

Abstract
The Notch signaling pathway is essential for embryonic vascular development in vertebrates. Here we show that mouse embryos heterozygous for a targeted mutation in the gene encoding the DLL4 ligand exhibit haploinsufficient lethality because of defects in vascular remodeling. We also describe vascular defects in embryos homozygous for a mutation in the Rbpsuh gene, which encodes the primary transcriptional mediator of Notch signaling. Conditional inactivation of Rpbsuh function demonstrates that Notch activation is essential in the endothelial cell lineage. Notch pathway mutant embryos exhibit defects in arterial specification of nascent blood vessels and develop arteriovenous malformations. These results demonstrate that vascular remodeling in the mouse embryo is sensitive to Dll4 gene dosage and that Notch activation in endothelial cells is essential for embryonic vascular remodeling.
Keywords: Notch signaling, haploinsufficiency, vascular remodeling, arteriovenous malformation, Dll4, RBP-J
The Notch signaling pathway is an evolutionarily conserved intercellular signaling mechanism. Mutations in Notch pathway components disrupt embryonic development in diverse multicellular organisms and cause inherited disease syndromes in humans (Gridley 2003; Lai 2004; Schweisguth 2004). Genes of the Notch family encode large transmembrane receptors that interact with membrane-bound ligands encoded by genes of the Delta and Serrate/Jagged families. The signal induced by ligand binding is transmitted intracellularly by a process involving proteolytic cleavage of the receptor and nuclear translocation of the intracellular domain of the Notch family protein (Notch-IC). Once in the nucleus, Notch-IC forms a complex with the RBP-J protein, which is encoded by the Rbpsuh gene. The RBP-J protein is a sequence-specific DNA-binding protein that is the primary transcriptional mediator of Notch signaling. The Notch-IC/RBP-J complex then activates transcription of downstream target genes.
Recent work has shown that Notch signaling is essential for embryonic vascular development in vertebrates (Lawson and Weinstein 2002; Rossant and Howard 2002; Shawber and Kitajewski 2004). Here we demonstrate that mouse embryos heterozygous for a targeted mutation in the gene encoding the DLL4 ligand exhibit haploinsufficient lethality because of defects in vascular remodeling. We describe vascular defects in embryos homozygous for a mutation in the Rbpsuh gene and show that Rbpsuh-null (-/-) embryos do not express several arterial-specific endothelial cell markers. Conditional inactivation of Rpbsuh function demonstrates that Notch activation is essential in the endothelial cell lineage. Notch pathway mutant embryos also exhibit arteriovenous malformations, likely as a consequence of an inability to establish and maintain distinct arterial-venous vascular beds. These results demonstrate that the Dll4 gene encodes the predominant Notch ligand for early vascular development, that vascular remodeling in the mouse embryo is sensitive to the Dll4 gene dosage, and that Notch activation in endothelial cells is required for embryonic vascular remodeling.
Results and Discussion
Haploinsufficient lethality of Dll4+/- embryos
Previous gene-expression analysis suggested that the Dll4 gene encoded the Notch ligand most likely signaling to the NOTCH1 and NOTCH4 receptors during early vascular development in mice (Krebs et al. 2000; Shutter et al. 2000). To analyze whether the Dll4 gene is required for vascular development, we created a Dll4 deletion allele by gene targeting (Supplementary Fig. 1). Three independent Dll4+/- embryonic stem (ES) cell clones were generated and were used to make chimeric mice, which then were mated to outbred Black Swiss or inbred C57BL/6J mice. Despite germ-line transmission of the ES cell genome (assessed by inheritance of agouti coat color), only a single viable Dll4+/- heterozygote, from a mating to Black Swiss mice, was obtained in 180 adult agouti progeny. This included 115 progeny in California from matings to Black Swiss mice and 65 progeny in Bar Harbor from matings to C57BL/6J mice. Normally, 50% of the agouti progeny (∼90 mice) should be Dll4+/- heterozygotes. Although the single Dll4+/- heterozygous