

Abstract
The surface functionalization of TiO2-based materials with alkylsilanes is attractive in several cutting-edge applications, such as photovoltaics, sensors, and nanocarriers for the controlled release of bioactive molecules. (3-Aminopropyl)triethoxysilane (APTES) is able to self-assemble to form monolayers on TiO2 surfaces, but its adsorption geometry and solar-induced photodegradation pathways are not well understood. We here employ advanced experimental (XPS, NEXAFS, AFM, HR-TEM, and FT-IR) and theoretical (plane-wave DFT) tools to investigate the preferential interaction mode of APTES on anatase TiO2. We demonstrate that monomeric APTES chemisorption should proceed through covalent Si–O–Ti bonds. Although dimerization of the silane through Si–O–Si bonds is possible, further polymerization on the surface is scarcely probable. Terminal amino groups are expected to be partially involved in strong charge-assisted hydrogen bonds with surface hydroxyl groups of TiO2, resulting in a reduced propensity to react with other species. Solar-induced mineralization proceeds through preferential cleavage of the alkyl groups, leading to the rapid loss of the terminal NH2 moieties, whereas the Si-bearing head of APTES undergoes slower oxidation and remains bound to the surface. The suitability of employing the silane as a linker with other chemical species is discussed in the context of controlled degradation of APTES monolayers for drug release and surface patterning.
1. Introduction
Alkylsilanes are known to self-assemble at the surface of oxide materials. This ability has been widely exploited, for example, to tune the wetting and electronic transport properties of the surface.1−3 Furthermore, alkylsilanes are often used as coupling agents for attaching organic moieties4 or nanoparticles to the oxide surface,5 forming tailored three-dimensional supramolecular arrays. In this respect, aminoalkylsilanes, such as (3-aminopropyl)triethoxysilane (APTES, Scheme 1), have been extensively employed.6
Scheme 1.

The ability to control the surface properties of technologically relevant materials, such as TiO2,7,8 opens new perspectives in cutting-edge industrial sectors, such as microelectronics,9 sensors,10,11 and photovoltaics.12,13 Furthermore, surface functionalization of TiO2 has been recently employed to immobilize specific biomolecules, paving the way toward the design of novel chemical sensors and biosensors,14 drug release systems,15,16 and materials with increased biocompatibility.17,18 To this end, alkylsilanes such as APTES are ideal linker candidates because they can be selectively photodegraded to prompt the controlled release of proteins, enzymes, and bioactive molecules.15,16 Moreover, by irradiating the silane-functionalized TiO2 surface through a patterning mask,19 a site-selective degradation of the organic layer can be obtained, for example producing patterns with wetting contrast;20 the resulting patterns can be used to assemble complex, hierarchical structures where metal nanoparticles, polymers, or biological molecules are chemisorbed to specific surface sites. Light irradiation of silane-functionalized TiO2 can also be used as a trigger to activate the on-demand controlled release of active substances from stimulus-responsive Pickering emulsions.21,22
Despite the increasing interest in APTES-functionalized TiO2, no general consensus exists on the structure of the organic–inorganic interface. Several models have been proposed, mostly in the case of SiO2 substrates, such as hydrogen bond-mediated coupling of −NH2 (or −NH3+) and surface −OH (or – O–) groups or direct chemisorption via the silane headgroup, with the further complication of the possible formation, to various extents, of −O–Si–O–Si–O– polymeric bonds between neighboring APTES molecules at the surface.23−25 Qualitative estimates of the relative number of various APTES···surface interaction modes are usually drawn by comparing the intensities of XPS peaks in the N 1s region. However, the presence of surface contaminants25,26 makes an accurate quantification on this basis a very difficult task. Also, the exact attribution of the measured XPS chemical shift to the putative state of the surface nitrogen species is a debated aspect.23,24,27 The silane–surface interaction mode is a crucial aspect for the development of advanced composite materials because it determines whether −NH2 terminal groups are available to react with electrophilic species, allowing further functionalization through standard chemical procedures, and it has been shown to depend crucially on the substrate type and functionalization conditions.24,28
In this work, we aim to shed light on the preferred interaction modes of APTES molecules with the TiO2 surface. Synchrotron XPS and NEXAFS experiments were complemented by AFM, HR-TEM, and FT-IR measurements on pristine and APTES-functionalized nanocrystalline TiO2. High-level plane-wave (PW) density functional theory (DFT) calculations were also performed to simulate possible arrangements of the chemisorbed APTES molecules at the TiO2 surface. Finally, AFM, FTIR, XPS, and NEXAFS measurements on ex situ solar-irradiated samples on the time scale of an hour allowed us to go a step further with respect to a purely static picture, providing information on the photoinduced oxidation mechanism of the organic layer. It is worth noting that the silane oxidation pathway has been scarcely investigated in the literature, whereas its study represents a crucial issue for a series of applications involving a light-triggered response, such as the use of these hybrids as nanocarriers for drug release.
2. Materials and Methods
2.1. Materials
All chemicals were purchased from Sigma-Aldrich with reagent-grade purity and used as received; doubly distilled water passed through a Milli-Q apparatus was utilized.
2.2. Sample Preparation
The TiO2 substrate film was deposited on a commercial ITO layer (Sigma-Aldrich, 8–12 Ω/sq) as reported elsewhere.29 The procedure was repeated two times to obtain a 150-nm-thick TiO2 film.11 As specified elsewhere,11 the grazing incidence X-ray diffraction confirmed that it consisted mainly of the anatase polymorph. To remove surface contaminants by photocatalytic oxidation30 and promote extensive surface hydroxylation,19,31 the bare TiO2 film underwent extensive photo-oxidation by UV irradiation through an iron-halogenide lamp (Jelosil HG500, effective power density 58 mW cm–2 between 300 and 400 nm). Water contact angle measurements served as a probe for the completeness of the oxidation process: UV irradiation was stopped when complete wetting was observed. Finally, the film was placed, together with a Teflon cup filled with APTES diluted in toluene (150 μL of 2 M solution), in a glass bottle under a dry N2 atmosphere to avoid APTES gelation. The bottle was kept in an oven at 80 °C for 2 h. Subsequently, the film was sonicated in toluene for 15 min and then blown dry with an N2 gas stream. The final water contact angle, as determined by a Krüss Easy Drop apparatus, was 55 ± 5°.
2.3. Photocatalytic Degradation
The photocatalytic degradation of the APTES layer was studied by irradiating APTES-TiO2 films with simulated solar light using a halogen lamp (Lot Oriel, effective power density 15 mW cm–2 in the range of 300–800 nm). The degradation of the hydrophobic layer was monitored by water contact angle measurements (Krüss Easy Drop). Three different irradiation times (t = 0, 30, and 60 min, corresponding to initial, intermediate, and ca. 0° water contact angle values) were selected as relevant benchmarks to study the reaction. To ensure consistency, all of the irradiated samples were obtained from the same functionalized TiO2 film, which was cut in pieces, and each piece was irradiated for a different time.
2.4. Surface Characterization
The bare and APTES-functionalized TiO2 films were analyzed by atomic force microscopy (AFM) with a Ntegra Aura (NT-MDT) device in tapping mode, equipped with NSC35/AIBS tips (μmasch). The adsorbed species at the surface of APTES-functionalized anatase TiO2 during photocatalytic degradation were analyzed by Fourier transform infrared (FT-IR) spectroscopy using a PerkinElmer Spectrum 100 FT-IR (ATR) spectrometer.
2.4.1. Experiments with Synchrotron Light
X-ray spectroscopy (XPS) and near-edge X-ray absorption fine structure (NEXAFS) experiments were carried out at the bending magnet source of the Materials Science Beamline at the Elettra Synchrotron in Trieste. Full technical details are reported in Section S1.1 of the Supporting Information, SI.
2.5. Computational Setup
The adsorption geometries of APTES molecules at the most stable (101) anatase TiO2 surface32 were investigated by DFT + U calculations33 using the generalized gradient approximation (GGA)34 with the Perdew–Burke–Ernzerhof (PBE) parametrization of the exchange and correlation functional,34 as implemented by the VASP plane-wave program.35,36 See the Supporting Information (Section S1.2) for full details.
3. Results and Discussion
3.1. Surface Morphology
AFM scans on unfunctionalized TiO2 (Figure 1a) show a homogeneous surface with low roughness (estimated root-mean-square, rms, on a 10 × 10 μm2 area <1 nm). APTES-bearing surfaces (Figure 1b) show a homogeneous coverage consistent with the formation of an APTES monolayer (see also Section 3.2). The few nanometric aggregates in Figure 1b are attributable to contamination and residues from the preparation process. The effect of simulated solar light irradiation was also investigated (Figure 1c,d). After 30 min, the surface appears less homogeneous, with appreciable aggregates (Figure 1c) whose presence might be related to the occurrence of either vertical polymerization among physisorbed APTES or their degradation residues. The increased hydrophilicity of TiO2 upon irradiation can easily result in the presence of adsorbed water molecules,31 which might promote such polymerization. Prolonged irradiation (Figure 1d) results instead in a more homogeneous and polished surface, as shown by RMS values, which might be indicative of (almost) complete APTES degradation.
