

Abstract
Growth factor withdrawal has been studied across different species and has been shown to have dramatic consequences on cell survival. In the nervous system, withdrawal of nerve growth factor (NGF) from sympathetic and sensory neurons results in substantial neuronal cell death, signifying a requirement for NGF for the survival of neurons in the peripheral nervous system (PNS). In contrast to the PNS, withdrawal of central nervous system (CNS) enriched brain-derived neurotrophic factor (BDNF) has little effect on cell survival but is indispensible for synaptic plasticity. Given that most early events in neuropsychiatric disorders are marked by a loss of synapses, lack of BDNF may thus be an important part of a cascade of events that leads to neuronal degeneration. Here we review reports on the effects of BDNF withdrawal on CNS neurons and discuss the relevance of the loss in disease.
Keywords: BDNF, Neuronal degeneration, Synaptic plasticity, Synapse loss, Neuropsychiatric disorders
1. Introduction
Neurotrophins play a pivotal role in modulating the survival and function of neurons in the nervous system. Evidence for the profound effects of trophic support has been demonstrated in both in vitro and in vivo systems, largely by treating neurons with exogenous neurotrophins and has been expansively reviewed. In addition to gain of function, loss of function studies have also unraveled critical roles of neurotrophins. Neurotrophic factor withdrawal has been studied extensively in sensory and sympathetic neurons where it has resulted in dramatic cell loss through transcription-dependent programmed cell death mechanisms (Deshmukh and Johnson, 1997; Levi-Montalcini, 1964; Levi-Montalcini and Angeletti, 1963; Oppenheim et al., 1990).
Studies on growth factor withdrawal were pioneered more than six decades ago with the discovery of NGF (Levi-Montalcini, 1964). Experiments conducted in new-born mice deprived of NGF demonstrated a significant role of NGF as a bona fide survival factor for peripheral neurons (Levi-Montalcini and Booker, 1960). This idea sparked interest in identifying other brain specific neurotrophins and investigating their survival effects on neuronal subtypes in the CNS. It soon became apparent that neurotrophins were not essential for promoting neuronal survival in the CNS (Johnson et al., 1986; Rauskolb et al., 2010). Evidence has now emerged that a lack of trophic factor support in the central nervous system has profound effects on neuronal morphology, synaptic integrity and physiology (Cohen-Corey and Fraser, 1995; Jeanneteau et al., 2010; Korte et al., 1995). BDNF in particular has been widely studied in CNS neurons due to its prevalent expression and has been shown to mediate many morphologic and synaptic functions of CNS neurons.
In this review, we will summarize evidence from recent studies that address the consequences of a lack of BDNF in the CNS. We will consider cell-type specific effects as well as structural, behavioral and molecular consequences of a lack of trophic factor support. We will conclude by addressing the clinical relevance of the changes associated with reduced BDNF in brain disorders and propose strategies for restoring BDNF in diseased neurons to ameliorate neuronal integrity.
2. Region specific effects of BDNF sequestration on neuronal morphology
Although the effects of NGF on cell differentiation and proliferation are well recognized in the PNS, the global deprivation of BDNF in the CNS has appreciable differences on different brain regions. Prenatally, BDNF is required for survival of neurons, as BDNF−/− mice die shortly after birth (Maisonpierre et al., 1990). Conditional deletion of BDNF has region-specific effects on dendritic morphology. While most cell types are spared, striatal neurons are the most vulnerable and succumb to a lack of postnatal BDNF, which manifest in reduced dendritic complexity and spine density (Rauskolb et al., 2010). Moreover, a decrease in cortical BDNF in the Emx-Cre conditional mouse resulted in a loss of anterograde transport of BDNF to the striatum, dendritic deficits and neuronal loss (Baquet et al., 2004).
Another vulnerable region is the midbrain-hindbrain area, where a lack of BDNF leads to aberrant development of dopaminergic neurons in the substantia nigra pars compacta (Baquet et al., 2005). Baquet and others used the Wnt-Cre mouse line to selectively delete BDNF in midbrain-hindbrain region. Targeted deletion of BDNF adversely affected expression of tyrosine hydroxylase and proper establishment of dopamine neurons in the substantia nigra. Interestingly, a loss of BDNF selectively affected dopaminergic neurons as total neuron number in the substantia nigra remained unchanged (Baquet et al., 2005). In the cortex, single cell gene knockout experiments also demonstrated that a lack of BDNF results in a reduced number of functional Garbaergic synapses in layer II/III of the cortex (Kohara et al., 2007). Impaired dendritic arborization of cortical neurons also occurs in the absence of BDNF (Cohen-Corey and Fraser, 1995). Similarly, maintenance of the cortical dendritic structure requires BDNF. Restricted deletion of BDNF in the mouse forebrain affects maintenance of cortical dendritic arbors; layer II/III pyramidal neurons initially develop normal soma size and dendritic tree complexity, which retards gradually resulting in loss of dendrite complexity by three weeks of age (Gorski et al., 2003). This finding indicates that BDNF has an essential role of supporting the survival and maturation of established dendritic structures. Retinal ganglion cells also suffer appreciable morphological changes due to a lack of BDNF. Reduced size of ganglionic cells and hypomyelination has been reported early in development in the optic nerve of BDNF−/− mice (Cellerino et al., 1997), further emphasizing the importance of BDNF in neuronal morphology.
3. Physiology and behavior
The spatial and temporal changes in BDNF expression contribute to changes in synaptic efficacy and plasticity. BDNF modulates synapse formation and development; BDNF−/− mice exhibit substantial synaptic fatigue at CA1 synapses as well as reduced synaptic vesicle docking, which can be reversed by applying exogenous BDNF (Pozzo-Miller et al., 1999). Re-expression of BDNF in BDNF−/− hippocampal neurons has also been reported to increase synapse number, thus rescuing the loss of synapses due to a lack of BDNF (Singh et al., 2006).
Enhancement of LTP at Schaffer collateral synapses is dependent upon BDNF. Homozygous and heterozygous BDNF knockout mice display significant deficits in hippocampal long-term potentiation (Korte et al., 1995). BDNF−/− mice show deficits in hippocampal LTP, which is rescued by application of exogenous BDNF (Patterson et al., 1996). Synaptic plasticity is also compromised at mossy fiber synapses in BDNF+/− mice. Mossy fiber LTP is reduced in hippocampal slices from BDNF+/− mice to comparable levels with wild-type hippocampal slices treated with TrkB-Fc to sequester BDNF (Schildt et al., 2013). The effects of BDNF on synaptic plasticity are further illustrated by behavioral paradigms of learning and memory conducted in BDNF knockout mice. Age-dependent deficits in fear learning have been reported in heterozygous BDNF knock-out mice where reduced levels of BDNF in the amygdala correlate strongly with deficits in consolidation of fear memory (Endres and Lessmann, 2012). Although cued fear learning is normal in young BDNF+/− mice, significant deficits become apparent with age manifesting three months after birth, highlighting the importance of BDNF in driving mechanisms that underlie learning and memory in the adult CNS (Endres and Lessmann, 2012).
