

Abstract
The ubiquitous occurrence of emerging micropollutants (EMPs) in water is an issue of growing environmental-health concern worldwide. However, there remains a paucity of data regarding their levels and occurrence in water. This study determined the occurrence of EMPs namely: carbamazepine (CBZ), galaxolide (HHCB), caffeine (CAF), tonalide (AHTN), 4-nonylphenol (NP), and bisphenol A (BPA) in water from Gauteng, Mpumalanga, and North West provinces, South Africa using comprehensive two-dimensional gas chromatography coupled to high resolution time-of-flight mass spectrometry (GCxGC-HRTOFMS). Kruskal-Wallis test and ANOVA were performed to determine temporal variations in occurrence of the EMPs. Principal component analysis (PCA) and Surfer Golden Graphics software for surface mapping were used to determine spatial variations in levels and occurrence of the EMPs. The mean levels ranged from 11.22 ± 18.8 ng/L for CAF to 158.49 ± 662 ng/L for HHCB. There was no evidence of statistically significant temporal variations in occurrence of EMPs in water. Nevertheless, their levels and occurrence vary spatially and are a function of two principal components (PCs, PC1 and PC2) which controlled 89.99% of the variance. BPA was the most widely distributed EMP, which was present in 62% of the water samples. The detected EMPs pose ecotoxicological risks in water samples, especially those from Mpumalanga province.
Keywords: drinking water, emerging micropollutants, GCxGC-HRTOFMS, spatial variations, wastewater
1. Introduction
Emerging micropollutants (EMPs) are ubiquitous in aquatic environments and are a matter of growing concern worldwide [1,2,3,4,5,6,7,8,9,10]. The EMPs comprise of a wide range of natural and synthetic organic compounds, which include pharmaceuticals and personal care products (PPCPs), detergents, steroid hormones, industrial chemicals, pesticides, and many other contaminants of emerging concern [2,3,5,7,9]. Compared to other contaminants of anthropogenic origin, the EMPs have largely been outside the scope of monitoring and regulation worldwide, and there is paucity of data on their levels, occurrence, and fate in water [1,2,4,6,8,9,10,11,12,13,14,15,16,17,18,19,20]. Nevertheless, the ubiquity occurrence of EMPs has been increasingly identified in both surface and ground water sources as a result of influx of effluents from wastewater treatment plants (WWTPs), on-site wastewater disposal systems, runoff from roadways wash off, agriculture fields, recreational activities, atmospheric deposition, animal feeding operations, leaking sewer lines, and landfill and septic tank leachate [8,10,21,22,23,24,25,26,27,28,29].
The majority of the existing WWTPs are not particularly constructed with the intention to remove EMPs from wastewater streams [10,30,31,32]. Consequently, the EMPs pass through WWTPs owing to their hydrophilicity, partial degradation, persistency, and the continuous introduction into the drinking water value chains of the hydrological cycle coupled with lack of well-established precautionary measures and monitoring actions [10,30,31,32]. Once the EMPs find their way into ground and surface water environments, they may re-enter the biosphere where they pose a threat to plant and animal life in addition to posing challenges to the potable water treatment facilities [10,33]. Despite their occurrence at low concentrations (i.e., from pg/L to μg/L) in water environments, the EMPs are associated with several ecotoxicological effects, such as chemo-sensitization, endocrine disruption, disruption of the production of platelets, red and white blood cells, short and long term toxicity, antibiotic resistance of microorganisms, and insomnia among others [1,3,4,6,8,9,10,11,12,13,14,15,16,17,18,19]. Severe ecotoxicological effects of EMPs have been reported to be as a result of the interactions of EMPs with human and/or aquatic life usually after long-term exposure to the EMPs [13,15,34]. Among these EMPs, Bisphenol A (BPA) (C15H16O), 4-Nonylphenol (NP) (C15H24O), carbamazepine (CBZ) (C15H12N2O), caffeine (CAF) (C8H10N4O2), galaxolide (HHCB) (C18H26O), and tonalide (AHTN) (C18H26O), present significant research interests due to their extensive use in several products, their ecotoxicological effects, as well as their physicochemical properties which allow their persistent presence in water [7,8,11,13,15,16] (Table 1). While BPA and NP are endocrine disrupting compounds, HHCB and AHTN are chemo-sensitizers (they have the potential to interfere with transportation of proteins (P-glycoprotein) and inhibition of cellular defence mechanisms), CBZ has the potential to disrupt the production of red blood cells, white blood cells, and platelets, and CAF has the potential to disrupt sleep (insomnia) (Table 1).
Table 1.
Some of applications, effects, and properties of Bisphenol A (BPA), 4-Nonylphenol (NP), caffeine (CAF), galaxolide (HHCB), tonalide (AHTN), and carbamazepine (CBZ) in wastewater and drinking water.
| Compound | Application | Ecotoxicological Effects | CAS-No | pKow |
|---|---|---|---|---|
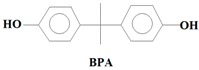 |
Component in plastics (resins, poly-carbonate), food-can liners, thermal paper (like the kind used for some receipts), and some dental sealants | Endocrine disruptor (estrogenic) | 80-05-7 | 3.32 |
| Formula: C15H16O2 | ||||
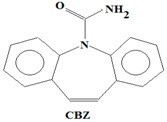 |
Anti-epileptic drug | Disruption in the production of red blood cells, white blood cells, and platelets | 298-46-4 | 2.45 |
| Formula: C15H12N2O | ||||
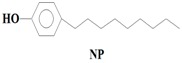 |
Constituent of detergents, anti-oxidant | Endocrine disruptor, interaction with estrogenic receptor | 104-40-5 | 5.76 |
| Formula: C15H24O | ||||
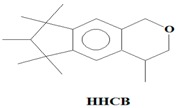 |
Polycyclic musk (fragrance used in consumer products) | Chemo-sensitizer (interference with transporter proteins (P-glycoprotein) leads to inhibition of the cellular defence mechanism) | 1222-05-5 | 5.9 |
| Formula: C18H26O | ||||
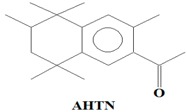 |
Polycyclic musk (fragrance used in consumer products) | Chemo-sensitizer (interference with transporter proteins (P-glycoprotein) leads to inhibition of the cellular defence mechanism) | 1506-02-1 | 5.7 |
| Formula: C18H26O | ||||
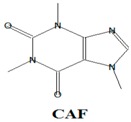 |
Stimulant, indicator for untreated wastewater inputs | Sleep disruption or insomnia | 58-08-2 | −0.07 |
| Formula: C8H10N4O2 |
According to Musolff et al. [8], an understanding of spatial and temporal variations in concentration heterogeneities of EMPs is one of the most important aspects in the study of EMPs. Nevertheless, the levels and occurrence of EMPs in the environment are largely ill understood in many parts of the world [20]. Generally, surface waters and wastewaters have been assumed to contain diverse and higher levels of EMPs compared to groundwater [20]. Lapwrorth et al. [20] noted that this assumption and the resulting paucity of data on occurrence of EMPs in water might simply be a function of limitations in capacity of several analytical methods and equipment, as well as the limited number of research studies rather than actual levels and occurrence of EMPs in water. With the advent of novel and highly sensitive analytical equipment capable of detecting chemical constituents at very low levels in complex sample matrices, the detection of EMPs is now possible, and has thus attracted the attention of researchers worldwide [1,3,4,6,8,9,10]. Traditionally, mass spectrometry (MS) or selective detectors coupled with gas or liquid chromatography had to be used for the identification of EMPs. However, one of the problems associated with these analytical techniques in the identification of these organic compounds, including EMPs, has been the co-elution of several compounds and their inability to quantify the compounds below traditionally reported limits of detection [35]. Another limitation of these traditional techniques is the additional time required and material needed for analysis when repeating the analysis on another column with different polarity [35].
The LECO Pegasus® 4D comprehensive two-dimensional gas chromatography coupled to high resolution time-of-flight mass spectrometry (GC × GC-HRTOFMS) is a recently developed robust state-of-the-art analytical technique that offers a solution to the co-elution problem with minimal time and material requirements for analysis [36]. The GC × GC-HRTOFMS (two-dimensional gas chromatography coupled to high resolution time-of-flight mass spectrometry) has proven to be one of the most powerful analytical techniques for the analysis of environmental samples and plays a significant role in the determination of organic compounds, including EMPs, in environmental samples [36,37,38,39]. According to LECO Corporation (St. Joseph, MI, USA) [36], the GC × GC-HRTOFMS system utilises four dimensions of separation and resolution, namely: (1) chromatographic resolution in first dimension; (2) chromatographic resolution in second dimension; (3) exceptional mass accuracy of 1 ppm as well as amplified mass resolution; and (4) high resolution deconvolution® (HRD®) available in the ChromaTOF-HRT® software package (LECO Corporation, St. Joseph, MI, USA) which is used to operate the GC × GC-HRTOFMS system. The ChromaTOF-HRT® software package coupled with increased peak capacity of the GC × GC at least two times greater than any other product on the market allows environmental samples to be handled in such a way that compound identification and quantification are not compromised. In addition, the GC × GC-HRTOFMS produces data with much improved separation capacity, signal-to-noise (S/N) ratio, chemical selectivity, and sensitivity [36,37,39,40,41,42].
In principle, the method consists of two GC systems equipped with columns of different polarity connected by an interface with an integrated cryogenic trap [35,36]. The cryogenic trap repeatedly condenses compounds eluting from the primary column and releases them periodically as short pulses to the secondary column [35]. Parameters like duration and frequency of both condensation and injection pulses are variable and allow precise tuning of the instrument according to the requirements of the analysis. Since GC × GC produces very narrow peaks (down to 50 ms, depending on the frequency of cryogenic modulation) a HRTOFMS detector capable of mass resolutions of up to 50,000 with a high acquisition rate (up to 200 spectra/s) is utilised [36]. The HRTOFMS system utilises a chemical ionisation source (HR-CI) which further enhances the system accuracy as well as high resolution on pseudo-molecular ions, which substantiates the conventional electron ionisation source (HR-EI) which provides the comprehensive characterisation of unknown compounds [36].