Figure 1.
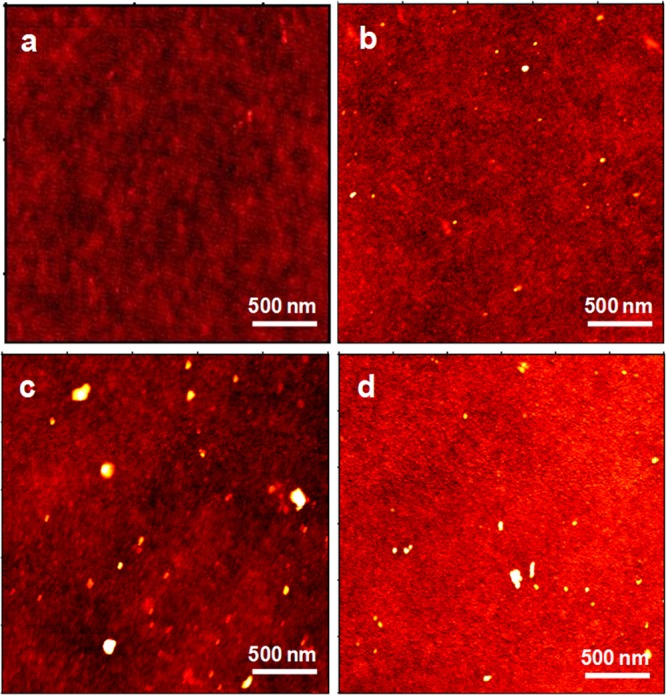
AFM topography images of bare TiO2 (a), APTES-functionalized TiO2 (b), and APTES-TiO2 after 30 min (c) and 60 min (d) of light irradiation. The corresponding RMS roughness estimates are (a) 0.56, (b) 0.75, (c) 1.46, and (d) 1.18 nm.
3.2. XPS Results
Reference unfunctionalized TiO2 was first analyzed to investigate the effect of surface contaminants on the recorded signals. To this end, XPS spectra were collected before and after sputtering by Ar+ ions for 10 min. Comparing the XPS spectra of the as-synthesized and sputtered TiO2 layers shows that adventitious surface C and N species are indeed present, which are almost entirely removed after sputtering (Figure S2). These species are likely ascribable to the fast adsorption of atmospheric contaminants at the bare TiO2 surface because of its well-known photoinduced amphiphilic behavior.19
In the unfunctionalized substrate, before sputtering (Figure 2a) the Ti 2p region is characteristic of Ti(IV) (Ti 2p3/2 458.7 eV, Δ = 5.7 eV). The O/Ti ratio is close to the theoretical value of 2 (Table 1). Upon surface functionalization with APTES (Figure 2a), the Ti signals shift to a slightly lower B.E. (Δ = −0.3 eV), which is indicative of more electron-rich Ti atoms. Furthermore, in agreement with previous reports,28 the Ti peaks also show a slight decrease in intensity. If significant, the latter might be attributed to the attenuation of the signal due to the limited inelastic mean free path of electrons, i.e., the probe depth of XPS, because in functionalized samples they have to cross the adsorbed alkylsilane layer before reaching the detector.37 It should be underlined that the detected XPS signals contain information on both the APTES layer and the underlying oxide because they show both organic species and surface Ti ions. This is expected on the basis of the probe depth of the emitted photoelectrons considered here (∼5.3–6.5 Å for kinetic energies in the range of 50–170 eV). Note that these probe depths are comparable to the monolayer thickness in the limiting case of fully grafted APTES molecules normal to the surface at the maximum extension of the linear hydrocarbon chain (see also Section 3.5). These findings, together with the homogeneity of the AFM image of APTES-TiO2 (Figure 1a), are consistent with the formation of a monolayer.
Figure 2.

High-resolution XP spectra of the pristine (dotted line) and APTES-functionalized TiO2 (solid line) as a function of irradiation time (0, 30, and 60 min): Ti 2p (a) and O 1s (b).
Table 1. Ratios between Peak Areas of XPS Survey Spectra, Corrected for the Intensity of the Photon Beam and Atomic Sensitivities, for Bare and APTES-Functionalized TiO2 Filmsa.
| pristine TiO2 | APTES-TiO2, 0 min | APTES-TiO2, 30 min | APTES-TiO2, 60 min | |
|---|---|---|---|---|
| O/Ti | 2.2(1) | 2.7(1) | 3.0(1) | 3.0(1) |
| C/N | 23(2) | 4.9(3) | 7.1(5) | 6.2(4) |
| N/Si | 0.75(5) | 0.50(4) | 0.34(2) | |
| Si/Ti | 0.61(4) | 0.45(3) | 0.53(4) |
Estimated standard deviations are given in parentheses.
Upon ex situ light irradiation, a progressive shift of the Ti 2p peaks toward higher binding energy (B.E.) takes place along with an increase in Ti relative content, i.e., the effects of APTES functionalization are reversed. After 60 min of irradiation, an almost full recovery of the chemical states of near-surface transition-metal ions is observed.
As for the O 1s signal (Figure 2b), surface O atoms undergo a very similar negative B.E. shift (∼−0.2 eV) upon functionalization with APTES. This is likely the signature of surface band bending, which determines the formation of a negative space charge layer and implies that Ti4+ ions achieve a more reduced state, also according to PAW-DFT outcomes (Section 3.5.2).38 Furthermore, a marked increase in the component at higher B.E. is appreciable, which seems to be related to a component at 532.3 eV (Table S1) possibly ascribable to C–O bonds of surviving ethoxy moieties. Accordingly, the O/Ti ratio increases upon functionalization with APTES from 2.2 to 2.7 (Table 1). This is due to the silane stoichiometry (Scheme 1), which brings 3 atoms of oxygen per Si center, and implies an incomplete condensation of the ethoxy functionalities with surface hydroxyl groups. However, a higher O content might imply either the presence of unhydrolyzed ethoxy groups or uncondensed Si–OH moieties derived from Si-OEt hydrolysis. FTIR results (vide infra) support the latter hypothesis.
Furthermore, upon light irradiation the O/Ti ratio increases to 3.0 (Table 1) and the intensity of the components ascribable to OH and C–O species also increases with respect to the pure oxide (Table S1). These observations might be related to the expected increase in surface hydroxylation upon light irradiation31 as well as to the progressive oxidation of the alkyl chain of APTES. Moreover, the presence of organic C–O bonds in functionalized samples is confirmed by typical C–O features in the C 1s region, whose relative weight decreases upon irradiation (Table S1) with respect to the C=O peak (Figure S5 and Table S3).
The Si 2p region (Figure 3a) in APTES-TiO2 shows an ∼1.85 eV-wide peak at 102.2 eV, which could be indicative of different environments around the Si atom. The Si/Ti ratio is ∼0.6 (Table 1), in agreement with previous reports of APTES monolayers on TiO2.28 Upon solar irradiation, the signal shifts to higher B.E. as a result of further oxidation of the adsorbate. Interestingly, the Si/Ti ratio remains almost unchanged with respect to the nonirradiated sample. Figure 3b shows the high-resolution XP spectra of the N 1s region for the pristine and functionalized TiO2 samples. Partial surface contamination due to N-containing species is unavoidable,26 and indeed a weak N 1s peak at around 400.3 eV is already present in the unfunctionalized TiO2 film (feature marked as I in Figure 3b). Literature studies report several conflicting attributions to this component,27 e.g., surface N22 or amides.23 When APTES is added, however, organic nitrogen due to the aliphatic amine moiety is clearly recognizable because two components neatly distinguishable from the contaminant species appear (marked as II and III in Figure 3b, curve at t = 0 min), with a relative weight of ∼60:40. In agreement with previous reports on APTES-functionalized oxides, these features can be attributed to free NH2 (399.6 eV) and to either H-bonded NH2 or NH3+ (401.3 eV).23,25,28,39 The presence of amides (B.E. 400.3 eV) due to the reaction of the amine group with atmospheric CO2 has been previously reported for amine-containing monolayers.23 In the present case, however, the absence of significant amounts of amides is confirmed by the very low C=O content observed in the C 1s XP spectrum of the nonirradiated sample (Table S3) as well as by FTIR results (vide infra).
Figure 3.

High-resolution XP spectra of the pristine (dotted line) and APTES-functionalized TiO2 (full line) as a function of irradiation time (0, 30, 60 min): Si 2p (a) and N 1s (b).
