

Abstract
Two named reactions of fundamental importance and paramount utility in organic synthesis have been reinvestigated, the Barton decarboxylation and Giese radical conjugate addition. N-hydroxyphthalimide (NHPI) based redox-active esters were found to be convenient starting materials for simple, thermal, Ni-catalyzed radical formation and subsequent trapping with either a hydrogen atom source (PhSiH3) or an electron-deficient olefin. These reactions feature operational simplicity, inexpensive reagents, and enhanced scope as evidenced by examples in the realm of peptide chemistry.
Keywords: nickel catalysis, redox-active, esters, decarboxylation, conjugate addition
Graphical abstract
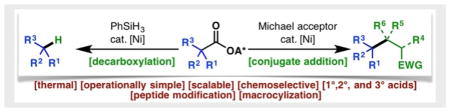
Simple, thermal and practical Barton-type decarboxylation and decarboxylative Giese addition have been developed based on nickel catalyzed fragmentation of N-hydroxyphthalimide redox active esters. The operational simplicity and chemoselectivity of these reactions allow for expedient preparations of various building blocks and late stage modifications of complex molecules.
Sir Derek Barton’s interactions with Schering-Plough in the 1970s radically expanded the paradigm of organic chemistry.1 The acclaimed Barton-McCombie deoxygenation was born out of this experience, enlisting radical reactivity to selectively remove secondary hydroxyl groups in aminoglycosides for improved antibacterial activity.2 Subsequent efforts to utilize carboxylate groups in similar radical reactions had an equally profound impact, culminating in the invention of Barton esters (1, Figure 1A) in 1983.3 These thiohydroxamic anhydrides can be conveniently obtained when ubiquitous carboxylic acids are coupled with pyrithione (readily accessible from industrial feedstock). They undergo visible light-induced fragmentation to liberate carbon dioxide while furnishing alkyl radicals that can be converted to a myriad of functional groups.4 Reductive decarboxylation with tin hydride or an alkyl thiol (1 to 2) and Giese reaction with Michael acceptors (1 to 3) are two notable examples. The thiocarbonyl group, in Barton’s own words, acts as a “disciplinary group”, conferring the process chemoselectivity and functional group compatibility, leading to numerous applications over four decades. Although photoinduced electron transfer processes with carboxylic acid derivatives have been surveyed as an alternative to Barton esters beginning with Okada’s work in 1988,5 these methods require specialized equipment, expensive catalysts, and prolonged reaction times. Notwithstanding the need for a photochemical setup (for both decarboxylation and conjugate addition) and the use of toxic and odorous reductants (for decarboxylation), chemists remain beholden to the venerable Barton reaction as evidenced by its use in many recent total syntheses.6,7 The low stability of Barton esters stemming from their photo- and thermal-sensitivity adds another layer of difficulty. Although alternative reductants such as tris(trimethylsilyl)silane8 or chloroform have been introduced for the decarboxylation reaction,9,10 the other problems have remained largely unmitigated. Thus, thermally-driven and operationally-simple means of Barton decarboxylation and decarboxylative Giese reactions remain highly desirable.
Figure 1.

(A) Use of redox active esters in reductive decarboxylation and Giese reactions. (B) Optimization of Ni-catalyzed decarboxylation and conjugate additions. NHPI=N-hydroxyphthalimide, TCNHPI=tetrachloro-N-hydroxyphthalimide.
Efforts in our laboratory beginning in 2015 led to the realization that Barton esters (1) are redox-active species which readily accept electrons from metal complexes through a purely thermal single electron transfer (SET) process.11–14 This property is shared by various activated carboxylates commonly employed in amide coupling. Collectively termed redox-active esters (RAEs), these compounds (exemplified by NHPI ester 4) undergo facile SET reduction with inexpensive nickel11–13 or iron catalysts.14 The ensuing radical anion fragments to afford alkyl radicals under thermal conditions. Combination of the alkyl radical with a metal center by virtue of the persistent radical effect (PRE) enables cross-coupling of various modalities. Herein, reductive decarboxylation (4 to 2) and Giese reaction (4 to 3) of NHPI RAEs are disclosed, exhibiting broad substrate scope, chemoselectivity, and scalability while obviating the need for toxic reductants and photochemical irradiation. The bench-stability of 4 offers another advantage although as shown below its discreet isolation is not necessary.
