

Abstract
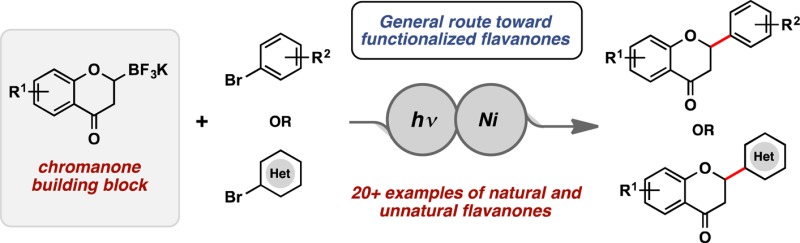
Utilizing photoredox/nickel dual catalysis, diverse flavanones have been synthesized by coupling novel 2-trifluoroboratochromanone building blocks with aryl and heteroaryl bromide partners. The newly reported trifluoroboratochromanones can be easily accessed from the corresponding chromones on multigram scale. This represents a general route for accessing natural and unnatural flavanones that were previously formed through a synthetically more restrictive ring closure route from chalcone precursors.
Over the past 15 years, the number of known flavanones has increased significantly, to the point of where they can be considered a class of their own, alongside the related flavones.1 Flavanones are readily found in nature (e.g., citrus fruits), and their role in flavonoid biosynthetic pathways has been extensively studied.2 Chalcone cores are typically the biosynthetic precursors to flavanones (Figure 1), accessed through a cyclization of the reactive chalcone moiety. In turn, flavanone cores are intermediates in the synthesis of a wide range of flavonoids (Figure 1).3 Because of the natural abundance of the flavonoids, many practical uses for these materials have been explored. For decades, flavanones and flavonoids have been used as dietary supplements and also as potent therapeutic agents (e.g., as antianxiety agents2c and inhibitors of HIV-1 reverse transcriptase4). Additionally, substituted flavones have recently been used as fluorescent scaffolds.5
Figure 1.
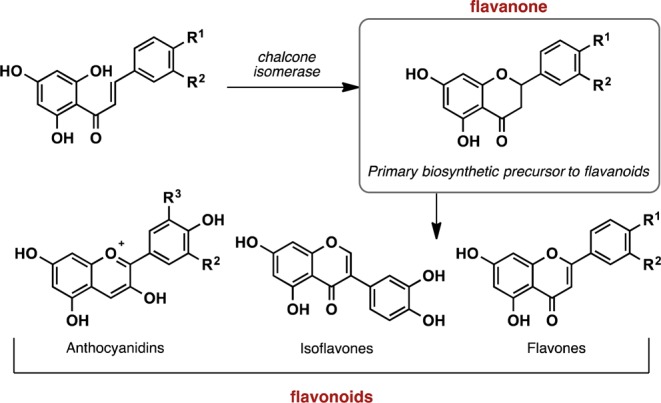
Biosynthetic pathway for accessing flavanones and various flavonoids.
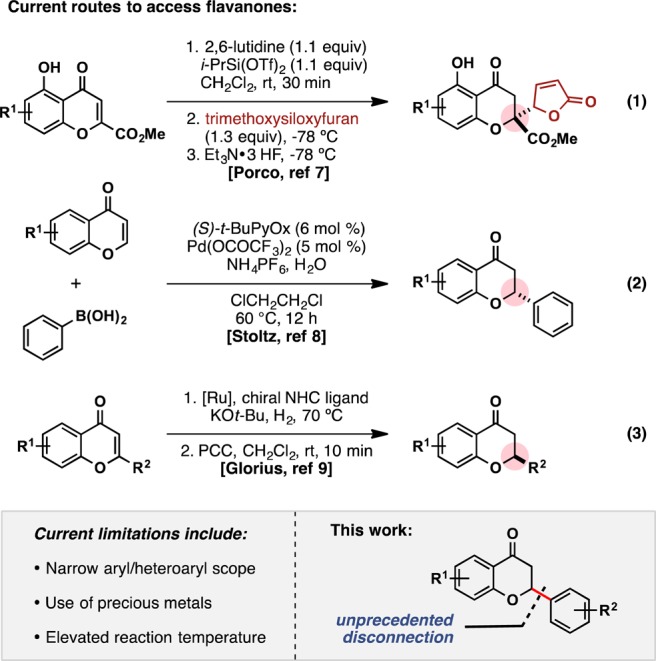
Currently, the primary synthetic route to access functionalized flavanones has traversed through chalcones, in analogy to the biosynthetic pathway.6 Although there have been robust methods developed, synthesis of the chalcone precursor is not necessarily straightforward. Limitations become even more apparent when attempting to move to more complex cores. As a result, there has been significant effort devoted toward alternative synthetic routes. For example, the Porco group disclosed a vinylogous addition to specifically functionalized chromones, synthesizing a variety of chromanone butenolides (Scheme 1, eq 1).7 The Stoltz group also published a palladium-catalyzed conjugate addition of arylboronic acids to chromone cores, accessing 2-aryl flavanones (Scheme 1, eq 2).8 In a complementary approach, Glorius and co-workers reported a ruthenium-catalyzed asymmetric hydrogenation of flavones to the corresponding enantioenriched flavanols, which were subsequently oxidized by PCC to the optically active flavanones (Scheme 1, eq 3).9
Scheme 1. Synthetic Routes toward Flavanones.
