

Abstract
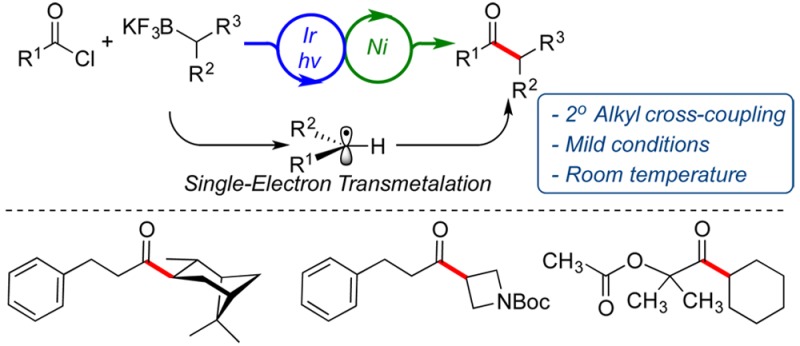
Visible light photoredox/nickel dual catalysis has been employed in the cross-coupling of acyl chlorides with potassium alkyltrifluoroborates. This protocol, based on single-electron-mediated alkyl transfer, circumvents the restriction of using reactive alkylmetallic nucleophiles in transition-metal-catalyzed acylation and achieves a mild and efficient method for the synthesis of unsymmetrical alkyl ketones. In this approach, a variety of acyl chlorides have been successfully coupled with structurally diverse potassium alkyltrifluoroborates, generating the corresponding ketones with good yields.
Ketones, widespread in nature and in pharmaceuticals,1 are among the most important and versatile functional groups in organic molecules and have long been used as important intermediates for the synthesis of complex structures. Because of their ubiquity, the development of diverse synthetic methods to prepare these compounds has been the subject of intense research.
In principle, one of the most straightforward methods for the synthesis of alkyl ketones is the reaction of acyl halides with alkylmetallic reagents.2 However, the addition of highly reactive alkylmetallic reagents (e.g., alkyllithium or Grignard reagents) typically generates tertiary alcohols, and the enolization of the produced ketones in these reactions cannot be repressed completely. The use of low temperatures or less reactive nucleophiles such as alkylcopper, alkylmanganese, alkylzinc, or alkylzirconium reagents can be used to generate the ketones without undesired over-reaction, but many of these reagents are prepared from alkyllithium or Grignard reagents and thus are often minimally functionalized.3 Furthermore, these reagents are not shelf-stable, and many alkylzinc and alkylstannane reagents lack atom economy because only one alkyl group on the metal is transferred.
To overcome these problems, significant progress has been made in the development of transition-metal-catalyzed acylation of alkyl nucleophiles using acyl halides.4 In general, however, metal-catalyzed ketone formation employing aliphatic nucleophiles with carboxylic acid chlorides has been the least explored, perhaps owing to the slow transmetalation of the alkylmetallic nucleophiles and the susceptibility of the resulting transition metal intermediates to undergo β-hydride elimination.5
Recently, our group has developed an efficient photoredox/Ni dual catalysis paradigm for transition-metal-catalyzed cross-couplings of alkylboron reagents with halide electrophiles based on a single-electron transfer (SET) transmetalation pathway.6 This synergistic catalysis approach via the simultaneous activation and engagement of two separate coupling partners with two distinct catalysts through low-barrier, open-shell processes, has had a profound impact on C(sp3)–C(sp2) bond formations under mild conditions. In this protocol, complications associated with the transmetalation of alkylmetallic reagents in conventional cross-coupling methods, which are mainly a consequence of the two-electron nature of the transmetalation step, are avoided. The Doyle group has employed a related strategy for acylation using anhydride electrophiles (eq 1).7 This method was restricted to α-amino radical precursors, and in fact acyl chlorides could not be used in the protocol developed. Our group subsequently investigated the photoredox/Ni dual catalyzed approach to α-alkoxy ketones using α-alkoxyalkyltrifluoroborates as the radical precursor in conjunction with carboxylic acid chloride electrophiles (eq 2).8
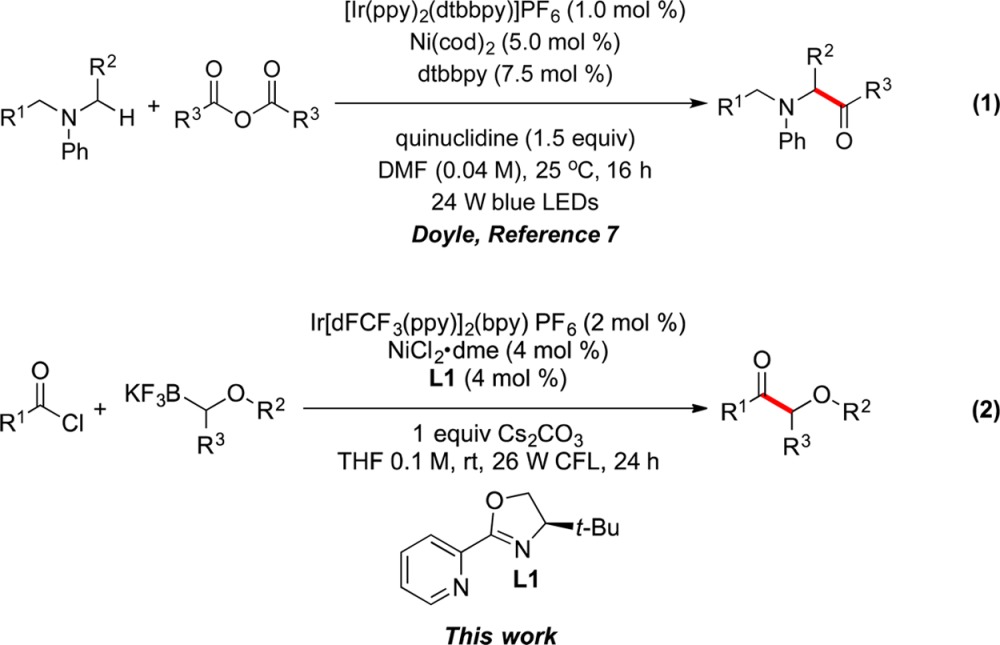 |
1 |
To achieve an efficient protocol for the general synthesis of aliphatic ketones and in an effort to broaden the range of electrophiles to which the photoredox/Ni dual catalysis activation mode could be applied, we investigated the potential of this method for the cross-coupling of acyl chlorides with potassium secondary alkyltrifluoroborates to generate alkyl–alkyl and alkyl–aryl ketones (Figure 1).
Figure 1.
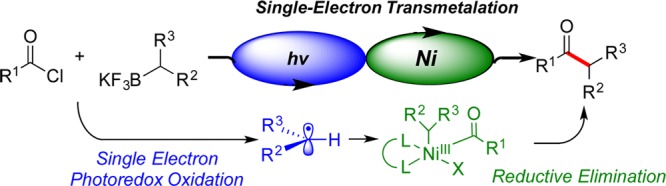
Photoredox/Ni dual catalysis cross-coupling of acyl chlorides with secondary R–BF3K.
To validate the strategy outlined in Figure 1 through the conversion of acyl chlorides to aliphatic ketones, the cross-coupling of hydrocinnamoyl chloride with i-PrBF3K was evaluated as a model reaction under a variety of conditions (e.g., varying solvents, Ni catalysts, ligands, and additives). The investigation commenced with the evaluation of an extensive array of Ni sources and ligands (L1–L8) by means of microscale high-throughput experimentation (HTE, Figure 2).9 After addition of an internal standard to the reaction mixtures in these screens, the results were analyzed by UPLC. Then, to determine the relative amount of product formed in each microscale reaction, the product to internal standard (P/IS) ratios were calculated. The P/IS ratios of the first screen are shown in Figure 2.