The C/N ratio is about 5, i.e., higher than the theoretical value of 3 expected in fully grafted APTES-TiO2; this might be related to adventitious carbon as well as to unhydrolyzed ethoxy groups. Another possible reason for the higher-than-expected C/N ratio could be that some APTES molecules might present a reversed or folded configuration. As a result, the nitrogen signal would be attenuated by the limited electron mean free path. The same effect can explain the N/Si ratio lower than 1. The latter hypothesis is also supported by DFT calculations and FTIR results (vide infra). As the solar irradiation proceeds, the N/Si ratio diminishes (Table 1). After 30 min, no further changes are detectable in the N 1s XPS signal (compare the curves at t = 30 and 60 min in Figure 3b), and the residual peaks are very broad and weak, similar to that due to adsorbed impurities in pristine TiO2. No nitrate (B.E. > 405 eV), imine, or nitrile (B.E. < 399 eV) groups are observed. These findings are likely related to the disappearance of the APTES nitrogen (Figure S6).
The following conclusions can be drawn from XPS outcomes. (i) Before irradiation, ∼40% of amine groups are involved in hydrogen bonds or are protonated, which is in good agreement with previous reports about SiO2 and other oxides,25,28,40 showing a higher proportion of free amine with respect to H-bonded/protonated NH2. (ii) Upon irradiation, simulated solar light promotes extensive oxidation of the APTES alkyl chain. Mineralization does not affect the Si headgroup, as observed for other silanes,20 whereas the alkyl chain degradation leads to the cleavage of the amino group.
3.3. FT-IR Results
The degradation pathway of APTES molecules adsorbed at the TiO2 surface under simulated solar irradiation was also studied via FT-IR spectroscopy. The spectrum of the bare TiO2 reference can be found in Figure S7. To disentangle the FT-IR background due to the TiO2 substrate from the APTES signature, difference spectra were analyzed as a function of the irradiation time (Figure 4).
Figure 4.

Differential absorbance FT-IR spectra obtained by subtracting the curve of the pristine samples from those of APTES-functionalized TiO2 at different irradiation times: 4000–2500 cm–1 spectral range (a) and 1900–900 cm–1 spectral range (b). Curves 1–13, exposure time 0–60 min, in increments of 5 min. Curves 14, 15:120 and 180 min, respectively. Vertical lines are a guide to the eye and indicate the main peak positions at 0 min.
Despite a rather unfavorable signal-to-noise ratio, the changes observed as a function of the irradiation time are systematic and have physical significance. The as-synthesized sample (t = 0 min) shows a clear envelope in the 3000–2850 cm–1 spectral region, attributable to the C–H stretching bands of −CHn– species. In particular, bands at 2920 and 2864 cm–1 are due to the asymmetric and symmetric stretches of −CH2 groups on the alkyl chain, whereas no clear indication of CH3 asymmetric modes is discerned. The corresponding bending counterparts are located in the 1500–1350 cm–1 spectral region. Previous studies of surface functionalization by alkylsilanes of TiO2 and other oxides41,42 reported the occurrence of negative peaks in the −OH (surface hydroxyls) and HOH (undissociated water molecules) regions, which were attributed to the formation of Si–O–Si bonds between the surface and alkylsilane molecules with a consequent loss of surface hydroxyl groups and physisorbed water. On the contrary, in the present case no negative peaks in these ranges are observed. Chong et al.43 reported a slight increase in the width of the broad peak at 3500 cm–1 for APTES-functionalized zeolites, which was attributed to the overlap of the symmetric N–H stretching. It is worth noting that the same band was reported at ∼3346 cm–1 for the free amine and at about 3305 cm–1 for terminal amine groups cross-linked with silanol groups.44 As for the low-frequency branch of the spectrum, the absorption bands at ∼1562 and ∼1628 cm–1 were assigned to the scissor modes of −NH2 groups and to the asymmetric −NH3+ deformation mode, respectively.40,43 The corresponding symmetric −NH3+ deformation mode can be related to the component at ca. 1490 cm–1.40 The band at ca. 1320 cm–1 falls in the characteristic region of C–N stretching in amines. The overlap of the stretching bands of Si–O–Si, Si–O–Ti, and Si–O–C prevents a complete attribution of the peaks falling in the 950–1250 cm–1 region.45,46 The stretching mode of Si–O–Si bonds has been reported in the range of 1010–1150 cm–1.13,40,47,48 The stretching mode of the Ti–O–Si moieties is expected to fall at 1024 cm–1,49 and the Si–O–C stretching modes were also assigned at about 1050 cm–1.45 Other modes reported in this region are the Si–CH2–R vibrational modes, usually observed in the 1250–1200 cm–1 spectral range.43
Solar-light irradiation (Figure 4) leads to the disappearance of the bands related to −CHx moieties (especially the bands at 3000–2850 cm–1), in agreement with XPS results. Also, the peaks associated with −NH2 bending disappear as the irradiation time increases, while a peak at 1645 cm–1 strengthens. This peak can be assigned to the in-plane HOH bending mode of water molecules. An alternative attribution of this peak to the C=O stretching mode of amides is contradicted by the concomitant disappearance of NH2 bending signals. Peaks in the 1600–1200 cm–1 region disappear in the first 20 min of irradiation, and a new peak appears at ∼1427 cm–1 after a longer irradiation, which might be attributed to C–O–H bending. It is noteworthy that the two broad peaks at ca. 1100 and 1015 cm–1 are still appreciable even after prolonged irradiation (180 min), although they shift to higher wavenumbers as the degradation proceeds and at the same time their relative intensities change. This evidence suggests that the amine group is degraded very rapidly (in the first 20 min), as also supported by XPS analyses, whereas the degradation of alkyl chains requires longer times. The silane headgroup is instead still appreciable after prolonged irradiation, as confirmed by the persistence of the two main peaks in the 1150–1000 cm–1 region, even though the observed shift in the peak position and the change in relative intensity suggest the occurrence of oxidation (i.e., loss of the contributions due to Si–C–O and Si–CH2–R).
3.4. NEXAFS Results
Figures 5 and 6 show the NEXAFS spectra of TiO2-functionalized APTES surfaces at both grazing and normal incidence geometries across the C Kα and N Kα edges. Prior to irradiation, the C K-edge signals (Figure 5a) are dominated by two main peaks, namely, a sharp resonance at 288.4 eV and a broader one at ∼292.5 eV. The latter can be attributed to excitations to C–C (or C–N) σ* orbitals,23 and the former could be due to C 1s → σ*(C–H).50 It should be mentioned, however, that a C 1s → π*(C=O) resonance might be present at ∼289 eV, probably because of X-ray-induced oxidation of the alkyl chain.23 Indeed, the sample after 30 min of irradiation (Figure 5b) shows a more intense peak at 288.6 eV, which could be related to the presence of C 1s → π*(C=O) transitions, as also supported by XPS results showing an increased content of C=O species. Even though some artifacts are still appreciable in the pre-edge region, probably because of Auger transitions, the σ* resonances at ∼292 eV in NI geometry are invariably stronger than in GI geometry, which suggests a preferential horizontal orientation of APTES alkyl chains with respect to the surface.
Figure 5.

C K-edge NEXAFS spectra of APTES adsorbed on TiO2, measured at normal (NI, solid lines) and grazing (GI, dashed lines) X-ray incidence as a function of different ex situ solar light irradiation times: t = 0 (a) and 30 min (b).
Figure 6.
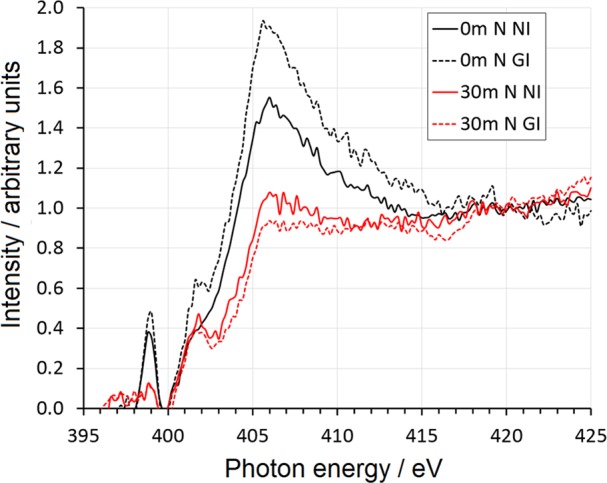
N K-edge NEXAFS spectra of APTES adsorbed on TiO2, measured at normal (NI, full lines) and grazing (GI, dashed lines) X-ray incidence as a function of different ex situ solar irradiation times (t = 0, in black, and t = 30 min, in red).
