AM-labeled HL-60 cells differentiated into neutrophil-like cells were added to the upper chambers on top of the HUVEC monolayers in the presence of 1.5 μM NDSK-II without or with 0.5 μM RAP. The cells migrated into the lower chambers were collected and quantified as described in Materials and methods. The number of cells migrated in the absence of NDSK-II (control) were subtracted and the results were expressed as percentage of the cells migrated in the presence of NDSK-II. The graph shows combined data obtained from 2 independent experiments performed in triplicate; error bars denote means ± SD; ***P < 0.001. Insert shows the levels of VLDLR (top panel) and β-tubulin (lower panel) in HUVECs cultured in 2% or 10% FBS as determined by immunoblotting; upper and lower bands in the top panel represent mature VLDLR and underglycosylated VLDLR precursor, respectively.
Figure 5A shows the results of transmigration experiments indicating that control IgG1 had no effect on NDSK-II-induced leukocyte transmigration while mAb 1H10 inhibited this process in a concentration-dependent manner with the saturation at about 500 nM. The inhibitory effect of mAb 1H10 at this and higher concentration (2 μM) was found to be about 80% indicating that in this in vitro model NDSK-II-induced leukocyte transmigration is carried out mainly through the fibrin-VLDLR-dependent pathway. The results obtained with mAb 1H5 were very similar (Figure 5B). Thus, mAb 1H10 and 1H5 both efficiently inhibited transendothelial migration of leukocytes in these in vitro experiments.
Figure 5. Inhibitory effect of the anti-VLDLR monoclonal antibodies on NDSK-II-induced transendothelial migration of leukocytes (neutrophils) in vitro.
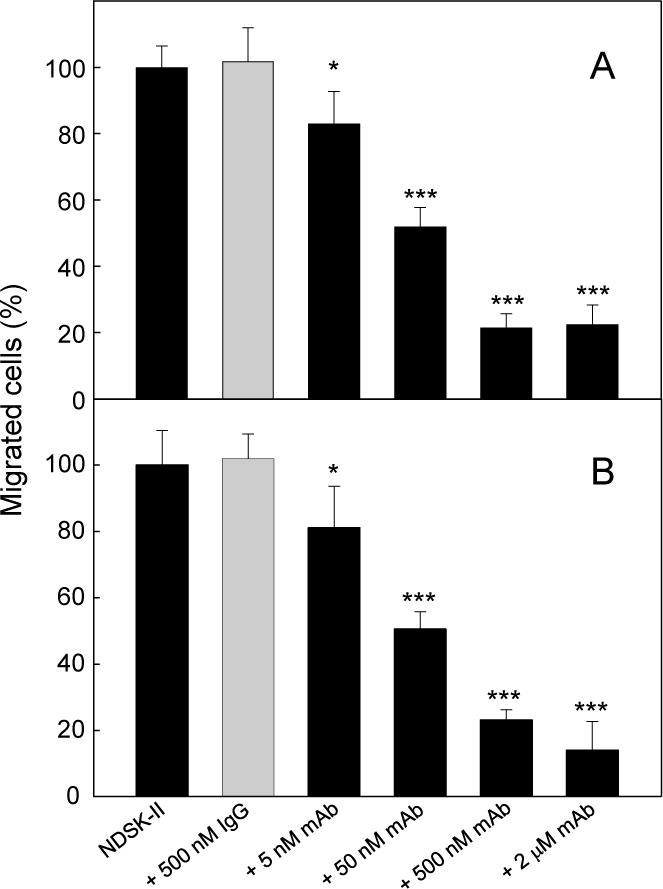
HUVECs were cultured in the medium containing 10% FBS for 48 hours before transmigration assays and then were grown to confluency on gelatin-coated cell culture inserts. Calcein AM-labeled HL-60 cells differentiated into neutrophil-like cells were added to the upper chambers on top of the HUVEC monolayers in the presence of 1.5 μM NDSK-II without or with IgG1, or with increasing concentrations (from 5 nM to 2 μM) of mAb 1H10 (panel A) or mAb 1H5 (panel B). Mouse IgG1 was used as a negative IgG isotype control. The cells migrated into the lower chambers were collected and quantified as described in Materials and methods. The number of cells migrated in the absence of NDSK-II (control) were subtracted and the results were expressed as percentage of the cells migrated in the presence of NDSK-II. Each graph shows combined data obtained from 2 independent experiments performed in triplicate; error bars denote means ± SD. *P < 0.05; ***P < 0.001.