Though conceptually straightforward, both transformations were only realized after considerable experimentation. Figure 1B provides the optimal reaction parameters along with a simplified picture of the optimization process on piperidine derivative 5. Zinc (0.5 equiv.) (entry 1, Figure 1B1) was necessary to reduce the Ni(II) species in the decarboxylation reaction while PhSiH3 was found to be the most effective source of hydrogen (entry 3, 10, and 14). The ternary mixture comprising THF, DMF and iPrOH delivers the highest yield whereby iPrOH is believed to provide activation of the silane.15 A 1:2 mixture of NiCl2•6H2O and L1 provided the best catalyst system (entry 9). Addition of tBuSH or LiCl as additives had deleterious effects on the yield.
Meanwhile, LiCl was identified as an essential additive in the conjugate addition (entry 3, Figure 1B2). In the absence of a terminal silane reductant, zinc was required in stoichiometric quantities. Ni(ClO4)2•6H2O ($120/mol) performed the best for this reaction; addition of Lewis acids, bipyridine ligands or use of other metal salts all led to diminished yields. In both transformations, the choice of RAE was critical; while NHPI esters reacted efficiently, use of TCNHPI esters led to lower yields. Zinc (ca. $5/mol) could be used straight out of the bottle, as the use of activated zinc had minimal impact in either reaction. Overall, decarboxylation was complete within 1 h at 40 °C, while the conjugate addition proceeded smoothly at ambient temperature.
RAEs derived from primary (10, 13, 15, and 17), secondary (6, 12, and 14), and tertiary (8, 9, 11, 16, 18, and 19a) carboxylic acids are all competent substrates in the decarboxylation reaction. Though 10 and 12 are also accessible from the corresponding Barton esters, the classical conditions enlist stoichiometric amounts of pungent tBuSH.16 Conversely, 8 can be made in comparable yield using Tsanaktsidis and Williams’s modified protocol with chloroform as the hydrogen donor; the need for photo-irradiation, however, still presents an inconvenience (especially on large scale).9 Densely functionalized natural products were decarboxylated in high yields;17 indole (13), hydroxyl (15, 18a, and 19a), ketones (17 and 19a), and olefins (18a and 19a) were left unscathed. The alternative route to 15 involves dissolving metal reduction of the corresponding nitrile.18 Although 19a can be furnished in good yields through PET mediated decarboxylation, long reaction times (24 h), an expensive solvent (TFE), and Fukuzumi’s photosensitizer (more expensive than the starting carboxylic acid) are required.5d Barton decarboxylation has been applied numerous times to access pyrroloindoline alkaloids from tryptophan derivatives.19 Substrate 14, though not natural product-derived per se, showcases the viability of this methodology to synthesize these alkaloids in a highly practical fashion.
Similarly, the conjugate addition exhibited broad scope with respect to both reaction partners. Michael acceptors bearing assorted electron withdrawing groups (EWGs) such as esters, ketones, sulfones, and nitriles were all shown to react in good yields. These functional groups open up many opportunities for product diversification. It is also of note that the addition is unencumbered by α or β substitutions on the Michael acceptor (26–27, 28–29). Whereas similar conjugate additions with Barton esters oftentimes require large excesses of Michael acceptors, two equivalents of these activated olefins was deemed sufficient in the nickel catalyzed reaction.20
As with the decarboxylation, RAEs of primary, secondary and tertiary acids, bearing multiple functional groups, can be used in this Giese-type reaction (32–44).21 Good levels of stereoceontrol were observed on several substrates (36, 40–42). Many substrates represent derivatives of natural products which are difficult to access through alternative routes. For example, a previous synthesis of the acid corresponding to 42 requires a laborious 6-step sequence from the same starting material.22 Moreover, attempts to make 35 and structural analogs of 40 through the decarboxylative conjugate addition of Barton esters were reportedly complicated by the competing coupling of the 2-thiopyridone byproduct with the intermediate β radical, requiring a separate step of thiopyridyl removal.20 To this end, the use of an NHPI redox-active ester circumvents this lingering problem in classical Barton chemistry.