Despite the enhanced detectability and reliability of the GC × GC-HRTOFMS in the identification and quantification of EMPs in complex environmental samples owing to the two retention times and well-ordered bands of compound groups in the GC × GC system [36,43], there has been very little, if any, focus on the monitoring and determination of EMPs in groundwater and surface water sources used by thousands of people for their domestic needs in Gauteng, Mpumalanga, and North West provinces. However, these water sources are also at risk of contamination by a variety of contaminants including EMPs. As of yet, no lasting solutions have been proposed to address the problems associated with EMP contamination in many parts of the world, including Gauteng, Mpumalanga, and North West provinces in South Africa.
Due to the aforementioned widespread use, occurrence, distribution, fate, and effects of EMPs, sensitive and selective multi-residue analytical methods and techniques are required that will allow detection in environmental samples. The objectives of this study were therefore to: (1) determine the levels and occurrence of the analytes (i.e., BPA, CAF, CBZ, HHCB, NP, and AHTN) in the solid phase extraction (SPE) extracts using comprehensive GC × GC-HRTOFMS in water samples from Gauteng, Mpumalanga, and North West provinces, South Africa; (2) determine the limit of detection (LOD) and limit of quantification (LOQ) for BPA, NP, CAF, HHCB, AHTN, and CBZ by using comprehensive GC × GC-HRTOFMS; and (3) determine the temporal and spatial variations in the occurrence of the analytes during the study period.
2. Materials and Methods
2.1. Materials and Preparation of Reagents
The chemicals, BPA standard, NP standard, CAF standard, HHCB standard, AHTN standard, CBZ standard, methanol LC-MS CHROMASOLV®, and dichloromethane LC-MS CHROMASOLV®, hydrochloric acid (HCl), sodium hydroxide (NaOH), and standard pH buffers (for pH 4 and 7) were obtained from Sigma-Aldrich, Johannesburg, South Africa. All chemicals were used without further purification.
2.2. Study Area and Water Sample Collection
2.2.1. Study Area
The areas selected for study were Mpumalanga, Gauteng, and North West provinces in South Africa (Figure 1). Gauteng is the smallest province in South Africa, which covers an area of 18,178 square kilometres. Gauteng province is bordered by the North West, Limpopo, Free State, and Mpumalanga provinces. It is the most populated province with a total population of 12,272,263 [44]. Mpumalanga province is located to the eastern part of South Africa and is surrounded by Swaziland and Mozambique on the eastern side, and Gauteng province on the western side (Figure 1). Mpumalanga province covers an area of 79,487 square kilometres [44]. Mpumalanga province has a total population of 4,039,939 [44]. The North West province is located at the central part of South Africa and bordered by the Northern Cape on the southwestern side, the Free State to the southern part, Gauteng to the eastern side, and Limpopo to the northeastern side, with Botswana on its northern border (Figure 1). North West province has a total surface area of 116,231 square kilometres [44]. North West has a total population of 3,509,953 [44]. The increase in population in all the three provinces has resulted in increased water consumption, which aggravates pressure on South Africa’s existing water resources. In addition, Mpumalanga and North West provinces are faced with acute water resource constraints because they are largely arid provinces. Furthermore, based on both the Blue Drop Report of 2012 and the Green Drop Report of 2012, the majority of water supply sources in North West and Mpumalanga provinces were not fit for human consumption [45,46].
Figure 1.

Map of South Africa showing location of Mpumalanga, Gauteng, and North West provinces and sampling points.
2.2.2. Water Sample Collection and Onsite Water Sample Analyses
In this study, water samples were collected once every two months between June 2014 and April 2016. A total of 44 quasi-randomly selected locations within drinking water and wastewater sources in Mpumalanga, Gauteng, and North West provinces were utilized. Sampling sites were selected taking into account the variations in physiography and anthropogenic activities around the selected sites in each of the three provinces. In Mpumalanga province, groundwater and surface water samples were collected from school boreholes, shallow wells, the Eerstehoek water treatment plant (WTP), effluent from the Eerstehoek wastewater treatment plant (WWTP), and rivers located in the low-income areas situated in the Chief Albert Luthuli municipality in Mpumalanga, close to the Oshoek border between Swaziland and South Africa (Figure 1). In Gauteng and North West provinces, water samples were collected from rivers, Roodeplat and Hartbeespoort dams, and the Schoemansville water treatment plant. For analysis of EMPs (BPA, NP, CAF, HHCB, AHTN, and CBZ) the sample bottles were rinsed twice with water from a particular sampling site before obtaining the final sample. Grab water samples were collected using 1 L glass bottles in triplicate using standard sampling procedures. The water samples were analyzed for levels of pH, electrical, conductivity (EC), and total dissolved solids (TDS) in the field immediately after sampling, using a Hanna Instrument (Woonsocket, RI, USA) model HI-9828 multi-meter. Deionized water was used to rinse the electrode of the meter prior to determination of the levels of TDS, EC, temperature, and pH of any successive sample to avoid inter-sample contamination.
2.3. Preparation of Standard Solutions, Sample Preparation, and Solid Phase Extraction of Emerging Micropollutants
2.3.1. Preparation of Standard Solutions
The 1000 mg/L stock solutions for BPA, NP, HHCB, AHTN, and CBZ were prepared by weighing 1 mg of each into a vial (1.5 mL) and dissolving the sample in methanol (LC-MS CHROMASOLV® grade) (1 mL) under vortex and ultrasonication for 10 min. In addition, 1000 mg/L stock solutions for CAF were prepared by weighing 1 mg of CAF into 1.5 mL vial followed by dissolving the sample in dichloromethane (LC-MS CHROMASOLV® grade) (1 mL) under vortex and ultrasonication for 10 min. The stock solutions were stored in a refrigerator at a temperature of below 4 °C. From each of the stock solutions, 100 µL was pipetted and placed into one 1.5 mL vial and made up to the 1.0 mL mark with methanol (LC-MS CHROMASOLV® grade) to prepare a 100 mg/L mixed standard solution from which all working standards of different concentrations (10, 20, 30, 40, and 50 parts per million (ppm)) were prepared for the preparation of the calibration curve.
2.3.2. Sample Preparation and Solid Phase Extraction of Emerging Micropollutants
The water samples were kept in cooler boxes under ice and transported to the laboratory where they were filtered using a 1.2 μm GF/C Whatman filter paper to remove suspended matter prior to autotrace solid phase extraction (SPE) treatment and stored in a refrigerator below 4 °C until analysis. Solid phase extraction was used to extract, clean, and enrich/pre-concentrate the analytes in water samples using Dionex autotrace 280 by Thermo Scientific. The autotrace-SPE system was optimized with regard to initial analyte dose, sample pH, and sample volume. The EmporeTM Styrene Divinyl Benzene (SDB-RPS) autotrace-SPE disks were conditioned with methanol and deionised water. One hundred millilitres of water samples containing the target analytes were loaded onto the autotrace SPE and eluted through the SPE disks followed by washing with 5 mL of deionised water. The SPE disks were then subjected to vacuum drying in order to remove excess water before eluting the compounds with methanol (5 mL). The extracted solution was evaporated to dryness using nitrogen gas before reconstituting with methanol (1 mL) for GC × GC-HRTOFMS analysis [47].
The SPE system was optimized with regard to initial analyte dose, sample pH, and sample volume. The initial analyte dose of 5 µg/L, 10 µg/L, and 15 µg/L for each analyte into actual water samples as well as ultra-pure water samples was investigated via the standard addition method while keeping sample pH, sample volume, volume of methanol, and flow rate constant. The sample pH of the actual water samples and ultra-pure water was measured using Hanna model HI-9812 multi-meter (Hanna Instruments Limited, Bedfordshire, UK). The electrode of the meter was rinsed with deionised water before determining pH of any subsequent sample to prevent inter-sample contamination. Sample pH was optimized by varying the pH of actual water samples and ultra-pure water spiked with 1000 μg/L of each analyte. The pH was adjusted to 2, 4, 5, 7, 8, and 10 with HCl (1 mol/L) and NaOH (0.6 mol/L) [48].
The effect of sample volume was investigated by passing different volumes of actual water samples as well as ultra-pure water spiked with 1000 μg/L of each compound through the SDB-RPS autotrace-SPE disks while keeping the volume of methanol LC-MS CHROMASOLV® grade (5 mL), the volume of ultra-pure water (5 mL), and the flow rate (1 mL/min) constant [48]. The volumes of spiked ultra-pure water were in the range of 10 mL to 200 mL. The compounds retained by the SDB-RPS autotrace-SPE disks were eluted with methanol (5 mL) before evaporating to dryness using nitrogen gas before reconstituting with methanol (1 mL). For quality assurance, the percentage recoveries of the SPE extracted samples were calculated by comparing the recovered levels with the standard dose levels expressed as a percentage.
2.4. Determination of Levels and Occurrence of BPA, CAF, CBZ, HHCB, NP, and AHTN in Water Samples
The SPE extracts were analysed using the GCxGC-HRTOFMS (LECO Corporation, St. Joseph, MI, USA) equipped with a thermal modulator and a split/splitless injector using liquid nitrogen. A low-polarity phase Rxi-5SilMS column (30 m × 0.25 mm i.d., 0.25 µm film thickness) was used for GC in the first dimension analysis (1D GC). The second dimension analysis (2D GC) was performed on a polar Rxi-17SilMS (1 m × 0.25 mm i.d., 0.25 µm film thickness). Helium was used as the carrier gas at a constant linear velocity of 1.9 mL/min. Using an autosampler (Agilent 7890A Series, Santa Clara, CA, USA), splitless injection mode was used with the vaporised sample moving through the injection port liner. The oven temperature was programmed as follows: 50 °C (held for 1 min) ramped to 210 °C at 10 °C/min (held for 2 min) and then ramped to 250 °C at 15 °C/min and held for 10 min. The injector and interface temperatures were set at 220 °C, with the MS quad temperature set at 150 °C and the MS source at 230 °C. The secondary oven was operated at a temperature 5 °C higher than that of the primary oven and was operated in an iso-ramping mode. The modulation period, the hot-pulse duration, and the cool time between stages were set at 3.0 s, 0.4 s, and 1.1 s, respectively. The transfer line to HRTOFMS detector source was operated at 250 °C. The source temperature was 230 °C with a filament bias voltage of −70 eV. The MS mass range was 45 to 550 atomic mass units (amu), with the data acquisition rate at 200 spectra/s, while the detector voltage was 1750 V. The inlet temperature was 200 °C with a modulator offset temperature of 40 °C, and the purge time was 60 s. The mass spectrometer was operated in the positive ion mode with an ionisation voltage of 70 eV using selected ion monitoring (SIM). Prior to injection, the syringe was cleaned five times with n-hexane and once with the sample. An external standard mixture was measured after each batch of five samples to verify instrument measurement performance. Data were processed and consecutively visualised on 2D and 3D chromatograms using LECO ChromaTOF-HRT® software (LECO Corporation, St. Joseph, MI, USA).