Figure 2.

Graphical chart of P/IS ratios of the cross-coupling reactions using different Ni sources and ligands. Reaction conditions: 0.01 mmol of hydrocinnamoyl chloride, 1.5 equiv of i-PrBF3K, 3 mol % Ir[dF(CF3)ppy]2(bpy)PF6, 6 mol % Ni source, 6 mol % ligand, 1.0 equiv of lutidine, rt, blue LEDs, 24 h in THF (0.1 M).
In this screen, with hydrocinnamoyl chloride and i-PrBF3K (1.5 equiv) as the coupling partners, using Ir[dF(CF3)ppy]2(bpy)PF6 (1) [dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine, bpy = 2,2′-bipyridine] as the photocatalyst, NiCl2·dme (6 mol %)/4,4′-di-tert-butyl-2,2′-dipyridyl, L1, (6 mol %) as the cross-coupling catalyst, and lutidine (1 equiv) as a base in THF at room temperature provided the best yield.
Control experiments revealed that all of the reaction parameters (photocatalyst, Ni source, and ligand) are essential for efficient coupling, in agreement with our mechanistic proposal. Additionally, without added base, the product to internal standard ratio noticeably decreased. Considering the important role of the base, which is ascribed to sequestration of the BF3 generated upon oxidation of the alkyltrifluoroborate, an extensive screening of various bases in different solvents was carried out (Figure 3). The investigations revealed that using KF instead of lutidine and DME instead of THF provided a substantial improvement and afforded the highest P/IS ratio.
Figure 3.

Graphical chart of P/IS ratios of the cross-coupling reactions using different bases and solvents. Reaction conditions: 0.01 mmol of hydrocinnamoyl chloride, 1.5 equiv of i-PrBF3K, 3 mol % Ir[dF(CF3)ppy]2(bpy)PF6, 6 mol % NiCl2·dme, 6 mol % L1, 1.0 equiv of base, rt, blue LEDs, 24 h (0.1 M).
Further investigations demonstrated that lower concentrations (0.05 M) increased the yield of the reaction, while higher molarities (0.2 M) resulted in diminished yields. To explore the generality of this transformation, the developed conditions were applied to the cross-coupling of hydrocinnamoyl chloride with a variety of structurally diverse, secondary alkyltrifluoroborates. As demonstrated in Table 1, all the secondary alkyltrifluoroborates function efficiently in this transition-metal-catalyzed acylation protocol. Isopropyl- and sec-butyltrifluoroborates were acylated in 78% and 77% isolated yields, respectively (entries 1 and 2). In these reactions only the desired regioisomers were obtained. Although alkyltrifluoroborates possessing various ring sizes were coupled in good yields (entries 3–5), the reaction with potassium cyclopropyltrifluoroborate did not produce the desired product owing to the high s character of the hybrid orbitals, which destabilizes the incipient radical and engenders an unfavorable redox potential on the trifluoroborate. The products derived from sterically hindered structures such as 2-methylcyclopentyl and pinanyl substrates were also isolated in good yields (entries 6 and 8). In contrast to the reported procedures using Pd catalysis,10 no isomerization was observed for these substrates, and trans-diastereomers were the major products formed in these cross-coupling reactions. The reaction with aliphatic, heterocyclic compounds containing oxygen or protected nitrogen atoms produced the coupled products with moderate to good yields under this protocol (entries 9–12), illustrating the versatility of this method. Moreover, β-trifluoroboratoketones and -esters were successfully coupled to generate the corresponding 1,4-diketones (entries 13–15), which are useful precursors to generate substituted furans, pyrroles, and thiophenes via the Paal–Knorr synthesis.11 The scalable nature of this coupling was demonstrated by performing the reaction on 6.0 mmol scale (entry 3). In this reaction, using half of the catalyst loadings (1.5 mol % of I and 3 mol % of NiCl2·dme/L1) afforded the desired product in 69% yield.
Table 1. Scope of Secondary R–BF3K in Cross-Coupling with Hydrocinnamoyl Chloride.

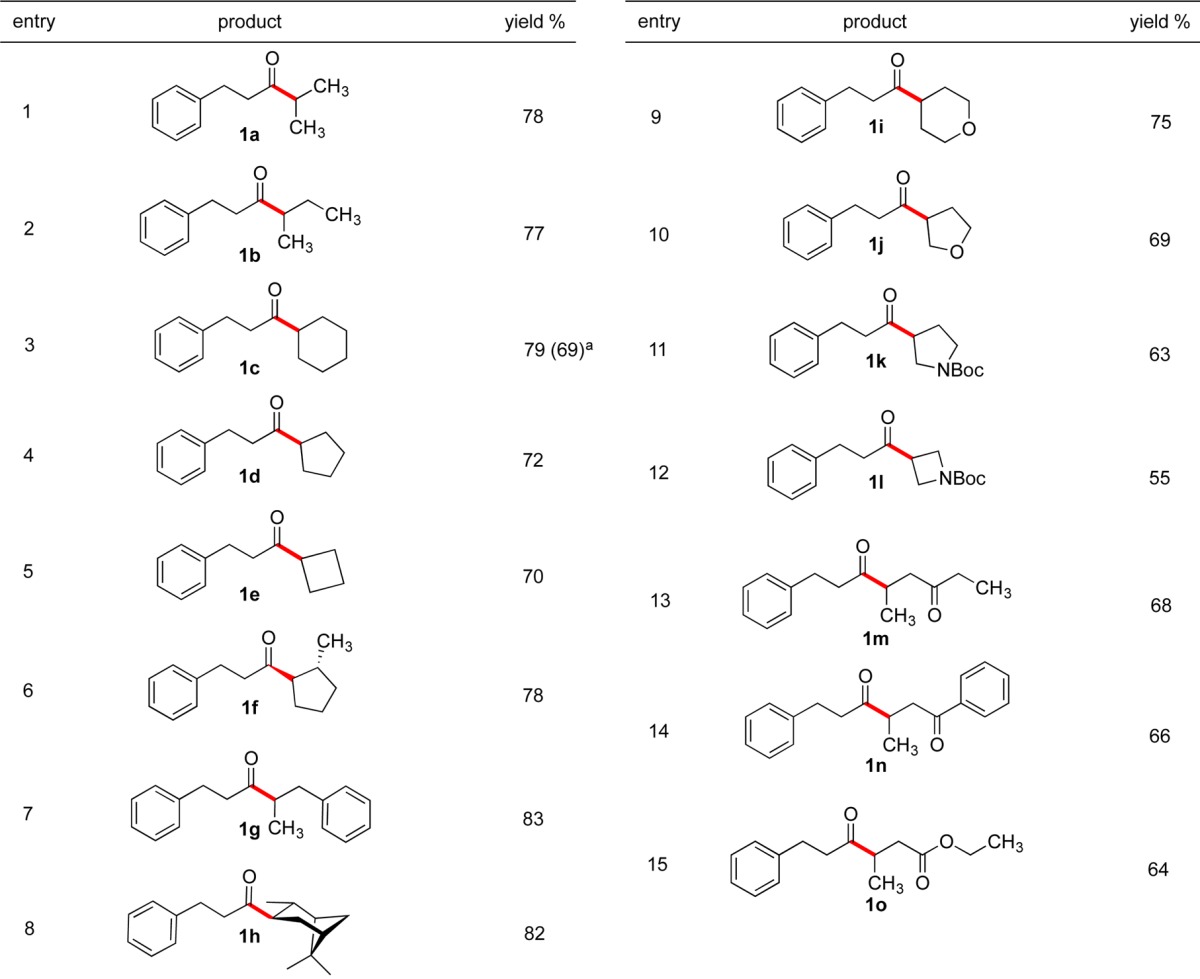
Reaction completed on 6.0 mmol scale with 1.5 mol % Ir photocatalyst 1 and 3 mol % NiCl2·dme/L1.