In vivo study of the anti-inflammatory effect of mAb 1H10 and 1H5
To test the effect of mAb 1H10 and 1H5 on leukocyte transmigration in vivo, we used a mouse model of peritonitis in which leukocyte migration from the circulation into the peritoneum is stimulated by intraperitoneal injection of thioglycollate, and leukocyte (neutrophil) accumulation is evaluated after 4 hours by counting the cells in the peritoneal lavage. In our experiments, each mice was injected intravenously with mAb 1H10 or mAb 1H5; control mice received an intravenous injection of non-specific IgG1. The dose for injection, 100 μg, was selected based on our transmigration assay experiments in which the inhibition plateau was observed at mAb concentration of 0.5–2.0 μM (Figure 5). At this dose, the estimated concentration of mAb in the blood of mice was approximately 0.5 μM. The experiments revealed that the number of neutrophils accumulated in the peritoneum of mAb-treated mice was reduced by about 2-fold (Figure 6). This indicates that both monoclonal antibodies, 1H10 and 1H5, efficiently inhibited leukocyte transmigration and thereby inflammation in vivo.
Figure 6. Inhibitory effect of the monoclonal antibodies 1H10 and 1H5 on neutrophil infiltration in a mouse model of peritonitis.
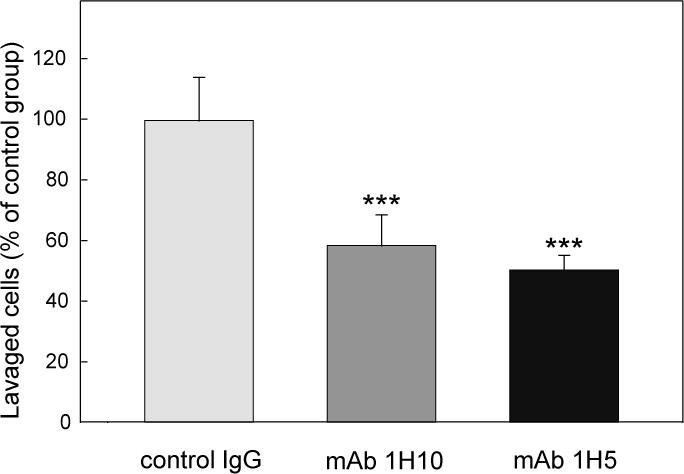
Monoclonal antibody 1H10 or 1H5, 100 μg each in 200 μL PBS, was injected intravenously in each mouse prior to thioglycollate injection; control mice were injected with the same amount and volume of IgG1. Four hours after thioglycollate injection, the number of infiltrated neutrophils was estimated as described in Materials and methods. The graph shows combined data from 2 independent experiments, and the results are means ± SD (n = 6–8); ***P < 0.001.
Testing the cardioprotective effect of mAb 1H10 and 1H5
It was previously shown that fibrin-derived β15–42 containing peptides, which inhibit transmigration of leukocytes across the endothelium, substantially reduce leukocyte infiltration and infarct size in rodent models of myocardial ischemia-reperfusion injury and thereby are potential candidates for myocardial reperfusion therapy in humans (3, 20). To evaluate the potential of the inhibitory anti-VLDLR mAbs for myocardial infarction therapy, we used a mouse model of myocardial ischemia-reperfusion injury. In this model, ischemia is achieved by ligation of the left coronary artery, reperfusion is initiated after 30 min, and the size of infarcted area is evaluated after 2 hours of reperfusion. The injected dose of mAbs, 100 μg, was the same as in the peritonitis model experiments; however, the injection was made twice to insure sufficient amount of mAbs for the inhibition. Using this model, we tested the cardioprotective effect of both mAbs, 1H10 and 1H5. The results presented in Figure 7 indicate that infarct size in mice treated with both mAbs was reduced by about two-fold in comparison with that in control mice. Thus, in this in vivo model both monoclonal antibodies, 1H10 and 1H5, exhibited significant cardioprotective effect.