male mouse was runted, it was at least partially fertile, yielding 12 wild-type but no Dll4+/- heterozygous progeny in matings to Black Swiss females. This pattern of transmission is suggestive of haploinsufficient lethality (Carmeliet et al. 1996; Ferrara et al. 1996), so we isolated embryos at different gestational ages from matings of the chimeras to wild-type C57BL/6J mice. At embryonic days 9.5 and 10.5 (E9.5 and E10.5), Dll4+/- embryos exhibited characteristic vascular remodeling defects, such as a mottled avascular yolk sac, growth retardation, and pericardial effusions (Supplementary Fig. 2). Staining of endothelial cells with anti-PECAM-1 antibody revealed a complete absence of vascular remodeling in the Dll4+/- yolk sacs at E9.5, similar to what we observed previously in Notch1-/- mutant and Notch1-/- Notch4-/- double mutant yolk sacs (Krebs et al. 2000). The Dll4+/- mutants failed to remodel the yolk sac primary vascular plexus to form the large and small blood vessels of the mature yolk sac (Fig. 1a,b). In Dll4+/- embryos (Fig. 1d), the capillary network was less extensive and more primitive than the capillary network of wild-type control littermates. Some vessels in the Dll4+/- embryos were reduced in diameter or were atretic (Fig. 1d,f). In contrast to the yolk sacs, vascular remodeling defects in Dll4+/- embryos in general were less severe than those observed in Notch1-/- and Notch1-/- Notch4-/- mutant embryos. This may reflect the fact that the Dll4+/- embryos retain a functional copy of the Dll4 gene, but may also reflect a differential sensitivity to Dll4 gene dosage between the yolk sac and embryo, or a role for other Notch ligands such as JAG1 (Xue et al. 1999) or DLL1 (Hrabé de Angelis et al. 1997) during vascular development in the embryo.
Figure 1.

Vascular defects in Dll4+/- embryos. PECAM-1-stained yolk sacs (a,b) and embryos (c,d) at E9.5. (b) The Dll4+/- mutant yolk sac has failed to remodel the primary vascular plexus to form the large vitelline blood vessels. (d) The vascular network in the Dll4+/- embryo appears less intricate and more primitive than the capillary network of the control littermate. (e,f) Histological sections of PECAM-1-stained E9.5 embryos at the level of the otic vesicle (asterisk). (f) The dorsal aorta of this Dll4+/- embryo is reduced in diameter (arrowhead) on one side and is atretic (i.e., contains no lumen) on the other side (arrow).
Vascular remodeling defects in Rbpsuh-/- embryos
The RBP-J protein, encoded by the Rbpsuh gene, is the primary transcriptional mediator of the Notch signal (Kato et al. 1997; for reviews, see Gridley 2003; Lai 2004; Schweisguth 2004). Although the vascular defects present in embryos mutant for Notch family receptors (Krebs et al. 2000), ligands (Hrabé de Angelis et al. 1997; Xue et al. 1999), and downstream effectors (Fischer et al. 2004) have been described, vascular defects of Rbpsuh-null mutant embryos have not been reported. The vascular defects of Rbpsuh-/- embryos (Fig. 2b,d) were more severe than those of Dll4+/- embryos and were similar to defects observed previously in Notch1-/- Notch4-/- double mutant embryos (Krebs et al. 2000). Histological analysis of the placentas of the Rbpsuh-/- embryos also revealed that, similar to Notch1-/- Notch4-/- embryos (Krebs et al. 2000), Rbpsuh-/- mutant blood vessels did not efficiently penetrate the labyrinthine layer of the placenta. However, the histological analysis also suggested that there may be a reduction of the spongiotrophoblast layer of the placenta in the Rbpsuh-/- embryos (data not shown). This may be due to a role for Notch signaling during placental development that is independent of its role in the vasculature, as previous work has shown that the Notch2 gene is expressed in the spongiotrophoblast and trophoblast giant cell layers of the developing placenta (Nakayama et al. 1997). In addition to the observed defects in vascular remodeling, Rbpsuh-/- embryos exhibit multiple additional defects that have been described previously. These include defects in somite formation (del Barco Barrantes et al. 1999) and in heart looping morphogenesis, caused by Notch pathway regulation of leftright asymmetry determination (Krebs et al. 2003; Raya et al. 2003).
Figure 2.