2.5. Determination of the Mass Accuracies, Limits of Quantification, Limits of Detection, Linearity, and S/N Ratios for BPA, NP, CAF, HHCB, AHTN, and CBZ
Linear regression analysis was used to determine LOD and LOQ for BPA, NP, CAF, HHCB, AHTN, and CBZ based on GC × GC-HRTOFMS’ linear calibration curves for each analyte. It was assumed from the obtained linear calibration curves for each analyte that the GC × GC-HRTOFMS response matrix Y was linearly related to the descriptor matrix X for a limited range of concentrations. The limit of quantification (LOQ) and limit of detection (LOD) for each analyte were thus determined based on the signal-to-noise (S/N) ratios of 10 and 3 based on the residual standard deviation of the response or the standard deviation (SD) of the y-intercept of the regression line of the calibration curve and the sensitivity or slope of the regression line, as shown in Equations (1) and (2):
| (1) |
| (2) |
The LOQ and LOD and tests were performed in triplicate to confirm the accuracy regarding each of the detected EMPs at varying concentrations. The mass accuracies for BPA, NP, CAF, HHCB, AHTN, and CBZ were also obtained directly from GC × GC-HRTOFMS analyses.
2.6. Determination of the Temporal and Spatial Variations in the Occurrence of the Analytes during the Study Period
The R statistical software was used to compute descriptive statistics correlation studies, Kruskal Wallis test, ANOVA, and principal component analysis (PCA) utilizing data obtained from both onsite and GC × GC-HRTOFMS analyses of all the 44 water samples over the studied period. Kruskal Wallis test and analysis of variance (ANOVA) were performed to determine temporal variations in the occurrence of analytes in the water samples at 95% confidence level. The PCA and correlation studies were used as quantitative and independent approaches for water classification, allowing the grouping of the water samples and the establishment of correlations between chemical parameters and water samples, respectively. The principal components (PCs) of the PCA were extracted using the Varimax rotation. The total number of PCs to retain was based on the Kaiser criterion, wherein only PCs with eigenvalues >1 were retained and the parameters were retained if their p-value < 0.05 at 95% confidence level. Equation (3) shows the R-mode PCA model that was used to compute factor scores:
| (3) |
where fr were the rth common factors, p was the specified number of factors, j was the random variation unique to the original hydrochemical variable Xj, and ajr was the loading of the jth variate on the rth factor. The PCA model corresponded to the loading or weights on the extracted PCs. The new factor was expressed as shown using Equation (4)
| (4) |
where ai was the loading of i index; Ii was the standardized data of I index.
The factor score loadings for each water sample were utilised to model spatial variations in the occurrence of the EMPs using Surfer Golden Graphics software for surface mapping (version 8). Specifically, the value of each factor score represented the importance of a given factor at the sampled site. A factor score >+1 reflected sampling areas significantly influenced of EMPs highly loaded in a particular PC. Factor scores <−1 reflected sampling areas virtually unaffected by EMPS highly loaded in a particular PC, whereas near-zero scores reflected areas moderately influenced by EMPs highly loaded in a particular PC. The spatial variations of the occurrence of EMPs highly loaded in a particular PC were assessed by surface mapping contour plots of the factor scores representing each factor.
3. Results
3.1. Solid Phase Extraction Optimisation Results
3.1.1. Initial Analyte Dose
Autotrace-SPE of actual water samples as well as ultra-pure water samples spiked with initial analyte concentrations of 5 µg/L, 10 µg/L, and 15 µg/L, in triplicate, were investigated. From the results (Table 2), it was observed that the mean percent recovery (n = 3) of all the analytes was higher for the samples with an analyte concentration of 10 µg/L than that of samples containing the initial analyte concentration of 5 µg/L. Although the mean percent recovery of analytes for a 15 µg/L initial dose was higher than that of the 5 µg/L initial dose, they were actually found to be lower than those for the 10 µg/L initial dose, possibly due to interferences from other sample matrices, especially in actual water samples. It was thus observed that the 10 µg/L initial dose was the optimal initial dose for autotrace-SPE of all the analytes.
Table 2.
The effect of initial dose of analytes on autotrace-SPE recovery of BPA, NP, CAF, HHCB, AHTN, and CBZ (n = 3).
| Analyte | Initial Dose (5 µg/L) | Initial Dose (10 µg/L) | Initial Dose (15 µg/L) | |||
|---|---|---|---|---|---|---|
| Recovery Levels (µg/L) | Mean Recovery (%) | Recovery Levels (µg/L) | Mean Recovery (%) | Recovery Levels (µg/L) | Mean Recovery (%) | |
| BPA | 3.9 | 78 ± 0.9 | 9.6 | 96 ± 2.5 | 8.2 | 82 ± 0.82 |
| NP | 3.6 | 72 ± 1.1 | 9.4 | 94 ± 1.2 | 8.5 | 85 ± 0.56 |
| CAF | 3.5 | 70 ± 2.3 | 8.2 | 82 ± 1.5 | 7.4 | 74 ± 1.2 |
| HHCB | 3.0 | 60 ± 2.4 | 8.9 | 89 ± 1.3 | 7.1 | 71 ± 1.8 |
| AHTN | 3.1 | 62 ± 0.5 | 9.1 | 91 ± 2.7 | 8.3 | 83 ± 0.76 |
| CBZ | 3.2 | 64 ± 2.5 | 8.3 | 83 ± 2.1 | 6.8 | 68 ± 1.2 |
3.1.2. Sample pH
The highest mean percent autotrace-SPE recoveries for the analytes, measured in triplicate, were recorded at pH 7 (Figure 2). Therefore, the optimum pH for the autotrace-SPE recoveries of the analytes was selected as pH 7, the neutral pH. These results are in line with those reported by Santos et al. [48] who obtained a mean recovery of 80% for ketoprofen at neutral pH. On the other hand, Madikizela et al. [47] observed that a low pH is required for the analysis of acidic pharmaceuticals to prevent the dissociation of acidic compounds. However, Madikizela et al. [47] also stated that the sample pH during SPE must not be too low because acidic compounds that interfere in wastewater treatment processes may also be co-extracted and could interfere in the analysis if the sample pH is too low.
Figure 2.

Effect of pH on the mean percent autotrace SPE recovery (n = 3) of BPA, NP, CAF, HHCB, AHTN, and CBZ (n = 3).
3.1.3. Sample Volume
In this study it was observed that the mean percent autotrace SPE recoveries of all analytes were affected by the volume of the actual water samples as well as ultra-pure water samples loaded into the SDB-RPS autotrace-SPE disks. Specifically, an improvement in the mean percent autotrace-SPE recoveries of each of the analytes was observed when increasing the loading from 10 mL to 100 mL and then a decline was observed when increasing the loading from 100 mL to 200 mL (Figure 3). Therefore, a sample volume of 100 mL was selected as the optimum sample volume for the highest mean percent autotrace-SPE recoveries of the analytes. This is in line with the findings of Madikizela et al. [47] who observed that this trend is as a result of the capacity of the sorbent being exceeded and that higher volumes tend to overload the SPE cartridge and target compounds end up competing for the adsorbent material with matrix interferences. Madikizela et al. [47] further noted that high sample volume may result in the saturation of the SPE sorbent thereby leading to poor percent recoveries.
Figure 3.

Effect of sample volume of spiked ultra-pure water onto Oasis® HLB cartridge on the mean percent autotrace SPE recoveries of NP, CAF, HHCB, AHTN, and CBZ (n = 3).
3.2. Mass Accuracies, Limits of Quantification, Limits of Detection, Linearity, and S/N Ratios for BPA, NP, CAF, HHCB, AHTN, and CBZ
For all analytes, good linearity and reproducibility of analyses (R2 > 0.99) was achieved (Table 3). All the analytes registered desirable S/N ratios that were much higher than 100:1 (Table 3). The computed LOD, which according to Glaser et al. [49] corresponds to the lowest concentration that can be reliably detected and readily distinguished from zero with a certain degree of confidence, ranged from 0.25 ng/L for HHCB to 1.1 ng/L for CBZ (Table 3). On the other hand, the computed LOQ, which is the lowest amount of analyte that can be quantitatively determined at a definite level of accuracy and/or precision, ranged from 1.2 ng/L for HHCB to 3.9 ng/L for CAF (Table 3). All the analytes registered mass accuracies ranging from −0.97 to 0.52 (Table 3), which were below 1 ppm, the maximum allowable exceptional mass accuracy for GC × GC-HRTOFMS [36].
Table 3.
Computed mass accuracies, limits of quantification (LOQ), limits of detection (LOD), linearity, and S/N ratios for BPA, NP, CAF, HHCB, AHTN, and CBZ.
| Analyte | S/N Ratio | Mass Accuracy (ppm) | LOQ (ng/L) | LOD (ng/L) | Linearity, R2 (n = 3) |
|---|---|---|---|---|---|
| BPA | 4543 | −0.97 | 1.3 | 0.45 | 0.999 |
| NP | 6497 | 0.42 | 2.8 | 0.94 | 0.999 |
| CAF | 6276 | 0.52 | 3.9 | 1.1 | 0.999 |
| HHCB | 8650 | −0.07 | 1.2 | 0.25 | 0.999 |
| AHTN | 9459 | 0.33 | 1.3 | 0.39 | 0.998 |
| CBZ | 1758 | 0.07 | 3.13 | 1.0 | 0.997 |
3.3. The GCxGC-HRTOFMS Mass Spectra and Chromatograms, Levels, and Occurence of BPA, NP, CAF, HHCB, AHTN, and CBZ
3.3.1. The GC × GC-HRTOFMS Mass Spectra and Chromatograms of BPA, NP, CAF, HHCB, AHTN, and CBZ
The identification criteria for the analytes (BPA, NP, CAF, HHCB, AHTN, and CBZ) were two-fold in that both a first dimension retention time deviation (±2 s) and a second dimension retention time deviation (±0.5 s) were utilized, with the second taking into account the mass spectra with a similarity factor higher than 600 based on the library search. The similarity factor describes how well the library hit matches the peak when using all small molecule mass spectra in the U.S. National Institute of Standards and Technology (NIST) mass spectral libraries [40]. In the NIST mass spectra search algorithm the similarity is computed using numerical functions [40]. Confirmation of target analytes (BPA, NP, CAF, HHCB, AHTN, and CBZ) was based on the retention time, the accurate mass measurement of the molecular ion, the isotopic pattern, and by automated mass spectral library searches with GC × GC-HRTOFMS (Table 4).