Figure 7. Cardioprotective effect of mAb 1H10 and 1H5 during myocardial ischemia-reperfusion injury.
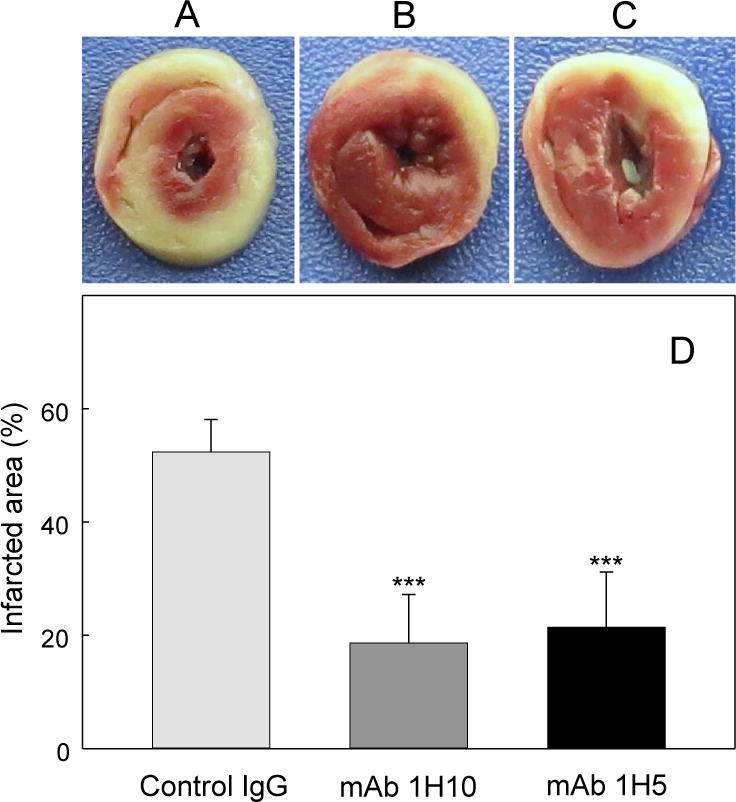
Representative mouse heart slices after myocardial ischemia-reperfusion in mice treated with control IgG1 (A), mAb 1H10 (B), or mAb 1H5 (C). The size of infarcted areas, which appear pale in color in (A-C), was determined with ImageJ (NIH), and the results are presented in (D) as a percentage of total area of the slices. The results are means ± SD (n = 5). ***P < 0.001.
Discussion
Our discovery of a novel interaction between fibrin and the endothelial VLDL receptor, which promotes transendothelial migration of leukocytes (4), opened a perspective for the development of antagonists of this interaction that may be potent inhibitors of fibrin-dependent leukocyte transmigration and thereby inflammation. As the first step towards the development of such antagonists, we localized the VLDLR-binding site in fibrin and the complementary fibrin-binding site in the VLDL receptor (4, 19). In the present study, we made our next step, namely, we identified two monoclonal antibodies whose epitopes overlap with the fibrin-binding site of VLDLR and found that they completely inhibit fibrin-VLDLR interaction. Furthermore, we demonstrated the inhibitory effect of these antibodies on leukocyte transmigration in vitro, and their anti-inflammatory properties and cardioprotective effect in the in vivo models.