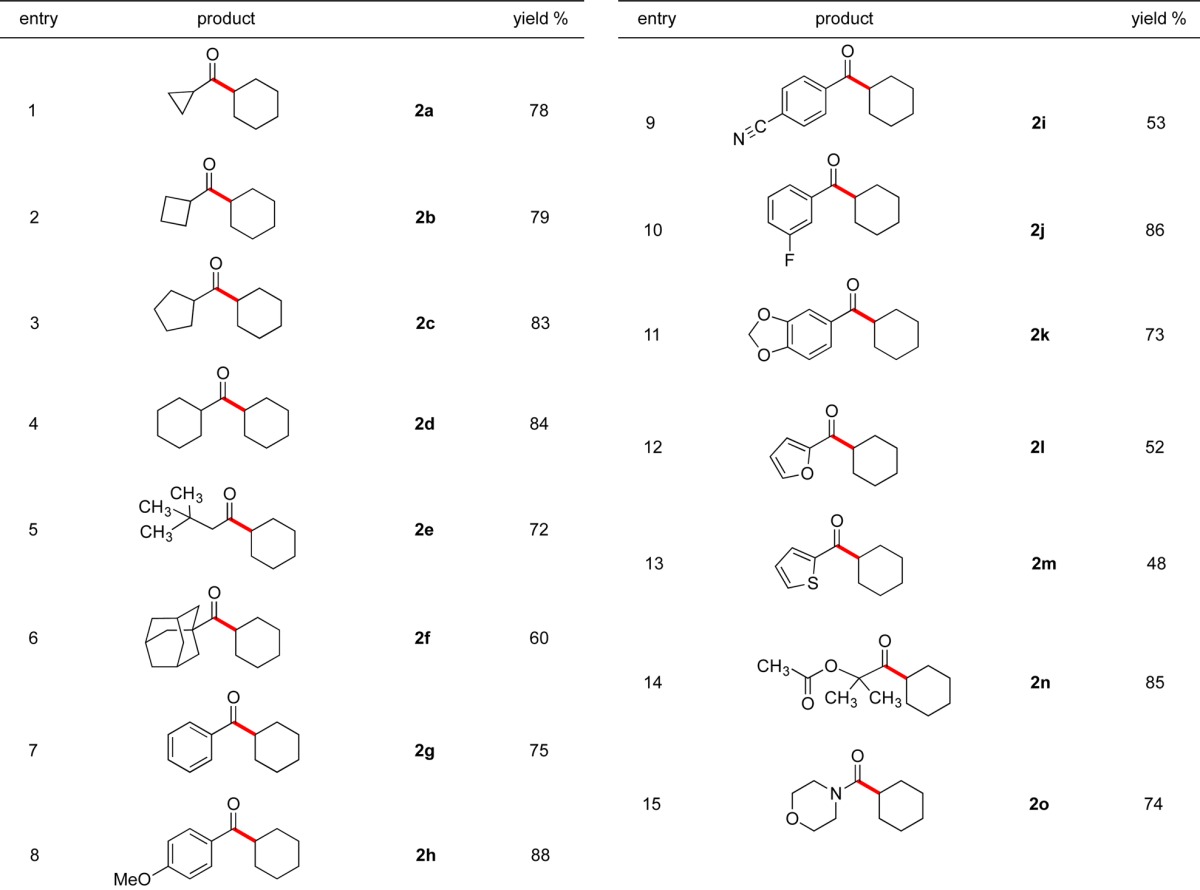
To investigate the applicability of this method to other electrophiles, potassium cyclohexyltrifluoroborate was coupled with various acyl halides (Table 2). We found that a broad range of aliphatic and aromatic acyl chlorides participated in the cross-coupling reaction with great efficiency. In these reactions, the desired products 2a–o were obtained with yields up to 88%. The reaction proceeded well with cyclopropane- and cyclobutanecarbonyl chlorides, resulting in 78% and 79% yields, respectively (entries 1 and 2). Both electron-rich and electron-poor aromatic acyl chlorides were coupled using the optimal conditions with 53–88% yields (entries 8–10). Remarkably, the sterically hindered 1-adamantanecarbonyl chloride provided the desired cross-coupled product with 60% yield (entry 6). The success of the reaction with 4-cyanobenzoyl chloride (entry 9) and 1-chlorocarbonyl-1-methylethyl acetate (entry 14) further demonstrates the functional group compatibility of this method. Finally, the reaction with 4-morpholinecarbonyl chloride to generate cyclohexyl(morpholino)methanone in 74% yield (entry 15) shows the extension of this protocol to carbamoyl chloride cross-coupling.
Table 2. Scope of the Cross-Coupling with Acyl Chlorides.
It is important to note that primary alkyltrifluoroborates are not suitable substrates for these processes owing to an unfavorable redox potential. Additionally, preliminary results indicate that a different set of conditions are required for reaction of tertiary alkyltrifluoroborates, and research along this line of investigation continues.
In conclusion, the first visible light photoredox cross-coupling of acyl chlorides with potassium secondary alkyltrifluoroborates has been developed to access a variety of dialkyl- and alkyl-aryl ketones in high yields. This efficient protocol establishes one of the few catalytic methods to generate 2° alkyl ketones from acyl chlorides. It extends photoredox/Ni dual catalytic cross-coupling of secondary alkylboron reagents to catalytic acylation approaches. Finally, the ability to employ bench-stable, storable nucleophiles, the mild conditions, high functional group tolerance, and the operational simplicity of this protocol lend themselves favorably to the facile synthesis of unsymmetrical alkyl ketones.
Experimental Section
General Considerations
All reactions were carried out under an inert atmosphere of nitrogen or argon unless otherwise noted. THF was dried over activated alumina. DME (99.5%, anhydrous) was used as received. IrCl3·xH2O and NiCl2·dme were purchased from commercial sources. All other reagents were purchased commercially and used as received. Photoredox reactions were irradiated with two or three standard 26 W compact fluorescent light bulbs. Melting points (°C) are uncorrected. NMR spectra were recorded on a 400 or 500 MHz spectrometer. Data are presented as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, br = broad), coupling constant J (Hz), and integration. Analytical thin-layer chromatography (TLC) was performed on TLC silica gel plates (0.25 mm) precoated with a fluorescent indicator. Visualization of the TLC plates was effected with ultraviolet light. HRMS data were obtained by either ESI or CI using a TOF mass spectrometer.
Synthesis of Potassium Secondary Alkyltrifluoroborates
Most of the potassium alkyltrifluoroborates were purchased commercially. In cases where the desired potassium organotrifluoroborate was not available, the corresponding boronic acid derivative was converted to the trifluoroborate by the following procedure.
General Procedure for Conversion of Boronic Acids to Trifluoroborates
To a solution of boronic acid derivative in MeOH (0.1 M) at 0 °C saturated aq KHF2 (4.5 M) was added dropwise over 30 min. After completion of the reaction, followed by 11B NMR, the resulting suspension was concentrated under reduced pressure. H2O was removed by lyophilizer. The remaining solid was suspended in hot acetone (3 × 100 mL) and filtered. The filtrate was concentrated to a minimal volume (5–20 mL), and hexane or Et2O (∼200 mL) was added to yield a white precipitate. The precipitate was isolated by filtration, washing with hexanes (∼30 mL) and CH2Cl2 (∼30 mL), to afford the desired secondary alkyltrifluoroborate.