Table 4.
Retention times, similarity, and m/z fragments for BPA, NP, CAF, HHCB, AHTN, and CBZ.
| Analyte | Mass | Unique Mass * | Retention Time (s) | Similarity | m/z or Adduct Ion | Fragment |
|---|---|---|---|---|---|---|
| BPA | 228.1143 | 213.091 | 1381.83, 2.58972 | 889 | 228.1143 | Molecular ion (M+) |
| 213.0910 | Unique mass | |||||
| 119.0492 | (M+ − C7H7O)-H2 | |||||
| 107.0494 | (C7H7O)+ | |||||
| 99.0337 | (M+ − C9H5O) | |||||
| 77.0385 | (Ar) | |||||
| 65.0386 | (M+ − C10H11O2) | |||||
| CBZ | 236.0694 | 193.089 | 1574.12, 0.66629 | 889 | 236.0944 | Molecular ion (M+) |
| 193.0886 | Unique mass | |||||
| 165.0701 | (M+ − H)-C2H2N2O | |||||
| 73.0469 | (M+ + H)-C8H6N2 | |||||
| 63.0231 | (M+ + H)-C10H8N2O | |||||
| NP | 220.1822 | 107.049 | 1102.40, 1.33257 | 894 | 220.1822 | Molecular ion (M+) |
| 107.0491 | Unique mass | |||||
| 77.0386 | (Ar) | |||||
| CAF | 194.0799 | 109.064 | 1093.39, 2.08686 | 942 | 194.0799 | Molecular ion (M+) |
| 109.0635 | Unique mass | |||||
| 137.0584 | (M+ − CH2COH3) | |||||
| 82.0526 | (M+ − CH2CH2NHNHO2) | |||||
| 55.0417 | CH2COCH | |||||
| 42.0341 | –CH3CH2CH- | |||||
| HHCB | 258.1978 | 243.175 | 1099.40, 1.37029 | 906 | 258.1978 | Molecular ion (M+) |
| 243.1745 | Unique mass | |||||
| 213.1639 | (M+ − H)-C3H8 | |||||
| 185.1326 | (M+ − Ar) + H2 | |||||
| 171.1169 | (M+ − H)-C6H14 | |||||
| 157.1102 | (M+ − Ar)-C2H2 | |||||
| 153.0697 | ((M+ − H)-Ar)-H2CH3 | |||||
| 128.0622 | (Ar-CO-CH2-CH-) | |||||
| 115.0544 | (Ar + C3H3)-H | |||||
| 57.0700 | (CH3CHOCH2) | |||||
| 41.0389 | -CH2CH2CH- | |||||
| AHTN | 258.1978 | 43.0179 | 1108.41, 1.34514 | 840 | 258.1978 | Molecular ion (M+) |
| 43.01798 | Unique mass | |||||
| 243.1745 | (M+ − CH3) | |||||
| 187.1118 | (M+ − Ar) + 3H+ | |||||
| 159.1169 | (M+ − Ar)-CH5O |
* quantifier ion.
In this study, all the analytes (BPA, NP, CAF, HHCB, AHTN, and CBZ) registered first dimension retention time deviations of ±2 s, second dimension retention time deviations of ±0.5 s, and mass spectra with similarity factors greater than 850, which were much higher than the set 600 identification criterion (Table 4). To this effect all the analytes (BPA, NP, CAF, HHCB, AHTN, and CBZ) met both GG × GC-HRTOFMS identification criteria, and their mass spectra surface plots are presented in Figure 4. In addition, the mass spectra of all analytes obtained from GC × GC-HRTOFMS are presented in Figure 5a–f. The ChromaTOF-HRT® software allowed peak deconvolution of each co-eluting peak, defined a unique ion m/z ratio, and extracted the pure mass spectra of individual analytes across the unresolved area (Figure 4 and Figure 5a–f). Caffeine was observed to be the most polar and volatile EMP of all the analytes and CBZ was the least. The m/z fragments for each analyte are presented in Table 4.
Figure 4.

HRTOFMS surface plots of BPA, NP, CAF, CBZ, HHCB, and AHTN spectra obtained from GC × GC-HRTOFMS (two-dimensional gas chromatography coupled to high resolution time-of-flight mass spectrometry) with modulation.
Figure 5.




HRTOFMS mass spectra of BPA (a); CBZ (b); NP (c); CAF (d); HHCB (e); and AHTN (f) obtained from GC × GC-HRTOFMS with modulation.
The obtained GC × GC-HRTOFMS chromatograms for the analytes BPA, NP, CAF, AHTN, HHCB, and CBZ are presented in the contour plot 2D version (Figure 6). Each spot or peak on the two images represents an individual compound for which a full mass spectrum was available. The retention times in the 1D GC (GC × GC-HRTOFMS without modulation) and 2D GC (GC × GC-HRTOFMS with modulation) and their coordinates in the contour plot were used to identify peaks, or spots in the contour plot. The contour plot of the GC × GC-HRTOFMS chromatogram typically demonstrates the main advantage of a high resolution 2D GC, namely the unique feature of “structured” chromatograms; this structured nature separates compounds into distinct groups for easy identification. It clearly shows the different groups of analytes in certain bands along the 2D plane. The analysis of GC × GC-HRTOFMS contour plot group types is powerful and a major advantage for GC × GC analysis.
Figure 6.

Contour plot of a GC × GC chromatogram of BPA, NP, CAF, AHTN, HHCB and CBZ with the retention times on column 1 (indicating volatility) and column 2 (indicating polarity) on the x- and y-axes, respectively.
Inspection of the 3D surface plot images of the GC × GC-HRTOFMS chromatograms compared with the traditional one-dimensional (1D) version (without modulation) chromatograms indicates that the GC × GC-HRTOFMS has better separation capability and the plot identifies the locations in the 2D separation plane (Figure 6). This also demonstrates the other advantages of GC × GC-HRTOMS, namely “structured” chromatograms, high sensitivity through peak sharpening and creating additional peak capacity as the chromatographic plane is expanded [43,50,51,52,53]. It was further observed that some peaks, such as those of nonylphenol, were invisible in the conventional 1D GC chromatogram (without modulation) but visible in 2D GC (i.e., GCxGC chromatogram with modulation).
3.3.2. Levels and Occurrence of BPA, NP, CAF, HHCB, AHTN, and CBZ in Water
The mean concentrations (measured in triplicate) of BPA, NP, CAF, HHCB, AHTN, and CBZ in water samples from Gauteng, Mpumalanga, and North West provinces have been presented in Table 5. The levels of BPA ranged from a not detectable limit (n.d.) to 181 ± 8.3 ng/L (recorded at Spring 1—Sisukumile Secondary School and Lower Lochiel Community) (Table 5). Elsewhere, studies by Regnery and Püttmann [54], Reinstorf et al. [6], Kim et al. [55], and Peng et al. [56] reported BPA levels in the ranges of 192–215, 192–215, 7.5–334, and 6–881 ng/L, respectively. Generally, the recorded levels of BPA in this study were lower than those reported by Regnery and Püttmann [54], Reinstorf et al. [6], Kim et al. [55], and Peng et al. [56]. The observed occurrence of BPA in the majority of the water sources in this study was attributed to the influence of municipal wastewater. The results are similar to those reported by Furhacker et al. [57]; Fromme et al. [58], and Musolff et al. [8] who also observed that the potential sources of BPA in water are municipal wastewater as well as industrial wastewater. The Minnesota Department of Health (MDH) [59] developed a guidance value of 20 µg/L for BPA. To this effect, a person drinking water with a BPA concentration at or below 20 µg/L would have little or no risk of any health effects from BPA. However, BPA has been reported to be one of the endocrine disrupting compounds with estrogenic receptors in human and animal life even at lower concentrations [8] (Table 1). In this study, all samples were found to have mean BPA concentrations below the lowest guidance value of 20 µg/L BPA [59]. Although the levels of BPA were generally low, the presence of increased BPA levels in some water samples such as the spring at Sisukumile School in the Lochiel Community is a major concern since it is the major source of drinking water, and water used for other domestic purposes (i.e., cooking and washing) for the Sisukumile Secondary School and Lower Lochiel Community. The spring is centrally located and therefore the spring water is most often used by the Lochiel community; however, a lot of litter in the form of plastics, food can liners, and paper was observed in and around the spring. The school (Sisukumile) is located near the spring and there are some agricultural activities and outside pit latrines in the area.
Table 5.
Mean concentration (measured in triplicate) of a given emerging micro-pollutant in wastewater and drinking water from Gauteng, Mpumalanga, and North West provinces.