Previous studies reported a lack of polarization dependence of APTES monolayers, which was attributed to a completely disordered layer structure.51 Our C K-edge NEXAFS results seem instead to support the formation of a low-density monolayer with APTES molecules tilted toward the surface, in agreement also with DFT calculations, which will be reported in Section 3.5. Such a difference with literature reports could be related to the preparation procedure adopted here: chemical vapor deposition has been previously reported to lead to a lower degree of cross-linking with respect to wet procedures42 as a result of the absence of a prepolymerization step in solution. As for N K-edge spectra (Figure 6), at t = 0 min a sharp peak is present at ∼398–399 eV, whose attribution is debated: it has been assigned to either N=C or N–C=C π* resonances originating from radiation damage23 or to N 1s → σ*(N–H) resonances of NH3+.52 Interestingly, this feature vanishes in samples degraded for 30 min, consistent with the disappearance of the APTES N 1s XPS peak upon solar irradiation. The large shoulder at 401–402 eV could be attributed to either σ* N–H transitions or to π* N≡C resonances arising from radiation damage; however, it is still present after the degradation of the silane (red curves in Figure 6) and is therefore attributable to either APTES or adventitious nitrogen-adsorbed species. The peak above 405 eV can be assigned to a 1s → N–C σ* transition.
No clear preferential orientation is observed for N groups, especially after irradiation. This is not surprising because the amine end group is expected to be conformationally more free than the whole hydrocarbon chain, resulting in less preferred orientations for the nitrogen-bearing groups. The observed changes in the spectra upon irradiation indicate possible changes in the intermolecular H bonding or even fragmentation of the molecules, in agreement with XPS and FT-IR results (Sections 3.2 and 3.3). Accordingly, the N signal is more isotropic (red curves in Figure 6) because there are fewer differences between the NI and GI K-edge spectra.
3.5. Plane-Wave DFT Calculations
3.5.1. Geometry
The APTES adsorption on a 11.5 × 21.0 Å2 (101) anatase TiO2 surface was simulated by plane-wave (PW) DFT calculations. PW-DFT has already been successfully employed to study the adsorption of amino-terminated silanes on a different class of substrates.53 The (101) anatase TiO2 surface was selected according to HR-TEM images (Figure S8) showing that the thermodynamic {101} facets are predominant in our anatase TiO2. This was expected on the basis of our synthesis conditions, which allow crystallites to nucleate and grow under thermodynamic control, as it is well known that the most stable surface on anatase has a (101) orientation.54,55 Because the VASP program suite always applies 3-D periodic boundary conditions, to reproduce a 2-D periodic slab, we introduced a vacuum layer along the third dimension that was thick enough (14 Å) to avoid unphysical interactions between neighboring slabs (Figure S1).
The accuracy of the functional and of the chosen computational setup was verified through calculations on isolated APTES molecules, i.e., by comparing the VASP-predicted molecular geometries and electronic structure in vacuo with geometries optimized through atom-centered linear combinations of Gaussian-type functions (LC-GTF). The NWCHEM package56 was used to perform such benchmark calculations at the 6-311G(p,d)/PBE theory level, i.e., using the same exchange-correlation functional employed in PW simulations (Table S4 and Figure S9). The relative absolute differences in bond lengths among the PW and LC-GTF models were invariably lower than 1% with very similar electronic structures, so we feel confident that our plane-wave simulations are able to reproduce with satisfactory accuracy the highly localized states associated with the adsorbed silanes. Given the good agreement between the two types of calculations, we conclude that our computational procedure is able to reproduce the correct features of APTES molecules in both gas and gas–surface arrangements. A detailed description is reported in Section S5.
The adsorption of one to three molecules was modeled. The theoretical coverage corresponds to 0.41 (0.82) molecules/nm2 when the APTES monomer (dimer) is adsorbed (to be compared with an average monolayer density on glass or silicon substrates of 2.1–4.2 molecules/nm2).57,58 To better simulate the adsorption process, a large slab of TiO2 anatase (4 × 4 × 4 unit cell) was modeled, i.e., four O–Ti–O trilayers thick. The surface was hydroxylated, simulating a dissociative water chemisorption process59 (Scheme 2, Figure 7). This picture is probable for defective surfaces such as our nanostructured system, where there are numerous acid Ti(V) sites.60,61 Physisorbed water was not included in our model because it is reversibly bound to the surface and its interactions with APTES molecules are intrinsically dynamic, i.e., they produce an effect on a time-averaged basis. On the other hand, strongly bound chemisorbed water likely has a more significant influence because it can form stable hydrogen bond networks also involving the organic layer.
Scheme 2.

Figure 7.
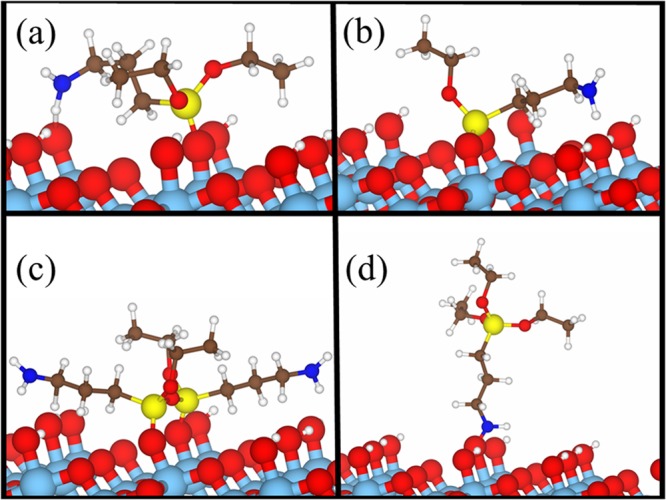
Chemisorption geometries of APTES molecules (a–d, Scheme 2) at a (101) anatase surface. Only the upper layers of the 4 × 4 × 4 supercell are shown. Color code: Ti (light blue), O (red), H (white), Si (yellow), N (deep blue), and C (brown).
The two top O–Ti–O layers were relaxed to model the surface region perturbed by the interface and by the APTES molecular adsorption. The two bottom layers were instead kept fixed in order to simulate the bulk TiO2. Indeed, silane binding onto anatase has been reported to induce local conformational changes in the TiO2 structure, including small geometry rearrangements of the Ti4+ and O2– ions next to the molecular adsorbate.62 Six binding modes between the silane group and the surface were modeled (Scheme 2). The single molecule was chemisorbed with either one (Scheme 2a), two (Scheme 2b), or three Si–O–Ti covalent bonds, implying the loss of one, two, or three ethoxy groups. The adsorption of oligomers was also taken into account: both a covalently bonded Si–O–Si dimer (Scheme 2c) and a trimer were also considered. Finally, reversed adsorption geometries, i.e., where the silane molecule interacts with the oxide surface via the amino group, were considered: one geometry involving the formation of covalent interactions (Scheme 2d).
APTES physisorption mediated by a H-bond between the amine group and a surface silanol was not simulated because of the intrinsic limit of the DFT procedure in modeling correlations among distant electrons responsible for dispersive interactions. It is also worth noting that, upon solar irradiation, the Si-containing headgroups seem to remain grafted onto the surface, as shown by XPS (Table 1, rows 4 and 5) and FTIR (as appreciable from peaks at 1150–1000 cm–1 in Figure 4) results. This implies that physisorption, if any, should be just a minority interaction mode. Hence, the chemisorption modes shown in Scheme 2a–c should adequately represent the APTES–TiO2 interface. For each system, the corresponding isolated APTES derivative with three, two, or one ethoxy group (EtO−), i.e., Si[(CH2)3NH2](OEt)3, Si[(CH2)3NH2](OEt)2OH, or Si[(CH2)3NH2]OEt(OH)2, was considered for gas-phase benchmark calculations (vide supra). By comparing gas-phase and adsorbed analogues (Table 2), the molecular distortion taking place upon surface adsorption can be determined for the different geometries.
Table 2. Relevant Bond Distances in APTES Molecules, Both in the Gas Phase and after Adsorption at the Anatase TiO2 Surface, As Retrieved from the PW-DFT Optimized Geometriesa.
| simulated system | dSi–Ob | dSi–Oc | dSi–Od | dSi–Oe | dN–Hf | dH···Og | |
|---|---|---|---|---|---|---|---|
| Si[(CH2)3NH2](OEt)3 | gas phase | 1.6563 | 1.0209 | ||||
| Si[(CH2)3NH2](OEt)2OH | 1.6563 | 1.6569 | 1.0229 | ||||
| [−O0.5–Si(R)EtO]2(OH)2 | 1.6566 | 1.7100 | 1.6769 | 1.0249 | |||
| 1.5714 | |||||||
| Si[(CH2)3NH2]OEt(OH)2 | 1.6556 | 1.6572 | 1.0204 | ||||
| (a)h | adsorbed | 1.5945 | 1.6550 | 1.1318 | 1.4207 | ||
| (b)h | 1.6059 | 1.6322 | 1.0486 | 1.7348 | |||
| 1.6181 | |||||||
| (c)h | 1.5999 | 1.6651 | 1.6148 | 1.0298 | 2.1637 | ||
| 1.6030 | 1.6669 | 1.0265 | 1.9371 | ||||
| (d)h | 1.6474 | i |
Values are given in angstroms.