Synthesis of Ir[dF(CF3)ppy]2(bpy)PF6 as the Photocatalyst 1
Photocatalyst 1 was synthesized according to the literature procedure.6
High-Throughput Experiments in the Design and Optimization of the Photoredox Cross-Coupling Reaction of Hydrocinnamoyl Chloride with Potassium Isoropyltrifluoroborate as Model Coupling Partners
High Throughput Experimentation (HTE) was performed at the Penn/Merck Center for High Throughput Experimentation at the University of Pennsylvania. The screens were performed on a 10 μmol scale. To reaction vials equipped with a Teflon coated magnetic stir bar in a glovebox was added a solution of the Ni source and ligand [1:1] dissolved in THF. The solvent was removed in vacuo under an inert atmosphere. Then a solution of a desired additive, potassium isopropyltrifluoroborate, hydrocinnamoyl chloride, and photocatalyst 1 in a desired solvent was added to each vial. The vials were sealed and stirred over blue LED lights. After 24 h the reactions were opened to air, 1 μmol of 4,4′-di-tert-butylbiphenyl (500 μL of a 0.002 μM solution in MeCN) was added to each vial as an internal standard, and the reaction mixtures were diluted with MeCN. The reaction mixtures were then analyzed by UPLC to obtain product-to-internal standard (P/IS) ratios for different conditions.
General Procedure for the Photoredox Cross-Coupling Reaction of Acyl Chlorides with Potassium Alkyltrifluoroborates
To a long, thin glass vial (∼20 mL) equipped with a Teflon-coated magnetic stir bar was added NiCl2·dme (6.6 mg, 0.03 mmol) and 4,4′-di-tert-butyl-2,2′-bipyridine L1 (8.1 mg, 0.03 mmol). The vial was sealed, evacuated under vacuum, and purged with Ar three times. Anhydrous degassed THF (∼1 mL) was added by syringe under an inert atmosphere, and the resulting mixture was stirred until it appeared as a pale green suspension. Then, the solvent was removed under vacuum. Once dry, potassium alkyltrifluoroborate (0.75 mmol, 1.5 equiv), Ir[dFCF3ppy]2(bpy)PF6 (15.1 mg, 0.015 mmol), and KF (29.1 mg, 0.5 mmol) were added. Next, the vial was sealed, evacuated, and purged four times. Anhydrous degassed DME (10 mL) was then added by syringe under an inert atmosphere followed by the corresponding acyl halide (0.5 mmol). The resulting mixture was stirred for 24 h in the presence of two 26 W fluorescent light bulbs while a fan was blown across the reaction setup to suppress the heat and maintain an ambient temperature of 24 °C. After completion, the crude reaction mixture was filtered through a plug of silica and rinsed with EtOAc (20 mL). The resulting solution was concentrated, and the residue was purified by column chromatography on silica gel, eluting with EtOAc and hexanes, to obtain products in pure form.
Gram Scale Reaction
1-Cyclohexyl-3-phenylpropan-1-one (1c)
To a long, thin-walled vacuum flask equipped with a Teflon-coated magnetic stir bar was added NiCl2·dme (39.5 mg, 0.18 mmol, 3.0 mol %) and 4,4′-di-tert-butyl-2,2′-bipyridine L1 (48.3 mg, 0.18, 3.0 mol %). Anhydrous degassed THF (∼5 mL) was added by syringe under Ar, and the resulting mixture was stirred until it appeared as a pale green suspension. The solvent was then removed under vacuum. Once dry, potassium cyclohexyltrifluoroborate (1.71 g, 9.0 mmol, 1.5 equiv), Ir[dFCF3ppy]2(bpy)PF61 (90.8 mg, 0.09 mmol, 1.5 mol %), and anhydrous KF (348.6 mg, 6.0 mmol, 1.0 equiv) were added. The vial was then capped with a rubber septum and purged and evacuated four times. Under an inert atmosphere, DME (120 mL, 0.05 M) was introduced followed by hydrocinnamoyl chloride (1.012 g, 6.0 mmol). The resulting mixture was stirred vigorously for 48 h in the presence of three 26 W fluorescent light bulbs while a fan was blown across the reaction setup to maintain an ambient temperature of 24 °C. After completion, the crude reaction mixture was filtered through an approximately 4 cm × 2 cm cylindrical plug of silica, washing with EtOAc (60 mL). The resulting solution was concentrated, and the residue was purified by column chromatography on silica gel, eluting with EtOAc and hexanes, to obtain 895.5 mg (69% yield) of the product as a colorless liquid.
4-Methyl-1-phenylpentan-3-one (1a).12
The title compound was obtained as a liquid in 78% yield (0.5 mmol scale, 70.5 mg). 1H NMR (500 MHz, CDCl3): δ 7.30–7.27 (m, 2H), 7.20–7.18 (m, 3H), 2.90 (t, J = 8.0 Hz, 2H), 2.77 (t, J = 8.0 Hz, 2H), 2.60–2.54 (m, 1H), 1.08 (d, J = 6.5 Hz, 6H); 13C NMR (125.8 MHz, CDCl3): δ 213.9, 141.6, 128.7, 128.5, 126.2, 42.2, 41.2, 30.0, 18.3.
4-Methyl-1-phenylhexan-3-one (1b).12
The title compound was obtained as a liquid in 77% yield (0.5 mmol scale, 73.3 mg). 1H NMR (500 MHz, CDCl3): δ 7.29–7.26 (m, 2H), 7.20–7.17 (m, 3H), 2.90 (t, J = 7.5 Hz, 2H), 2.81–2.70 (m, 2H), 2.47–2.40 (m, 1H), 1.71–1.62 (m, 1H), 1.41–1.33 (m, 1H), 1.04 (d, J = 7.0 Hz, 3H), 0.85 (t, J = 7.5 Hz, 3H); 13C NMR (125.8 MHz, CDCl3): δ 213.9, 141.6, 128.7, 128.6, 126.3, 48.2, 43.0, 29.9, 26.1, 16.0, 11.9.
1-Cyclohexyl-3-phenylpropan-1-one (1c).13
The title compound was obtained as a liquid in 79% yield (0.5 mmol scale, 85.4 mg). 1H NMR (500 MHz, CDCl3): δ 7.29–7.23 (m, 2H), 7.17–7.15 (m, 3H), 2.86 (t, J = 7.5 Hz, 2H), 2.74 (t, J = 7.5 Hz, 2H), 2.31–2.26 (m, 1H), 1.80–1.73 (m, 4H), 1.65–1.62 (m, 1H), 1.33–1.22 (m, 5H); 13C NMR (125.8 MHz, CDCl3): δ 213.4, 141.7, 128.7, 128.6, 126.2, 51.2, 42.5, 30.0, 28.6, 26.1, 25.9.
1-Cyclopentyl-3-phenylpropan-1-one (1d).14
The title compound was obtained as a liquid in 72% yield (0.5 mmol scale, 72.8 mg). 1H NMR (500 MHz, CDCl3): δ 7.29–7.26 (m, 2H), 7.20–7.17 (m, 3H), 2.91 (t, J = 7.5 Hz, 2H), 2.87–2.81 (m, 1H), 2.78 (t, J = 7.5 Hz, 2H), 1.82–1.54 (m, 8H); 13C NMR (125.8 MHz, CDCl3): δ 212.5, 141.6, 128.7, 128.6, 126.2, 51.8, 43.6, 30.1, 29.0, 26.2.
