

Abstract
As microbial genome sequencing becomes more widespread, the capacity of microorganisms to produce an immense number of metabolites has come into better view. Utilizing a metabolite/gene cluster correlation platform, the biosynthetic origins of a new family of natural products, the rimosamides, were discovered. The rimosamides were identified in Streptomyces rimosus and associated with their NRPS/PKS-type gene cluster based upon their high frequency of co-occurrence across 179 strains of actinobacteria. This also led to the discovery of the related detoxin gene cluster. The core of each of these families of natural products contains a depsipeptide bond at the point of bifurcation in their unusual branched structures, the origins of which are definitively assigned to nonlinear biosynthetic pathways via heterologous expression in Streptomyces lividans. The rimosamides were found to antagonize the antibiotic activity of blasticidin S against Bacillus cereus.
Graphical abstract
Microorganisms have evolved the ability to produce complex molecules known generally as natural products (or secondary metabolites) that include numerous high-value compounds with applications in the food, agricultural, chemical, and pharmaceutical industries.1 As microbial genome sequencing has increased over the past decade, the vastness of chemical space occupied by natural products has become more clear and quantifiable.2–4 Current estimates for the numbers of natural product producing gene clusters far outstrip the number of known compounds. Because natural products comprise a rich source of new chemistry and drug leads,5–8 the availability of thousands of microbial genomes presents plentiful opportunities for the discovery of novel compounds and biosynthetic pathways using a variety of technologies. Knowledge of the biosynthetic origin of natural products has seen increased interest (beyond an academic concentration) with the development of novel expression approaches for the production of secondary metabolites for industrial purposes.9 As such, identifying natural product families and their corresponding gene clusters via correlative methods, such as pattern-based genome mining,10 protein/metabolite profiling,11,12 and gene/metabolite co-occurrences13,14 have the potential to complement a growing field.
In a recent study, we sequenced and examined the genomes of 178 actinobacteria, leading to the detection and classification of thousands of natural product biosynthetic gene clusters (BGCs).13 Similar BGCs were then grouped into gene cluster families (GCFs) likely to produce members of the same family of natural products. Metabolomics data were also obtained using liquid chromatography coupled with sensitive and highly accurate mass spectrometry (LC-MS) to identify hundreds of discrete metabolites for each strain. We then combined the genomic and metabolomic data to generate a simple correlation score that indicates the likelihood that an observed natural product and GCF are associated, based upon their co-occurrence across multiple strains. This method of combining metabolomics and genomics is known as metabologenomics.13,14
Metabologenomics data can be mined in a variety of ways: to identify metabolites that represent new members of interesting structural classes, to determine the biosynthetic origin of an “orphan” natural product (metabolites for which no BGC has been discovered), or to identify secondary metabolites that represent entirely new structural classes altogether.13,14 When metabologenomics is applied to actinomycetes, a rich source of microbial natural products,15 the method provides a new screen for active secondary metabolism.13 Thus, despite the extensive efforts of the natural products research community to characterize actinomycetes natural products, these organisms still contain a trove of new scaffolds and enzymes. The continued rate of new compound discovery from gene clusters for which no natural product have been associated strongly supports this view.16,17
Here, we use gene cluster/metabolite correlations14 to identify the biosynthetic origin of a new family of compounds produced by Streptomyces rimosus NRRL B-2659, the rimosamides, and the BGC for a related family of natural products, the detoxins (Figure 1). The gene clusters responsible for producing the rimosamides and detoxins include both nonribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) components and reflect the branched structure of these compounds. Notably, the gene cluster responsible for the production of the detoxins had remained a mystery since the discovery of the compounds 50 years ago. The discovery of new natural products produced by S. rimosus, a prolific oxytetracycline producer and an extensively studied source of antibiotics, demonstrates both the value of metabologenomics to find new compounds and the depth of the undiscovered natural product reservoir in actinobacteria.13
Figure 1.