O: oxo ion on the TiO2 surface. When two Si–O–Ti bonds are present, both distances are given.
O of the hydroxyl group in benchmark isolated silane calculations. (If more than one Si–OH bond is present, then the average is given.)
O: bridging atom in the Si–OEt bond. (If more than one Si–OEt bond is present, then the average is given.)
O: bridging atom in the Si–O–Si system in the APTES dimer (c). Both symmetry-independent distances are given.
The H atom involved in the H-bonding interaction is always considered. If no relevant hydrogen bonds are present, then the average value is given. For the isolated APTES molecules, the average N–H distance is given.
Distance between the amine H atom and the oxide O atom on the TiO2 surface acting as a hydrogen bond acceptor. When two NH···O contacts are present, both distances are given.
In the adduct (d), the amine hydrogen is directly linked to the surface through a N–O bond, with dN–O = 1.2894 Å.
A first important result is that, according to the calculations, not all of the considered adsorption geometries are feasible. In particular, the simulation of APTES chemisorption via three hydroxyl groups did not lead to a converged electronic structure result, and we conclude that a tripodal link to the surface is not feasible. The formation of three Si–O–Ti bonds between a single APTES molecule and the oxide surface thus seems to be prevented by the bond lengths/angles in the silane molecules in conjunction with the structure of the TiO2 surface. It is worth noting that such findings are in agreement with previous FT-IR studies.62Figure 7 shows the corresponding fully relaxed configurations of the geometries that reached a converged electronic structure result.
When only one Si–O–Ti bond forms (Figure 7a), the APTES molecules lie horizontally on the surface because of the formation of a hydrogen bond interaction between the H of the APTES amine group and an O of a surface hydroxyl group (NH···O distance 1.42 Å, Table 2). This is a surprisingly short contact, with a donor–acceptor N···O distance comparable to the length of charge-assisted bonds found between charged groups in metalorganic complexes.63 It also implies a significant lengthening of the N–H covalent bond in the grafted molecule (dN–H = 1.13 Å) with respect to the isolated one (dN–H = 1.02 Å) while at the same time the Si–O bond with the surface shortens and strengthens (Table 2). When two Si–O–Ti bonds form between the Si headgroup and the surface (Figure 7b), the APTES molecule shows similar folding, with the amino group approaching the surface similarly to what was observed with a single Si–O–Ti bond. However, in this case, the distance between the N–H group and the surface O is higher (1.73 Å). As for the reversed attachment configuration (Scheme 2d, Figure 7d), the direct interaction between the N of the APTES molecule and a surface oxygen (Figure 7d) was taken into account. In this configuration, the APTES molecule is perpendicular to the TiO2 surface, showing no interactions between the silane head and the surface.
The adsorption of partially hydrolyzed oligomers was also modeled. In this case, a direct silane chemisorption was always considered. As done for the isolated monomers, we performed benchmark calculations on the isolated dimer, as reported in Section S5. Figure 7c reports the adsorption geometries of a representative APTES dimer at the TiO2 surface. When two APTES molecules bond with neighboring surface hydroxyls, the formation of an intermolecular Si–O–Si bond results in a 20° tilt of the APTES alkyl chain with respect to the surface. The interaction of the amino group with the surface is still substantial, although the hydrogen-acceptor distance (dH···O = 1.94–2.16 Å) is now much longer than in the case of a single monomer (Figure 7a,b). Moreover, the Si–O–Si bond is highly strained, as seen from bond angle values (167°).
Some attempts were also carried out to simulate larger oligomers chemisorbed on the surface, e.g., APTES trimers in various configurations. For example, we tried to simulate an APTES trimer similar to the system shown in Scheme 2c, where three R–SiO3 headgroups were attached to the TiO2 surface via Si–O–Ti bonds while being held together by two bridging Si–O–Si bonds. However, these calculations invariably led to APTES trimers bound to the surface only through two Si–O–Ti bonds. We believe that this evidence is consistent with the high bond strain detected from geometrical bond angles associated with the Si center, very far from those expected for an ideal sp3 geometry. Such strain could be related to the reported limited occurrence of covalent interactions between silane headgroups in ordered monolayers and hydroxyl groups on the oxide surface.1 This high strain might also favor the chemisorption of APTES molecules at the TiO2 surface as monomers or very short oligomers.
3.5.2. Electronic Structure
Figure 8 reports the density of states (DOS) of systems (a–d). For the sake of comparison, the corresponding plot for the bare, fully hydroxylated TiO2 (101) surface is reported in Figure S10. In agreement with previous literature findings,64 the only effect that hydroxylation has on the anatase band structure is the generation of a few empty states just above the valence band (VB) edge. Figure 8a shows that in the single-bonded monomer structure (Scheme 2a, Figure 7a), oxygen atoms inject new states at the top of the VB, which are likely contributed from the silane moiety, whereas N is close enough to the surface to be able to add shallow midgap empty states just above the Fermi level (Figure 8a, magenta line). The electronic structure of the double-bonded monomer (Scheme 2b, Figure 7b), reported in Figure 8b, is pretty similar, although the different folding and chemical state of the organic system lead to a few differences. In particular, the amino group is now more distant from the surface (Table 2), so N midgap states are less dense, although still significant in terms of the DOS intensity. Second, the contribution to deep VB states from oxygen increases, as two oxygen atoms are now acting as bridges between Si and Ti.
Figure 8.
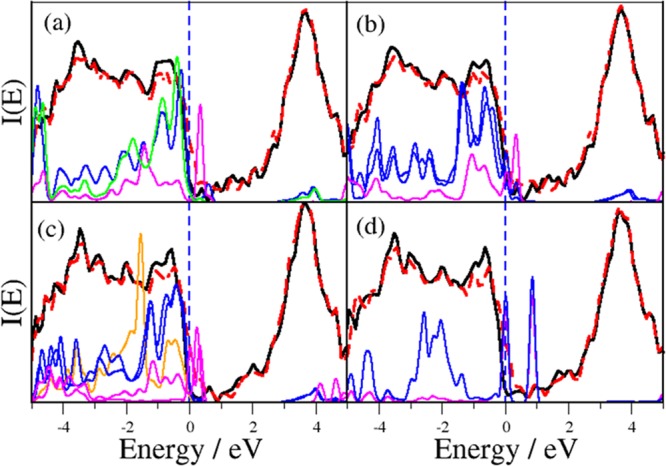
Simulated PW-DFT DOS of the hybrid APTES-TiO2 system. Total-spin-polarized DOS in red and black. (a) Single-bonded monomer (Scheme 2a and Figure 7a). (b) Double-bonded monomer (Scheme 2b and Figure 7b). (c) APTES dimer (Scheme 2c and Figure 7c). (d) Reversed (N–O–Ti) chemisorption (Scheme 2d and Figure 7d). The vertical dashed blue line represents the Fermi energy, which is set as the zero of the energy. Light curves are enhanced atom-projected partial DOSs (PDOSs); magenta, N; blue, O atoms bound to Si (a–c) or N (d); green, (a) oxide O interacting with N through the hydrogen bond; orange, (c) bridging oxygen between two APTES monomers (main text). The PDOS of all atoms is enhanced by a factor of 100 in order to aid the interpretation of the results.
In the APTES dimer (Scheme 2c, Figure 7c), the general picture is still the same (Figure 8c). It is worth noting, however, that the oxygen atom involved in the Si–O–Si bridge (orange line in Figure 8c) provides states more deeply buried in the VB with respect to those involved in Si–O–Ti interactions (blue line in Figure 8c). Finally, as expected, the electronic structure is markedly different when the reversed chemisorption geometry (Scheme 2d, Figure 7d) is considered. Figure 8d shows that two shallow states, due to the overlap of localized states on the bridging N–O system, appear in the vicinity of the top of the VB. They are very close in energy (<1 eV), and indeed they are most likely understandable as a bonding/antibonding couple associated with localized N–O orbitals. This implies that the reversed configuration should be immediately destroyed at finite temperature by visible/thermal electron excitation.