We were interested in establishing a complementary route, allowing access to both 2-aryl- and 2-heteroaryl-substituted flavanones. Inspired by recent work in our group,10 novel trifluoroboratochromanones were envisioned to serve as radical precursors in the photoredox/Ni dual catalytic cross-coupling with a variety of aryl and heteroaryl bromides (Figure 2). By synthesizing such unprecedented 2-trifluoroboratochromanone building blocks, a large library of natural and unnatural flavanones could quickly be accessed.
Figure 2.
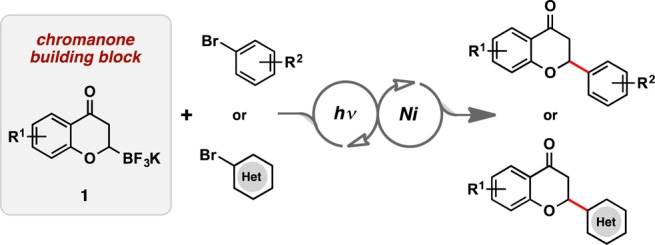
Proposed route toward accessing diversified flavonoids.
In the past decade, photoredox catalysis has experienced renewed popularity in the organic synthesis community, largely because of the mild and robust conditions typically used.11 Our group disclosed a dual catalytic photoredox/nickel paradigm, making previously challenging cross-couplings possible at room temperature.10a More recently, there have been reports of α-alkoxy couplings, creating precedent for the transformation proposed herein.10c,10d Previous examples were composed exclusively of 1° alkoxymethyltrifluoroborates and acyclic 2° alkoxyalkyltrifluoroborates,12 the latter of which would be anticipated to exhibit a similar or even more favorable redox profile as their 1° counterpart (Ered = +1.11 V vs SCE). Before exploring the feasibility of the proposed transformation, a robust route to the desired, but previously unknown, 2-trifluoroboratochromanones (1) was needed.
In a first effort toward the synthesis of 1, a copper-catalyzed method reported by our group was used.13 Although there were numerous methods for β-borylation of α,β-unsaturated carbonyl substrates,14 these conditions were ultimately chosen because they incorporated an inexpensive, readily available copper catalyst and the atom economical bisboronic acid. Gratifyingly, little optimization was required, for a 75% yield of 1a was achieved using published conditions. Slightly higher yields were achieved by increasing the CuCl and ligand loadings to 2 mol % from 1 mol % (Table 1). Alkyl substituents were tolerated as demonstrated in 1b. Chloride moieties were also preserved during the borylation (1c), thus providing a handle for further decoration.
Table 1. Chromone β-Borylation Scopea.