Among three previously generated anti-VLDLR monoclonal antibodies, 1H5, 1H10, and 5F3, the epitopes for two of them were partially characterized (18). Namely, it was shown that mAb 1H5 and 1H10 both recognize recombinant VLDLR fragments containing CR-domains 1–8, 3–6, and 5–8, while mAb 1H10 failed to recognize a fragment containing CR-domains 1–4 and mAb 1H5 showed reduced binding to this fragment (18). Based on these findings it was concluded that both antibodies prefer domains located within the C-terminal region of the VLDLR ligand binding region which includes domains 1–8 (18). In the present study, we confirmed binding of mAb 1H10 and 1H5 to CR-domains 1–8, 3–6, and 5–8; however, we also observed a very strong binding of both mAbs to CR-domains 1–4 (Figure 1A). Such a discrepancy may be connected with different methods of detection of the binding or restricted number of VLDLR fragment used in the previous study. Whatever the reason for the discrepancy is, our binding experiments with smaller VLDLR fragments allowed us to perform more precise localization of the epitopes for mAb 1H10 and 1H5 as well as to localize the epitope for mAb 5F3.
The present study localized the epitope for mAb 1H10 to CR-domains 3–6. In contrast, the epitope for mAb 1H5 was localized to CR-domains 1–2 and 5–6, although the first CR-domain may not be a part of the epitope as mentioned in Results. The fact that these two pairs of CR-domains are not contiguous may suggest that they are closely spaced in the 3D structure. Alternatively, mAb 1H5 may recognize identical sequences in these highly homologous CR-domains. The second alternative seems to be less probable since our control ELISA experiments revealed that both mAbs exhibit no interaction with another member of the LDL receptor family, LRP, which has 31 homologous CR-domains (not shown), suggesting that mAb 1H10 and mAb 1H5 are highly specific to VLDLR.
Since among domains containing the epitope for mAb 1H10 and 1H5 (Figure 1B), domains 2–4 were shown to be involved in fibrin binding (19), we expected that both mAbs should inhibit the interaction of fibrin with VLDLR. This was directly confirmed by our inhibition experiments. Furthermore, our in vitro experiments revealed that mAb 1H10 and 1H5 both efficiently inhibited transendothelial migration of leukocytes. The inhibitory effect of these antibodies on the NDSK-II-induced leukocyte transmigration was very significant (~80%). The in vivo inhibitory effect of both mAbs on neutrophil infiltration into the peritoneum was less pronounced (~50%) but still very considerable. Thus, the present study revealed that mAb 1H10 and 1H5 both are potent inhibitors of the fibrin-VLDLR-dependent pathway of leukocyte transmigration and thereby inflammation.
Our previous conclusion that fibrin-VLDLR interaction promotes transendothelial migration of leukocytes was mainly based on the ability of RAP and the (β15–66)2 fragment to inhibit leukocyte transmigration in the in vitro experiments and on the lack of such inhibition by (β15–66)2 in the in vivo experiments with VLDLR-deficient mice (4). However, the facts that RAP and (β15–66)2 are not specific inhibitors of fibrin-VLDLR interaction left some doubts about the validity of such conclusion. Indeed, RAP inhibits interaction of other members of the LDL receptor family with their ligands (21, 25) and (β15–66)2 also interacts with heparin and proteoglycans (26), VE-cadherin (10), and possibly some other endothelial receptor(s). In the present study, we demonstrated in direct experiments that highly specific inhibitors of fibrin-VLDLR interaction, mAb 1H10 and 1H5, inhibit leukocyte transmigration in both in vitro assay and in vivo models. These findings confirm and further reinforce the above mentioned conclusion.
Our experiments using mouse model of myocardial ischemia-reperfusion injury revealed that treatment with either mAb 1H10 or 1H5 of mice subjected to ischemia-reperfusion reduced myocardial infarct size by about two-fold, indicating that these mAbs have significant cardioprotective effect. We previously observed a similar cardioprotective effect with the synthetic (β15–44)2 fragment, which corresponds to the N-terminal halves of a pair of fibrin βN-domains and has high affinity to VE-cadherin (20). However, that effect was attributed to the inhibition of the fibrin-VE-cadherin pathway of leukocyte transmigration by (β15–44)2 (20). In contrast, monoclonal antibodies 1H10 and 1H5 both compete for fibrin-VLDLR interaction and thus should presumably inhibit only the fibrin-VLDLR dependent pathway of leukocyte transmigration.