| Sampling Site | Province | Mean Concentrations (ng/L) ± SD | |||||
|---|---|---|---|---|---|---|---|
| BPA | CAF | CBZ | HHCB | AHTN | NP | ||
| Treated water—Eerstehoek WTP | Mpumalanga | 4.0 ± 1 | 6.4 ± 1.2 | n.d. | n.d. | n.d. | n.d. |
| Raw water—Eerstehoek WTP | Mpumalanga | 20 ± 0.2 | 9.1 ± 0.5 | n.d. | n.d. | n.d. | n.d. |
| Effluent—Eerstehoek WWTP | Mpumalanga | 20 ± 4 | 11 ± 0.5 | 58 ± 0.2 | 19 ± 3 | 96 ± 0.9 | 98 ± 1.4 |
| Mkomazane River | Mpumalanga | 13 ± 0.8 | 82.41 ± 5.1 | 29 ± 0.9 | 9 ± 0.6 | 17 ± 0.3 | 12 ± 0.2 |
| Spring 1—Sisukumile Secondary School and Lower Lochiel Community | Mpumalanga | 181 ± 8.3 | n.d. | n.d. | 3477 ± 35 | 26 ± 1.0 | n.d. |
| Spring 2—Upper Lochiel Community | Mpumalanga | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Masakhane Primary School | Mpumalanga | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Lipoponyane River | Mpumalanga | 9.3 ± 0.6 | 2.5 ± 0.3 | 24 ± 0.5 | 7.2 ± 1.4 | 13 ± 0.1 | n.d. |
| Sisukumile water tank (JoJo) | Mpumalanga | 34 ± 1.8 | n.d. | n.d. | n.d. | 22 ± 0.7 | n.d. |
| Schoemansville—raw water | North West | 0.8 ± 0.2 | 1.1 ± 0.0 | 2.3 ± 0.1 | n.d. | n.d. | n.d. |
| Schoemansville—polymer dosing point | North West | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Schoemansville—after flocculation | North West | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Schoemansville—treated water | North West | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| Roodeplat dam 10 | Gauteng | 5.1 ± 0.5 | n.d. | 1.75 ± 0.3 | n.d. | 7.77 ± 2.2 | 4.27 ± 0.5 |
| Roodeplat dam 15 | Gauteng | 5.0 ± 0.8 | n.d. | 49.4 ± 8.6 | 0.3 ± 0.01 | 7.24 ± 3.2 | 1.35 ± 0.02 |
| Roodeplat dam 20 | Gauteng | 5.2 ± 0.8 | n.d. | 24.27 ± 5.2 | 1.03 ± 0.3 | n.d. | 2.23 ± 0.6 |
| Roodeplat dam 25 | Gauteng | 5.8 ± 1 | 2.23 ± 0.2 | 46.87 ± 9.3 | 59.74 ± 8.7 | 11.18 ± 3.2 | 5.38 ± 0.8 |
| Roodeplat dam 30 | Gauteng | 6.1 ± 0.2 | 11.31 ± 5.1 | 10.83 ± 2.2 | 17.93 ± 4.9 | 19.73 ± 1.3 | 28.72 ± 5.2 |
| Roodeplat dam 35 | Gauteng | 6.4 ± 0.3 | 4.72 ± 1.0 | n.d. | 18.26 ± 2.9 | 56.64 ± 8.8 | 24.82 ± 9.7 |
| Roodeplat dam 40 | Gauteng | 4.1 ± 0.5 | 8.72 ± 0.56 | 35.25 ± 6.4 | 6.49 ± 1.8 | 14.02 ± 3.2 | 2.87 ± 0.9 |
| Roodeplat dam 50 | Gauteng | 3.0 ± 0.8 | 10.6 ± 1.7 | 23.69 ± 4.5 | 36.2 ± 8.3 | 72.71 ± 10.1 | n.d. |
| Roodeplat dam 55 | Gauteng | 5.2 ± 0.8 | 1.55 ± 0.6 | 37.45 ± 7.1 | 37.77 ± 5.6 | 82.69 ± 7.3 | 5.47 ± 1.4 |
| Roodeplat dam 60 | Gauteng | 6.8 ± 1.2 | n.d. | 11.04 ± 1.2 | 46.88 ± 9.0 | 20.92 ± 7.5 | 19.56 ± 2.5 |
| Roodeplat dam 65 | Gauteng | n.d. | n.d. | 9.66 ± 2.4 | 65.59 ± 4.8 | 57.09 ± 8.2 | 5.13 ± 0.9 |
| Roodeplat dam 70 | Gauteng | n.d. | n.d. | 38.08 ± 3.1 | 100.94 ± 10.4 | 3.09 ± 0.75 | n.d. |
| Roodeplat dam 75 | Gauteng | n.d. | n.d. | 6.78 ± 0.81 | 26.97 ± 4.57 | 5.62 ± 1.4 | 8.33 ± 13.2 |
| Roodeplat dam 80 | Gauteng | n.d. | n.d. | 1.6 ± 0.02 | 2.37 ± 1.2 | 64.75 ± 6.78 | 1.22 ± 0.06 |
| Roodeplat dam 85 | Gauteng | 30.34 ± 8.3 | 5.84 ± 3.7 | 7.7 ± 0.08 | 3.02 ± 1.1 | 27.59 ± 5.7 | n.d. |
| Hartbeespoort 1-Krokodil River | North West | 81.24 ± 3.2 | n.d. | 8.58 ± 2.8 | 18.08 ± 1.9 | 98.1 ± 7.11 | n.d. |
| Hartbeespoort 2-HBD Inlet-Krokodil River | North West | 36.67 ± 7.3 | 5.14 ± 1.1 | 6.76 ± 3.8 | 9.83 ± 3.2 | 63.58 ± 0.5 | 2.57 ± 0.5 |
| Hartbeespoort 3-Hartbeerspoort dam Wall | North West | 12.31 ± 5.7 | n.d. | 1.74 ± 0.66 | 69.52 ± 14.8 | 10.95 ± 3.23 | n.d. |
| Hartbeespoort 4-HBD/Krokodil River | North West | 5.11 ± 1.22 | n.d. | 13.99 ± 5.1 | n.d. | 85.41 ± 9.76 | n.d. |
| Hartbeespoort 5-Snake Park | North West | n.d. | n.d. | 1.28 ± 0.4 | 129.37 ± 15.9 | 3.65 ± 1.1 | n.d. |
| Hartbeespoort 6-HBD/Krokodil/Megalies | North West | 7.34 ± 2.1 | n.d. | n.d. | 0.45 ± 0.005 | 75.84 ± 7.93 | n.d. |
| Hartbeespoort 7 | North West | n.d. | 2.49 ± 0.6 | 25.71 ± 6.32 | 2.86 ± 1.32 | 54.46 ± 9.7 | n.d. |
| Hartbeespoort 8-Megalies River | North West | 3.15 ± 2.3 | 24.52 ± 2.6 | 52.35 ± 8.6 | n.d. | 27.44 ± 7.4 | 3.43 ± 1.3 |
| Hartbeespoort 9-HBD inlet-Megalies River | North West | 1.52 ± 0.8 | 1.43 ± 0.9 | 30.52 ± 11.5 | 13.74 ± 5.34 | 27.18 ± 6.35 | n.d. |
| Hartbeespoort 11-Harties Boat Park | North West | n.d. | n.d. | 1.3 ± 0.32 | 29.91 ± 7.72 | 46.85 ± 7.8 | n.d. |
| Hartbeespoort 12-Mangwanani | North West | n.d. | n.d. | 9.22 ± 2.53 | 82.43 ± 5.13 | 13.96 ± 3.89 | n.d. |
| Hartbeespoort 14 | North West | n.d. | 5.73 ± 0.7 | 18.79 ± 5.71 | n.d. | 69.82 ± 8.8 | 1.35 ± 0.05 |
| Hartbeespoort 15 | North West | n.d. | n.d. | n.d. | n.d. | 75.84 ± 18.4 | n.d. |
The mean CAF concentrations ranged from n.d. to 82.41 ± 5.1 ng/L (Table 5), with the highest mean concentration (i.e., 82.41 ± 5.1 ng/L) registered in the Mkomazane River, into which the effluent from the Eerstehoek (Elukwatini) WWTP is discharged. Elsewhere, studies by Ternes et al. [60], Musolff et al. [8], and Spongberg et al. [61] reported CAF concentrations in the range of 190 ± 90, 48, and 241, 121, 446 ng/L, respectively. The maximum levels of CAF recorded in this study were generally lower than those reported by Ternes et al. [60] and Spongberg et al. [61]. Nevertheless, the levels of CAF observed in this study could be attributed to an additional constant source of untreated wastewater [60]. In terms of ecotoxicological effects, studies have shown that CAF leads to insomnia (sleep disruption) in humans and other animals, even at trace concentrations [8] (Table 1). The levels of CBZ were highest (i.e., 58 ± 0.2 ng/L) in Eerstehoek WWTP effluent (Table 5). Elsewhere, studies by Regnery and Püttmann [54] and Reinstorf et al. [6] reported CBZ levels in the range of 102–1194 ng/L. The highest levels of CBZ reported by Regnery and Püttmann [54] and Reinstorf et al. [6] were generally higher than those observed in this study. Studies have shown that CBZ has the virtue of disrupting the production of red blood cells, white blood cells, and platelets in humans and other animals, even at lower concentrations [8].
The mean levels of NP ranged from n.d to 98 ± 1.4 ng/L observed in the Eerstehoek WWTP effluent (Table 5). A study by Peng et al. [56] reported NP levels in the range of 36–33,231 ng/L. The recorded levels of NP in this study were generally lower than the maximum levels of NP as reported by Peng et al. [56]. It is worth noting that NP is a constituent of detergents and an anti-oxidant, and it was therefore considered that municipal wastewater would be its main source in water. According to Musolff et al. [8], NP has an endocrine disrupting effect in humans and other animals, even in trace concentrations. The mean HHCB levels were highest in Spring 1—Sisukumile Secondary School, Lower Lochiel Community with the mean concentration ranging from n.d. to 3477 ± 35 ng/L (Table 5). The mean levels AHTN levels were highest (i.e., 98.1 ± 7.11 ng/L) in the Krokodil River at Hartbeespoort 1 (Table 5), although high levels of AHTN were also observed in the effluent, Eerstehoek WWTP. Elsewhere, studies by Regnery and Püttmann [54] and Reinstorf et al. [6] reported HHCB and AHTN levels in the ranges of 35–1814 ng/L and 5–273 ng/L, respectively. The maximum levels of AHTN and HHCB recorded in this study were generally lower than those observed by Regnery and Püttmann [54] and Reinstorf et al. [6]. According to Standley et al. [62], a combination of musk fragrances such as AHTN and HHCB with CAF is a unique tracer for the impact of wastewater and wastewater treatment plant effluents on water resource. Both AHTN and HHCB are both polycyclic musks commonly used in fragrances, and they are known to have a chemo-sensitisation effect, a phenomenon characterised by the inhibition of proper functioning of particular cellular glycoprotein and the production of tumour cells in humans and other animals [8].
To the best of our knowledge, there are no reported guidelines for CAF, NP, CBZ, HHCB, and AHTN for water and wastewater. However, research has shown that among the EMPs, CBZ, HHCB, and AHTN are very resistant to biodegradation [7]. In particular, Schirmer and Schirmer [7] reported that only around 30% of CBZ were bio-transformed when sewage sludge was used as a source of microorganisms for biodegradation of CBZ in long-term (two-year) batch experiments done by UFZ-Department of Analytical Chemistry. It is thus clear that plant and animal life, as well as some communities (especially those living in close proximity to contaminated water sources) are at risk of the effects of the identified EMPs since all the EMPs were present in all types of water samples even though found to be at different concentrations.