The effects of BDNF on synaptic plasticity are also evident in the development of the visual system. BDNF regulates the maturation of inhibitory inputs and the visual cortex during critical periods of visual development in rodents (Huang et al., 1999). Moreover, BDNF overexpression can rescue visual deprivation that results from dark rearing (Gianfranceschi et al., 2003); phenomenon that is often associated with prolonging the critical period for ocular dominance plasticity. Recent studies have demonstrated that recovery of visual acuity in adult amblyopic rats by environmental enrichment is also associated with an increase in expression of BDNF (Tognini et al., 2012). Although other cellular proteins also change due to environmental enrichment, changes in BDNF reinforce the earlier studies that report the modulatory role of BDNF in critical period plasticity. Furthermore, BDNF is also effective in preserving visual function in the DBA/2J animal model of glaucoma (Domenici et al., 2014). Thus, activity-dependent release of BDNF plays an important role in promoting development of the visual cortex.
4. Relevance in disease
Given the diverse functions of BDNF in the nervous system, it is inevitable that lack of BDNF impacts brain health. Reduced expression of BDNF has been widely reported in neurodegenerative and neuropsychiatric disorders.
4.1. Alzheimer’s disease
Experimental data from post mortem human Alzheimer’s disease (AD) brains and in several animal models of AD reflect reduced expression of BDNF (Durany et al., 2000; Hock et al., 2000; Phillips et al., 1991). Cell types that are vulnerable to degeneration show reduced expression of BDNF and its receptor TrkB (Ginsberg et al., 2010; Phillips et al., 1991). Reduced BDNF levels have been reported in different brain regions particularly in the cortex, and the Meynert nucleus basalis, which is a major source of cholinergic innervation (Murer et al., 2001). In patients with AD, BDNF levels are significantly low in the dentate gyrus and in neurons that harbor neurofibrillary tangles, a hallmark of AD (Murer et al., 1999; Narisawa-Saito et al., 1996). However, conflicting evidence also exists that BDNF is increased in the hippocampus in AD patients and APP models of AD (Burbach et al., 2004; Murer et al., 1999). In spite of these observations, a lack of BDNF is associated with many pathological and behavioral deficits associated with AD. For instance, in rodent and primate models of AD, BDNF gene therapy ameliorates synapse loss, gene expression and improves learning and memory deficits associated with AD (Nagahara et al., 2009). These rescue experiments solidify the important roles of BDNF in neurodegenerative disorders. The effect of BDNF on reversal of neurodegeneration in AD models is independent of Aβ clearance as levels of Aβ are unchanged upon BDNF therapy (Blurton-Jones et al., 2009; Nagahara et al., 2009). Thus, BDNF could be exerting its effects primarily through promoting synapse formation and repair.
4.2. Parkinson’s disease
Although reports on reduced trophic factor levels in Parkinson’s disease (PD) are limited; a few studies to date indicate that BDNF may also be important in PD pathogenesis. Reduced BDNF mRNA has been reported in the substantia nigra, pars compacta, a region that is selectively vulnerable to degeneration in PD (Murer et al., 2001; Parain et al., 1999). Moreover, there is evidence associating reduced BDNF production with pathogenic mutations in α-synuclein that are associated with familial PD (Kohno et al., 2004; Zuccato and Cattaneo, 2009). A number of BDNF targeted deletion studies resulted in lower BDNF levels in PD mouse models. These studies culminated in symptomatic features that are parallel to human PD, which include loss of dopaminergic neurons in the substantia nigra and reduction in striatal dopamine output (Baquet et al., 2004; Fumagalli et al., 2003; Porritt et al., 2005). Thus, BDNF display neuroprotective effects on dopaminergic neurons that are selectively vulnerable in PD.
4.3. Huntington’s disease
Substantial and compelling evidence has been accumulated for the role of BDNF in preventing neurodegeneration in Huntington’s disease (HD). Striatal medium spiny neurons are selectively vulnerable to degeneration in HD. BDNF produced and anterogradely transported to the striatum is well known to support survival of medium spiny neurons (Altar et al., 1997) and loss of these neurons can be the consequence of mutant huntingtin protein interfering with BDNF transport (Gauthier et al., 2004).
Several reports also implicate the huntingtin protein in affecting activity-dependent release of BDNF in the striatum. Mechanistically, huntingtin has been shown to transcriptionally modulate BDNF expression by acting on exon II on the BDNF promoter (Ferrer et al., 2000; Zuccato et al., 2001). Thus, mutations in the huntingtin protein greatly impact BDNF levels in striatal neurons. This is demonstrated in studies that reported low expression of BDNF in mouse models of HD and in post-mortem human HD brains (Zuccato and Cattaneo, 2007; Zuccato et al., 2001, 2008). Furthermore, mice that are genetically manipulated to have low levels of BDNF in cortical areas show morphological and behavioral deficits that are similar to clinical symptoms of HD, underscoring the positive effects of BDNF on survival of striatal neurons (Baquet et al., 2004; Strand et al., 2007). Gene expression profiling indicated that depletion of BDNF in the cortex most closely resembles early grade human HD (Strand et al., 2007). These results suggest that striatal-specific atrophy in HD may be a consequence of a decrease in cortical BDNF by mutant huntingtin.
4.4. Mood disorders
Pre-clinical and clinical evidence suggest the involvement of BDNF in mood disorders. BDNF signaling is believed to be a downstream target of anti-depressant treatments (Hashimoto et al., 2004). This is supported by experimental evidence in animal models of depression where infusion of BDNF into the midbrain resulted in anti-depressant like effects (Shirayama et al., 2002; Siuciak et al., 1997). Additional evidence demonstrating a reduction in hippocampal BDNF mRNA in response to forced swim test in animal models of depression further emphasizes the importance of BDNF in the therapeutic response to antidepressant treatment (Russo-Neustadt et al., 1999). Furthermore, heterozygous BDNF knockout mice and TrkB mutant mice exhibit considerable resistance to anti-depressant treatment while undergoing the forced swim test (Saarelainen et al., 2003) suggesting that anti-depressants exert their effects through modulating BDNF-TrkB signaling.