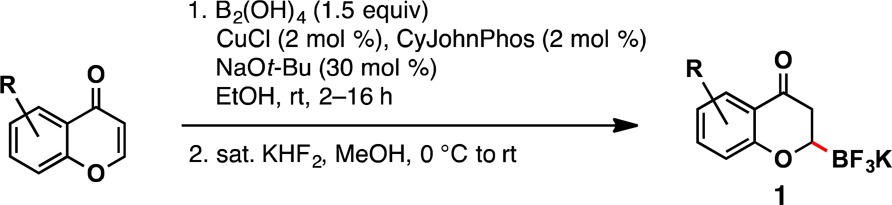
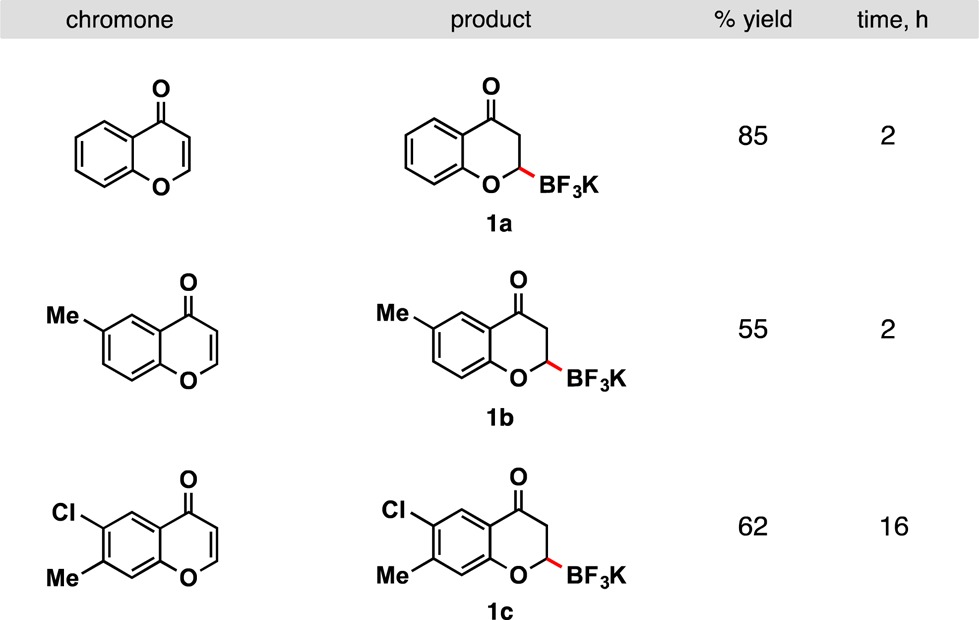
Reactions were run with chromone (1.0 equiv), B2(OH)4 (1.5 equiv), CuCl (2 mol %), CyJohnPhos (2 mol %), NaOt-Bu (30 mol %) in freshly distilled EtOH on 2.0–4.0 mmol scale.
With the trifluoroborate building blocks in hand, the viability of the coupling was explored. Using conditions that were appropriate for 2° alkyltrifluoroborate cross-couplings using Ir catalyst 3 (2.5 mol %), NiCl2·dme (5 mol %), dtbbpy (5 mol %), Cs2CO3 (1.5 equiv), and dioxane,10b a modest 24% yield was achieved. To assess more suitable conditions quickly, high throughput experimentation15 was utilized to screen various nickel sources, bases, and solvents. Although Ni(COD)2 was found to yield slightly higher conversions, NiCl2·dme was ultimately chosen because of its stability and consequent ease of handling. K2HPO4 was found to be the preferred additive to sequester the BF3 generated in the oxidation of the trifluoroborate, and dioxane was a suitable solvent.
Our attention was next directed to the photocatalyst (Table 2). It was noted that the ultimate practicality of the reaction was hampered by the high cost of the iridium photocatalyst 3 (∼$1/mg). Other, less expensive photocatalysts were compared, including organophotocatalysts Eosin Y16 and MesAcr11c (2 and 4, respectively). Although the oxidation potential for MesAcr is more than sufficient (Ered = +2.06 V vs SCE) to oxidize the trifluoroborate,11c the reduction potential (Ered = +0.49 V vs SCE)11c was not adequate to turn over the nickel cycle (Ered = +1.10 V vs SCE).15 Conversely, Eosin Y has insufficient oxidation potential to induce a single electron oxidation (Ered = +0.83 V vs SCE) of 1. During the optimization process, the Zhang group disclosed the synthesis of a rationally designed organophotocatalyst amenable to photoredox/nickel dual catalytic manifolds.17 At merely $6/g, 4CzIPN (5) was a highly attractive alternative. Just as Zhang and co-workers observed in their systems, 4CzIPN outperformed the iridium photocatalyst (entry 4) and was therefore used in the remainder of the study.
Table 2. Photoredox/Nickel Optimizationa.