Vascular defects and loss of arterial marker expression in Rbpsuh-/- embryos. (a-d) PECAM-1-stained E9.5 embryos and yolk sacs. (b) The Rbpsuh-/- embryo exhibits severe growth retardation and a very primitive vascular network. (d) The Rbpsuh-/- yolk sac exhibits no vascular remodeling. (e-h) Efnb2-tau-lacZ expression in E9.5 embryos and yolk sacs. In the wild-type embryo, Efnb2-taulacZ is expressed in the dorsal aorta (arrow) and intersomitic arteries of the embryo (e) and the vitelline arteries of the yolk sac (g). Expression is also observed in somites, nephrogenic mesoderm, branchial arches and hindbrain. Arterial Efnb2-tau-lacZ expression is lost in the Rbpsuh-/- embryo (f) and yolk sac (h). (i-l) Whole mount immunohistochemistry of E9.5 embryos. CD44 (i,j) and αSMA (k,l) expression in the dorsal aorta (arrow) of wild-type embryos is down-regulated in Rbpsuh-/- embryos.
Loss of arterial marker expression in Rbpsuh-/- embryos
Recent studies have revealed that endothelial tubes are specified as arteries or veins at the earliest stages of vessel formation, prior to the onset of blood flow (Wang et al. 1998; Adams et al. 1999; Gerety et al. 1999; for reviews, see Lawson and Weinstein 2002; Rossant and Howard 2002; Shawber and Kitajewski 2004). In addition, studies in zebrafish have indicated that Notch signaling regulates arterial-venous specification (Lawson et al. 2001, 2002; Zhong et al. 2001). We therefore examined expression of several arterial markers in Rbpsuh-/- embryos. EphrinB2 (Efnb2), a ligand for the EphB family of tyrosine kinase receptors, is one of the earliest arterial-specific endothelial cell markers. To most sensitively examine Efnb2 expression, we crossed an Efnb2-tau-lacZ knockin allele (Wang et al. 1998) into the Rbpsuh null background. In Rbpsuh+/+ or Rbpsuh+/- embryos heterozygous for the Efnb2-tau-lacZ allele, whole mount staining for β-galactosidase enzymatic activity revealed Efnb2-tau-lacZ expression in arterial endothelium of embryos and their yolk sacs, as well as in the somites, nephrogenic mesoderm, and hindbrain (Fig. 2e,g). In Rbpsuh-/- embryos heterozygous for the Efnb2-tau-lacZ allele, β-galactosidase expression in the arterial endothelium was lost whereas expression in all other tissues was retained (Fig. 2f,h). CD44 protein expression in the dorsal aorta (Wheatley et al. 1993), another arterial marker, was down-regulated in Rbpsuh-/- embryos (Fig. 2j). Recruitment of pericytes and smooth muscle cells to support the walls of nascent blood vessels occurs earlier in arteries than in veins. We therefore examined expression of α smooth muscle actin (αSMA) around the dorsal aorta in Rbpsuh-/- embryos and found that αSMA protein expression was down-regulated (Fig. 2l). Examination of the expression of these arterial markers indicates that, as in zebrafish (Lawson et al. 2001, 2002; Zhong et al. 2001), loss of Notch signaling leads to loss of arterial specification of the forming blood vessels.
Notch activation is essential in the endothelial cell lineage
Gain-of-function experiments expressing an activated form of the NOTCH4 receptor have shown that constitutive activation of Notch signaling in the endothelial cell lineage leads to embryonic vascular defects (Uyttendaele et al. 2001). However, a requirement for Notch signaling in endothelial cells has not been demonstrated by loss-of-function analysis. To test this, we deleted the Rbpsuh gene in endothelial cells by crossing mice containing a floxed Rbpsuh allele (Rbpsuhfl) (Tanigaki et al. 2002) to mice expressing Cre recombinase under control of the Tek (also known as Tie-2) promoter (Tek-Cre) (Koni et al. 2001). Tek-Cre Rbpsuhfl/null embryos isolated at E9.5 exhibited vascular remodeling defects similar to those observed in Notch pathway mutants such as Notch1-/-, Notch1-/- Notch4-/-, and Dll4+/- embryos. The Tek-Cre Rbpsuhfl/null mutant embryos exhibited an avascular yolk sac, growth retardation, and pericardial effusion (Fig. 3b). PECAM-1 antibody staining revealed a complete absence of vascular remodeling in the yolk sac (Fig. 3d), and histological analysis of the Tek-Cre Rbpsuhfl/null mutant embryos showed reductions in diameter of some vessels, such as the dorsal aortae (Fig. 3f). These data demonstrate that Notch pathway activation in the endothelial cell lineage is essential for embryonic vascular development. Taken together, the vascular defects exhibited by the Notch receptor gain-of-function mutant embryos (Uyttendaele et al. 2001) and the haploinsufficient lethality and vascular remodeling defects observed in Dll4+/- embryos indicate that the levels of Notch activation in endothelial precursors of the forming vasculature must be precisely regulated for normal vascular development.