4.5. Amyotrophic lateral sclerosis
Evidence exist that points to the beneficial role of BDNF in slowing down the progressive loss of motor neuron function in Amyotrophic lateral sclerosis (ALS). BDNF has been reported to slow progression of motor neuron atrophy in the wobbler mice, an animal model of ALS (Mitsumoto et al., 1994). Additionally, the TrkB agonist 7,8-dihydroxyflavone (7,8-DHF) ameliorated motor neuron deficits in the SOD1 (G93A) ALS mouse model (Korkmaz et al., 2014), although efforts to replicate the positive effects of 7,8-DHF upon TrkB activity have been unsuccessful (Todd et al., 2014). Despite the therapeutic potential of BDNF, previous clinical trials in ALS patients have failed due to difficulties in administered BDNF to reach degenerating neurons (Anon., 1999). Alternative approaches such as monoclonal antibody-based therapies that can promote BDNF signaling and thus motor neuron function can circumvent these challenges.
5. Gene expression changes in BDNF-deprived hippocampal neurons and neurodegeneration
Reduced BDNF in central neurons also results in changes in expression of distinct classes of genes. BDNF sequestration in cultured neurons leads to significant decreases in genes involved in synaptic function, vesicular trafficking, endosomal function and MAP kinase signaling (Mariga et al., 2015b). The changes in these classes of genes are relevant as they have also been reported in Alzheimer’s disease and aging where BDNF expression is low (Berchtold et al., 2013). A comparison of the genes changing in AD and BDNF-deprived hippocampal neurons highlight similarities in genes involved in vesicular trafficking and synaptic function (Table 1). Gene classes related to synaptic vesicle trafficking and transmission are also predominantly down-regulated in CA1 pyramidal neurons of post-mortem AD patients relative to age-matched post-mortem controls (Ginsberg et al., 2012). Synaptic loss is believed to be one of the early events in neurodegeneration (Selkoe, 2002; Shankar and Walsh, 2009). Thus, changes in synaptic genes following BDNF withdrawal correlates with early stages of neurodegeneration. Loss of BDNF contributes to decreases in expression of synaptic proteins, which gradually leads to synapse loss. The loss of synaptic connections may compromise the ability of neurons to adapt to environmental changes resulting in increased susceptibility to degeneration.
Table 1.
Comparison of changes in synaptic gene expression in Alzheimer’s disease and BDNF-deprived hippocampal neurons. Column 3 shows relative decreases in expression of BDNF and synaptic proteins in hippocampal tissue from post-mortem AD brain dataset, (Berchtold et al., 2013). Column 4 shows comparable decreases of similar synaptic proteins from cultured hippocampal neurons deprived of BDNF for 3 h (Mariga et al., 2015a,b).
| Gene symbol | Gene name | Alzheimer’s disease hippocampal tissue | BDNF-deprived hippocampal neurons (3 h) |
|---|---|---|---|
| BDNF | Brain-derived neurotrophic factor | 0.28 | – |
| STX6 | Syntaxin 6 | 0.76 | 0.38 |
| NPTXII | Neuronal Pentraxin 2 | 0.43 | 0.69 |
| VAMP4 | Vesicular-associated membrane protein 4 | 0.62 | 0.50 |
| Rab8b | Ras-related protein Rab-8b | – | 0.48 |
Furthermore, changes in endosomal genes following BDNF deprivation supports a model where endosomal dysfunction is a cell biological feature that occurs during neurodegeneration (Ginsberg et al., 2010). In single-cell gene expression profiling studies, changes in expression of endosomal genes was evident in the CA1 region of the hippocampus suggesting impaired LTP and consequent decrease in synaptic efficacy. Some of the genes that change in both BDNF-deprived neurons and in aging and neurodegeneration are mechanistically regulated by BDNF and important for BDNF mediated synaptic functions. For instance, mRNA and protein expression of the synaptic protein Narp is significantly reduced in both AD postmortem hippocampal tissue and BDNF deprived neurons (Berchtold et al., 2013; Mariga et al., 2015b). Narp is highly regulated by BDNF and mediates the effects of BDNF on mossy fiber synapses (Mariga et al., 2015a). The involvement of Narp in modulating BDNF-dependent synaptic processes relevant to learning and memory (Fig. 1) offers a good example of how BDNF can change the function of synapses through regulating levels of synaptic proteins that orchestrate the plasticity process.
Fig. 1.
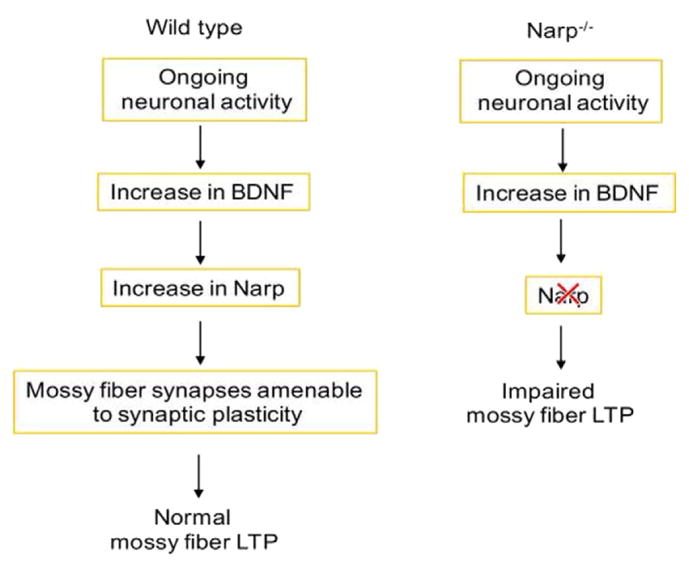
Role of ongoing BDNF-Narp signaling in mossy fiber LTP. BDNF, a trophic factor that is regulated by ongoing neuronal activity exerts its housekeeping functions on the mossy fiber synapses by inducing Narp, which is necessary for mossy fiber LTP. A lack of Narp results in an impairment of activity-induced LTP in the mossy fiber pathway.
6. The therapeutic potential of BDNF
BDNF has been proposed as a treatment strategy for AD, PD, amyotrophic lateral sclerosis (ALS) and peripheral neuropathy. A BDNF clinical trial conducted in a small cohort of ALS patients showed a promising increase in survival, delayed loss of pulmonary function, and slowed the decline in walking speed in ALS patients. However, these effects were not reproducible in follow up studies with larger patient populations (Beck et al., 2005). Similarly the majority of attempted clinical trials on other neurodegenerative diseases have been met with disappointing results due to difficulties in delivery, poor diffusion of neurotrophins and side effects (Thoenen and Sendtner, 2002). There are many obstacles for directly administering neurotrophins to human subjects. The problems in managing the dose and pharmacokinetics of these proteins have hindered the application of neurotrophic factors as a therapeutic intervention for neurodegenerative diseases.