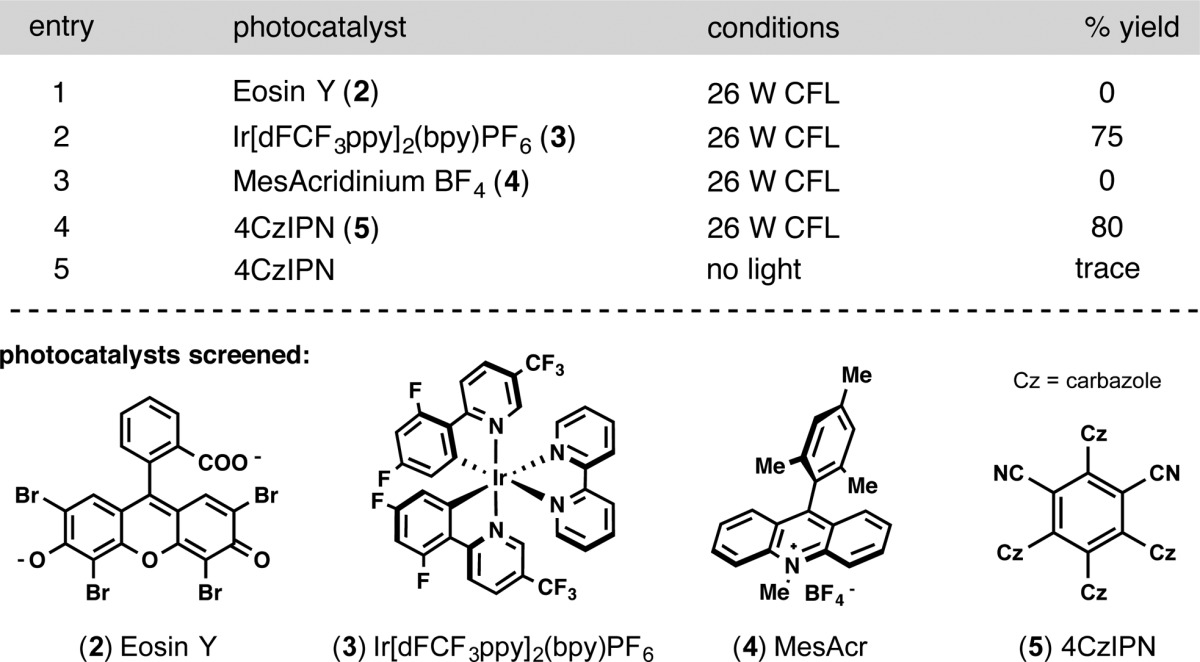
Optimization reactions were run with aryl bromide (1.0 equiv), trifluoroborate (1.5 equiv), photocatalyst (2.5 mol %), NiCl2·dme (5 mol %), dtbbpy (5 mol %), and KH2PO4 (2.0 equiv) in dioxane on 0.1 mmol scale. Yields were obtained via HPLC using a calibration curve.
After establishing suitable conditions, the scope of the reaction in terms of aryl/heteroaryl bromide partners was explored (Scheme 2). Initially, aryl bromides with various functional groups were investigated (2a–2d). The activated 4-bromocyanobenzene used for optimization afforded a 70% yield, but when steric pressure was applied at the ortho position (2d), the yield was significantly reduced. An aldehyde functional group was tolerated, providing a respectable 67% yield (2b). Chloride handles would allow further functionalization; thus, 2c is appropriately functionalized for additional cross-coupling, and 2i is primed for subsequent SN2 reactions. Trifluoromethyl substituents, commonly used in medicinal chemistry, were well tolerated (2f, 2h, 2o). Additionally, protic functional groups as in the halide partner leading to 2j also proved viable.
Scheme 2. Aryl/Heteroaryl Halide Substrate Scope.