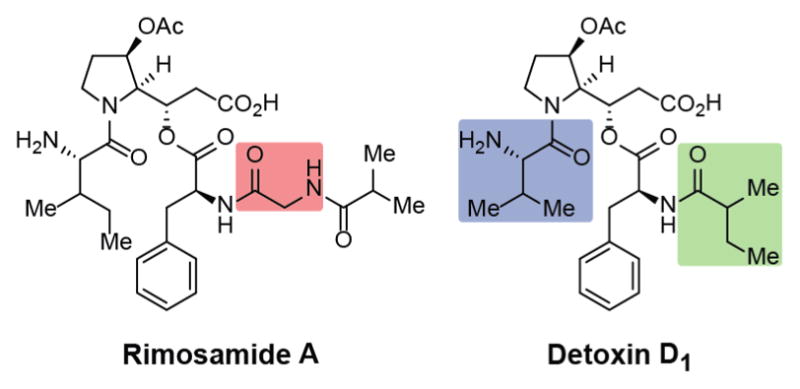
Representative structures of the rimosamides and the detoxins. Key differences between the rimosamides and detoxins are highlighted above and include the addition of a glycine residue (red), incorporation of an isoleucine residue (blue), and incorporation of an iso-butyrate (green).
RESULTS AND DISCUSSION
Metabolomics Screening of Actinobacteria and Gene Cluster Correlation
We have previously shown that a secondary metabolite can be associated with a BGC by correlating large-scale metabolomics with interpreted genome sequences.13,14 To implement the metabologenomics workflow for identification of candidate BGCs for new natural products, the co-occurrence frequencies of specific molecular masses and specific gene cluster families were counted across the extracts and annotated genomes for 178 strains of actinobacteria. The metabolomics experiments for each strain were analyzed by HPLC coupled to positive-mode electrospray ionization mass spectrometry. Mass spectral features that were observed in multiple strains were matched by accurate mass (10 ppm cutoff) and similar LC retention times (3 min cutoff). During the metabolomics screening of S. rimosus NRRL B-2659, we observed a highly abundant mass spectral feature (m/z 605.3184), which was not found in publicly accessible natural product databases such as Dictionary of Natural Products or AntiBase. Of the 178 strains, the m/z 605 ion was detected in 14 strains. The results for correlation of this ion to the top scoring GCFs are presented in Table 1. Six GCFs (NRPS_83, NRPS_424, NRPS 436, NRPS_502, NRPS_590, and NRPS_599) co-occur with the detection of m/z 605 in all 14 strains. However, this ion was not observed in some other strains that contain only the lowest scoring GCFs (NRPS_424, NRPS_590, NRPS_599, or NRPS_83; Table 1, bottom row). NRPS_436 and NRPS_502, the two highest scoring GCFs, had identical correlation scores of 237. To distinguish between these two GCFs, we selected one additional strain, NRRL WC-3876, whose genome encodes GCF NRPS_502 but not NRPS_436. Detection of the target ion in this strain improved the correlation score to 258 for gene cluster NRPS_502, strongly implicating this GCF as being responsible for production of m/z 605. Nearly all of the strains that encode this GCF are found within the S. rimosus clade (Supporting Information Figure S1).
Table 1.
Data Enabling the Metabologenomic Identification of the Rimosamides and Their Biosynthetic Gene Clustera
| Strain ID |
m/z 605.3184 |
Selected Ion Intensity | NRPS Gene Cluster Families | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NRRL B-2659 | yes | 2.6E+08 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 432 | ||
| NRRL B-2661 | yes | 2.7E+07 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 488 | 549 | 432 |
| NRRL B-2775 | yes | 2.4E+06 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 488 | 549 | 432 |
| NRRL B-3501 | yes | 6.2E+07 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 488 | 549 | 432 | |
| NRRL B-8076 | yes | 5.5E+06 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 488 | 549 | 432 |
| NRRL WC-3927 | yes | 2.9E+08 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 488 | 549 | 432 |
| NRRL WC-3930 | yes | 9.1E+06 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 488 | 549 | 432 |
| NRRL WC-3874 | yes | 2.8E+07 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 549 | ||
| NRRL WC-3560 | yes | 1.4E+08 | 502 | 590 | 436 | 599 | 424 | 83 | 465 | 488 | 432 | ||
| NRRL WC-3703 | yes | 1.1E+08 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 488 | 549 | 432 |
| NRRL WC-3868 | yes | 3.6E+06 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 488 | 549 | 432 |
| NRRL WC-3877 | yes | 7.3E+07 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 488 | 549 | 432 |
| NRRL WC-3532 | yes | 5.9E+06 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 488 | 549 | 432 |
| NRRL WC-3929 | yes | 5.2E+07 | 502 | 590 | 436 | 599 | 424 | 83 | 570 | 465 | 488 | 549 | 432 |
| NRRL ISP5539 | no | 0 | 590 | ||||||||||
| NRRL WC-3773 | no | 0 | 590 | 599 | 424 | 83 | 465 | 432 | |||||