Across the many substrates examined, both reactions were found to be impervious to moisture and required minimal precautions. This robustness, in combination with the inexpensive reagents employed bode well for large scale reactions. Indeed, gram-scale decarboxylation and Giese addition using 5 delivered 6 and 7 in high yield.
While RAEs employed in these transformations are readily diversifiable building blocks which can be conveniently purified and stored without special precautions, further experimentations revealed that isolation of the RAEs was not necessary–decarboxylation and conjugate addition can be performed on in situ generated RAEs. For example, a simple activation of acid 45, followed by the decarboxylation or conjugate addition protocols furnished 6 and 7 in comparable yields. Owing to the ubiquity and availability of carboxylic acids, such one-pot procedures may have a tangible impact on medicinal chemistry to expedite the radical diversification of acids. The presence of radical intermediates was confirmed via rupture of the cyclopropyl ring in 46 to afford cyclopropane-opened product 47 as the exclusive conjugate addition product of the reaction.
Much fame has been accorded to the Barton decarboxylative and deoxygenative functionalizations because of their unparalleled chemoselectivity. Over the years, these reactions have been shown to tolerate a host of Lewis basic functionalities.6 The operationally simple variants disclosed herein have the potential to enable chemoselective modifications on substrates of even greater complexity. Application of the decarboxylative conjugate addition to complex peptide modification, both on solid phase and in solution, epitomizes this notion. Accordingly, the coupling of resin-bound acids and Michael acceptors was demonstrated (48 to 49 and 50, Figure 2A); such processes facilitate the late-stage introduction of designer amino acids23 while the sulfone and nitrile groups thus appended can serve as convenient handles for further functionalizations. Alternatively, alkyl groups can be introduced onto resin-bound peptides containing a Michael acceptor (51 to 52). It is noteworthy that products are isolated in synthetically useful yields after as many as 8–14 steps from the resin loading, further testifying to the robustness of the reaction. Moreover, an intramolecular variant of the Giese addition could also be performed on an unprotected peptide substrate,24 affording the coveted macrocycle (53 to 54). Macrocyclic peptides have demonstrated tremendous therapeutic potential owing to their resistance towards both exo- and endoproteases and their potential for exquisite target selectivity.25 However, such macrocycles remain challenging synthetic targets for which there are limited available disconnection strategies. This radical conjugate addition approach opens new opportunities to explore the biological activities of cyclic peptides and may serve as a gateway for peptide stapling.26
Figure 2.

Use of Ni-catalyzed decarboxylative conjugate addition in complex peptide modification (See SI for experimental details.).
Nickel catalyzed decarboxylative fragmentation of redox-active NHPI esters has enabled thermal reductive decarboxylation and Giese reactions. These operationally simple protocols circumvent several lingering shortcomings of classic Barton chemistry, avoiding the use of toxic reductants, photochemical irradiation, unstable starting materials, and in the case of conjugate addition, the formation of thiopyridyl side products. It is worth emphasizing that the total reaction cost, including solvent, for a typical 1 mmol-scale decarboxylation runs about $1.4 without any further optimization or reduction in catalyst loading. The simple reagents and broad spectrum of substrates demonstrated should make these methods amenable to both medicinal chemistry and process development. Finally, applications to peptide syntheses illustrate their potential to effect chemoselective late-stage modifications in a particularly challenging context.
Supplementary Material
Scheme 1.