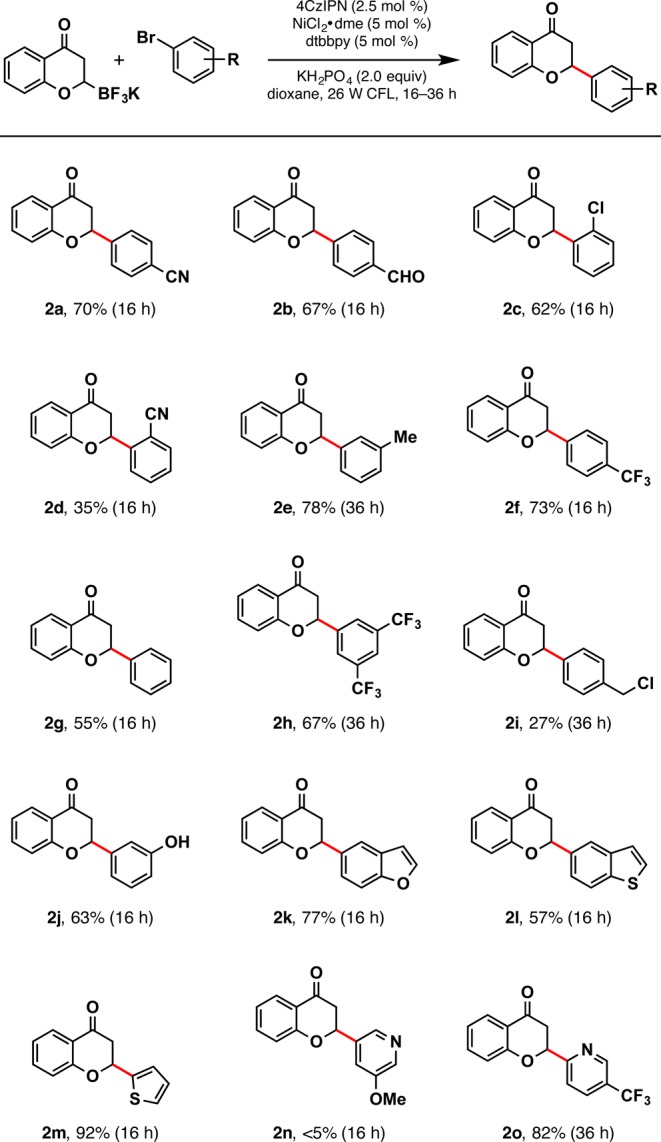
Reactions were run with aryl bromide (1.0 equiv), trifluoroborate (1.5 equiv), photocatalyst (2.5 mol %), NiCl2·dme (5 mol %), dtbbpy (5 mol %), and KH2PO4 (2.0 equiv) in dioxane on 0.5 mmol scale.
Next, the heteroaryl halide scope was probed. Suprisingly, heteroarenes such as thiophene (2m), benzofuran (2k), and pyridine (2o) had markedly higher yields than their aryl counterparts. Although electron-deficient pyridines were well tolerated, electron-donating groups led to diminished reactivity (2n). Additionally, bromo-substituted indoles and pyrroles were not suitable partners, presumably because of their electron-rich nature. Lastly, it is worth noting that although they are relatively simple derivatives, only 2a–c, 2e–g, and 2j have been previously reported in the literature, highlighting the utility of this protocol for accessing a wide range of novel, functionalized flavanones.
Subsequently, the scope for functionalized 2-trifluoroboratochromanones was explored (Scheme 3). The yield for an alkyl-substituted chromanone (3a) was found to be comparable to the unsubstituted example (2a). The chloro-chromanone 3c demonstrates the feasibility for further diversification within the heteroaryl core. Although unexplored at this time, we predict halide substituents at different positions of the aryl ring would yield similar results. Finally, nitro substituents were not tolerated, as observed by other groups in photoredox-catalyzed processes.18
Scheme 3. 2-Trifluoroboratochromanone Substrate Scope.
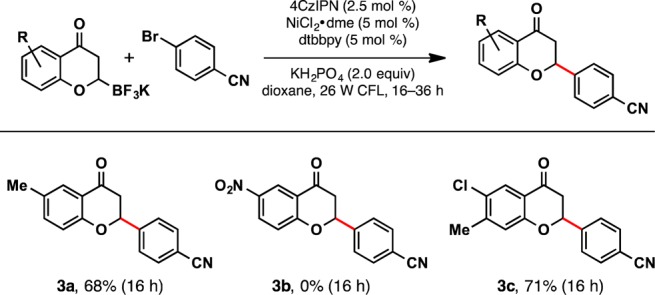
Reactions were run with aryl halide (1.0 equiv), trifluoroborate (1.5 equiv), photocatalyst (2.5 mol %), NiCl2·dme (5 mol %), dtbbpy (5 mol %), and KH2PO4 (2.0 equiv) in dioxane on 0.5 mmol scale.