3.4. Temporal and Spatial Variations in the Levels and Occurrence of BPA, NP, CAF, HHCB, AHTN, and CBZ in Water Samples
3.4.1. Temporal Variations in the Levels and Occurrence of BPA, NP, CAF, HHCB, AHTN, and CBZ in Water Samples
Except for pH (p-value = 0.000), the results of the Kruskal-Wallis test and ANOVA revealed that there was no statistically significant temporal variation in the occurrence of BPA, NP, CAF, HHCB, AHTN, and CBZ (p-value > 0.05) at the 95% confidence level (Table 6), despite an overall increase in the mean bi-monthly concentrations of all analytes in all water samples between June 2014 and April 2016. It was observed that the levels of pH, TDS, and EC were uniformly and significantly correlated with each other (p-value < 0.05), but weakly and statistically insignificantly correlated with the EMPs, BPA, CAF, CBZ, HHCB, AHTN, and NP (p-value > 0.05) at the 95% confidence level (Table 7). This suggested that, regardless of time of sample collection, the occurrence and distribution of the EMPs in water collected from Gauteng, Mpumalanga, and North West provinces were largely independent of mineralisation processes in water, as shown by the statistically insignificant correlations with measures of the level of mineralization in water (i.e., TDS and EC). Nevertheless, there was a significant correlation between BPA and HHCB (r2 = 0.905, p-value = 0.000), CAF and NP (r2 = 0.607, p-value = 0.043), CBZ and CAF (r2 = 0.610, p-value = 0.046), and HHCB and AHTN (r2 = 0.692, p-value = 0.014) (Table 7), suggesting that the sources of these EMPs were similar.
Table 6.
Descriptive statistics showing the p-value (at 95% confidence level), minimum, maximum, and mean concentration of a given emerging micro-pollutant in wastewater and drinking water from Gauteng, Mpumalanga, and North West provinces over the study period.
| Analyte | Minimum | Maximum | Mean | p-Value |
|---|---|---|---|---|
| pH | 6.00 | 9.20 | 7.48 ± 0.82 | 0.113 |
| EC (µS/cm) | 20.43 | 993.00 | 310.49 ± 285 | 0.000 |
| TDS (mg/L) | 3.00 | 569.00 | 130.01 ± 112 | 0.065 |
| BPA (ng/L) | n.d. | 181.00 | 28.55 ± 44.7 | 0.686 |
| CAF (ng/L) | n.d. | 82.41 | 11.22 ± 18.8 | 0.638 |
| CBZ (ng/L) | n.d. | 58.35 | 19.63 ± 16.8 | 0.187 |
| HHCB (ng/L) | n.d. | 3477.00 | 158.49 ± 662 | 0.057 |
| AHTN (ng/L) | n.d. | 98.10 | 40.44 ± 28.9 | 0.199 |
| NP (ng/L) | n.d. | 98.33 | 13.75 ± 23.6 | 0.669 |
Table 7.
Correlation studies of the emerging micropollutants and selected physicochemical water quality parameters.
| Parameter/r2/Sig. | pH | EC | TDS | BPA | CAF | CBZ | HHCB | AHTN | NP | |
|---|---|---|---|---|---|---|---|---|---|---|
| pH | r2 | 1 | 0.605 ** | 0.540 ** | 0.037 | 0.007 | 0.001 | 0.071 | −0.076 | 0.223 |
| Sig. | 0.000 | 0.004 | 0.892 | 0.979 | 0.995 | 0.725 | 0.681 | 0.0390 | ||
| EC | r2 | 0.605 ** | 1 | 0.574 ** | −0.082 | −0.222 | −0.036 | 0.170 | 0.085 | −0.167 |
| Sig. | 0.000 | 0.002 | 0.764 | 0.375 | 0.853 | 0.396 | 0.645 | 0.523 | ||
| TDS | r2 | 0.540 ** | 0.574 ** | 1 | −0.145 | −0.029 | −0.201 | −0.047 | 0.093 | −0.278 |
| Sig. | 0.004 | 0.002 | 0.591 | 0.909 | 0.296 | 0.815 | 0.614 | 0.280 | ||
| BPA | r2 | 0.037 | −0.082 | −0.145 | 1 | −0.048 | −0.321 | 0.905 ** | −0.088 | −0.110 |
| Sig. | 0.892 | 0.764 | 0.591 | 0.896 | 0.336 | 0.000 | 0.775 | 0.890 | ||
| CAF | r2 | 0.007 | −0.222 | −0.029 | −0.048 | 1 | 0.610 | −0.050 | −0.237 | 0.607 ** |
| Sig. | 0.979 | 0.375 | 0.909 | 0.896 | 0.046 | 0.872 | 0.395 | 0.043 | ||
| CBZ | r2 | 0.001 | −0.036 | −0.201 | −0.321 | 0.610 | 1 | −0.158 | −0.103 | −0.265 |
| Sig. | 0.995 | 0.853 | 0.296 | 0.336 | 0.046 | 0.462 | 0.609 | 0.321 | ||
| HHCB | r2 | 0.071 | 0.170 | −0.047 | 0.905 ** | −0.050 | −0.158 | 1 | 0.692 ** | 0.101 |
| Sig. | 0.725 | 0.396 | 0.815 | 0.000 | 0.872 | 0.462 | 0.014 | 0.731 | ||
| AHTN | r2 | −0.076 | 0.085 | 0.093 | −0.088 | −0.237 | −0.103 | 0.692 ** | 1 | −0.444 |
| Sig. | 0.681 | 0.645 | 0.614 | 0.775 | 0.395 | 0.609 | 0.014 | 0.085 | ||
| NP | r2 | 0.223 | −0.167 | −0.278 | −0.110 | 0.607 ** | −0.265 | 0.101 | −0.444 | 1 |
| Sig. | 0.390 | 0.523 | 0.280 | 0.890 | 0.043 | 0.321 | 0.731 | 0.085 | ||
** Pearson correlation (r2) is significant at the 95% confidence level (two-tailed).
3.4.2. Spatial Variations in the Levels and Occurrence of BPA, NP, CAF, HHCB, AHTN and CBZ in Water Samples
Table 8 presents PCs and variable loadings generated by the PCA model. Two PCs, which accounted for 89.99% of the total variance were extracted (Table 8). PC1 explained 51.46% of the variance and accounted for the majority of the variance in the original dataset for levels and occurrence of EMPs in water samples from the studied area. PC2 explained 38.53% the variance in the original dataset for levels and occurrence of EMPs in water samples from the studied area. PC1 registered high positive loadings in CAF, CBZ, and BPA and high negative loadings in pH, TDS, and EC. This suggested that CAF, CBZ, and BPA might have originated from similar sources and that their levels and occurrence in water are weakly affected by mineralization processes in water. This also suggested that the occurrence of CAF, CBZ, and BPA increased with decreases in pH of water. On the other hand, PC2 registered high positive loadings in AHTN, HHCB, and NP. This suggested that AHTN, HHCB, and NP might have originated from similar sources. This is not surprising considering that AHTN, HHCB, and NP are major ingredients of personal care products and detergents.
Table 8.
Principal components and variable loadings extracted using the principal component analysis using Varimax rotation.
| Variable | Principal Component 1 Loadings | Principal Component 2 Loadings |
|---|---|---|
| pH | −0.998 | |
| Total dissolved solids | −0.992 | |
| Electrical Conductivity | −0.962 | |
| Caffeine | 0.980 | |
| Carbamazepine | 0.935 | |
| Tonalide | 0.997 | |
| Galaxolide | 0.961 | |
| Bisphenol A | 0.906 | |
| Nonylphenol | 0.902 | |
| % Variance | 51.46 | 38.53 |
| Cumulative % | 51.46 | 89.99 |
It was observed that the concentrations of AHTN, CBZ, HHCB, CAF, NP, and BPA in water varied spatially, with BPA being the most widely distributed EMP that was present in 62% of the sampled sites (Table 5). Figure 7 shows the surface mapping contour plots of the PCA factor score model. The surface mapping contour plot showing the spatial distribution of PC1 factor scores has been presented in Figure 7a. Positive PC1 factor scores (i.e., >+1) were observed in water samples collected from Mpumalanga province. The majority of water samples from Gauteng and North West provinces registered negative PC1 factor scores and therefore were unaffected by EMPs highly loaded in PC1. Similarly, the surface mapping contour plot showing the spatial distribution of PC2 factor scores has been presented in Figure 7b. Positive PC2 factor scores (i.e., >+1) were observed in water samples collected from Mpumalanga province. The majority of water samples from Gauteng and North West provinces registered negative PC2 factor scores and therefore were unaffected by EMPs highly loaded in PC2, with a few samples moderately affected by EMPs with high loadings in PC2.
Figure 7.

Surface mapping contour plots showing spatial distribution of the PCA analysis factor scores and spatial variations in the occurrence of EMPs with high loadings in (a) PC1 and (b) PC2.
It was clear from the surface mapping contour plots of the PCA factor model that the levels and occurrence of EMPs with water samples collected from Mpumalanga province were higher than the levels of the EMPs registered for their Gauteng and North West counterparts. These higher levels of EMPs in Mpumalanga were attributed to the location, proximity of the sources to sanitary facilities, and open nature of the water sources in Mpumalanga which made them prone to EMP input through litter in the form of plastics, food can liners, and paper in and around the water sources in addition to water sources being prone to wastewater input. Some agricultural activities and outside pit latrines could also have contributed to the higher levels of EMPs in the water sources in Mpumalanga. It was clear that the communities especially in Mpumalanga province were at risk of the effects of the identified EMPs since all the EMPs were present in all types of water samples, though at different concentrations. There is need for better scientific understanding of occurrence, distribution, and environmental fate of EMPs, as well as effective EMP removal technologies in order to protect human health and the environment. These findings agree with those by Reinstorf et al. [6], Musolff et al. [9], Luo et al. [10], Huerta-Fontela et al. [18] and You et al. [63] who reported widespread spatial variations in the occurrence of EMPs, most of which have not been well studied despite the studies on spatial variations and linkages of the same EMPs with existing environmental factors having the virtue of enriching the scientific understanding of behaviour, distribution, and fate of the EMPs in water bodies in addition to assisting direct future management and water pollution safeguards.