There are indications that these obstacles can be overcome. Higher order functions, such as the circuits involved in pain, anxiety, depression, obesity and other maladaptive behaviors are modulated by changing the levels of trophic factors, such as NGF and BDNF. Decreased levels of BDNF are associated with depression and become enhanced following antidepressant treatment (Hashimoto et al., 2004). Increased neuronal activity or exercise can lower the risk of these conditions through increases in trophic factors (Cotman et al., 2007). New cell-based methods with BDNF are successful in preventing cochlear spiral ganglion degeneration and deafness (Pinyon et al., 2014). BDNF regulates the formation and maintenance of neuronal networks associated with psychiatric disorders (Autry and Monteggia, 2012; Martinowich et al., 2007). BDNF provides trophic support and increases in synaptogenesis and dendritic and axonal branching (Nagahara et al., 2013, 2009; Park and Poo, 2013; Wang et al., 2015). Because neurotrophin signaling is germane for many neurodegenerative and psychiatric disorders where structural plasticity is compromised (Lu et al., 2013), one approach is to increase the levels of neurotrophins or signaling through its Trk receptors.
7. Modulating BDNF signaling in the central nervous system
7.1. Electroconvulsive therapy as a method to increase BDNF?
Electroconvulsive treatment has been used as a therapy for several mood disorders, although the mechanism by which it relieves depressive symptoms is unknown. A key observation that linked neurotrophins to plasticity was increased secretion of neurotrophins by neuronal activity, which reinforces and stabilizes synaptic connections (Thoenen, 1995). In addition, neurotrophins can increase neurotransmitter release from neurons during activity that can result in a dramatic increase in mRNA encoding NGF and BDNF in the dentate gyrus, CA1 and CA3 regions after seizure (Gall and Isackson, 1989; Isackson et al., 1991). These results indicated that activity-dependent regulation of BDNF frequently occurs and suggests that other physiological stimuli, such as depolarization, neurotransmitters, light, hormones and exercise can also influence the expression and levels of trophic factors.
Electroconvulsive shocks (ECS) in rodent models and electroconvulsive therapy in humans has been reported to increase BDNF levels (Kim et al., 2010; Zetterstrom et al., 1998). Studies have shown that ECS in rats increases the expression of hippocampal and amygdala BDNF mRNA, as well as BDNF protein levels (Altar et al., 2003; Nibuya et al., 1995; Pan et al., 1998). Although there are many genes activated by ECS, BDNF and other growth factors are likely candidates for activity-dependent regulation. It is likely that the efforts to use deep brain stimulation in PD, depression and Rett syndrome (Hao et al., 2015; Herrington et al., 2015) may reflect in increases in neurotrophic factors after stimulation, which provides beneficial outcomes in these disorders.
In Alzheimer’s disease, ECS has been applied in treatment of AD-related severe agitation. ECS therapy improved severe agitation without compromising cognitive function in an early-onset AD patient (Aksay et al., 2014). Although this study had one subject, it highlights the potential of ECS as a treatment modality for behavioral symptoms of AD. Increases in serum levels of BDNF following ECS have also been reported in patients with treatment resistant major depression disorder (MDD) (Bocchio-Chiavetto et al., 2006; Marano et al., 2007). Although other studies were not able to replicate this finding, (Fernandes et al., 2009; Gedge et al., 2012), meta-analysis studies suggest a strong correlation of serum levels of BDNF and severity of depression (Brunoni et al., 2008). Furthermore, depression severity levels decrease following ECS (Gedge et al., 2012). Also, the studies that examined the levels of BDNF, conducted the measurements after different time frames of treatment, thus this difference could account for the variability. With further standardization of the timeframe of treatment and additional large patient cohort studies, ECS can offer a non-pharmacological alternative that can increase BDNF and decrease disease severity in mood disorders.
7.2. Transactivation of Trk receptors
Transactivation represents an alternative approach to use small molecules to elicit neurotrophic effects for the treatment of a variety of neurodegenerative and psychiatric diseases (Lee and Chao, 2001; Thoenen and Sendtner, 2002; Wiese et al., 2007). Several ligands that interact with G protein-coupled receptors (GPCR), such as adenosine and PACAP (Lee and Chao, 2001; Lee et al., 2002); dopamine (Iwakura et al., 2008); and glucocorticoids (Jeanneteau et al., 2008) possess the ability to transactivate Trk receptors. Transactivation of Trk receptors resulted in neuroprotective effects through Akt activity. The ability of adenosine to rescue motor neurons from cell death required the activity of TrkB, since motor neurons devoid of TrkB (TrkB−/− mice) were not rescued by adenosine (Wiese et al., 2007). TrkB is even a target of transactivation by zinc (Huang et al., 2008). Based upon the observation that the EGF receptor and other receptor tyrosine kinases are capable of being activated through G protein-coupled receptors (GPCR) signaling (Daub et al., 1996), several GPCR ligands have been shown to transactivate TrkA and TrkB receptors in cultured cells (Lee and Chao, 2001). Treatment of hippocampal neurons or PC12 cells with adenosine resulted in an increase in Trk receptor autophosphorylation. The effect of adenosine upon TrkA receptor activity occurred in a low concentration range. A time course of adenosine action showed that the increase in TrkA activation was slow and required at least 60 min, which is delayed compared to treatment with NGF. The phosphorylation of Trk substrates, Shc and PLC-γ required at least 60 min time course (Lee and Chao, 2001; Lee et al., 2002). This is in contrast to other transactivation events, such as the activation of EGF receptors by GPCR ligands angiotensin, bradykinin or isoproterenol, which occurs rapidly, within minutes of treatment. Also, in contrast to other transactivation events that lead to transient increases in MAP kinase activity, adenosine signaling through Trk neurotrophin receptors leads to selective activation of the PI3-kinase/Akt pathway over a sustained time course of hours and days.