In summary, the present study has identified two anti-VLDLR monoclonal antibodies, mAb 1H10 and mAb 1H5, whose epitopes overlap with fibrin-binding domains of the VLDL receptor. Binding experiments revealed that both mAbs have high affinities to the fibrin-binding fragment of VLDLR and efficiently inhibit interaction of fibrin with this receptor. Furthermore, both monoclonal antibodies efficiently inhibited transendothelial migration of leukocytes in the in vitro experiments and significantly reduced infiltration of leukocytes into the peritoneum in the in vivo experiments. Finally, both antibodies exhibited significant cardioprotective effect in the experiments using a mouse model of myocardial ischemia-reperfusion injury. Thus, monoclonal antibodies 1H10 and 1H5 are novel specific inhibitors of the fibrin-VLDLR dependent leukocyte transmigration pathway that may be developed as potent therapeutics for treatment of inflammation-related cardiovascular diseases including myocardial ischemia-reperfusion injury.
Acknowledgments
Financial Support: This work was supported by National Institutes of Health R01 Grants HL056051 to L.M., HL114379 and HL120388 to D.K.S, and NS082607 to L.Z.
Footnotes
Conflicts of interest
None declared.
Disclaimer
This work was prepared while Alexey Belkin was employed at the University of Maryland School of Medicine. The opinions expressed in this article are the author’s own and do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
References
- 1.Clark RA. Fibrin and wound healing. Ann NY Acad Sci. 2001;936:355–367. doi: 10.1111/j.1749-6632.2001.tb03522.x. [DOI] [PubMed] [Google Scholar]
- 2.Altieri DC. Regulation of leukocyte-endothelium interaction by fibrinogen. Thromb Haemost. 1999;82:781–786. [PubMed] [Google Scholar]
- 3.Petzelbauer P, Zacharowski PA, Miyazaki Y, et al. The fibrin-derived peptide Bβ15–42 protects the myocardium against ischemia-reperfusion injury. Nat Med. 2005;11:298–304. doi: 10.1038/nm1198. [DOI] [PubMed] [Google Scholar]
- 4.Yakovlev S, Mikhailenko I, Cao C, et al. Identification of VLDLR as a novel endothelial cell receptor for fibrin that modulates fibrin-dependent transendothelial migration of leukocytes. Blood. 2012;119:637–644. doi: 10.1182/blood-2011-09-382580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost. 2005;3:1894–1904. doi: 10.1111/j.1538-7836.2005.01365.x. [DOI] [PubMed] [Google Scholar]
- 6.Medved L, Weisel JW, on behalf of Fibrinogen and Factor XIII Subcommittee of Scientific Standardization Committee of International Society on Thrombosis and Haemostasis Recommendations for nomenclature on fibrinogen and fibrin. J Thromb Haemost. 2009;7:355–359. doi: 10.1111/j.1538-7836.2008.03242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blomback B, Blomback M, Henschen A, et al. N-terminal disulphide knot of human fibrinogen. Nature. 1968;218:130–134. doi: 10.1038/218130a0. [DOI] [PubMed] [Google Scholar]
- 8.Bach TL, Barsigian C, Yaen CH, et al. Endothelial cell VE-cadherin functions as a receptor for the β15–42 sequence of fibrin. J Biol Chem. 1998;273:30719–30728. doi: 10.1074/jbc.273.46.30719. [DOI] [PubMed] [Google Scholar]
- 9.Martinez J, Ferber A, Bach TL, Yaen CH. Interaction of fibrin with VE-cadherin. Ann N Y Acad Sci. 2001;936:386–405. doi: 10.1111/j.1749-6632.2001.tb03524.x. [DOI] [PubMed] [Google Scholar]