Figure 3.

Notch signal activation is required in endothelial cells. The Rbpsuh gene was conditionally inactivated in the endothelial cell lineage by crossing mice containing a floxed Rbpsuh allele to mice expressing Cre recombinase under control of the Tek promoter. (a,b) Whole mounts of E9.5 embryos. (b) The Tek-Cre Rbpsuhfl/null embryo exhibits growth retardation and pericardial effusion. (c,d) PECAM-1-stained E9.5 embryos and yolk sacs. (d) The Tek-Cre Rbpsuhfl/null mutant yolk sac has failed to remodel the primary vascular plexus to form large vitelline blood vessels. (e,f) Sections of PECAM-1-stained E9.5 embryos. The dorsal aortae (arrows) of the Tek-Cre Rbpsuhfl/null mutant are reduced in diameter.
Formation of arteriovenous malformations in Dll4+/- and Tek-Cre Rbpsuhfl/null embryos
Reduction of Notch signaling in zebrafish embryos results in formation of arteriovenous malformations (AVMs) (Lawson et al. 2001, 2002; Zhong et al. 2001), the fusion of arteries and veins without an intervening capillary bed. We examined Dll4+/- and Tek-Cre Rbpsuhfl/null embryos for the presence of AVMs by ink injection into the hearts of E9.5 embryos. All mutant embryos exhibited the presence of AVMs. In wild-type embryos (n = 40), ink injected into the proximal outflow tract of the heart exited through the branchial arch arteries, entered the paired dorsal aortae, and traversed the entire length of the embryo (Fig. 4a,d). In Dll4+/- (n = 5) (Fig. 4b,e) and Tek-Cre Rbpsuhfl/null (n = 8) (Fig. 4c,f) embryos, injected ink exited the distal outflow tract, then entered the venous circulation via small-diameter anastamoses with the anterior cardinal vein. Histological analysis of Dll4+/- (Fig. 5b,d) and Tek-Cre Rbpsuhfl/null (Fig. 5c,e) embryos confirmed the presence of AVMs. Some Dll4+/- and Tek-Cre Rbpsuhfl/null embryos also exhibited AVMs more caudally that were caused by fusion of the dorsal aorta with the common cardinal vein. Although the small size and multiple defects present in Rbpsuh-/- embryos prevented successful intracardiac ink injections, histological analysis of Rbpsuh-/- embryos revealed the presence of AVMs similar to those observed in Dll4+/- and Tek-Cre Rbpsuhfl/null embryos (Fig. 5g).
Figure 4.