Transactivation also represents an alternative approach to use trophic factor signaling for neurodegenerative diseases, without using neurotrophic factors. Treatment of basal forebrain neurons with PACAP, a neuropeptide, resulted in neuroprotection of cholinergic neurons after axotomy (Takei et al., 2000). PACAP can also transactivate Trk receptors with a time course similar to that of adenosine (Lee et al., 2002). The activity of PACAP is likely due to transactivation of Trk receptors expressed in basal forebrain neurons. Akt activity can also be stimulated by PACAP in a TrkA-dependent manner. These observations are significant since cholinergic neurons in the basal forebrain degenerate in Alzheimer’s disease, and these neurons are normally dependent upon NGF for survival. In addition, there is much evidence to support a causal role of BDNF in Huntington’s disease (Zuccato and Cattaneo, 2009). In fact, intraperitoneal injection of CGS21680 has been shown to ameliorate the symptoms in a Huntington’s disease transgenic mouse model (Chou et al., 2005). Therefore, small molecules that transactivate the TrkB receptor could also be used for the treatment of Huntington’s disease, in lieu of using BDNF.
7.3. Physical exercise
Aerobic exercise has been shown to increase levels of BDNF and other genes that are important for learning and memory (Cotman and Berchtold, 2002; Neeper et al., 1995, 1996) and is associated with improving cognitive function particularly in old age (Blomquist and Danner, 1987; Laurin et al., 2001; Rogers et al., 1990). The increase in BDNF is believed to promote behavioral changes that underlie learning. Rats subjected to voluntary exercise show improvement in performance in the Morris water maze test, which is mediated by increased BDNF expression (Vaynman et al., 2004). In this study, the benefit of BDNF on cognition was inhibited with TrkB-Fc (Vaynman et al., 2004).
Recent reports have also linked exercise to increased BDNF gene expression in animal models of neurodegenerative disorders. In a recent study, a 5-months long treadmill exercise increased levels of BDNF-positive cells and spatial memory in the APPswe/PS1dE9 AD mouse model (Xiong et al., 2015). Another study has also reported increase in BDNF and improved long-term potentiation following treadmill exercise in a rat model of AD (Aβ rats) (Dao et al., 2015). Improvement in cognition in models of AD exposed to physical exercise is consistent with effects of exercise on cognition that have been reported in AD-related exercise trials in human subjects (de Andrade et al., 2013; Hernandez et al., 2010). Increasing evidence suggest physical exercise as a strategy to reduce development of PD (Chen et al., 2005). In PD exercise also increases BDNF and the increase is associated with reducing damage to dopaminergic neurons and promoting motor function (Wu et al., 2011). Thus, physical exercise can provide a practical alternative for improving levels of BDNF, which can have tremendous effects on overall brain function.
7.4. Stem cell transplantation
Another approach for modulating BDNF signaling is through neural stem cell transplantation. Transplantation of hippocampal neural stem cells in AD mice can rescue learning and memory deficits by inducing a BDNF-dependent increase in synaptic density that is independent of levels of tau pathology (Blurton-Jones et al., 2009). Similarly, striatal transplantation of neural stem cells into a mouse model of dementia with Lewy bodies restores BDNF levels, thus improving AD-related motor and cognitive decline. In this study, transplanting BDNF-depleted neural stem cells did not improve behavior, while BDNF delivery via bilateral injection of adeno-associated virus mimicked the benefits of BDNF-expressing stem cells (Goldberg et al., 2015). Stem cell therapy has also been reported in mouse models of HD where mesenchymal stem cells benefits have been investigated (Olson et al., 2012). Mesenchymal stem cells secrete BDNF, which has been linked to promoting in vitro survival of cortical neurons deprived of trophic support or exposed to nitric oxide (Wilkins et al., 2009). Additionally, intrastriatal transplantation of mesenchymal stem cells genetically modified to over-express BDNF in the striatum slowed neuronal degeneration and improved behavior in YAC 128 mouse model of HD (Dey et al., 2010). Indications of improved motor function and neuronal survival in HD through BDNF-related stem cell strategies have been demonstrated in R6/2 huntingtin mutant mice. Viral delivery of BDNF and noggin in R6/ 2 mice triggered active recruitment of subependymal progenitor cells and formation of medium spiny neurons that achieved proper maturation and circuit integration (Benraiss et al., 2013). These findings suggest a potential benefit of stem cell-based therapies in modulating neural processes such as synaptic plasticity that are highly dependent on BDNF.
8. Concluding remarks
BDNF signaling is fundamental for proper functioning of neurons and lack of BDNF support has profound negative molecular, behavioral and plasticity effects in neurons. Loss of BDNF reduces expression of synaptic proteins and thus synapse integrity. These changes have manifested in various brain disorders where BDNF levels have been reported to be low. Although BDNF protein or mimetics delivery have had limited success as drug therapies, there is promise in new strategies such as physical exercise and deep brain stimulation (ECS) that can improve serum levels of BDNF (summarized in Fig. 2). Because levels of BDNF are low in neurodegenerative and neuropsychiatric disorders, BDNF can be a biomarker for the progressive decline in neuronal function. One of the hallmarks of neurodegenerative disease is reduced expression of synaptic proteins and loss of synaptic contacts, which is consistent with effects of reduced BDNF support in neurons. This idea presents additional ways to ameliorate synaptic efficacy through increasing levels of BDNF and its downstream signaling. Another approach for activating BDNF signaling is through transactivation of Trk receptors with pharmacological agents, which can restore synaptic integrity by modulating expression and function of synaptic proteins. Thus modulating BDNF signaling presents a series of powerful mechanistic approaches for reversing synaptic loss associated with neurodegenerative and neuropsychiatric disorders.
Fig. 2.

Summary of strategies to modulate BDNF signaling. Improving BDNF signaling enhances processes that repair synapses and overall brain function.