Scope of the Ni-catalyzed decarboxylation and conjugate additions of NHPI redox-active esters. Reaction conditions for decarboxylation: RAE (1.0 equiv), NiCl2•6H2O (10 mol %), L1 (20 mol%), PhSiH3 (1.5 equiv), Zn (0.5 equiv), THF/DMF/iPrOH (10:2:1), 0.15 M, 40 °C, 1 h. Reaction conditions for Giese conjugate addition: RAE (1.0 equiv), Ni(ClO4)2•6H2O (20 mol%), Michael acceptor (2.0 equiv), LiCl (3.0 equiv), Zn (2.0 equiv), MeCN, 12 h, RT. a = using 3.0 equiv PhSiH3, 60 °C. b = using 4.0 equiv Michael acceptor. c = reaction run at 50 °C.
Scheme 2.

A) One-pot decarboxylation and conjugate addition from carboxylic acids. B) Radical ring-opening of a cyclopropyl methylene redox active ester.
Acknowledgments
Financial support for this work was provided by NIH (grant number GM-118176 and F32GM117816 postdoctoral fellowship to L. R. M), Bristol-Myers Squibb, Department of Defense (NDSEG fellowship to J. T. E.), and Bayer Science and Education Foundation (Otto Bayer Scholarship to A. N.). We also thank Michael Schmidt for helpful discussions. We are grateful to D.-H. Huang and L. Pasternack (The Scripps Research Institute) for assistance with nuclear magnetic resonance spectroscopy. We also thank Dr. Craig M. Williams at Queensland University and Dr. John Tsanaktsidis at CSIRO Materials Science and Engineering for insightful discussions and a generous sample of cubane dimethyl ester. We are appreciative of Drs. Chao Li and Josep Cornella for initial investigations and Dr. Javier Lopez Ogalla for technical assistance.
Contributor Information
Dr. Tian Qin, The Scripps Research Institute (TSRI), North Torrey Pines Road, La Jolla, California, 92037, United States
Dr. Lara R. Malins, The Scripps Research Institute (TSRI), North Torrey Pines Road, La Jolla, California, 92037, United States
Jacob T. Edwards, The Scripps Research Institute (TSRI), North Torrey Pines Road, La Jolla, California, 92037, United States
Rohan R. Merchant, The Scripps Research Institute (TSRI), North Torrey Pines Road, La Jolla, California, 92037, United States
Alexander J. E. Novak, The Scripps Research Institute (TSRI), North Torrey Pines Road, La Jolla, California, 92037, United States
Jacob Z. Zhong, The Scripps Research Institute (TSRI), North Torrey Pines Road, La Jolla, California, 92037, United States
Riley B. Mills, The Scripps Research Institute (TSRI), North Torrey Pines Road, La Jolla, California, 92037, United States
Ming Yan, The Scripps Research Institute (TSRI), North Torrey Pines Road, La Jolla, California, 92037, United States.
Dr. Changxia Yuan, Chemical Development, Bristol-Myers Squibb, One Squibb Drive, New Brunswick, New Jersey 08903, United States
Dr. Martin D. Eastgate, Chemical Development, Bristol-Myers Squibb, One Squibb Drive, New Brunswick, New Jersey 08903, United States
Prof. Phil S. Baran, The Scripps Research Institute (TSRI), North Torrey Pines Road, La Jolla, California, 92037, United States
References
- 1.Barton DHR. In: Some Recollections of Gap Jumping: Profiles, Pathways and Dreams. Seeman JI, editor. American Chemical Society; Washington, DC: 1991. [Google Scholar]
- 2.Barton DHR, McCombie SW. J Chem Soc, Perkin Trans 1. 1975:1574. [Google Scholar]
- 3.(a) Barton DHR, Crich D, Motherwell WB. J Chem Soc, Chem Commun. 1983:939. [Google Scholar]; (b) Barton DHR, Crich D, Motherwell WB. Tetrahedron. 1985;41:3901. [Google Scholar]
- 4.For C–Cl, C–Br, C–I, C–S, C–Se, and C–O bond-forming reactions via fragmentation of Barton esters, see: Zhu J, Klunder AJH, Zwanenburg B. Tetrahedron. 1995;51:5099.For radical addition onto protonated heterocycles, see: Barton DHR, Garcia B, Togo H, Zard SZ. Tetrahedron Lett. 1986;27:1327.Castagnino E, Corsano S, Barton DHR, Zard SZ. Tetrahedron Lett. 1986;27:6337.For Giese-type radical additions onto Michael acceptors, see: Barton DHR, Crich D, Kretzschmar G. Tetrahedron Lett. 1984;25:1055.Barton DHR, Chern CY, Jaszberenyi JC. Tetrahedron Lett. 1992;33:5013.