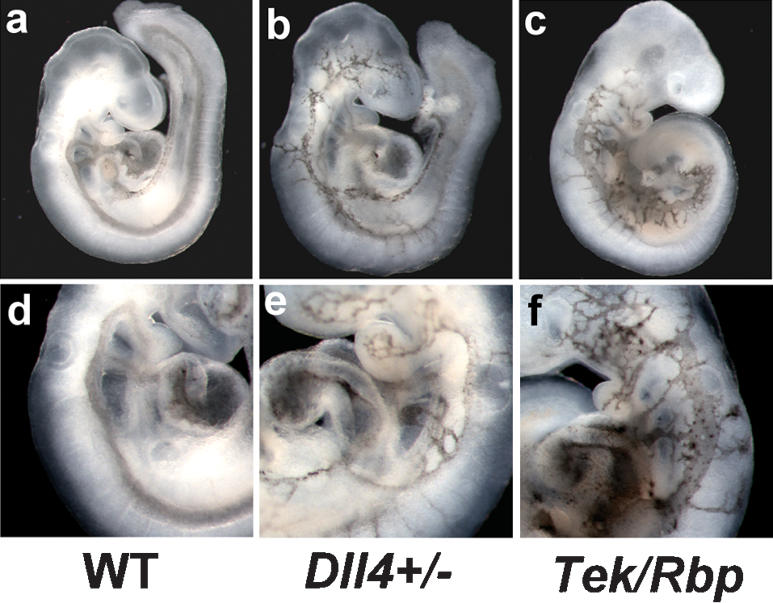
Arteriovenous malformations in Notch pathway mutant embryos. India ink was injected into the proximal outflow tract of the heart in order to visualize blood flow and arteriovenous malformations. (a,d) In wild-type embryos, ink injected into the heart exited through the branchial arch arteries, entered the paired dorsal aortae, and traversed the entire length of the embryo. In Dll4+/- (b,e) and Tek-Cre Rbpsuhfl/null (c,f) embryos, injected ink exited the distal outflow tract, then entered the venous circulation via small-diameter anastamoses with the anterior cardinal vein. The Dll4+/- and Tek-Cre Rbpsuhfl/null embryos shown in b and c are different embryos than those shown in e and f. (a-c) Low-magnification views. (d-f) High-magnification views.
Figure 5.
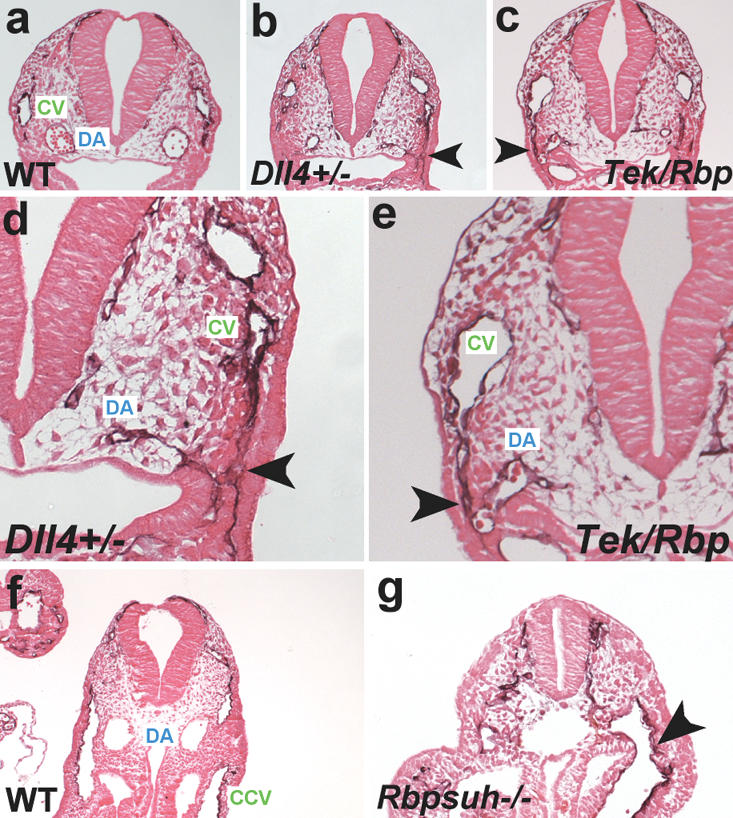
Histological analysis of arteriovenous malformations in Notch pathway mutants. (a-c) Sections of wild-type, Dll4+/- and Tek-Cre Rbpsuhfl/null PECAM-1-stained embryos at E9.5. (d,e) Higher-magnification views of the embryos in b and c. In each mutant, an arteriovenous malformation (arrowhead) consisting of a small-diameter anastomosis connecting the cardinal vein (CV) with the dorsal aorta (DA) can be observed. (f,g) Sections of wild-type and Rbpsuh-/- PECAM-1-stained embryos at E9.5. (g) The mutant embryo exhibits an arteriovenous malformation (arrowhead) caused by the fusion of the dorsal aorta with the common cardinal vein (CCV).