4. Conclusions
The EMPs, BPA, NP, CAF, HHCB, AHTN, and CBZ were successfully extracted and pre-concentrated using autotrace-SPE prior to determination using the GCxGC-HRTOFMS system in water samples collected from Mpumalanga, Gauteng, and North West provinces, South Africa at better S/N ratios as well as lower LOD and LOQ. Although no statistically significant temporal variation in occurrence of the analytes was observed in water samples at the 95% confidence level, all the analytes were detected, at different levels concentrations, in the different sample types analysed across a broad spectrum of wastewater effluent, surface water, groundwater, and treated water, with BPA found to be present in 62% of the sampling sites and thus identified as the most widely distributed EMP in these water systems. It was also observed that the levels and occurrence of EMPs varied spatially and were a function of two PCs (PC1 and PC2) which controlled 89.99% of the observed variance. The results indicated that the identified EMPs pose ecotoxicological risks to aquatic life as well as communities, especially in Mpumalanga province which was largely influenced by EMPs with high loadings in the two PCs. The results of this study will thus contribute to the body of knowledge on levels and occurrence of EMPs in water, especially in considering the case of Gauteng, North West, and Mpumalanga provinces in South Africa. An understanding of temporal and spatial variations in the levels and occurrence of EMPs in water is critical for enrichment of the scientific understanding of behaviour, distribution, and fate of the EMPs in water bodies necessary for informed decision making on direct future water resources management, water pollution safeguards, as well as regulation of the EMPs in water.
Acknowledgments
The authors are grateful to Nanotechnology for Water Sustainability Research Unit, University of South Africa for funding and Mzuzu University, Malawi, for granting study leave to Elijah M. M. Wanda.
Author Contributions
Elijah M. M. Wanda collected and prepared the water samples for analysis. Elijah M. M. Wanda and Hlengilizwe Nyoni developed and optimized the method for analysis of water samples. Bhekie B. Mamba and Titus A. M. Msagati provided high level support to Elijah M. M. Wanda during method development, data analysis, and write-up of the manuscript. The manuscript was approved for publication by all authors.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Drangert J.O., Cronin A.A. Use and abuse of the urban groundwater resource: Implications for a new management strategy. Hydrogeol. J. 2004;12:94–102. doi: 10.1007/s10040-003-0307-z. [DOI] [Google Scholar]
- 2.Ellis J.B. Pharmaceutical and personal care products (PPCPs) in urban receiving waters. Environ. Pollut. 2006;144:184–189. doi: 10.1016/j.envpol.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 3.Osenbrück K., Gläser H.R., Knöller K., Weise S.M., Möder M., Wennrich R., Schirmer M., Reinstorf F., Busch W., Strauch G. Source and transport of selected organic micropollutants in urban groundwater underlying the city of Halle (Saale), Germany. Water Res. 2007;41:3259–3270. doi: 10.1016/j.watres.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 4.Verliefde A., Cornelissen E., Amy G., van der Bruggen B., van Dijk H. Priority organic micropollutants in water sources in Flanders and the Netherlands and assessment of removal possibilities with nanofiltration. Environ. Pollut. 2007;146:281–289. doi: 10.1016/j.envpol.2006.01.051. [DOI] [PubMed] [Google Scholar]
- 5.Ikehata K., Gamal El-Din M., Snyder S. Ozonation and advanced oxidation treatment of emerging organic pollutants in water and wastewater. Ozone Sci. Eng. 2008;30:21–26. doi: 10.1080/01919510701728970. [DOI] [Google Scholar]
- 6.Reinstorf F., Strauch G., Schirmer K., Gläser H.R., Möder M., Wennrich R., Osenbrück K., Schirmer M. Mass fuxes and spatial trends of xenobiotics in the waters of the city of Halle, Germany. Environ. Pollut. 2008;152:452–460. doi: 10.1016/j.envpol.2007.05.033. [DOI] [PubMed] [Google Scholar]
- 7.Schirmer K., Schirmer M. Who is chasing whom? A call for a more integrated approach to reduce the load of micro-pollutants in the environment. Water Sci. Technol. 2008;57:145–150. doi: 10.2166/wst.2008.826. [DOI] [PubMed] [Google Scholar]
- 8.Musolff A., Leschik S., Möder M., Strauch G., Reinstorf F., Schirmer M. Temporal and spatial patterns of micropollutants in urban receiving waters. Environ. Pollut. 2009;157:3069–3077. doi: 10.1016/j.envpol.2009.05.037. [DOI] [PubMed] [Google Scholar]
- 9.Wille K., de Brabander H.F., de Wulf E., van Caeter P., Janssen C.R., Vanhaecke L. Coupled chromatographic and mass-spectrometric techniques for the analysis of emerging pollutants in the aquatic environment. Trends Anal. Chem. 2012;35:87–108. doi: 10.1016/j.trac.2011.12.003. [DOI] [Google Scholar]
- 10.Luo Y., Guo W.S., Ngo H.H., Nghiem L.D., Hai F.I., Zhang J., Liang S., Wang X.C. A review on the occurrence of micropollutants in the aquatic environment and their fate and removal during wastewater treatment. Sci. Total Environ. 2014;473–474:619–641. doi: 10.1016/j.scitotenv.2013.12.065. [DOI] [PubMed] [Google Scholar]
- 11.Daughton C.G., Ternes T.A. Pharmaceuticals and personal care products in the environment: Agents of subtle change? Environ. Health Perspect. 1999;107:907–938. doi: 10.1289/ehp.99107s6907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pera-Titus M., Garcia-Molina V., Banos M.A., Gimenez J., Esplugas S. Degradation of chlorophenols by means of advanced oxidation processes: A general review. Appl. Catal. B Environ. 2004;47:219–256. doi: 10.1016/j.apcatb.2003.09.010. [DOI] [Google Scholar]
- 13.Luckenbach T., Epel D. Nitromusk and polycyclic musk compounds as longterm inhibitors of cellular xenobiotic defense systems mediated by multidrug transporters. Environ. Health Perspect. 2005;113:17–24. doi: 10.1289/ehp.7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwarzenbach R.P., Escher B.I., Fenner K., Hofstetter T.B., Johnson C.A., von Gunten U., Wehrli B. The challenge of micropollutants in aquatic systems. Science. 2006;313:1072–1077. doi: 10.1126/science.1127291. [DOI] [PubMed] [Google Scholar]
- 15.Pomati F., Orlandi C., Clerici M., Luciani F., Zuccato E. Effects and interactions in an environmentally relevant mixture of pharmaceuticals. Toxicol. Sci. 2008;10:129–137. doi: 10.1093/toxsci/kfm291. [DOI] [PubMed] [Google Scholar]
- 16.Strauch G., Möder M., Wennrich R., Osenbrück K., Gla¨ser H.R., Schladitz T., Müller C., Schirmer K., Reinstorf F., Schirmer M. Indicators for assessing anthropogenic impact on urban surface and groundwater. J. Soils Sediments. 2008;8:23–33. doi: 10.1065/jss2007.06.234. [DOI] [Google Scholar]
- 17.Klavarioti M., Mantzavinos D., Kassinos D. Removal of residual pharmaceuticals from aqueous systems by advanced oxidation processes. Environ. Int. 2009;35:402–417. doi: 10.1016/j.envint.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 18.Huerta-Fontela M., Galceran M.T., Ventura F. Occurrence and removal of pharmaceuticals and hormones through drinking water treatment. Water Res. 2011;45:1432–1442. doi: 10.1016/j.watres.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 19.Schirmer M., Reinstorf F., Leschik S., Musolff A., Krieg R. Mass fluxes of xenobiotics below cities: Challenges in urban hydrogeology. Environ. Earth Sci. 2012 doi: 10.1007/s12665-010-0880-0. [DOI] [Google Scholar]
- 20.Lapworth D.J., Baran N., Stuart M.E., Ward R.S. Emerging organic contaminants in groundwater: A review of sources, fate and occurrence. Environ. Pollut. 2012;163:287–303. doi: 10.1016/j.envpol.2011.12.034. [DOI] [PubMed] [Google Scholar]
- 21.Loeffler D., Rombke J., Meller M., Ternes T.A. Environmental fate of pharmaceuticals in water/sediment systems. Environ. Sci. Technol. 2005;39:5209–5218. doi: 10.1021/es0484146. [DOI] [PubMed] [Google Scholar]
- 22.Nakada N., Komori K., Suzuki Y., Konishi C., Houwa I., Tanaka H. Occurrence of 70 pharmaceutical and personal care products in Tone River basin in Japan. Water Sci. Technol. 2007;56:133–140. doi: 10.2166/wst.2007.801. [DOI] [PubMed] [Google Scholar]
- 23.Lin A.Y., Yu T.H., Lin C.F. Pharmaceutical contamination in residential, industrial, and agricultural waste streams: Risk to aqueous environments in Taiwan. Chemosphere. 2008;74:131–141. doi: 10.1016/j.chemosphere.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 24.Caliman F.A., Gavrilescu M. Pharmaceuticals, personal care products and endocrine disrupting agents in the environment—A review. CLEAN Soil Air Water. 2009;37:277–303. doi: 10.1002/clen.200900038. [DOI] [Google Scholar]
- 25.Veach A.M., Bernot M.J. Temporal variation of pharmaceuticals in an urban and agriculturally influenced stream. Sci. Total Environ. 2011;409:4553–4563. doi: 10.1016/j.scitotenv.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 26.Bernot M.J., Smith L., Frey J. Human and veterinary pharmaceutical abundance and transport in a rural central Indiana stream influenced by confined animal feeding operations (CAFOs) Sci. Total Environ. 2013;445–446:219–230. doi: 10.1016/j.scitotenv.2012.12.039. [DOI] [PubMed] [Google Scholar]
- 27.Ferguson P.J., Bernot M.J., Doll J.C., Lauer T.E. Detection of pharmaceuticals and personal care products (PPCPs) in near-shore habitats of southern Lake Michigan. Sci. Total Environ. 2013;458–460:187–196. doi: 10.1016/j.scitotenv.2013.04.024. [DOI] [PubMed] [Google Scholar]
- 28.Tijani J.O., Fatoba O.O., Petrik L.F. A review of pharmaceuticals and endocrine disrupting compounds: Sources, effects, removal, and detections. Water Air Soil Pollut. 2013;224:1770. doi: 10.1007/s11270-013-1770-3. [DOI] [Google Scholar]
- 29.Tran N.H., Li J., Hu J., Ong S.L. Occurrence and suitability of pharmaceuticals and personal care products as molecular markers for raw wastewater contamination in surface water and groundwater. Environ. Sci. Pollut. Res. Int. 2014;21:4727–4740. doi: 10.1007/s11356-013-2428-9. [DOI] [PubMed] [Google Scholar]
- 30.Bolong N., Ismail A.F., Salim M.R., Matsuura T. A review of the effects of emerging contaminants in wastewater and options for their removal. Desalination. 2009;239:229–246. doi: 10.1016/j.desal.2008.03.020. [DOI] [Google Scholar]
- 31.Grassi M., Kaykioglu G., Belgiorno V., Lofrano G. Removal of emerging contaminants from water and wastewater by adsorption process. In: Lofrano G., editor. Emerging Compounds Removal from Wastewater, Springer Briefs in Molecular Science. Springer; Berlin/Heidelberg, Germany: 2012. pp. 15–37. [Google Scholar]
- 32.Petrie B., Barden R., Kasprzyk-Hordern B. A review on emerging contaminants in wastewaters and the environment: Current knowledge, understudied areas and recommendations for future monitoring. Water Res. 2015;72:3–27. doi: 10.1016/j.watres.2014.08.053. [DOI] [PubMed] [Google Scholar]
- 33.Snyder S.A., Adham S., Redding A.M., Cannon F.S., DeCarolis J., Oppenheimer J., Wert E.C., Yoon Y. Role of membranes and activated carbon in the removal of endocrine disruptors and pharmaceuticals. Desalination. 2007;202:156–181. doi: 10.1016/j.desal.2005.12.052. [DOI] [Google Scholar]
- 34.Fent K., Weston A.A., Caminada D. Ecotoxicologyof human pharmaceuticals. Aquat. Toxicol. 2006;76:122–159. doi: 10.1016/j.aquatox.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 35.Kalinová B., Jiros P., Zd’árek J., Wen X., Hoskovec M. GC × GC/TOFMS technique—A new tool in identification of insect pheromones: Analysis of the persimmon bark borer sex pheromone gland. Talanta. 2006;69:542–547. doi: 10.1016/j.talanta.2005.10.045. [DOI] [PubMed] [Google Scholar]
- 36.LECO Corporation LECO’s Pegasus® GC-HRT 4D. Form No. 209-245, 5/5-REV0, 2015. [(accessed on 6 June 2016)]. Available online: http:// photos.labwrench.com/equipmentManuals/21641-7524.pdf.