References
- Aksay SS, et al. Severe agitation in severe early-onset Alzheimer’s disease resolves with ECT. Neuropsychiatr Dis Treat. 2014;10:2147–2151. doi: 10.2147/NDT.S71008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar CA, et al. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Altar CA, et al. Effects of electroconvulsive seizures and antidepressant drugs on brain-derived neurotrophic factor protein in rat brain. Biol Psychiatry. 2003;54:703–709. doi: 10.1016/s0006-3223(03)00073-8. [DOI] [PubMed] [Google Scholar]
- Anonymous. A controlled trial of recombinant methionyl human BDNF in ALS: the BDNF Study Group (Phase III) Neurology. 1999;52:1427–1433. doi: 10.1212/wnl.52.7.1427. [DOI] [PubMed] [Google Scholar]
- Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev. 2012;64:238–258. doi: 10.1124/pr.111.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquet ZC, et al. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquet ZC, et al. Brain-derived neurotrophic factor is required for the establishment of the proper number of dopaminergic neurons in the substantia nigra pars compacta. J Neurosci. 2005;25:6251–6259. doi: 10.1523/JNEUROSCI.4601-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, et al. Autonomic dysfunction in ALS: a preliminary study on the effects of intrathecal BDNF. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:100–103. doi: 10.1080/14660820510028412. [DOI] [PubMed] [Google Scholar]
- Benraiss A, et al. Sustained mobilization of endogenous neural progenitors delays disease progression in a transgenic model of Huntington’s disease. Cell Stem Cell. 2013;12:787–799. doi: 10.1016/j.stem.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold NC, et al. Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer’s disease. Neurobiol Aging. 2013;34:1653–1661. doi: 10.1016/j.neurobiolaging.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomquist KB, Danner F. Effects of physical conditioning on information-processing efficiency. Percept Mot Skills. 1987;65:175–186. doi: 10.2466/pms.1987.65.1.175. [DOI] [PubMed] [Google Scholar]
- Blurton-Jones M, et al. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 2009;106:13594–13599. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchio-Chiavetto L, et al. Electroconvulsive therapy (ECT) increases serum brain derived neurotrophic factor (BDNF) in drug resistant depressed patients. Eur Neuropsychopharmacol. 2006;16:620–624. doi: 10.1016/j.euroneuro.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Brunoni AR, et al. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11:1169–1180. doi: 10.1017/S1461145708009309. [DOI] [PubMed] [Google Scholar]
- Burbach GJ, et al. Induction of brain-derived neurotrophic factor in plaque-associated glial cells of aged APP23 transgenic mice. J Neurosci. 2004;24:2421–2430. doi: 10.1523/JNEUROSCI.5599-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellerino A, et al. Reduced size of retinal ganglion cell axons and hypomyelination in mice lacking brain-derived neurotrophic factor. Mol Cell Neurosci. 1997;9:397–408. doi: 10.1006/mcne.1997.0641. [DOI] [PubMed] [Google Scholar]
- Chen H, et al. Physical activity and the risk of Parkinson disease. Neurology. 2005;64:664–669. doi: 10.1212/01.WNL.0000151960.28687.93. [DOI] [PubMed] [Google Scholar]
- Chou SY, et al. CGS21680 attenuates symptoms of Huntington’s disease in a transgenic mouse model. J Neurochem. 2005;93:310–320. doi: 10.1111/j.1471-4159.2005.03029.x. [DOI] [PubMed] [Google Scholar]
- Cohen-Corey S, Fraser SE. Effects of brain-derived neurotrophic factor on optic axon branching and remodelling in vivo. Nature. 1995;378:192–196. doi: 10.1038/378192a0. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25:295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- Cotman CW, et al. Exercise builds brain health: key roles of growth factor cascades and inflammation. Trends Neurosci. 2007;30:464–472. doi: 10.1016/j.tins.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Dao AT, et al. Comparison of the effect of exercise on late-phase LTP of the dentate gyrus and CA1 of Alzheimer’s disease model. Mol Neurobiol. 2015 doi: 10.1007/s12035-015-9612-5. [DOI] [PubMed] [Google Scholar]
- Daub H, et al. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- de Andrade LP, et al. Benefits of multimodal exercise intervention for postural control and frontal cognitive functions in individuals with Alzheimer’s disease: a controlled trial. J Am Geriatr Soc. 2013;61:1919–1926. doi: 10.1111/jgs.12531. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Johnson EM. Programmed cell death in neurons: focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Mol Pharmacol. 1997;51:897–906. doi: 10.1124/mol.51.6.897. [DOI] [PubMed] [Google Scholar]
- Dey ND, et al. Genetically engineered mesenchymal stem cells reduce behavioral deficits in the YAC 128 mouse model of Huntington’s disease. Behav Brain Res. 2010;214:193–200. doi: 10.1016/j.bbr.2010.05.023. [DOI] [PubMed] [Google Scholar]
- Domenici L, et al. Rescue of retinal function by BDNF in a mouse model of glaucoma. PLoS One. 2014;9:e115579. doi: 10.1371/journal.pone.0115579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durany N, et al. Brain-derived neurotrophic factor and neurotrophin-3 levels in Alzheimer’s disease brains. Int J Dev Neurosci. 2000;18:807–813. [PubMed] [Google Scholar]
- Endres T, Lessmann V. Age-dependent deficits in fear learning in heterozygous BDNF knock-out mice. Learn Mem. 2012;19:561–570. doi: 10.1101/lm.028068.112. [DOI] [PubMed] [Google Scholar]
- Fernandes B, et al. Serum brain-derived neurotrophic factor (BDNF) is not associated with response to electroconvulsive therapy (ECT): a pilot study in drug resistant depressed patients. Neurosci Lett. 2009;453:195–198. doi: 10.1016/j.neulet.2009.02.032. [DOI] [PubMed] [Google Scholar]
- Ferrer I, et al. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000;866:257–261. doi: 10.1016/s0006-8993(00)02237-x. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, et al. BDNF gene expression is reduced in the frontal cortex of dopamine transporter knockout mice. Mol Psychiatry. 2003;8:898–899. doi: 10.1038/sj.mp.4001370. [DOI] [PubMed] [Google Scholar]
- Gall CM, Isackson PJ. Limbic seizures increase neuronal production of messenger RNA for nerve growth factor. Science. 1989;245:758–761. doi: 10.1126/science.2549634. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Gedge L, et al. Effects of electroconvulsive therapy and repetitive transcranial magnetic stimulation on serum brain-derived neurotrophic factor levels in patients with depression. Front Psychiatry. 2012;3:12. doi: 10.3389/fpsyt.2012.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianfranceschi L, et al. Visual cortex is rescued from the effects of dark rearing by overexpression of BDNF. Proc Natl Acad Sci U S A. 2003;100:12486–12491. doi: 10.1073/pnas.1934836100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol Psychiatry. 2010;68:885–893. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, et al. Gene expression levels assessed by CA1 pyramidal neuron and regional hippocampal dissections in Alzheimer’s disease. Neurobiol Dis. 2012;45:99–107. doi: 10.1016/j.nbd.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg NR, et al. Neural stem cells rescue cognitive and motor dysfunction in a transgenic model of dementia with Lewy bodies through a BDNF-dependent mechanism. Stem Cell Rep. 2015;5:791–804. doi: 10.1016/j.stemcr.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, et al. Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci. 2003;23:6856–6865. doi: 10.1523/JNEUROSCI.23-17-06856.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, et al. Forniceal deep brain stimulation rescues hippocampal memory in Rett syndrome mice. Nature. 2015;526:430–434. doi: 10.1038/nature15694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, et al. Critical role of brain-derived neurotrophic factor in mood disorders. Brain Res Brain Res Rev. 2004;45:104–114. doi: 10.1016/j.brainresrev.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Hernandez SS, et al. Effects of physical activity on cognitive functions, balance and risk of falls in elderly patients with Alzheimer’s dementia. Rev Bras Fis. 2010;14:68–74. [PubMed] [Google Scholar]