In summary, our results demonstrate that, as was suggested by our previous gene expression studies (Krebs et al. 2000; Shutter et al. 2000), the Dll4 gene encodes the predominant Notch ligand during early vascular development in mice. Further, vascular remodeling in the mouse embryo is sensitive to Dll4 gene dosage, as Dll4+/- embryos exhibit haploinsufficient lethality. Notch signaling has been shown to be downstream of VEGF-A signaling in both zebrafish (Lawson et al. 2002) and mammalian endothelial cells in culture (Liu et al. 2003). Strikingly, mouse embryos heterozygous for a targeted null mutation of the Vegfa gene also exhibit haploinsufficient lethality (Carmeliet et al. 1996; Ferrara et al. 1996), suggesting that the Dll4 gene may be a target for regulation by VEGF-A and may be at least part of the mechanism causing the haploinsufficient lethality exhibited by Vegfa+/- mouse embryos.
This work also demonstrates that, as in zebrafish, loss of Notch activation in mice leads to defects in arterial specification of endothelial cells forming nascent blood vessels. This finding is supported by recent work demonstrating that expression of arterial markers is down-regulated in mouse embryos doubly homozygous for mutations of the Hey1 and Hey2 genes (Fischer et al. 2004), which encode basic helix-loop-helix (bHLH) transcription factors that are downstream Notch pathway effectors. As an apparent consequence of the loss of arterial-venous specification, Notch pathway mutant embryos form arterial-venous vascular shunts, or AVMs. Similar AVMs have been observed in Notch pathway mutants in zebrafish (Lawson et al. 2001), as well as in mouse mutants in the Transforming Growth Factor β (TGFβ) pathway components endoglin (Eng) and activin receptor-like kinase I (Acvrl1) (Sorensen et al. 2003). These AVMs appear to arise as a consequence of an inability to establish and maintain distinct arterial-venous vascular beds. Future work will examine whether there is any cross-talk between the Notch pathway and the TGFβ pathway, or between these pathways and other signaling pathways involved in arterial-venous specification, such as the ephrin/Eph receptor pathway.
Materials and methods
Dll4 gene targeting and genotyping
The Dll4 targeting vector (Dll4d1) contained a 4.9-kb genomic fragment subcloned upstream of a PGK-neo expression cassette, and a 3.2-kb fragment subcloned downstream. This resulted in the deletion of part of exon 1 and exons 2-6 of the Dll4 gene, including the predicted translation initiation codon and the exons encoding the DSL domain of the DLL4 protein. A thymidine kinase cassette was introduced for negative selection. The targeting vector was electroporated into 129x1/SvJ-derived Gsi-1 ES cells (Genome Systems). Dll4 chimeras were crossed to outbred Black Swiss (Taconic) or inbred C57BL/6J (Jackson Laboratory) mice. Yolk sacs or progeny were genotyped for the Dll4d1 allele using the oligonucleotide primers CGACATCCCTAACAAGCAG and TCGCCTTC TATCGCCTTCTTG. The Rbpsuhfl/null allele was genotyped by Southern blotting as previously described (Tanigaki et al. 2002). The Rbpsuh null allele was genotyped using primers TAGACCTTGGTTTGTTGTTTGG and CCATAGGAAAACATCCACAGC, which span the deleted region. Tek-Cre mice (Koni et al. 2001) were obtained from the Jackson Laboratory and were genotyped with the primers TGATGAGGTTCG CAAGAACC and CCATGAGTGAACGAACCTGG. Efnb2-tau-lacZ mutant mice were genotyped as previously described (Wang et al. 1998).
Immunohistochemistry
Immunohistochemistry was performed as previously described (Krebs et al. 2000). Primary antibodies included rat monoclonal anti-PECAM-1 (BD Biosciences Pharmingen), rat monoclonal anti-CD44 (BD Biosciences Pharmingen), and mouse monoclonal anti-α smooth muscle actin (clone 1A4; Sigma). Horseradish peroxidase-coupled secondary antibodies were from Jackson ImmunoResearch.
Cardiac ink injections
Embryos from timed mating were dissected in phosphate buffered saline (PBS) and immediately injected into the proximal outflow tract of the heart with 1% India ink in PBT (PBS with 0.1% Tween 20). Glass injection needles were made using a Model P-97 Flaming/Brown Micropipette Puller (Sutter Instruments). Embryos were fixed in 4% paraformaldehyde and photographed. For sectioning, embryos were dehydrated, embedded in paraffin, sectioned, and were counterstained with eosin.