1-Cyclobutyl-3-phenylpropan-1-one (1e)
The title compound was obtained as a liquid in 70% yield (0.5 mmol scale, 65.8 mg). 1H NMR (500 MHz, CDCl3): δ 7.29–7.26 (m, 2H), 7.20–7.17 (m, 3H), 3.26–3.19 (m, 1H), 2.90 (t, J = 7.5 Hz, 2H), 2.67 (t, J = 7.5 Hz, 2H), 2.22–2.07 (m, 4H), 1.99–1.90 (m, 1H), 1.83–1.76 (m, 1H); 13C NMR (125.8 MHz, CDCl3): δ 211.2, 141.6, 128.7, 128.6, 126.3, 45.7, 41.8, 30.0, 24.5, 18.0; FT-IR (neat): 1704, 1453, 1366, 1121, 978, 747, 698 cm–1; HRMS (ES+) m/z calcd for C13H16ONa [M + Na]+ 211.1099, found 211.1108.
1-(2-Methylcyclopentyl)-3-phenylpropan-1-one (1f)
The title compound was obtained as a liquid in 78% yield (0.5 mmol scale, 84.4 mg). 1H NMR (500 MHz, CDCl3): δ 7.30–7.27 (m, 2H), 7.20–7.18 (m, 3H), 2.91 (t, J = 7.5 Hz, 2H), 2.82–2.73 (m, 2H), 2.43–2.38 (m, 1H), 2.17–2.11 (m, 1H), 1.91–1.82 (m, 2H), 1.74–1.61 (m, 3H), 1.24–1.16 (m, 1H), 1.00 (d, J = 6.5 Hz, 3H); 13C NMR (125.8 MHz, CDCl3): δ 212.6, 141.6, 128.6, 128.5, 126.2, 59.8, 44.1, 37.9, 35.1, 30.0, 29.9, 24.9, 20.2; FT-IR (neat): 1705, 1453, 1374, 748, 698 cm–1; HRMS (ES+) m/z calcd for C15H20ONa [M + Na]+ 239.1412, found 239.1414.
2-Methyl-1,5-diphenylpentan-3-one (1g).15
The title compound was obtained as a liquid in 83% yield (0.5 mmol scale, 104.7 mg). 1H NMR (500 MHz, CDCl3): δ 7.25–7.21 (m, 4H), 7.18–7.13 (m, 2H), 7.09–7.08 (m, 4H), 2.95–2.91 (m, 1H), 2.82–2.66 (m, 4H), 2.56–2.51 (m, 2H), 1.03 (d, J = 7.0 Hz, 3H); 13C NMR (125.8 MHz, CDCl3): δ 213.2, 141.4, 139.9, 129.1, 128.6, 128.5, 126.4, 126.2, 48.5, 43.8, 39.3, 29.8, 16.5.
3-Phenyl-1-(2,6,6-trimethylbicyclo[3.1.1]heptan-3-yl)propan-1-one (1h)
The title compound was obtained as a liquid in 82% yield (0.5 mmol scale, 110.9 mg). 1H NMR (500 MHz, CDCl3): δ 7.30–7.27 (m, 2H), 7.21–7.18 (m, 3H), 2.96–2.70 (m, 5H), 2.43–2.39 (m, 1H), 2.30–2.26 (m, 1H), 2.20–2.14 (m, 1H), 1.95–1.91 (m, 1H), 1.84–1.79 (m, 2H), 1.21 (s, 3H), 1.03–1.01 (m, 6H), 0.87 (d, J = 10.0 Hz, 1H); 13C NMR (125.8 MHz, CDCl3): δ 212.4, 141.6, 128.7, 128.6, 126.3, 49.6, 47.4, 43.9, 41.0, 38.8, 36.8, 32.9, 30.4, 29.9, 28.1, 23.1, 22.6; FT-IR (neat): 1709, 1453, 1368, 909, 731, 698 cm–1; HRMS (ES+) m/z calcd for C19H26ONa [M + Na]+ 293.1881, found 293.1879.
3-Phenyl-1-(tetrahydro-2H-pyran-4-yl)propan-1-one (1i)
The title compound was obtained as a liquid in 75% yield (0.5 mmol scale, 81.9 mg). 1H NMR (500 MHz, CDCl3): δ 7.26–7.23 (m, 2H), 7.17–7.14 (m, 3H), 3.95–3.92 (m, 2H), 3.35 (dt, J = 11.5, 3.0 Hz, 2H), 2.87 (t, J = 7.5 Hz, 2H), 2.74 (t, J = 7.5 Hz, 2H), 2.49–2.44 (m, 1H), 1.69–1.58 (m, 4H); 13C NMR (125.8 MHz, CDCl3): δ 211.1, 141.3, 128.7, 128.5, 126.3, 67.4, 47.8, 42.1, 29.8, 28.2; FT-IR (neat): 1705, 1444, 1240, 1112, 1090, 1015, 749, 698 cm–1; HRMS (CI+) m/z calcd for C14H19O2 [M + H]+ 219.1385, found 219.1381.
3-Phenyl-1-(tetrahydrofuran-3-yl)propan-1-one (1j)
The title compound was obtained as a liquid in 69% yield (0.5 mmol scale, 70.5 mg). 1H NMR (500 MHz, CDCl3): δ 7.27–7.23 (m, 2H), 7.19–7.14 (m, 3H), 3.87–3.79 (m, 3H), 3.75–3.71 (m, 1H), 3.16–3.10 (m, 1H), 2.89 (t, J = 7.5 Hz, 2H), 2.79–2.74 (m, 2H), 2.03–1.99 (m, 2H); 13C NMR (125.8 MHz, CDCl3): δ 209.2, 141.1, 128.7, 128.5, 126.4, 69.4, 68.5, 51.3, 43.8, 29.9, 28.9; FT-IR (neat): 1709, 1453, 1102, 1066, 913, 749, 698 cm–1; HRMS (ES+) m/z calcd for C13H16O2Na [M + Na]+ 227.1048, found 227.1051.
tert-Butyl 3-(3-Phenylpropanoyl)pyrrolidine-1-carboxylate (1k)
The title compound was obtained as a liquid in 63% yield (0.5 mmol scale, 95.6 mg). 1H NMR (500 MHz, CDCl3): δ 7.25–7.22 (m, 2H), 7.16–7.12 (m, 3H), 3.49–3.40 (m, 3H), 3.30–3.25 (m, 1H), 3.06–3.00 (m, 1H), 2.92–2.84 (m, 2H), 2.81–2.70 (m, 2H), 1.99–1.96 (m, 2H), 1.43 (s, 9H); 13C NMR (125.8 MHz, CDCl3): δ 208.5, 154.6, 141.2, 128.8, 128.5, 126.5, 79.6, 47.4, 45.6, 43.5, 30.0, 28.8, 28.5, 28.1; FT-IR (neat): 1687, 1453, 1401, 1365, 1166, 1116, 879, 731, 699 cm–1; HRMS (ES+) m/z calcd for C18H25NO3Na [M + Na]+ 326.1732, found 326.1740.
tert-Butyl 3-(3-Phenylpropanoyl)azetidine-1-carboxylate (1l)
The title compound was obtained as a liquid in 55% yield (0.5 mmol scale, 79.6 mg). 1H NMR (500 MHz, CDCl3): δ 7.27–7.24 (m, 2H), 7.19–7.14 (m, 3H), 3.99–3.94 (m, 4H), 3.37–3.31 (m, 1H), 2.91 (t, J = 7.0 Hz, 2H), 2.72 (t, J = 7.0 Hz, 2H), 1.42 (s, 9H); 13C NMR (125.8 MHz, CDCl3): δ 207.1, 156.4, 140.9, 128.8, 128.5, 126.5, 79.9, 50.8, 42.5, 39.1, 29.8, 28.6; FT-IR (neat): 1694, 1454, 1392, 1365, 1131, 770, 730, 699 cm–1; HRMS (ES+) m/z calcd for C17H24NO3 [M + H]+ 290.1756, found 290.1761.