In conclusion, upon APTES functionalization, both Ti 2p and O 1s XPS signals shift toward lower B.E. (Figure 2), likely because of chemisorption-induced band bending.65 The silane molecule should act as an electron donor, implying charge transfer to the surface.66 As a consequence, surface Ti4+ ions formally achieve a less oxidized state. This is in fairly good agreement with the PW-DFT results: all of the Si–O–Ti grafted silanes (Scheme 2a–c) undergo a significant negative charge displacement toward the surface, as can be inferred from the N empty states that invariably appear near the top of the VB. This picture is also consistent with the optimized bond lengths (Table 2) calculated for APTES-TiO2 hybrids and, in particular, with (i) the significant shortening (lengthening) of the Si–O (N–H) bonds of the silane; (ii) the presence of very strong NH···O bonds, with H···O distances fully comparable to known cases of charge-assisted hydrogen bonds; and (iii) the significant rearrangement of the Ti–O coordination geometry close to the chemisorption site.
4. Conclusions
In this work, bare and APTES-functionalized TiO2 was extensively characterized both experimentally (XPS, NEXAFS, AFM, FT-IR, HR-TEM) and theoretically (PW-DFT) to clarify the structure of the organic layer at the TiO2 surface. AFM, FTIR, XPS, and NEXAFS analyses on ex situ solar-irradiated samples at various degrees of reaction extent also provided information on the photoinduced oxidation mechanism of the organic layer.
The combined experimental and theoretical evidence suggests that APTES chemisorption on TiO2 mainly implies one or two Si–O–Ti bonds involving the Si headgroup, whereas a tripod configuration seems unfeasible as highly strained. Other interaction modes, such as physisorption through terminal amino groups, are expected to be considerably more labile. Although dimerization of the silane at the surface through Si–O–Si bonds is possible, further polymerization appears to be less probable. Chemisorption of individual monomers leaves the APTES terminal amino groups available for hydrogen bonds with surface hydroxyl groups of TiO2. These interactions are rather strong because, on the basis of XPS and DFT results, they are reinforced by a probable charge displacement from the organic molecules toward the surface. In NEXAFS spectra, the strong C K-edge σ* resonances at normal incidence of the X-rays imply a preferential orientation, where alkyl chains are bent toward the surface. We therefore expect that NH···O interactions should constitute a preferential APTES–substrate secondary mode of interaction, especially at low surface coverage, as in our specimens ∼40% of the amine groups were estimated to be involved in some kind of strong hydrogen bond. The latter could hamper the availability of the amine as a nucleophilic center for further functionalization of the adsorbed alkylsilane molecules. We thus anticipate that the yield of reactions exploiting the APTES–NH2 chemical function should be lower at low surface coverage. Indeed, under these conditions strong NH···O interactions pose a kinetic barrier against the chemical availability of the amine.
We also found compelling evidence that solar irradiation promotes extensive oxidation of the APTES alkyl chain, rapidly leading to the cleavage of the amino group. On the contrary, the oxidation process does not destroy Si head moieties, which remain bound to the titania surface in a higher oxidation state. This implies that the TiO2 photocatalyst surface is not pristine at the end of the degradation process. Such an effect could hamper the TiO2 photocatalytic behavior,49 with notable implications for applications, but it could also be exploited, for instance, to produce SiO2/TiO2 composite surfaces for advanced optics as narrow bandpass filters.67 Moreover, amino group availability and lability are both essential factors for further functionalization of the oxide surface. Indeed, because of the rapid solar-induced N–C cleavage, chemical functionalization of APTES by small-to-medium organic molecules seems to be a suitable step toward the development of effective drug-carrier devices for disease prophylaxis and health care purposes. This topic will be explored in future works.
Acknowledgments
A. M. Ferretti is kindly acknowledged for HRTEM images. M.C. and G.D.L. acknowledge support from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. [647107]–SEMICOMPLEX–ERC-2014-CoG). The Elettra Synchrotron Light Laboratory at Trieste (Italy) is acknowledged for beam-time provision. The authors acknowledge the CINECA and the Regione Lombardia award under the LISA initiative (grant SURGREEN) for the availability of high-performance computing resources.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcc.6b10720.
Experimental details of synchrotron measurements and data analysis. Details of PAW-DFT calculations. High-resolution XP spectra of unfunctionalized TiO2 film after sputtering. Quantification of the signals in the XP spectra of pristine and APTES-functionalized TiO2. C 1s XP spectra of APTES-functionalized films as a function of irradiation time. Differential XP spectra of N 1s region. Reference FT-IR spectra of the bare TiO2 layer. HR-TEM results. Comparison between computed optimized geometries and electronic structures of isolated APTES molecules between VASP and NWCHEM calculations. PW-DFT DOS of the fully hydroxylated TiO2 anatase (101) surface. (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Onclin S.; Ravoo B. J.; Reinhoudt D. N. Engineering Silicon Oxide Surfaces Using Self-Assembled Monolayers. Angew. Chem., Int. Ed. 2005, 44, 6282–6304. 10.1002/anie.200500633. [DOI] [PubMed] [Google Scholar]
- Cappelletti G.; Ardizzone S.; Meroni D.; Soliveri G.; Ceotto M.; Biaggi C.; Benaglia M.; Raimondi L. Wettability of Bare and Fluorinated Silanes: A Combined Approach Based on Surface Free Energy Evaluations and Dipole Moment Calculations. J. Colloid Interface Sci. 2013, 389, 284–291. 10.1016/j.jcis.2012.09.008. [DOI] [PubMed] [Google Scholar]
- Meroni D.; Ardizzone S.; Cappelletti G.; Ceotto M.; Ratti M.; Annunziata R.; Benaglia M.; Raimondi L. Interplay between Chemistry and Texture in Hydrophobic TiO2 Hybrids. J. Phys. Chem. C 2011, 115, 18649–18658. 10.1021/jp205142b. [DOI] [Google Scholar]
- Haensch C.; Hoeppener S.; Schubert U. S. Chemical Modification of Self-assembled Silane Based Monolayers by Surface Reactions. Chem. Soc. Rev. 2010, 39, 2323–2334. 10.1039/b920491a. [DOI] [PubMed] [Google Scholar]
- Meroni D.; Ardizzone S.; Schubert U. S.; Hoeppener S. Probe-based Electrooxidative Lithography of OTS SAMs Deposited onto Transparent ITO Substrates. Adv. Funct. Mater. 2012, 22, 4376–4382. 10.1002/adfm.201200673. [DOI] [Google Scholar]
- Herzer N.; Haensch C.; Hoeppener S.; Schubert U. S. Orthogonal Functionalization of Silicon Substrates using Self-Assembled Monolayers. Langmuir 2010, 26, 8358–8365. 10.1021/la9047837. [DOI] [PubMed] [Google Scholar]
- Fujishima A.; Zhang X.; Tryk D. A. TiO2 Photocatalysis and Related Surface Phenomena. Surf. Sci. Rep. 2008, 63, 515–582. 10.1016/j.surfrep.2008.10.001. [DOI] [Google Scholar]