Acknowledgments
We thank David Anderson for providing the ephrinB2-tau-lacZ mice, Chris Norton and Kathleen Oram for technical assistance, Sue Ackerman and Tim O'Brien for comments on the manuscript, and Janet Rossant for exchanging unpublished results. This work was supported by grants to T.G. from the NIH (NS36437) and the March of Dimes Birth Defects Foundation, and by a grant from the National Cancer Institute (CA34196) to the Jackson Laboratory.
Supplemental material is available at http://www.genesdev.org.
Article published online ahead of print. Article and publication date are at http://www.genesdev.org/cgi/doi/10.1101/gad.1239204.
References
- Adams R.H., Wilkinson, G.A., Weiss, C., Diella, F., Gale, N.W., Deutsch, U., Risau, W., and Klein, R. 1999. Roles of ephrinB ligands and EphB receptors in cardiovascular development: Demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes & Dev. 13: 295-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P., Ferreira, V., Breier, G., Pollefeyt, S., Kieckens, L., Gertsenstein, M., Fahrig, M., Vandenhoeck, A., Harpal, K., Eberhardt, C., et al. 1996. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380: 435-439. [DOI] [PubMed] [Google Scholar]
- del Barco Barrantes I, Elia, A.J., Wunsch, K., De Angelis, M.H., Mak, T.W., Rossant, J., Conlon, R.A., Gossler, A., and de la Pompa, J.L. 1999. Interaction between Notch signalling and Lunatic fringe during somite boundary formation in the mouse. Curr. Biol. 9: 470-480. [DOI] [PubMed] [Google Scholar]
- Ferrara N., Carver-Moore, K., Chen, H., Dowd, M., Lu, L., O'Shea, K.S., Powell-Braxton, L., Hillan, K.J., and Moore, M.W. 1996. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380: 439-442. [DOI] [PubMed] [Google Scholar]
- Fischer A., Schumacher, N., Maier, M., Sendtner, M., and Gessler, M. 2004. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes & Dev. 18: 901-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerety S.S., Wang, H.U., Chen, Z.F., and Anderson, D.J. 1999. Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol. Cell 4: 403-414. [DOI] [PubMed] [Google Scholar]
- Gridley T. 2003. Notch signaling and inherited disease syndromes. Hum. Mol. Genet. 12 (Suppl. 1): R9-R13. [DOI] [PubMed] [Google Scholar]
- Hrabé de Angelis M., McIntyre II, J., and Gossler, A. 1997. Maintenance of somite borders in mice requires the Delta homologue Dll1. Nature 386: 717-721. [DOI] [PubMed] [Google Scholar]
- Kato H., Taniguchi, Y., Kurooka, H., Minoguchi, S., Sakai, T., Nomura-Okazaki, S., Tamura, K., and Honjo, T. 1997. Involvement of RBP-J in biological functions of mouse Notch1 and its derivatives. Development 124: 4133-4141. [DOI] [PubMed] [Google Scholar]
- Koni P.A., Joshi, S.K., Temann, U.A., Olson, D., Burkly, L., and Flavell, R.A. 2001. Conditional vascular cell adhesion molecule 1 deletion in mice: Impaired lymphocyte migration to bone marrow. J. Exp. Med. 193: 741-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs L.T., Xue, Y., Norton, C.R., Shutter, J.R., Maguire, M., Sundberg, J.P., Gallahan, D., Closson, V., Kitajewski, J., Callahan, R., et al. 2000. Notch signaling is essential for vascular morphogenesis in mice. Genes & Dev. 14: 1343-1352. [PMC free article] [PubMed] [Google Scholar]
- Krebs L.T., Iwai, N., Nonaka, S., Welsh, I.C., Lan, Y., Jiang, R., Saijoh, Y., O'Brien, T.P., Hamada, H., and Gridley, T. 2003. Notch signaling regulates left-right asymmetry determination by inducing Nodal expression. Genes & Dev. 17: 1207-1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai E.C. 2004. Notch signaling: Control of cell communication and cell fate. Development 131: 965-973. [DOI] [PubMed] [Google Scholar]