4-Methyl-1-phenyloctane-3,6-dione (1m)
The title compound was obtained as a liquid in 68% yield (0.5 mmol scale, 78.9 mg). 1H NMR (500 MHz, CDCl3): δ 7.28–7.25 (m, 2H), 7.19–7.15 (m, 3H), 3.05–3.00 (m, 1H), 2.96–2.85 (m, 5H), 2.46–2.33 (m, 3H), 1.04–1.01 (m, 6H); 13C NMR (125.8 MHz, CDCl3): δ 212.8, 210.3, 141.6, 128.7, 128.6, 126.2, 45.4, 43.2, 41.3, 36.1, 29.9, 16.8, 7.9; FT-IR (neat): 1707, 1454, 1376, 1361, 1112, 749, 699 cm–1; HRMS (ES+) m/z calcd for C15H20O2Na [M + Na]+ 255.1361, found 255.1358.
3-Methyl-1,6-diphenylhexane-1,4-dione (1n)
The title compound was obtained as a liquid in 66% yield (0.5 mmol scale, 92.5 mg). 1H NMR (500 MHz, CDCl3): δ 7.96–7.94 (m, 2H), 7.57–7.54 (m, 1H), 7.47–7.44 (m 2H), 7.31–7.28 (m, 2H), 7.25–7.18 (m, 3H), 3.57–3.52 (m, 1H), 3.26–3.19 (m, 1H), 3.03–2.91 (m, 5H), 1.14 (dd, J = 7.0, 0.5 Hz, 3H); 13C NMR (125.8 MHz, CDCl3): δ 212.7, 198.7, 141.6, 136.8, 133.4, 128.8, 128.7, 128.6, 128.2, 126.2, 43.3, 42.1, 41.4, 29.9, 16.9; FT-IR (neat): 1711, 1681, 1449, 1356, 1216, 1003, 748, 689 cm–1; HRMS (ES+) m/z calcd for C19H21O2 [M + H]+ 281.1542, found 281.1532.
Ethyl 3-Methyl-4-oxo-6-phenylhexanoate (1o).16
The title compound was obtained as a liquid in 64% yield (0.5 mmol scale, 79.4 mg). 1H NMR (500 MHz, CDCl3): δ 7.28–7.25 (m, 2H), 7.19–7.16 (m, 3H), 4.09 (q, J = 7.0 Hz, 2H), 3.00–2.94 (m, 1H), 2.92–2.89 (m, 2H), 2.88–2.81 (m, 2H), 2.78–2.73 (m, 1H), 2.30–2.26 (m, 1H), 1.22 (t, J = 7.0 Hz, 3H), 1.07 (d, J = 7.0 Hz, 3H); 13C NMR (125.8 MHz, CDCl3): δ 212.0, 172.4, 141.4, 128.6, 128.5, 126.2, 60.7, 43.0, 42.3, 37.2, 29.8, 16.7, 14.4.
Cyclohexyl(cyclopropyl)methanone (2a).17
The title compound was obtained as a liquid in 78% yield (0.5 mmol scale, 59.3 mg). 1H NMR (500 MHz, CDCl3): δ 2.46–2.42 (m, 1H), 1.94–1.84 (m, 3H), 1.74–1.72 (m, 2H), 1.63–1.60 (m, 1H), 1.35–1.13 (m, 5H), 0.92–0.89 (m, 2H), 0.78–0.75 (m, 2H); 13C NMR (125.8 MHz, CDCl3): δ 214.1, 51.7, 28.7, 26.2, 26.0, 19.1, 10.7.
Cyclobutyl(cyclohexyl)methanone (2b).17
The title compound was obtained as a liquid in 79% yield (0.5 mmol scale, 65.7 mg). 1H NMR (500 MHz, CDCl3): δ 3.39–3.32 (m, 1H), 2.32–2.27 (m, 1H), 2.23–2.15 (m, 2H), 2.09–2.02 (m, 2H), 1.97–1.87 (m, 1H), 1.79–1.71 (m, 5H), 1.63–1.61 (m, 1H), 1.32–1.14 (m, 5H); 13C NMR (125.8 MHz, CDCl3): δ 215.0, 49.1, 44.0, 28.8, 26.1, 26.0, 24.8, 18.1.
Cyclohexyl(cyclopentyl)methanone (2c).17
The title compound was obtained as a liquid in 83% yield (0.5 mmol scale, 74.8 mg). 1H NMR (500 MHz, CDCl3): δ 2.99–2.93 (m, 1H), 2.44–2.38 (m, 1H), 1.79–1.62 (m, 11H), 1.54–1.51 (m, 2H), 1.34–1.15 (m, 5H); 13C NMR (125.8 MHz, CDCl3): δ 216.9, 50.5, 49.7, 29.6, 28.9, 26.3, 26.1, 26.0.
Dicyclohexylmethanone (2d).17
The title compound was obtained as a liquid in 84% yield (0.5 mmol scale, 81.6 mg). 1H NMR (500 MHz, CDCl3): δ 2.47–2.41 (m, 2H), 1.74–1.72 (m, 8H), 1.64–1.61 (m, 2H), 1.32–1.14 (m, 10H); 13C NMR (125.8 MHz, CDCl3): δ 217.3, 49.4, 28.8, 26.1, 26.0.
1-Cyclohexyl-3,3-dimethylbutan-1-one (2e)
The title compound was obtained as a liquid in 72% yield (0.5 mmol scale, 65.6 mg). 1H NMR (500 MHz, CDCl3): δ 2.28–2.23 (m, 3H), 1.77–1.73 (m, 4H), 1.64–1.61 (m, 1H), 1.28–1.13 (m, 5H), 0.97 (s, 9H); 13C NMR (125.8 MHz, CDCl3): δ 214.4, 53.1, 52.6, 31.2, 30.0, 28.5, 26.1, 26.0; FT-IR (neat): 1705, 1449, 1364, 712 cm–1; HRMS (ES+) m/z calcd for C12H22ONa [M + Na]+ 205.1568, found 205.1569.
(Adamantan-1-yl)(cyclohexyl)methanone (2f)
The title compound was obtained as a white solid in 60% yield (0.5 mmol scale, 73.9 mg). Mp 80–81 °C; 1H NMR (500 MHz, CDCl3): δ 2.86–2.80 (m, 1H), 2.02 (s, 3H), 1.78–1.65 (m, 15H), 1.57–1.53 (m, 2H), 1.38–1.19 (m, 5H); 13C NMR (125.8 MHz, CDCl3): δ 218.7, 47.2, 44.3, 38.1, 37.0, 30.1, 28.3, 26.2; FT-IR (neat): 1695, 1450, 1244, 1196, 1162, 996, 967 cm–1; HRMS (CI+) m/z calcd for C17H26O [M]+ 246.1984, found 246.1978.
Cyclohexyl(phenyl)methanone (2g).18
The title compound was obtained as a liquid in 75% yield (0.5 mmol scale, 70.6 mg). 1H NMR (500 MHz, CDCl3): δ 7.92–7.91 (m, 2H), 7.52–7.49 (m, 1H), 7.44–7.41 (m, 2H), 3.26–3.21 (m, 1H), 1.88–1.79 (m, 4H), 1.73–1.69 (m, 1H), 1.46–1.20 (m, 5H); 13C NMR (125.8 MHz, CDCl3): δ 204.0, 136.5, 132.9, 128.8, 128.4, 45.8, 29.6, 26.2, 26.1.