| NRRL S-515 | no | 0 | 590 | ||||||||||
| NRRL WC-3908 | no | 0 | 590 | 599 | 424 | 83 | 432 | ||||||
| NRRL B-2120 | no | 0 | 590 | ||||||||||
| NRRL WC-3876 | yes | 8.6E+06 | 502 | 590 | |||||||||
| Correlation Score | 258 | 253 | 237 | 235 | 234 | 234 | 217 | 216 | 198 | 197 | 194 | ||
This table shows the co-occurrence data of rimosamide A with members of 11 gene cluster families for 20 Streptomycete strains. Strains are listed on the far left. Rimosamide detection by electrospray MS is shown in column 2 and ion intensity in column 3. Columns 4–14 show all NRPS GCFs that are present in 12 or more of this set of 20 strains. Note that data for the strains listed in the top 19 rows were obtained in the first pass application of LC-MS and automated data reduction. The strain listed in the row second from bottom (NRRL WC-3876) was interrogated in a targeted fashion for validation. The highest level of co-occurrence is between rimosamide and GCF NRPS_502. Numbers designating each gene cluster family appear as archived on the Web site at www.igb.illinois.edu/labs/metcalf/gcf. Correlation scores of the rimosamides against each GCF are shown in the bottom row.
Targeted Detection and Isolation of the Rimosamides
When S. rimosus NRRL B-2659 was grown in three separate culture conditions, m/z 605.3184 was observed at the highest level in arginine glycerol salts (AGS) medium. By monitoring other metabolites that exhibited increased signal intensities over a 7-day time course, three additional ion species (m/z 533.2976, 547.3131, 591.3029) were identified that also did not match any known metabolites. The four masses were spaced by 14.0158 or 58.0054 Da (unambiguously additions of a —CH2 or O-acetyl group, respectively), and their retention times increased slightly with mass. The four ion species generated similar peptide-like fragmentation patterns, resulting in a common amino acid sequence tag of [Ile/Val, Pro- (+C2H2O2), Phe, and C6H10NO2] (data in Supporting Information). This observation led to the expectation that the four species belong to the same class of compounds, all produced by the same BGC. We named these compounds the rimosamides A–D (m/z 605.3184, 591.3029, 547.3131, and 533.2976, respectively) after the strain from which they were discovered (Supporting Information Figure S2).
The targeted compounds were initially isolated as a mixture with 0.2 mg/L titers from the producing strain, S. rimosus NRRL B-2659. Further purification was carried out on the mixture with limited success. Two of the rimosamides species (rimosamides B and C) were isolated as a single fraction due to the difficulty of their separation; however, rimosamides A and D were purified separately with the highest yield of rimosamide A.
Structure Elucidation of the Rimosamides
To validate the amino acid assignments deduced by MS2 fragmentation, several metabolic feeding experiments were performed using stable isotope-labeled amino acids. LC–MS analyses of the labeled samples showed robust incorporation of proline and phenylalanine as well as isoleucine and/or valine. The incorporation of phenylalanine was confirmed across the four ion species. When supplemented with D7-proline, +6 Da mass shifts were observed for the m/z 605.3184 and 591.3029 ion species and +7 Da mass shifts for m/z 533.2976 and 547.3131, indicating that a modification is occurring in the proline residue in the larger mass compounds. Incorporation of isoleucine was only observed for two of the ion species (m/z 605.3184 and 547.3131). Supplementation with D8-valine led to +7 Da mass shifts for m/z 605.3184 and 547.3131, demonstrating a substitution of a single deuteron. The other ion species (m/z 591.3029 and 533.2976) exhibited +7 Da and +15 Da mass shifts, which led us to believe that these compounds likely incorporate two valine residues with a modification occurring for only one of the valine monomers. Isotope incorporation data for m/z 605.3184 is shown in Supporting Information Figure S3; data for the additional three species with differential incorporation is shown in Supporting Information Figure S4.