- Lawson N.D. and Weinstein, B.M. 2002. Arteries and veins: Making a difference with zebrafish. Nat. Rev. Genet. 3: 674-682. [DOI] [PubMed] [Google Scholar]
- Lawson N.D., Scheer, N., Pham, V.N., Kim, C.H., Chitnis, A.B., Campos-Ortega, J.A., and Weinstein, B.M. 2001. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128: 3675-3683. [DOI] [PubMed] [Google Scholar]
- Lawson N.D., Vogel, A.M., and Weinstein, B.M. 2002. Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev. Cell 3: 127-136. [DOI] [PubMed] [Google Scholar]
- Liu Z.J., Shirakawa, T., Li, Y., Soma, A., Oka, M., Dotto, G.P., Fairman, R.M., Velazquez, O.C., and Herlyn, M. 2003. Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: Implications for modulating arteriogenesis and angiogenesis. Mol. Cell. Biol. 23: 14-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H., Liu, Y., Stifani, S., and Cross, J.C. 1997. Developmental restriction of Mash-2 expression in trophoblast correlates with potential activation of the Notch-2 pathway. Dev. Genet. 21: 21-30. [DOI] [PubMed] [Google Scholar]
- Raya A., Kawakami, Y., Rodriguez-Esteban, C., Buscher, D., Koth, C.M., Itoh, T., Morita, M., Raya, R.M., Dubova, I., Bessa, J.G., et al. 2003. Notch activity induces Nodal expression and mediates the establishment of left-right asymmetry in vertebrate embryos. Genes & Dev. 17: 1213-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossant J. and Howard, L. 2002. Signaling pathways in vascular development. Annu. Rev. Cell Dev. Biol. 18: 541-573. [DOI] [PubMed] [Google Scholar]
- Schweisguth F. 2004. Notch signaling activity. Curr. Biol. 14: R129-R138. [PubMed] [Google Scholar]
- Shawber C.J. and Kitajewski, J. 2004. Notch function in the vasculature: Insights from zebrafish, mouse and man. Bioessays 26: 225-234. [DOI] [PubMed] [Google Scholar]
- Shutter J.R., Scully, S., Fan, W., Richards, W.G., Kitajewski, J., Deblandre, G.A., Kintner, C.R., and Stark, K.L. 2000. Delta4, a novel Notch ligand expressed in arterial endothelium. Genes & Dev. 14: 1313-1318. [PMC free article] [PubMed] [Google Scholar]
- Sorensen L.K., Brooke, B.S., Li, D.Y., and Urness, L.D. 2003. Loss of distinct arterial and venous boundaries in mice lacking endoglin, a vascular-specific TGFβ coreceptor. Dev. Biol. 261: 235-250. [DOI] [PubMed] [Google Scholar]
- Tanigaki K., Han, H., Yamamoto, N., Tashiro, K., Ikegawa, M., Kuroda, K., Suzuki, A., Nakano, T., and Honjo, T. 2002. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat. Immunol. 3: 443-450. [DOI] [PubMed] [Google Scholar]
- Uyttendaele H., Ho, J., Rossant, J., and Kitajewski, J. 2001. Vascular patterning defects associated with expression of activated Notch4 in embryonic endothelium. Proc. Natl. Acad. Sci. 98: 5643-5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.U., Chen, Z.F., and Anderson, D.J. 1998. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93: 741-753. [DOI] [PubMed] [Google Scholar]
- Wheatley S.C., Isacke, C.M., and Crossley, P.H. 1993. Restricted expression of the hyaluronan receptor, CD44, during postimplantation mouse embryogenesis suggests key roles in tissue formation and patterning. Development 119: 295-306. [DOI] [PubMed] [Google Scholar]
- Xue Y., Gao, X., Lindsell, C.E., Norton, C.R., Chang, B., Hicks, C., Gendron-Maguire, M., Rand, E.B., Weinmaster, G., and Gridley, T. 1999. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 8: 723-730. [DOI] [PubMed] [Google Scholar]
- Zhong T.P., Childs, S., Leu, J.P., and Fishman, M.C. 2001. Gridlock signalling pathway fashions the first embryonic artery. Nature 414: 216-220. [DOI] [PubMed] [Google Scholar]