Cyclohexyl(4-methoxyphenyl)methanone (2h).19
The title compound was obtained as a white solid in 88% yield (0.5 mmol scale, 96.1 mg). Mp 59–60 °C; 1H NMR (500 MHz, CDCl3): δ 7.92 (dd, J = 11.5, 2.5 Hz, 2H), 6.91 (dd, J = 11.5, 2.5 Hz, 2H), 3.84 (s, 3H), 3.22–3.17 (m, 1H), 1.86–1.80 (m, 4H), 1.72–1.70 (m, 1H), 1.52–1.44 (m, 2H), 1.41–1.32 (m, 2H), 1.29–1.22 (m, 1H); 13C NMR (125.8 MHz, CDCl3): δ 202.6, 163.4, 130.7, 129.4, 113.9, 55.6, 45.5, 29.8, 26.2, 26.1.
4-(Cyclohexanecarbonyl)benzonitrile (2i).20
The title compound was obtained as a white solid in 53% yield (0.5 mmol scale, 56.5 mg). Mp 60–61 °C; 1H NMR (500 MHz, CDCl3): δ 7.98–7.97 (m, 2H), 7.73–7.72 (m, 2H), 3.21–3.16 (m, 1H), 1.85–1.79 (m, 4H), 1.72–1.68 (m, 1H), 1.48–1.31 (m, 4H), 1.27–1.19 (m, 1H); 13C NMR (125.8 MHz, CDCl3): δ 202.7, 139.7, 132.7, 128.9, 118.3, 116.2, 46.2, 29.4, 26.1, 25.9.
Cyclohexyl(3-fluorophenyl)methanone (2j)
The title compound was obtained as a liquid in 86% yield (0.5 mmol scale, 88.7 mg). 1H NMR (500 MHz, CDCl3): δ 7.70 (dd, J = 7.5, 1.0 Hz, 1H), 7.61–7.58 (m, 1H), 7.44–7.39 (m, 1H), 7.24–7.20 (m, 1H), 3.21–3.15 (m, 1H), 1.87–1.81 (m, 4H), 1.74–1.70 (m, 1H), 1.51–1.33 (m, 4H), 1.29–1.23 (m, 1H); 13C NMR (125.8 MHz, CDCl3): δ 202.7, 164.1 (d, J = 247.7 Hz, 1C), 138.7 (d, J = 5.7 Hz, 1C), 130.4 (d, J = 7.8 Hz, 1C), 124.1 (d, J = 2.7 Hz, 1C), 119.9 (d, J = 21.5 Hz, 1C), 115.2 (d, J = 22.0 Hz, 1C), 46.0, 29.6, 26.1, 26.0 ; FT-IR (neat): 1681, 1587, 1440, 1254, 1226, 1164, 1154, 896, 768 cm–1; HRMS (ES+) m/z calcd for C13H15OFNa [M + Na]+ 229.1005, found 229.1001.
Benzo[d][1,3]dioxol-5-yl(cyclohexyl)methanone (2k)
The title compound was obtained as a white solid in 73% yield (0.5 mmol scale, 84.7 mg). Mp 68–69 °C; 1H NMR (500 MHz, CDCl3): δ 7.53 (dd, J = 8.0, 1.5 Hz, 1H), 7.40 (d, J = 1.5 Hz, 1H), 6.82 (d, J = 8.5 Hz, 1H), 6.00 (s, 2H), 3.17–3.11 (m, 1H), 1.84–1.79 (m, 4H), 1.72–1.68 (m, 1H), 1.50–1.42 (m, 2H), 1.39–1.30 (m, 2H), 1.27–1.9 (m, 1H); 13C NMR (125.8 MHz, CDCl3): δ 202.1, 151.7, 148.4, 131.3, 124.5, 108.4, 108.1, 102.0, 45.7, 29.8, 26.2, 26.1; FT-IR (neat): 1656, 1602, 1447, 1107, 1035, 936, 900, 743 cm–1; HRMS (ES+) m/z calcd for C14H17O3 [M + H]+ 233.1178, found 233.1168.
Cyclohexyl(furan-2-yl)methanone (2l).21
The title compound was obtained as a liquid in 52% yield (0.5 mmol scale, 46.3 mg). 1H NMR (500 MHz, CDCl3): δ 7.56–7.55 (m, 1H), 7.16–7.15 (m, 1H), 6.50–6.49 (m, 1H), 3.06–3.00 (m, 1H), 1.85–1.79 (m, 4H), 1.70–1.68 (m, 1H), 1.51–1.44 (m, 2H), 1.37–1.29 (m, 2H), 1.26–1.20 (m, 1H); 13C NMR (125.8 MHz, CDCl3): δ 193.1, 152.5, 146.4, 117.2, 112.3, 46.6, 29.2, 26.1, 26.0.
Cyclohexyl(thiophen-2-yl)methanone (2m).21
The title compound was obtained as a liquid in 48% yield (0.5 mmol scale, 46.6 mg). 1H NMR (500 MHz, CDCl3): δ 7.69 (dd, J = 3.5, 0.5 Hz, 1H), 7.59 (dd, J = 5.0, 0.5 Hz, 1H), 7.10 (dd, J = 3.5, 5.0 Hz, 1H), 3.10–3.04 (m, 1H), 1.89–1.80 (m, 4H), 1.72–1.69 (m, 1H), 1.56–1.48 (m, 2H), 1.39–1.30 (m, 2H), 1.28–1.20 (m, 1H); 13C NMR (125.8 MHz, CDCl3): δ 197.0, 144.1, 133.6, 131.7, 128.3, 47.7, 29.9, 26.1, 26.0.
1-Cyclohexyl-2-methyl-1-oxopropan-2-yl Acetate (2n)
The title compound was obtained as a liquid in 85% yield (0.5 mmol scale, 90.2 mg). 1H NMR (500 MHz, CDCl3): δ 2.69–2.63 (m, 1H), 2.02 (s, 3H), 1.74–1.62 (m, 5H), 1.45 (s, 6H), 1.40–1.32 (m, 2H), 1.24–1.17 (m, 3H); 13C NMR (125.8 MHz, CDCl3): δ 212.5, 170.4, 84.0, 45.1, 30.1, 26.0, 24.2, 21.5; FT-IR (neat): 1735, 1713, 1369, 1251, 1150, 1134, 1018, 990, 964 cm–1; HRMS (ES+) m/z calcd for C12H20O3Na [M + Na]+ 235.1310, found 235.1307.
Cyclohexyl(morpholino)methanone (2o).22
The title compound was obtained as a liquid in 74% yield (0.5 mmol scale, 73.0 mg). 1H NMR (500 MHz, CDCl3): δ 3.65–3.60 (m, 6H), 3.48 (s, 2H), 2.44–2.39 (m, 1H), 1.80–1.78 (m, 2H), 1.71–1.68 (m, 3H), 1.56–1.48 (m, 2H), 1.27–1.23 (m, 3H); 13C NMR (125.8 MHz, CDCl3): δ 174.8, 67.1, 66.9, 46.0, 42.0, 40.3, 29.4, 25.9.
Acknowledgments
This research was supported by the NIGMS (R01 GM-113878). Frontier Scientific is thanked for the donation of boron compounds used in this investigation. Johnson-Matthey is acknowledged for the donation of IrCl3 used for the synthesis of the photocatalyst. Dr. Rakesh Kohli (University of Pennsylvania) is acknowledged for obtaining HRMS data.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.6b02897.