Due to the likely high level of similarity between the rimosamides, we selected only rimosamide A for full structure elucidation by NMR spectroscopy. The molecular formula of rimosamide A was determined to be C30H45N4O9 by high-resolution mass spectrometry (m/z 605.3184 [M + H]+, calculated 605.3186). 1H NMR and 2D NMR (COSY, HSQC, and HMBC) spectroscopic data, recorded in D2O, confirmed a peptidic structure and the presence of the monomers that were identified in both MS2 fragmentation and stable isotope labeling experiments. Three signals in the aromatic region of the proton spectrum, H-9 (δH 7.26), H-10 (δH 7.37), and H-11 (δH 7.30), were found to represent a monosubstituted phenyl moiety. The HMBC couplings from methylene protons H-7 (δH 2.99 and 3.20) to C-8 and C-9 and C-12 and from proton H-6 (δH 4.70) to C-12, along with COSY correlations from H-6 to H-7 and H-9 to H-10 to H-11, confirmed the presence of a phenylalanine residue. Two overlapping methyl doublets (δH 1.07) that coupled to a methine at δH 2.50 along with HMBC couplings to a carbonyl carbon (δC 182.03) indicated the presence of an isobutyrl moiety, likely derived from valine, based on the results of the stable isotope labeling experiments. The presence of a pair of peaks from diastereotopic protons on C-4 (δH 3.79 and 3.83) led to the identification of a glycine residue as part of rimosamide A, which was further confirmed through HMBC analysis. Inspection of the MS2 fragmentation pattern indicated that the isobutyrl moiety was attached to the glycine nitrogen, which was confirmed by HMBC correlation across this amide bond. Finally, an HMBC cross peak from the α-proton of the phenyl alanine moiety to the carbonyl carbon of the glycine moiety established the peptide bond between these two units.
Two nonequivalent methyl groups, H-25 (δH 1.08) and H-27 (δH 0.95), provided an entry to the spin system representing the predicted isoleucine moiety. COSY correlations from H-25 to H-24 (δH 1.95), H-24 to H-26 (δH 1.20 and 1.51), and H-26 to H-27, in combination with HMBC correlations, confirmed the side chain of the isoleucine residue. The COSY and HMBC spectra further revealed a pyrrolidine moiety, consistent with the demonstrated incorporation of proline. Oxidation of the pyrrolidine, as had been proposed on the basis of stable isotope labeling, was confirmed at C-17 (δC 72.50). Additional HMBC correlations revealed that this oxidation site is acetylated, H-17 to C-18 (δC 174.02) and H-19 (δH 2.11) to C-18. The pyrrolidine is connected to a linear three-carbon side chain with a carboxylic acid terminus (C-15, δC 178.24), suggesting that this moiety is derived from condensation of a proline-derived precursor and a malonyl-derived two-carbon unit. Finally, an HMBC correlation from H-13 in the side chain of the pyrrolidine moiety to the carbonyl carbon of the phenylalanine moiety established the predicted depsipeptide bond. In-depth analysis of MS2 fragmentation patterns of rimosamide A further corroborated these NMR spectroscopic assignments. COSY and HMBC correlations are shown in Supporting Information Figure S5. Taken together, our data enabled detailed characterization of the structure of rimosamide A and to propose analogous structures for rimosamides B–D (Supporting Information Figure S1). The 13C and 1H NMR assignments are listed in Supporting Information Table S1, and all spectra are provided in the Supporting Information.
Searching the literature for any related structures revealed a family of compounds known as the detoxins, which feature the pyrrolidine moiety and depsipeptide bond present in rimosamide A (Figure 1).18,19 Based upon these structural similarities, we assigned the stereocenters within the rimosamides to match those in the detoxins, which provided a sensible match to all the NMR data. Moreover, an analysis of the sequence similarity of BGCs would likely result in matching stereochemistry (see below). The biosynthesis of the detoxins has remained unexplored since their discovery nearly 50 years ago. Although we did not detect any detoxins in our extracts from S. rimosus, we reasoned that they would originate from a pathway closely related to the rimosamides.