- 5.For examples of PET mediated radical decarboxylation and conjugate addition, see: Okada K, Okamoto K, Oda M. J Am Chem Soc. 1988;110:8736.Okada K, Okamoto K, Morita N, Okubo K, Oda M. J Am Chem Soc. 1991;113:9401.Cassani C, Bergonzini G, Wallentin CJ. Org Lett. 2014;16:4228. doi: 10.1021/ol5019294.Griffin JD, Zeller MA, Nicewicz DA. J Am Chem Soc. 2015;137:11340. doi: 10.1021/jacs.5b07770.Chu L, Ohta C, Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:10886. doi: 10.1021/ja505964r.Schnermann MJ, Overman LE. Angew Chem Int Ed. 2012;51:9576. doi: 10.1002/anie.201204977.Pratsch G, Lackner GL, Overman LE. J Org Chem. 2015;80:6025. doi: 10.1021/acs.joc.5b00795.Jamison CR, Overman LE. Acc Chem Res. 2016;49:1578. doi: 10.1021/acs.accounts.6b00284.Müller DS, Untiedt NL, Dieskau AP, Lackner GL, Overman LE. J Am Chem Soc. 2015;137:660. doi: 10.1021/ja512527s.
- 6.For selected reviews on Barton decarboxylation reactions, see: Barton DHR, Zard SZ. Pure Appl Chem. 1986;58:675.Saraiva MF, Couri MRC, Le Hyaric M, de Almeida MV. Tetrahedron. 2009;65:3563.Crich D, Quintero L. Chem Rev. 1989;89:1413.
- 7.For selected recent applications of Barton esters in total synthesis, see: Lathrop SP, Pompeo M, Chang WTT, Movassaghi M. J Am Chem Soc. 2016;138:7763. doi: 10.1021/jacs.6b04072.Fang C, Shanahan CS, Paull DH, Martin SF. Angew Chem Int Ed. 2012;51:10596. doi: 10.1002/anie.201205274.Asaba T, Katoh Y, Urabe D, Inoue M. Angew Chem Int Ed. 2015;54:14457. doi: 10.1002/anie.201509160.
- 8.Yamaguchi K, Kazuta Y, Abe H, Matsuda A, Shuto S. J Org Chem. 2003;68:9255. doi: 10.1021/jo0302206. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ko EJ, Savage GP, Williams CM, Tsanaktsidis J. Org Lett. 2011;13:1944. doi: 10.1021/ol200290m. [DOI] [PubMed] [Google Scholar]; (b) Ho J, Zheng J, Meana-Pañeda R, Truhlar DG, Ko EJ, Savage GP, Williams CM, Coote ML, Tsanaktsidis J. J Org Chem. 2013;78:6677. doi: 10.1021/jo400927y. [DOI] [PubMed] [Google Scholar]
- 10.For a review on hydrogen donors in radical reactions, see: Studer A, Amrein S. Synthesis. 2002;7:835.
- 11.Cornella J, Edwards JT, Qin T, Kawamura S, Wang J, Pan CM, Gianatassio R, Schmidt M, Eastgate MD, Baran PS. J Am Chem Soc. 2016;138:2174. doi: 10.1021/jacs.6b00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin T, Cornella J, Li C, Malins LR, Edwards JT, Kawamura S, Maxwell BD, Eastgate MD, Baran PS. Science. 2016;352:801. doi: 10.1126/science.aaf6123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Qin T, Chen TG, Wimmer L, Edwards JT, Cornella J, Vokits B, Shaw SA, Baran PS. Angew Chem Int Ed. 2016;55:9676. doi: 10.1002/anie.201605463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toriyama F, Cornella J, Wimmer L, Chen TG, Dixon DD, Creech G, Baran PS. J Am Chem Soc. 2016;138:11132. doi: 10.1021/jacs.6b07172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obradors C, Martinez RM, Shenvi RA. J Am Chem Soc. 2016;38:4962–4971. doi: 10.1021/jacs.6b02032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barton DHR, Hervé Y, Potier P, Thierry J. Tetrahedron. 1988;44:5479. [Google Scholar]
- 17.The remaining mass balance for both decarboxylation and Giese reactions largely comprise carboxylic acid resulting from hydrolysis of RAEs.