- Marchiori C.; Di Liberto G.; Soliveri G.; Loconte L.; Lo Presti L.; Meroni D.; Ceotto M.; Oliva C.; Cappelli S.; Cappelletti G.; et al. Unraveling the Cooperative Mechanism of Visible-light Absorption in Bulk N,Nb Codoped TiO2 Powders of Nanomaterials. J. Phys. Chem. C 2014, 118, 24152–24164. 10.1021/jp507143z. [DOI] [Google Scholar]
- Claridge S. A.; Liao W.-S.; Thomas J. C.; Zhao Y.; Cao H. H.; Cheunkar S.; Serino A. C.; Andrews A. M.; Weiss P. S. From the Bottom Up: Dimensional Control and Characterization in Molecular Monolayers. Chem. Soc. Rev. 2013, 42, 2725–2745. 10.1039/C2CS35365B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z.; Lisdat F.; Parak W. J.; Hickey S. G.; Tu L.; Sabir N.; Dorfs D.; Bigall N. C. Quantum-Dot-Based Photoelectrochemical Sensors for Chemical and Biological Detection. ACS Appl. Mater. Interfaces 2013, 5, 2800–2814. 10.1021/am3028662. [DOI] [PubMed] [Google Scholar]
- Soliveri G.; Pifferi V.; Panzarasa G.; Ardizzone S.; Cappelletti G.; Meroni D.; Sparnacci K.; Falciola L. Self-cleaning Properties in Engineered Sensors for Dopamine Electroanalytical Detection. Analyst 2015, 140, 1486–1494. 10.1039/C4AN02219J. [DOI] [PubMed] [Google Scholar]
- Luitel T.; Zamborini F. P. Covalent Modification of Photoanodes for Stable Dye-Sensitized Solar Cells. Langmuir 2013, 29, 13582–13594. 10.1021/la402256v. [DOI] [PubMed] [Google Scholar]
- Liu L.; Mei A.; Liu T.; Jiang P.; Sheng Y.; Zhang L.; Han H. Fully Printable Mesoscopic Perovskite Solar Cells with Organic Silane Self-Assembled Monolayer. J. Am. Chem. Soc. 2015, 137, 1790–1793. 10.1021/ja5125594. [DOI] [PubMed] [Google Scholar]
- Rajh T.; Dimitrijevic N. M.; Bissonnette M.; Koritarov T.; Konda V. Titanium Dioxide in the Service of the Biomedical Revolution. Chem. Rev. 2014, 114, 10177–10216. 10.1021/cr500029g. [DOI] [PubMed] [Google Scholar]
- Song Y.–Y.; Hildebrand H.; Schmuki P. Photoinduced Release of Active Proteins from TiO2 Surfaces. Electrochem. Commun. 2009, 11, 1429–1433. 10.1016/j.elecom.2009.05.022. [DOI] [Google Scholar]
- Song Y.-Y.; Roy P.; Paramasivam I.; Schmuki P. Voltage-Induced Payload Release and Wettability Control on TiO2 and TiO2 Nanotubes. Angew. Chem., Int. Ed. 2010, 49, 351–354. 10.1002/anie.200905111. [DOI] [PubMed] [Google Scholar]
- Sargeant T. D.; Rao M. S.; Koh C.-Y.; Stupp S. I. Covalent Functionalization of NiTi Surfaces with Bioactive Peptide Amphiphile Nanofibers. Biomaterials 2008, 29, 1085–1098. 10.1016/j.biomaterials.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardizzone S.; Biraghi I.; Cappelletti G.; Meroni D.; Spadavecchia F. In Surface Tailoring of Inorganic Materials for Biomedical Applications; Rimondini L., Ed.; Bentham Science Publishers, 2012; pp 3–42. [Google Scholar]
- Liu K.; Cao M.; Fujishima A.; Jiang L. Bio-inspired Titanium Dioxide Materials with special Wettability and their Applications. Chem. Rev. 2014, 114, 10044–10094. 10.1021/cr4006796. [DOI] [PubMed] [Google Scholar]
- Soliveri G.; Annunziata R.; Ardizzone S.; Cappelletti G.; Meroni D. Multiscale Rough TiO2 Films with Patterned Hydrophobic/Oleophobic Features. J. Phys. Chem. C 2012, 116, 26405–26413. 10.1021/jp309397c. [DOI] [Google Scholar]
- Zhang Q.; Bai R. X.; Guo T.; Meng T. Switchable Pickering Emulsions Stabilized by Awakened TiO2 Nanoparticle Emulsifiers Using UV/Dark Actuation. ACS Appl. Mater. Interfaces 2015, 7, 18240–18246. 10.1021/acsami.5b06808. [DOI] [PubMed] [Google Scholar]
- Bai R. X.; Xue L. H.; Dou R. K.; Meng S. X.; Xie C. Y.; Zhang Q.; Guo T.; Meng T. Light-Triggered Release from Pickering Emulsions Stabilized by TiO2 Nanoparticles with Tailored Wettability. Langmuir 2016, 32, 9254–9264. 10.1021/acs.langmuir.6b02329. [DOI] [PubMed] [Google Scholar]
- Graf N.; Yegen E.; Gross T.; Lippitz A.; Weigel W.; Krakert S.; Terfort A.; Unger W. E. S. XPS and NEXAFS Studies of Aliphatic and Aromatic Amine Species on Functionalized Surfaces. Surf. Sci. 2009, 603, 2849–2860. 10.1016/j.susc.2009.07.029. [DOI] [Google Scholar]
- Vandenberg E. T.; Bertilsson L.; Liedberg B.; Uvdal K.; Erlandsson R.; Elwing H.; Lundström I. Structure of 3-Aminopropyl Triethoxy Silane on Silicon Oxide. J. Colloid Interface Sci. 1991, 147, 103–118. 10.1016/0021-9797(91)90139-Y. [DOI] [Google Scholar]
- Acres R. G.; Ellis A. V.; Alvino J.; Lenahan C. E.; Khodakov D. A.; Metha G. F.; Andersson G. G. Molecular Structure of 3-Aminopropyltriethoxysilane Layers Formed on Silanol-Terminated Silicon Surfaces. J. Phys. Chem. C 2012, 116, 6289–6297. 10.1021/jp212056s. [DOI] [Google Scholar]
- Lo Presti L.; Ceotto M.; Spadavecchia F.; Cappelletti G.; Meroni D.; Acres R. G.; Ardizzone S. Role of the Nitrogen Source in Determining Structure and Morphology of N-doped Nanocrystalline TiO2. J. Phys. Chem. C 2014, 118, 4797–4807. 10.1021/jp412394e. [DOI] [Google Scholar]
- Spadavecchia F.; Ceotto M.; Lo Presti L.; Aieta C.; Biraghi I.; Meroni D.; Ardizzone S.; Cappelletti G. Second Generation Nitrogen doped Titania Nanoparticles: a Comprehensive Electronic and Microstructural Picture. Chin. J. Chem. 2014, 32, 1195–1213. 10.1002/cjoc.201400502. [DOI] [Google Scholar]
- Song Y.–Y.; Hildebrand H.; Schmuki P. Optimized Monolayer Grafting of 3-Aminopropyltriethoxysilane onto Amorphous, Anatase and Rutile TiO2. Surf. Sci. 2010, 604, 346–353. 10.1016/j.susc.2009.11.027. [DOI] [Google Scholar]
- Antonello A.; Soliveri G.; Meroni D.; Cappelletti G.; Ardizzone S. Photocatalytic Remediation of Indoor Pollution by Transparent TiO2 Films. Catal. Today 2014, 230, 35–40. 10.1016/j.cattod.2013.12.033. [DOI] [Google Scholar]
- Rimoldi L.; Ambrosi C.; Di Liberto G.; Lo Presti L.; Ceotto M.; Oliva C.; Meroni D.; Cappelli S.; Cappelletti G.; Soliveri G.; et al. Impregnation versus Bulk Synthesis: How the Synthetic Route Affects the Photocatalytic Efficiency of Nb/Ta:N Codoped TiO2 Nanomaterials. J. Phys. Chem. C 2015, 119, 24104–24115. 10.1021/acs.jpcc.5b06827. [DOI] [Google Scholar]
- Zuo J.; Torres E. Comparison of Adsorption of Mercaptopropyltrimethoxysilane on Amphiphilic TiO2 and Hydroxylated SiO2. Langmuir 2010, 26, 15161–15168. 10.1021/la102221v. [DOI] [PubMed] [Google Scholar]
- Yang H. G.; Sun C. H.; Qiao S. Z.; Zou J.; Liu G.; Smith S. C.; Cheng H. M.; Lu G. Q. Anatase TiO2 Single Crystals with a Large Percentage of Reactive Facets. Nature 2008, 453, 638–641. 10.1038/nature06964. [DOI] [PubMed] [Google Scholar]
- Dudarev S. L.; Botton G. A.; Savrasov S. Y.; Humphreys C. J.; Sutton A. P. Electron-Energy-loss Spectra and the Structural Stability of Nickel Oxide: An LSDA+U Study. Phys. Rev. B: Condens. Matter Mater. Phys. 1998, 57, 1505–1509. 10.1103/PhysRevB.57.1505. [DOI] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Furthmuller J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations using a Plane-Wave Basis Set. Phys. Rev. B: Condens. Matter Mater. Phys. 1996, 54, 11169–11186. 10.1103/PhysRevB.54.11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From Ultrasoft Pseudopotentials to the Projector Augmented-wave Method. Phys. Rev. B: Condens. Matter Mater. Phys. 1999, 59, 1758–1775. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- O’Boyle N. M.; Tenderholt A. L.; Langner K. M. CCLIB: A Library for Package-Independent Computational Chemistry Algorithms. J. Comput. Chem. 2008, 29, 839–845. 10.1002/jcc.20823. [DOI] [PubMed] [Google Scholar]