- 10.Gorlatov S, Medved L. Interaction of fibrin(ogen) with the endothelial cell receptor VE-cadherin: mapping of the receptor-binding site in the NH2-terminal portions of the fibrin β chains. Biochemistry. 2002;41:4107–4116. doi: 10.1021/bi0160314. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi S, Kawarabayasi Y, Nakai T, et al. Rabbit very low density lipoprotein receptor: a low density lipoprotein receptor-like protein with distinct ligand specificity. Proc Natl Acad Sci USA. 1992;89:9252–9256. doi: 10.1073/pnas.89.19.9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakai J, Hoshino A, Takahashi S, et al. Structure, chromosome location, and expression of the human very low density lipoprotein receptor gene. J Biol Chem. 1994;269:2173–2182. [PubMed] [Google Scholar]
- 13.Trommsdorff M, Gotthardt M, Hiesberger T, et al. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 14.Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci. 2006;7:850–859. doi: 10.1038/nrn2009. [DOI] [PubMed] [Google Scholar]
- 15.Hembrough TA, Ruiz JF, Swerdlow BM, et al. Identification and characterization of a very low density lipoprotein receptor-binding peptide from tissue factor pathway inhibitor that has antitumor and antiangiogenic activity. Blood. 2004;103:3374–3380. doi: 10.1182/blood-2003-07-2234. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi S, Sakai J, Fujino T, et al. The very low-density lipoprotein (VLDL) receptor: characterization and functions as a peripheral lipoprotein receptor. J Atheroscler Thromb. 2004;11:200–208. doi: 10.5551/jat.11.200. [DOI] [PubMed] [Google Scholar]
- 17.Lillis AP, Van Duyn LB, Murphy-Ullrich JE, et al. LDL receptor-related protein 1: unique tissue-specific functions revealed by selective gene knockout studies. Physiol Rev. 2008;88:887–918. doi: 10.1152/physrev.00033.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruiz J, Kouiavskaia D, Migliorini M, et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J Lipid Res. 2005;46:1721–1731. doi: 10.1194/jlr.M500114-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Yakovlev S, Medved L. Interaction of fibrin with the very low density lipoprotein receptor: Further characterization and localization of the fibrin-binding site. Biochemistry. 2015;54:4751–4761. doi: 10.1021/acs.biochem.5b00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yakovlev S, Gao Y, Cao C, et al. Interaction of fibrin with VE-cadherin and anti-inflammatory effect of fibrin-derived fragments. J Thromb Haemost. 2011;9:1847–1855. doi: 10.1111/j.1538-7836.2011.04438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams SE, Ashcom JD, Argraves WS, et al. A novel mechanism for controlling the activity of α2-macroglobulin receptor/low density lipoprotein receptor-related protein. Multiple regulatory sites for 39-kDa receptor-associated protein. J Biol Chem. 1992;267:9035–9040. [PubMed] [Google Scholar]
- 22.Collins SJ, Ruscetti FW, Gallagher RE, et al. Terminal differentiation of human promyelocytic leukemia cells induced by dimethyl sulfoxide and other polar compounds. Proc Natl Acad Sci USA. 1978;75:2458–2462. doi: 10.1073/pnas.75.5.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao C, Lawrence DA, Strickland DK, et al. A specific role of integrin Mac-1 in accelerated macrophage efflux to the lymphatics. Blood. 2005;106:3234–3241. doi: 10.1182/blood-2005-03-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hauert AB, Martinelli S, Marone C, et al. Differentiated HL-60 cells are a valid model system for the analysis of human neutrophil migration and chemotaxis. Int J Biochem Cell Biol. 2002;34:838–854. doi: 10.1016/s1357-2725(02)00010-9. [DOI] [PubMed] [Google Scholar]
- 25.Herz J, Goldstein JL, Strickland DK, et al. 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/α2-macroglobulin receptor. J Biol Chem. 1991;266:21232–21238. [PubMed] [Google Scholar]
- 26.Yakovlev S, Gorlatov S, Ingham K, et al. Interaction of fibrin(ogen) with heparin: further characterization and localization of the heparin-binding site. Biochemistry. 2003;42:7709–7716. doi: 10.1021/bi0344073. [DOI] [PubMed] [Google Scholar]