- 37.Pursch M., Sun K., Winniford B., Cortes H., Weber A., McCabe T., Luong J. Modulation techniques and applications in comprehensive two-dimensional gas chromatography (GC × GC) Anal. Bioanal. Chem. 2002;373:356–367. doi: 10.1007/s00216-002-1318-7. [DOI] [PubMed] [Google Scholar]
- 38.Frysinger G.S., Gaines R.B., Xu L., Reddy C.M. Resolving the unresolved complex mixture in petroleum-contaminated sediments. Environ. Sci. Technol. 2003;37:1653–1662. doi: 10.1021/es020742n. [DOI] [PubMed] [Google Scholar]
- 39.LECO Corporation LECO’s Pegasus® 4D GCxGC-TOFMS. Form No. 209-183, R1.55-REV4, 2014. [(accessed on 31 January 2015)]. Available online: http://uk.leco-europe.com/.../leco.../SEP-SCI-PEGASUS-4D-GCxGC-TOFMS-209-183.pdf.
- 40.Ong C.Y., Marriott P.J. A review of basic concepts in comprehensive two-dimensional gas chromatography. J. Chromatogr. Sci. 2002;40:276–291. doi: 10.1093/chromsci/40.5.276. [DOI] [PubMed] [Google Scholar]
- 41.Shellie R., Marriott P.J. Comprehensive two-dimensional gas chromatography with fast enantio-separation. Anal. Chem. 2002;74:5426–5430. doi: 10.1021/ac025803e. [DOI] [PubMed] [Google Scholar]
- 42.Wang B., Fang A., Heim J., Bogdanov B., Pugh S., Libardoni M., Zhang X. DISCO: Distance and spectrum correlation optimization alignment for two-dimensional gas chromatography time-of-flight mass spectrometry-based metabolomics. Anal. Chem. 2010;82:5069–5081. doi: 10.1021/ac100064b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zrostlíková J., Hajslová J., Cajka T. Evaluation of two-dimensional gas chromatography-time-of-flight mass spectrometry for the determination of multiple pesticide residues in fruit. J. Chromatogr. A. 2003;1019:173–186. doi: 10.1016/S0021-9673(03)01302-5. [DOI] [PubMed] [Google Scholar]
- 44.Statistics South Africa Census 2011 Statistical Release-P0301.4. Statistics South Africa, Pretoria, 2012. [(accessed on 31 January 2015)]; Available online: www.statssa.gov.za/publications/P03014/P030142011.pdf.
- 45.Department of Water Affairs (DWA) Blue Drop Report: South African Waste Water Quality Management Performance. Version 1. Pretoria, South Africa, 2012. [(accessed on 18 November 2016)]; Available online: https://www.dwa.gov.za/Documents/BD/BDIntro.pdf.
- 46.Department of Water Affairs (DWA) Green Drop Report: South African Waste Water Quality Management Performance. Version 1. Pretoria, South Africa, 2012. [(accessed on 18 November 2016)]; Available online: https://www.dwa.gov.za/Documents/GD/GDIntro.pdf.
- 47.Madikizela L.M., Muthwa S.F., Chimuka L. Determination of triclosan and ketoprofen in river water and wastewater by solid phase extraction and high performance liquid chromatography. S. Afr. J. Chem. 2014;67:143–150. [Google Scholar]
- 48.Santos J.L., Aparicio I., Alonso E., Callejón M. Simultaneous determination of pharmaceutically active compounds in wastewater samples by solid phase extraction and high-performance liquid chromatography with diode array and fluorescence detectors. Anal. Chim. Acta. 2005;550:116–122. doi: 10.1016/j.aca.2005.06.064. [DOI] [Google Scholar]
- 49.Glaser J.A., Foerst D.L., McKee G.D., Quave S.A., Budde W.L. Trace analyses for wastewaters. Environ. Sci. Technol. 1981;15:1426–1435. doi: 10.1021/es00094a002. [DOI] [Google Scholar]
- 50.Özel M.Z., Ward M.W., Hamilton J.F., Lewis A.C., Raventos-Duran T., Harrison R.M. Analysis of organic nitrogen compounds in urban aerosol samples using GC × GC-TOF/MS. Aerosol Sci. Technol. 2009;44:109–116. doi: 10.1080/02786820903410105. [DOI] [Google Scholar]
- 51.Phillips J.B., Beens J. Comprehensive two dimensional gas chromatography: A hyphenated method with strong coupling between the two dimensions. J. Chromatogr. A. 1999;856:331–347. doi: 10.1016/S0021-9673(99)00815-8. [DOI] [PubMed] [Google Scholar]
- 52.Adahchour M., Beens J., Brinkman U.A.T. Recent developments in the application of comprehensive two-dimensional gas chromatography. J. Chromatogr. A. 2008;1186:67–108. doi: 10.1016/j.chroma.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 53.Li M., Zhang S., Jiang C., Zhu G., Fowler M., Achal S., Milovic M., Robinson R., Larter S. Two-dimensional gas chromatograms as fingerprints of sour gas associated oils. Org. Geochem. 2008;39:1144–1149. doi: 10.1016/j.orggeochem.2008.02.015. [DOI] [Google Scholar]
- 54.Regnery J., Püttmann W. Occurrence and fate of organophosphorus flame retardants and plasticizers in urban and remote surface waters in Germany. Water Res. 2010;44:4097–4104. doi: 10.1016/j.watres.2010.05.024. [DOI] [PubMed] [Google Scholar]
- 55.Kim J.W., Jang H.S., Kim J.G., Ishibashi H., Hirano M., Nasu K. Occurrence of pharmaceutical and personal care products (PPCPs) in surface water from Mankyung River, South Korea. J. Health Sci. 2009;55:249–258. doi: 10.1248/jhs.55.249. [DOI] [Google Scholar]
- 56.Peng X., Yu Y., Tang C., Tan J., Huang Q., Wang Z. Occurrence of steroid estrogens, endocrine-disrupting phenols, and acid pharmaceutical residues in urban riverine water of the Pearl River Delta, South China. Sci. Total Environ. 2008;397:158–166. doi: 10.1016/j.scitotenv.2008.02.059. [DOI] [PubMed] [Google Scholar]
- 57.Furhacker M., Scharf S., Weber H. Bisphenol A: Emission from point sources. Chemosphere. 2000;41:751–756. doi: 10.1016/S0045-6535(99)00466-X. [DOI] [PubMed] [Google Scholar]
- 58.Fromme H., Küchler T., Otto T., Pilz K., Müller J., Wenzel A. Occurrence of phthalates and bisphenol A and F in the environment. Water Res. 2002;36:1429–1438. doi: 10.1016/S0043-1354(01)00367-0. [DOI] [PubMed] [Google Scholar]
- 59.Minnesota Department of Health Bisphenol A in Drinking Water. Drinking Water Contaminants of Emerging Concern Program, Environmental Health Division, 2014. [(accessed on 28 February 2015)]. Available online: http://www.health.state.mn.us/cec.
- 60.Ternes T.A., Bonerz M., Schmidt T. Determination of neutral pharmaceuticals in wastewater and rivers by liquid chromatography-electrospray tandem mass spectrometry. J. Chromatogr. A. 2001;938:175–185. doi: 10.1016/S0021-9673(01)01205-5. [DOI] [PubMed] [Google Scholar]
- 61.Spongberg A.L., Witter J.D., Acuña J., Vargas J., Murillo M., Umaña G. Reconnaissance of selected PPCP compounds in Costa Rican surface waters. Water Res. 2011;45:6709–6717. doi: 10.1016/j.watres.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 62.Standley L.J., Kaplan L.A., Smith D. Molecular tracers of organic matter sources to surface water resources. Environ. Sci. Technol. 2000;34:3124–3130. doi: 10.1021/es991381n. [DOI] [Google Scholar]
- 63.You L., Tung Nguyen V., Pal A., Chen H., He Y., Reinhard M., Karina Y.H.G. Investigation of pharmaceuticals, personal care products and endocrine disrupting chemicals in a tropical urban catchment and the influence of environmental factors. Sci. Total Environ. 2015;536:955–963. doi: 10.1016/j.scitotenv.2015.06.041. [DOI] [PubMed] [Google Scholar]