- Ceotto M.; Lo Presti L.; Cappelletti G.; Meroni D.; Spadavecchia F.; Zecca R.; Leoni M.; Scardi P.; Bianchi C. L.; Ardizzone S. About the Nitrogen Location in Nanocrystalline N-Doped TiO2: Combined DFT and EXAFS Approach. J. Phys. Chem. C 2012, 116, 1764–1771. 10.1021/jp2097636. [DOI] [Google Scholar]
- Kunze J.; Ghicov A.; Hildebrand H.; Macak J. M.; Traveira L.; Schmuki P. Challenges in the Surface Analytical Characterisation of Anodic TiO2 Films – a Review. Z. Phys. Chem. 2005, 219, 1561–1582. 10.1524/zpch.2005.219.11.1561. [DOI] [Google Scholar]
- Pasternack R. M.; Rivillon A. S.; Chabal Y. J. Attachment of 3-(Aminopropyl)triethoxysilane on Silicon Oxide Surfaces: Dependence on Solution Temperature. Langmuir 2008, 24, 12963–12971. 10.1021/la8024827. [DOI] [PubMed] [Google Scholar]
- Soliveri G.; Pifferi V.; Annunziata R.; Rimoldi L.; Aina V.; Cerrato G.; Falciola L.; Cappelletti G.; Meroni D. Alkylsilane-SiO2 Hybrids. A Concerted Picture of Temperature Effects in Vapor Phase Functionalization. J. Phys. Chem. C 2015, 119, 15390–15400. 10.1021/acs.jpcc.5b04048. [DOI] [Google Scholar]
- Soliveri G.; Meroni D.; Cappelletti G.; Annunziata R.; Aina V.; Cerrato G.; Ardizzone S. Engineered Organic/Inorganic Hybrids for Superhydrophobic Coatings by Wet and Vapour Procedures. J. Mater. Sci. 2014, 49, 2734–2744. 10.1007/s10853-013-7976-3. [DOI] [Google Scholar]
- Maria Chong A. S.; Zhao X. S. Functionalization of SBA-15 with APTES and Characterization of Functionalized Materials. J. Phys. Chem. B 2003, 107, 12650–12657. 10.1021/jp035877+. [DOI] [Google Scholar]
- White L. D.; Tripp C. P. Reaction of (3-Aminopropyl)dimethylethoxysilane with Amine Catalysts on Silica Surfaces. J. Colloid Interface Sci. 2000, 232, 400–407. 10.1006/jcis.2000.7224. [DOI] [PubMed] [Google Scholar]
- Zeitler V. A.; Brown C. A. The Infrared Spectra of Some Ti-O-Si, Ti-O-Ti and Si-O-Si Compounds. J. Phys. Chem. 1957, 61, 1174–1177. 10.1021/j150555a010. [DOI] [Google Scholar]
- Milanesi F.; Cappelletti G.; Annunziata R.; Bianchi C. L.; Meroni D.; Ardizzone S. Siloxane-TiO2 Hybrid Nanocomposites. The Structure of the Hydrophobic Layer. J. Phys. Chem. C 2010, 114, 8287–8293. 10.1021/jp1014669. [DOI] [Google Scholar]
- Young C. W.; Servais P. C.; Currie C. C.; Hunter M. J. Organosilicon Polymers. IV. Infrared Studies on Cyclic Disubstituted Siloxanes. J. Am. Chem. Soc. 1948, 70, 3758–3764. 10.1021/ja01191a064. [DOI] [PubMed] [Google Scholar]
- Tripp C. P.; Hair M. L. Reaction of Chloromethylsilanes with Silica: a Low-Frequency infrared Study. Langmuir 1991, 7, 923–927. 10.1021/la00053a019. [DOI] [Google Scholar]
- Ukaji E.; Furusawa T.; Sato M.; Suzuki N. The Effect of Surface Modification with Silane Coupling Agent on Suppressing the Photo-catalytic Activity of Fine TiO2 Particles as Inorganic UV Filter. Appl. Surf. Sci. 2007, 254, 563–569. 10.1016/j.apsusc.2007.06.061. [DOI] [Google Scholar]
- Retzko I.; Friedrich J. F.; Lippitz A.; Unger W. E. S. Chemical Analysis of Plasma-Polymerized Films: The Application of X-ray Photoelectron Spectroscopy (XPS), X-ray Absorption Spectroscopy (NEXAFS) and Fourier Transform Infrared Spectroscopy (FTIR). J. Electron Spectrosc. Relat. Phenom. 2001, 121, 111–129. 10.1016/S0368-2048(01)00330-9. [DOI] [Google Scholar]
- See refs (23) and (51) therein.
- Baio J. E.; Weidner T.; Brison J.; Graham D. J.; Gamble L. J.; Castner D. G. Amine Terminated SAMs: Investigating Why Oxygen is Present in These Films. J. Electron Spectrosc. Relat. Phenom. 2009, 172, 2–8. 10.1016/j.elspec.2009.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabacchi G.; Fois E.; Calzaferri G. Structure of Nanochannel Entrances in Stopcock-Functionalized Zeolite L Composites. Angew. Chem., Int. Ed. 2015, 54, 11112–11116. 10.1002/anie.201504745. [DOI] [PubMed] [Google Scholar]
- Shklover V.; Nazeeruddin M.-K.; Zakeeruddin S. M.; Barbé C.; Kay A.; Haibach T.; Steurer W.; Hermann R.; Nissen H.-U.; Grätzel M. Structure of Nanocrystalline TiO2 Powders and Precursor to Their Highly Efficient Photosensitizer. Chem. Mater. 1997, 9, 430–439. 10.1021/cm950502p. [DOI] [Google Scholar]
- Diebold U.; Ruzycki N.; Herman G. S.; Selloni A. One Step towards Bridging the Materials Gap: Surface Studies of TiO2 Anatase. Catal. Today 2003, 85, 93–100. 10.1016/S0920-5861(03)00378-X. [DOI] [Google Scholar]
- Valiev M.; Bylaska E. J.; Govind N.; Kowalski K.; Straatsma T. P.; Van Dam H. J. J.; Wang D.; Nieplocha J.; Apra E.; Windus T. L.; et al. NWChem: A Comprehensive and Scalable Open-Source Solution for Large Scale Molecular Simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. 10.1016/j.cpc.2010.04.018. [DOI] [Google Scholar]
- Dietrich P. M.; Streeck C.; Glamsch S.; Ehlert C.; Lippitz A.; Nutsch A.; Kulak N.; Beckhoff B.; Unger W. E. S. Quantification of Silane Molecules on Oxidized Silicon: Are there Options for a Traceable and Absolute Determination?. Anal. Chem. 2015, 87, 10117–10124. 10.1021/acs.analchem.5b02846. [DOI] [PubMed] [Google Scholar]
- Rathor N.; Panda S. Aminosilane Densities on Nanotextured Silicon. Mater. Sci. Eng., C 2009, 29, 2340–2345. 10.1016/j.msec.2009.06.003. [DOI] [Google Scholar]
- Islam M. M.; Calatayud M.; Pacchioni G. Hydrogen Adsorption and Diffusion on the Anatase TiO2(101) Surface: A First-Principles Investigation. J. Phys. Chem. C 2011, 115, 6809–6814. 10.1021/jp200408v. [DOI] [Google Scholar]
- Tilocca A.; Selloni A. Reaction Pathway and Free Energy Barrier for Defect-induced Water Dissociation on the (101) Surface of TiO2-Anatase. J. Chem. Phys. 2003, 119, 7445–7450. 10.1063/1.1607306. [DOI] [Google Scholar]
- Tilocca A.; Selloni A. Structure and Reactivity of Water Layers on Defect-Free and Defective Anatase TiO2(101) Surfaces. J. Phys. Chem. B 2004, 108, 4743–4751. 10.1021/jp037685k. [DOI] [Google Scholar]
- Iguchi N.; Cady C.; Snoeberger R. C.; Hunter B. M.; Sproviero E. M.; Schmuttenmaer C. A.; Crabtree R. H.; Brudvig G. W.; Batista V. S. Characterization of Siloxane Adsorbates Covalently Attached to TiO2. Proc. SPIE 2008, 70340C. 10.1117/12.798938. [DOI] [Google Scholar]
- Macetti G.; Rizzato S.; Beghi F.; Silvestrini L.; Lo Presti L. On the Molecular Basis of the Activity of the Antimalarial Drug Chloroquine: EXAFS-assisted DFT Evidence of a Direct Fe–N Bond with Free Heme in Solution. Phys. Scr. 2016, 91, 023001. 10.1088/0031-8949/91/2/023001. [DOI] [Google Scholar]
- Iacomino A.; Cantele G.; Ninno D.; Marri I.; Ossicini S. Structural, Electronic, and Surface Properties of Anatase TiO2 Nanocrystals from First Principles. Phys. Rev. B: Condens. Matter Mater. Phys. 2008, 78, 075405–075415. 10.1103/PhysRevB.78.075405. [DOI] [Google Scholar]
- Zhang Z.; Yates J. T. Jr. Band Bending in Semiconductors: Chemical and Physical Consequences at Surfaces and Interfaces. Chem. Rev. 2012, 112, 5520–5551. 10.1021/cr3000626. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Yates J. T. Jr. Effect of Adsorbed Donor and Acceptor Molecules on Electron Stimulated Desorption: O2/TiO2(110). J. Phys. Chem. Lett. 2010, 1, 2185–2188. 10.1021/jz1007559. [DOI] [Google Scholar]
- Pimenta S.; Cardoso S.; Miranda A.; De Beule P.; Castanheira E. M. S.; Minas G. Design and Fabrication of SiO2/TiO2 and MgO/TiO2 Based High Selective Optical Filters for Diffuse Reflectance and Fluorescence Signals Extraction. Biomed. Opt. Express 2015, 6, 3084–3098. 10.1364/BOE.6.003084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.