In conclusion, a simple and robust method has been established for accessing the increasingly important flavanone core. This study details the first synthesis of 2-boryl-substituted chromanones. With this procedure in place, practitioners are well equipped to access 2-aryl/heteroaryl flavanones quickly and efficiently, and these substructures can be decorated with a wide range of functional groups owing to the availability of literally thousands of diverse, commercially available (het)aryl bromide partners. The photoredox/Ni dual catalytic cross-coupling reactions proceed with a sustainable organic photocatalyst and a base-metal cross-coupling catalyst under extraordinarily mild conditions (weak base, ambient temperature, visible light). Lastly, the method highlights the applicability of photoredox/nickel dual catalysis for challenging coupling reactions that are typically prone to undesirable side reactions (e.g., β-hydride elimination), allowing access to unique structures by transformations that are complementary to more traditional approaches.
Acknowledgments
We thank the National Institutes of Health (NIGMS R01 GM113878, R01 GM081376) for support. We thank AllyChem for providing the bisboronic acid and Pfizer for the acridinium photocatalyst. Drew Heitz (University of Pennsylvania) and Dr. Christopher Kelly (University of Pennsylvania) prepared the 4CzIPN photocatalyst. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for collection of HRMS data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.orglett.6b03448.
Experimental procedures, HTE data, compound characterization data, and NMR spectra for all compounds (PDF)
The authors declare no competing financial interest.
In Scheme 2, compound 2c was corrected on February 3, 2017.
Supplementary Material
References
- Khan M. K.; Huma Z. E.; Dangles O. J. Food Compos. Anal. 2014, 33, 85. 10.1016/j.jfca.2013.11.004. [DOI] [Google Scholar]
- a Yan Y.; Kohli A.; Koffas A. G. Appl. Environ. Microbiol. 2005, 71, 5610. 10.1128/AEM.71.9.5610-5613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gaffield W.; Lundin R. E.; Gentili B.; Horowitz R. M. Bioorg. Chem. 1975, 4, 259. 10.1016/0045-2068(75)90036-X. [DOI] [Google Scholar]; c Leonard E.; Yan Y.; Lim K. H.; Koffas M. A. G. Appl. Environ. Microbiol. 2005, 71, 8241. 10.1128/AEM.71.12.8241-8248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Fowler Z. L.; Koffas M. A. G. Appl. Microbiol. Biotechnol. 2009, 83, 799. 10.1007/s00253-009-2039-z. [DOI] [PubMed] [Google Scholar]; e Fowler Z. L.; Gikandi W. W.; Koffas M. A. G. Appl. Environ. Microbiol. 2009, 75, 5831. 10.1128/AEM.00270-09. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Chandler I. M.; McIntyre C. R.; Simpson T. J. J. Chem. Soc., Perkin Trans. 1 1992, 2271. 10.1039/p19920002271. [DOI] [Google Scholar]; g Martens S.; Mithofer A. Phytochemistry 2005, 66, 2399. 10.1016/j.phytochem.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Forkmann G.; Martens S. Curr. Opin. Biotechnol. 2001, 12, 155. 10.1016/S0958-1669(00)00192-0. [DOI] [PubMed] [Google Scholar]
- Xu Z.-Q.; Buckheit R. W.; Stup T. L.; Flavin M. T.; Khilevich A.; Rizzo J. D.; Lin L.; Zembower D. E. Bioorg. Med. Chem. Lett. 1998, 8, 2179. 10.1016/S0960-894X(98)00380-1. [DOI] [PubMed] [Google Scholar]
- Miao J.; Cui H.; Jin J.; Lai F.; Wen H.; Zhang X.; Ruda G. F.; Chen X.; Yin D. Chem. Commun. 2015, 51, 881. 10.1039/C4CC06762B. [DOI] [PubMed] [Google Scholar]