- Herrington TM, et al. Mechanisms of deep brain stimulation. J Neurophysiol. 2015;00281:2015. doi: 10.1152/jn.00281.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock C, et al. Region-specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol. 2000;57:846–851. doi: 10.1001/archneur.57.6.846. [DOI] [PubMed] [Google Scholar]
- Huang ZJ, et al. BDNF regulates the maturation of inhibition and the critical period of plasticity in mouse visual cortex. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- Huang YZ, et al. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron. 2008;57:546–558. doi: 10.1016/j.neuron.2007.11.026. [DOI] [PubMed] [Google Scholar]
- Isackson PJ, et al. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- Iwakura Y, et al. Dopamine D1 receptor-induced signaling through TrkB receptors in striatal neurons. J Biol Chem. 2008;283:15799–15806. doi: 10.1074/jbc.M801553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanneteau F, et al. Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect. Proc Natl Acad Sci U S A. 2008;105:4862–4867. doi: 10.1073/pnas.0709102105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanneteau F, et al. The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nat Neurosci. 2010;13:1373–1379. doi: 10.1038/nn.2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JE, et al. Brain derived neurotrophic factor supports the survival of cultured rat retinal ganglion cells. J Neurosci. 1986;6:3031–3038. doi: 10.1523/JNEUROSCI.06-10-03031.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, et al. Effects of repeated minimal electroshock seizures on NGF, BDNF and FGF-2 protein in the rat brain during postnatal development. Int J Dev Neurosci. 2010;28:227–232. doi: 10.1016/j.ijdevneu.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohara K, et al. A local reduction in cortical GABAergic synapses after a loss of endogenous brain-derived neurotrophic factor, as revealed by single-cell gene knock-out method. J Neurosci. 2007;27:7234–7244. doi: 10.1523/JNEUROSCI.1943-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno R, et al. BDNF is induced by wild-type alpha-synuclein but not by the two mutants, A30P or A53T, in glioma cell line. Biochem Biophys Res Commun. 2004;318:113–118. doi: 10.1016/j.bbrc.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Korkmaz OT, et al. 7,8-Dihydroxyflavone improves motor performance and enhances lower motor neuronal survival in a mouse model of amyotrophic lateral sclerosis. Neurosci Lett. 2014;566:286–291. doi: 10.1016/j.neulet.2014.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M, et al. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurin D, et al. Physical activity and risk of cognitive impairment and dementia in elderly persons. Arch Neurol. 2001;58:498–504. doi: 10.1001/archneur.58.3.498. [DOI] [PubMed] [Google Scholar]
- Lee FS, Chao MV. Activation of Trk neurotrophin receptors in the absence of neurotrophins. Proc Natl Acad Sci U S A. 2001;98:3555–3560. doi: 10.1073/pnas.061020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FS, et al. Activation of Trk neurotrophin receptor signaling by pituitary adenylate cyclase-activating polypeptides. J Biol Chem. 2002;277:9096–9102. doi: 10.1074/jbc.M107421200. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R. Growth Control of Nerve Cells by a Protein Factor and its Antiserum Science. 1964:143. doi: 10.1126/science.143.3602.105. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R, Angeletti PU. Essential role of the nerve growth factor in the survival and maintenance of dissociated sensory and sympathetic embryonic nerve cells in vitro. Dev Biol. 1963;6:653–659. doi: 10.1016/0012-1606(63)90149-0. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R, Booker B. Destruction of the sympathetic ganglia in mammals by an antiserum to a nerve-growth protein. Proc Natl Acad Sci U S A. 1960;46:384–391. doi: 10.1073/pnas.46.3.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, et al. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat Rev Neurosci. 2013;14:401–416. doi: 10.1038/nrn3505. [DOI] [PubMed] [Google Scholar]
- Maisonpierre PC, et al. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Marano CM, et al. Increased plasma concentration of brain-derived neurotrophic factor with electroconvulsive therapy: a pilot study in patients with major depression. J Clin Psychiatry. 2007;68:512–517. doi: 10.4088/jcp.v68n0404. [DOI] [PubMed] [Google Scholar]
- Mariga A, et al. Definition of a bidirectional activity-dependent pathway involving BDNF and narp. Cell Rep. 2015a;13:1–10. doi: 10.1016/j.celrep.2015.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariga A, et al. Withdrawal of BDNF from hippocampal cultures leads to changes in genes involved in synaptic function. Dev Neurobiol. 2015b;75:173–192. doi: 10.1002/dneu.22216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, et al. New insights into BDNF function in depression and anxiety. Nat Neurosci. 2007;10:1089–1093. doi: 10.1038/nn1971. [DOI] [PubMed] [Google Scholar]
- Mitsumoto H, et al. Arrest of motor neuron disease in wobbler mice cotreated with CNTF and BDNF. Science. 1994;265:1107–1110. doi: 10.1126/science.8066451. [DOI] [PubMed] [Google Scholar]
- Murer MG, et al. An immunohistochemical study of the distribution of brain-derived neurotrophic factor in the adult human brain, with particular reference to Alzheimer’s disease. Neuroscience. 1999;88:1015–1032. doi: 10.1016/s0306-4522(98)00219-x. [DOI] [PubMed] [Google Scholar]
- Murer MG, et al. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog Neurobiol. 2001;63:71–124. doi: 10.1016/s0301-0082(00)00014-9. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Mateling M, Kovacs I, Wang L, Eggert S, Rockenstein E, Koo EH, Masliah E, Tuszynski MH. Early BDNF treatment ameliorates cell loss in the entorhinal cortex of APP transgenic mice. J Neurosci. 2013;33:15596–15602. doi: 10.1523/JNEUROSCI.5195-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narisawa-Saito M, et al. Regional specificity of alterations in NGF, BDNF and NT-3 levels in Alzheimer’s disease. Neuroreport. 1996;7:2925–2928. doi: 10.1097/00001756-199611250-00024. [DOI] [PubMed] [Google Scholar]
- Neeper SA, et al. Exercise and brain neurotrophins. Nature. 1995;373:109. doi: 10.1038/373109a0. [DOI] [PubMed] [Google Scholar]