- 18.Arapakos PG. J Am Chem Soc. 1967;89:6794. [Google Scholar]
- 19.For some applications of Barton decarboxylation in pyrroloindoline alkaloid synthesis, see: Foo K, Newhouse T, Mori I, Takayama H, Baran PS. Angew Chem Int Ed. 2011;50:2716. doi: 10.1002/anie.201008048.Lathrop SP, Movassaghi M. Chem Sci. 2014;5:333. doi: 10.1039/C3SC52451E.Espejo VR, Li XB, Rainier JD. J Am Chem Soc. 2010;132:8282. doi: 10.1021/ja103428y.Lucarini S, Bartoccini F, Battistoni F, Diamantini G, Piersanti G, Righi M, Spadoni G. Org Lett. 2010;12:3844. doi: 10.1021/ol101527j.Crich D, Pavlovic AB, Samy R. Tetrahedron. 1995;51:6379.
- 20.(a) Barton DHR, Chern CY, Jaszberenyi JC. Aust J Chem. 1995;48:407. [Google Scholar]; (b) Barton DHR, Gateau-Olesker A, Géro SD, Lacher B, Tachdjian C, Zard SZ. Tetrahedron. 1993;49:4589. [Google Scholar]
- 21.Competition experiment employing a secondary RAE (5, 1 equiv.), a primary RAE (SI-16, 1 equiv.), and benzylacrylate (1 equiv.) showed higher yields of the secondary radical adduct. See SI for details.
- 22.Zhang YN, Zhang W, Hong D, Shi L, Shen Q, Li JY, Li J, Hu LH. Bioorg Med Chem Lett. 2008;16:8697. doi: 10.1016/j.bmc.2008.07.080. [DOI] [PubMed] [Google Scholar]
- 23.For recent examples of late-stage amino acid diversification using radical 1,4-additions, see: Wright TH, Bower BJ, Chalker JM, Bernardes GJL, Wiewiora R, Ng W-L, Raj R, Faulkner S, Vallée MRJ, Phanumartwiwath A, Coleman OD, Thézénas M-L, Khan M, Galan SRG, Lercher L, Schombs MW, Gerstberger S, Palm-Espling ME, Baldwin AJ, Kessler BM, Claridge TDW, Mohammed S, Davis BG. Science. 2016 doi: 10.1126/science.aag1465.Yang A, Ha S, Ahn J, Kim R, Kim S, Lee Y, Kim J, Söll D, Lee HY, Park HS. Science. 2016;354:623–626. doi: 10.1126/science.aah4428.
- 24.A concurrent, photochemical approach to decarboxylative peptide macrocyclization employs an orthogonal side-chain protection strategy: McCarver SJ, Qiao JX, Carpenter J, Borzilleri RM, Poss MA, Eastgate MD, Miller M, MacMillan DWC. Angew Chem Int Ed. 2016 doi: 10.1002/anie.201608207.
- 25.For a review on peptide macrocyclization, see: White CJ, Yudin AK. Nature Chem. 2011;3:509. doi: 10.1038/nchem.1062.
- 26.For a review on macrocyclization-based peptide stapling, see: Lau YH, de Andrade P, Wu Y, Spring DR. Chem Soc Rev. 2015;44:91. doi: 10.1039/c4cs00246f.
- 27.Crystallographic data (excluding structure factors) for structures 18b and 19b reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no.CCDC-1510142 and CCDC-1510143.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.