- a Wang L.; Liu X.; Dong Z.; Fu X.; Feng X. Angew. Chem., Int. Ed. 2008, 47, 8670. 10.1002/anie.200803326. [DOI] [PubMed] [Google Scholar]; b Ahmed N.; Konduru N. K.; Praveen; Kumar A.; Kamaluddin J. Mol. Catal. A: Chem. 2013, 373, 135. 10.1016/j.molcata.2013.03.009. [DOI] [Google Scholar]; c Naik M. M.; Tilve S. G.; Kamat V. P. Tetrahedron Lett. 2014, 55, 3340. 10.1016/j.tetlet.2014.04.051. [DOI] [Google Scholar]; d Kabbe H.-J.; Widdig A. Angew. Chem., Int. Ed. Engl. 1982, 21, 247. 10.1002/anie.198202471. [DOI] [Google Scholar]; e Stermitz F. R.; Adamovics J. A.; Geigert J. Tetrahedron 1975, 31, 1593. 10.1016/0040-4020(75)87018-9. [DOI] [Google Scholar]; f Patonay T.; Varma R. S.; Vass A.; Levai A.; Dudas J. Tetrahedron Lett. 2001, 42, 1403. 10.1016/S0040-4039(00)02264-4. [DOI] [Google Scholar]; g Kavala V.; Lin C.; Kuo C.-W.; Fang H.; Yao C.-F. Tetrahedron 2012, 68, 1321. 10.1016/j.tet.2011.11.022. [DOI] [Google Scholar]
- Qin T.; Johnson R. P.; Porco J. A. J. Am. Chem. Soc. 2011, 133, 1714. 10.1021/ja110698n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shockley S. E.; Holder J. C.; Stoltz B. M. Org. Process Res. Dev. 2015, 19, 974. 10.1021/acs.oprd.5b00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D.; Beiring B.; Glorius F. Angew. Chem., Int. Ed. 2013, 52, 8454. 10.1002/anie.201302573. [DOI] [PubMed] [Google Scholar]
- a Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Karakaya I.; Primer D. N.; Molander G. A. Org. Lett. 2015, 17, 3294. 10.1021/acs.orglett.5b01463. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Amani J.; Sodagar E.; Molander G. A. Org. Lett. 2016, 18, 732. 10.1021/acs.orglett.5b03705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recent reviews on photoredox chemistry:; a Narayanam J. M. R.; Stephenson C. R. J. Chem. Soc. Rev. 2011, 40, 102. 10.1039/B913880N. [DOI] [PubMed] [Google Scholar]; b Prier C. K.; Rankic D. A.; MacMillan D. W. C. Chem. Rev. 2013, 113, 5322. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; Example with MesAcr photocatalyst:; c Nguyen T. M.; Manohar N.; Nicewicz D. A. Angew. Chem., Int. Ed. 2014, 53, 6198. 10.1002/anie.201402443. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tellis J. C.; Kelly C. B.; Primer D. N.; Jouffroy M.; Patel N. R.; Molander G. A. Acc. Chem. Res. 2016, 49, 1429. 10.1021/acs.accounts.6b00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Miyazawa K.; Yasu Y.; Koike T.; Akita M. Chem. Commun. 2013, 49, 7249. 10.1039/c3cc42695e. [DOI] [PubMed] [Google Scholar]; b Karimi-Nami R.; Tellis J. C.; Molander G. A. Org. Lett. 2016, 18, 2572. 10.1021/acs.orglett.6b00911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander G. A.; McKee S. A. Org. Lett. 2011, 13, 4684. 10.1021/ol201900d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lawson Y.; Lesly G.; Marder T.; Norman N.; Rice C. Chem. Commun. 1997, 7, 4674. [Google Scholar]; b Thorpe S. B.; Calderone J. A.; Santos W. L. Org. Lett. 2012, 14, 1918. 10.1021/ol300575d. [DOI] [PubMed] [Google Scholar]; c Lee J. C. H.; Hall D. G. J. Am. Chem. Soc. 2010, 132, 5544. 10.1021/ja9104057. [DOI] [PubMed] [Google Scholar]; d Schiffner J. A.; Muther K.; Oestreich M. Angew. Chem., Int. Ed. 2010, 49, 1194. 10.1002/anie.200906521. [DOI] [PubMed] [Google Scholar]
- a Dreher S. D.; Dormer P.G.; Sandrock D. L.; Molander G. A. J. Am. Chem. Soc. 2008, 130, 9257. 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Schmink J. R.; Bellomo A.; Berritt S. Aldrichimica Acta 2013, 46, 71. [Google Scholar]
- Hari D. P.; Konig B. Chem. Commun. 2014, 50, 6688. 10.1039/C4CC00751D. [DOI] [PubMed] [Google Scholar]
- Luo J.; Zhang J. ACS Catal. 2016, 6, 873. 10.1021/acscatal.5b02204. [DOI] [Google Scholar]
- Paria S.; Reiser O. Adv. Synth. Catal. 2014, 356, 557. 10.1002/adsc.201301069. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.