- Neeper SA, et al. Physical activity increases mRNA for brain-derived neurotrophic factor and nerve growth factor in rat brain. Brain Res. 1996;726:49–56. [PubMed] [Google Scholar]
- Nibuya M, et al. Regulation of BDNF and trkB mRNA in rat brain by chronic electro-convulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson SD, et al. Genetically engineered mesenchymal stem cells as a proposed therapeutic for Huntington’s disease. Mol Neurobiol. 2012;45:87–98. doi: 10.1007/s12035-011-8219-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim R, et al. Naturally occurring and induced neuronal death in the chick embryo in vivo requires protein and RNA synthesis: evidence for the role of cell death genes. Dev Biol. 1990;138:104–113. doi: 10.1016/0012-1606(90)90180-q. [DOI] [PubMed] [Google Scholar]
- Pan W, et al. Transport of brain-derived neurotrophic factor across the blood-brain barrier. Neuropharmacology. 1998;37:1553–1561. doi: 10.1016/s0028-3908(98)00141-5. [DOI] [PubMed] [Google Scholar]
- Parain K, et al. Reduced expression of BDNF protein in Parkinson’s disease substantia nigra. Neuroreport. 1999;10:557–561. doi: 10.1097/00001756-199902250-00021. [DOI] [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrn3379. [DOI] [PubMed] [Google Scholar]
- Patterson SL, et al. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Phillips HS, et al. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron. 1991;7:695–702. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- Pinyon JL, et al. Close-field electroporation gene delivery using the cochlear implant electrode array enhances the bionic ear. Sci Transl Med. 2014;6:233ra54. doi: 10.1126/scitranslmed.3008177. [DOI] [PubMed] [Google Scholar]
- Porritt MJ, et al. Inhibiting BDNF expression by antisense oligonucleotide infusion causes loss of nigral dopaminergic neurons. Exp Neurol. 2005;192:226–234. doi: 10.1016/j.expneurol.2004.11.030. [DOI] [PubMed] [Google Scholar]
- Pozzo-Miller LD, et al. Impairments in high-frequency transmission, synaptic vesicle docking, and synaptic protein distribution in the hippocampus of BDNF knockout mice. J Neurosci. 1999;19:4972–4983. doi: 10.1523/JNEUROSCI.19-12-04972.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauskolb S, et al. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci. 2010;30:1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL, et al. After reaching retirement age physical activity sustains cerebral perfusion and cognition. J Am Geriatr Soc. 1990;38:123–128. doi: 10.1111/j.1532-5415.1990.tb03472.x. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt A, et al. Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology. 1999;21:679–682. doi: 10.1016/S0893-133X(99)00059-7. [DOI] [PubMed] [Google Scholar]
- Saarelainen T, et al. Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant-induced behavioral effects. J Neurosci. 2003;23:349–357. doi: 10.1523/JNEUROSCI.23-01-00349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schildt S, et al. Acute and chronic interference with BDNF/TrkB-signaling impair LTP selectively at mossy fiber synapses in the CA3 region of mouse hippocampus. Neuropharmacology. 2013;71:247–254. doi: 10.1016/j.neuropharm.2013.03.041. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Walsh DM. Alzheimer’s disease: synaptic dysfunction and Abeta. Mol Neurodegener. 2009;4:48. doi: 10.1186/1750-1326-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama Y, et al. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J Neurosci. 2002;22:3251–3261. doi: 10.1523/JNEUROSCI.22-08-03251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, et al. Altered balance of glutamatergic/GABAergic synaptic input and associated changes in dendrite morphology after BDNF expression in BDNF-deficient hippocampal neurons. J Neurosci. 2006;26:7189–7200. doi: 10.1523/JNEUROSCI.5474-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuciak JA, et al. Antidepressant-like effect of brain-derived neurotrophic factor (BDNF) Pharmacol Biochem Behav. 1997;56:131–137. doi: 10.1016/S0091-3057(96)00169-4. [DOI] [PubMed] [Google Scholar]
- Strand AD, et al. Expression profiling of Huntington’s disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J Neurosci. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takei N, et al. Pituitary adenylate cyclase-activating polypeptide promotes the survival of basal forebrain cholinergic neurons in vitro and in vivo: comparison with effects of nerve growth factor. Eur J Neurosci. 2000;12:2273–2280. doi: 10.1046/j.1460-9568.2000.00118.x. [DOI] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- Thoenen H, Sendtner M. Neurotrophins: from enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat Neurosci. 2002;(5 Suppl):1046–1050. doi: 10.1038/nn938. [DOI] [PubMed] [Google Scholar]
- Todd D, et al. A monoclonal antibody TrkB receptor agonist as a potential therapeutic for Huntington’s disease. PLoS One. 2014;9:e87923. doi: 10.1371/journal.pone.0087923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognini P, et al. Environmental enrichment promotes plasticity and visual acuity recovery in adult monocular amblyopic rats. PLoS One. 2012;7:e34815. doi: 10.1371/journal.pone.0034815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaynman S, et al. Hippocampal BDNF mediates the efficacy of exercise on synaptic plasticity and cognition. Eur J Neurosci. 2004;20:2580–2590. doi: 10.1111/j.1460-9568.2004.03720.x. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. Autocrine action of BDNF on dendrite development of adult-born hippocampal neurons. J Neurosci. 2015;35:8384–8393. doi: 10.1523/JNEUROSCI.4682-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese S, et al. Adenosine receptor A2A-R contributes to motoneuron survival by transactivating the tyrosine kinase receptor TrkB. Proc Natl Acad Sci U S A. 2007;104:17210–17215. doi: 10.1073/pnas.0705267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins A, et al. Human bone marrow-derived mesenchymal stem cells secrete brain-derived neurotrophic factor which promotes neuronal survival in vitro. Stem Cell Res. 2009;3:63–70. doi: 10.1016/j.scr.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Wu SY, et al. Running exercise protects the substantia nigra dopaminergic neurons against inflammation-induced degeneration via the activation of BDNF signaling pathway. Brain Behav Immun. 2011;25:135–146. doi: 10.1016/j.bbi.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Xiong JY, et al. Long-term treadmill exercise improves spatial memory of male APPswe/PS1dE9 mice by regulation of BDNF expression and microglia activation. Biol Sport. 2015;32:295–300. doi: 10.5604/20831862.1163692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterstrom TS, et al. Repeated electroconvulsive shock extends the duration of enhanced gene expression for BDNF in rat brain compared with a single administration. Brain Res Mol Brain Res. 1998;57:106–110. doi: 10.1016/s0169-328x(98)00077-1. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington’s disease. Prog Neurobiol. 2007;81:294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol. 2009;5:311–322. doi: 10.1038/nrneurol.2009.54. [DOI] [PubMed] [Google Scholar]
- Zuccato C, et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, et al. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington’s disease. Brain Pathol. 2008;18:225–238. doi: 10.1111/j.1750-3639.2007.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]