NMR spectra for all compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Walter M. W. Nat. Prod. Rep. 2002, 19, 278. 10.1039/b100919m. [DOI] [PubMed] [Google Scholar]; b McDaniel R.; Thamchaipenet A.; Gustafsson C.; Fu H.; Betlach M.; Betlach M.; Ashley G. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 1846. 10.1073/pnas.96.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jabeen I.; Pleban K.; Rinner U.; Chiba P.; Ecker G. F. J. Med. Chem. 2012, 55, 3261. 10.1021/jm201705f. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Vooturi S. K.; Cheung C. M.; Rybak M. J.; Firestine S. M. J. Med. Chem. 2009, 52, 5020. 10.1021/jm900519b. [DOI] [PubMed] [Google Scholar]
- a Dieter R. K. Tetrahedron 1999, 55, 4177. 10.1016/S0040-4020(99)00184-2. [DOI] [Google Scholar]; b Fiandanese V.; Marchese G.; Martina V.; Ronzini L. Tetrahedron Lett. 1984, 25, 4805. 10.1016/S0040-4039(01)81525-2. [DOI] [Google Scholar]
- a Sato F.; Inoue M.; Oguro K.; Sato M. Tetrahedron Lett. 1979, 20, 4303. 10.1016/S0040-4039(01)86573-4. [DOI] [Google Scholar]; b Dubois J. E.; Boussu M.; Lion C. Tetrahedron Lett. 1971, 12, 829. 10.1016/S0040-4039(01)96567-0. [DOI] [Google Scholar]; c Dieter R. K.; Sharma R. R.; Yu H.; Gore V. K. Tetrahedron 2003, 59, 1083. 10.1016/S0040-4020(02)01526-0. [DOI] [Google Scholar]; d Cahiez G.; Laboue B. Tetrahedron Lett. 1989, 30, 7369. 10.1016/S0040-4039(00)70699-X. [DOI] [Google Scholar]; e Cahiez G.; Laboue B. Tetrahedron Lett. 1992, 33, 4439. 10.1016/S0040-4039(00)60104-1. [DOI] [Google Scholar]; f Cahiez G.; Razafintsalama L.; Laboue B.; Chau F. Tetrahedron Lett. 1998, 39, 849. 10.1016/S0040-4039(97)10747-X. [DOI] [Google Scholar]
- a Kosugi M.; Shimizu Y.; Migita T. Chem. Lett. 1977, 6, 1423. 10.1246/cl.1977.1423. [DOI] [Google Scholar]; b Labadie J. W.; Stille J. K. J. Am. Chem. Soc. 1983, 105, 669. 10.1021/ja00341a083. [DOI] [Google Scholar]; c Labadie J. W.; Tueting D.; Stille J. K. J. Org. Chem. 1983, 48, 4634. 10.1021/jo00172a038. [DOI] [Google Scholar]; d Grey R. A. J. Org. Chem. 1984, 49, 2288. 10.1021/jo00186a043. [DOI] [Google Scholar]; e Tamaru Y.; Ochiai H.; Nakamura T.; Yoshida Z. Angew. Chem., Int. Ed. Engl. 1987, 26, 1157. 10.1002/anie.198711571. [DOI] [Google Scholar]; f Harada T.; Kotani Y.; Katsuhira T.; Oku A. Tetrahedron Lett. 1991, 32, 1573. 10.1016/S0040-4039(00)74275-4. [DOI] [Google Scholar]; g Reddy C. K.; Knochel P. Angew. Chem., Int. Ed. Engl. 1996, 35, 1700. 10.1002/anie.199617001. [DOI] [Google Scholar]; h Coia N.; Mokhtari N.; Vasse J.-L.; Szymoniak J. Org. Lett. 2011, 13, 6292. 10.1021/ol202796w. [DOI] [PubMed] [Google Scholar]; i Cherney A. H.; Reisman S. E. Tetrahedron 2014, 70, 3259. 10.1016/j.tet.2013.11.104. [DOI] [Google Scholar]; j Benischke A. D.; Leroux M.; Knoll I.; Knochel P. Org. Lett. 2016, 18, 3626. 10.1021/acs.orglett.6b01677. [DOI] [PubMed] [Google Scholar]
- Jana R.; Pathak T. P.; Sigman M. S. Chem. Rev. 2011, 111, 1417. 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tellis J. C.; Primer D. N.; Molander G. A. Science 2014, 345, 433. 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Primer D. N.; Karakaya I.; Tellis J. C.; Molander G. A. J. Am. Chem. Soc. 2015, 137, 2195. 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joe C. L.; Doyle A. G. Angew. Chem., Int. Ed. 2016, 55, 4040. 10.1002/anie.201511438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amani J.; Sodagar E.; Molander G. A. Org. Lett. 2016, 18, 732. 10.1021/acs.orglett.5b03705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmink J. R.; Bellomo A.; Berritt S. Aldrichimica Acta 2013, 46, 71. [Google Scholar]
- a Dreher S. D.; Dormer P. G.; Sandrock D. L.; Molander G. A. J. Am. Chem. Soc. 2008, 130, 9257. 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li L.; Zhao S.; Joshi-Pangu A.; Diane M.; Biscoe M. R. J. Am. Chem. Soc. 2014, 136, 14027. 10.1021/ja508815w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Paal C. Ber. Dtsch. Chem. Ges. 1884, 17, 2756. 10.1002/cber.188401702228. [DOI] [Google Scholar]; b Knorr L. Ber. Dtsch. Chem. Ges. 1884, 17, 2863. 10.1002/cber.188401702254. [DOI] [Google Scholar]
- Erbing E.; Vazquez-Romero A.; Gjmez A. B.; Platero-Prats A. E.; Carson F.; Zou X.; Tolstoy P.; Martin-Matute B. Chem. - Eur. J. 2016, 22, 15659. 10.1002/chem.201603825. [DOI] [PubMed] [Google Scholar]
- Chen M.; Wang J.; Chai Z.; You C.; Lei A. Adv. Synth. Catal. 2012, 354, 341. 10.1002/adsc.201100782. [DOI] [Google Scholar]
- Gonzalez J.; Borchardt A. J.; Dragovich P. S.; Jewell T. M.; Linton M. A.; Zhou R.; Li H.; Tatlock J. H.; Abreo M. A.; Prins T. J. A. Patent WO2003/95441 A1, Nov. 20, 2003.
- Armesto D.; Esteban S.; Horspool W. M.; Martin J. A. F.; Martinez-Alcazar P.; Perez-Ossorio R. J. Chem. Soc., Perkin Trans. 1 1989, 751. 10.1039/P19890000751. [DOI] [Google Scholar]
- Fujimura T.; Aoki S.; Nakamura E. J. Org. Chem. 1991, 56, 2809. 10.1021/jo00008a043. [DOI] [Google Scholar]
- Krapcho A. P.; Kashdan D. S.; Jahngen E. G. E. Jr.; Lovey A. J. J. Org. Chem. 1977, 42, 1189. 10.1021/jo00427a019. [DOI] [Google Scholar]
- Imai S.; Togo H. Tetrahedron 2016, 72, 6948. 10.1016/j.tet.2016.09.019. [DOI] [Google Scholar]
- Ranu B. C.; Ghosh K.; Jana U. J. Org. Chem. 1996, 61, 9546. 10.1021/jo9615413. [DOI] [Google Scholar]
- Dohi S.; Moriyama K.; Togo H. Tetrahedron 2012, 68, 6557. 10.1016/j.tet.2012.05.059. [DOI] [Google Scholar]
- Kondo T.; Akazome M.; Tsuji Y.; Watanabe Y. J. Org. Chem. 1990, 55, 1286. 10.1021/jo00291a035. [DOI] [Google Scholar]
- Tillack A.; Rudloff I.; Beller M. Eur. J. Org. Chem. 2001, 2001, 523.. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.