Rimosamide Biosynthetic Gene Cluster
The GCF designated as NRPS_502 (http://www.igb.illinois.edu/labs/metcalf/gcf/gcfDisplay.php?gcf=NRPS_GCF.502) represents one of the most abundant gene clusters in our data set that lacks a known natural product. Association of this gene cluster with rimosamides led to the naming of the open reading frames (ORFs) as rmo, with annotation of the putative proteins summarized in Supporting Information Table S2. Encoded within the cluster are two modular NRPS proteins (RmoG and RmoH), one NRPS/PKS hybrid protein (RmoI), and a variety of tailoring enzymes (Figure 2A). RmoG, RmoH, and RmoI can be further divided into five NRPS/PKS biosynthetic modules (Figure 2B).20–22 Sequence analysis of RmoG identified a starter condensation domain (generally used for the incorporation of fatty acids), and the adenylation domain was predicted to incorporate a glycine residue. The adenylation domain of RmoH was predicted to incorporate a nonpolar amino acid and the specific Stachelhaus code had not been identified in any known systems. RmoH also contains a type I thioesterase domain. The NRPS/PKS hybrid protein, RmoI, contains two adenylation domains that were predicted to incorporate isoleucine and proline residues, respectively, as well as a ketosynthase, a ketoreductase, and another type I thioesterase domain. Of note is the lack of an acyltransferase in RmoI, which is necessary for the incorporation of the malonyl subunit. It has been previously reported that acyltransferase domains are capable of acting in trans to provide the charged monomer.23 The other noticeable feature of the cluster is the taurine dioxygenase (RmoL);24 it is likely that an RmoL is responsible for the oxidation of the modified proline residue,25 leading to the observed loss of a deuteron during stable isotope labeling experiments. On the basis of the enzymatic machinery present, this BGC matched up well with the rimosamide structures.
Figure 2.

Biosynthesis of the rimosamides. (A) The rimosamide biosynthetic gene cluster. Genes depicted as red arrows are NRPS. The purple arrow is NRPS/PKS hybrid. The green arrow is taurine dioxygenase. Blue arrows are various, and gray arrows are hypothetical ORFs. (B) NRPS and PKS domain predictions are shown for RmoG, RmoH, and RmoI. Functional annotations of additional ORFs in NPRS_502 can be found in Table S2. (C) Heterologous expression of the rimosamides in S. lividans 66.
To conclusively link the rimosamides with GCF NRPS_502, we heterologously expressed one of its member gene clusters in S. lividans 66 (whose genome does not encode any member of this GCF). A fosmid library consisting of ~1500 clones was constructed using genomic DNA from S. rimosus NRRL B-2659 and screened by PCR using primers from rmoB and rmoL (near the ends of the putative cluster).26 Among the clones, fosmid pRAM3 spanned a ~42 kb contiguous region of DNA as determined by end-sequencing and alignment with the genome sequence of S. rimosus. Further analysis of this fosmid indicated that it contained an insert covering the entire 30.5 kb gene cluster (GenBank accession number NZ_ANSJ01000026). To test whether this fosmid contained all of the genes required for rimosamide biosynthesis, the fosmid was recombined with pAE4 (Supporting Information Figure S6) using Gateway BP Clonase; the resulting recombinant plasmid pRAM4 was integrated into the S. lividans 66 chromosome via site-specific recombination at the ΦC31 attachment site26 to generate S. lividans 66-pRAM4. After growth in our four standard culture conditions, all four members of the rimosamide family were then detectable (and confirmed by MS2 analysis) in the extracted culture broth of S. lividans 66-pRAM4 (Figure 2C and Supporting Information Figure S7), showing that the cloned gene cluster is necessary for production of the rimosamides.
Proposed Biosynthesis of the Rimosamides
The genes responsible for the biosynthesis of the rimosamides and their ORF designations are shown in Figure 2A, along with functional predictions in Supporting Information Table S2. The structures of the rimosamides contain diverging peptide chains (e.g., the terminal isoleucine and isobutyrate residues), which indicates a nonlinear NRPS/PKS pathway as proposed in Figure 3. It is very likely that the starter condensation domain of RmoG is responsible for forming the amide bond between the isobutyrate (Ibu) monomer and the loaded glycine. Ibu-Gly is then extended onto the phenylalanyl-S-RmoH, itself a product of the adenylation domain of RmoH (Figure 3). Ibu-Gly-Phe is transferred to the active site serine of the thioesterase domain of RmoH for later use. RmoI builds the Ile-Pro dipeptidyl intermediate before the ketosynthase domain catalyzes the Claisen reaction between malonate and the Ile-Pro. The ketoreductase domain then reduces the ketone to an alcohol (highlighted in orange in Figure 3), which subsequently reacts with the thioesterase-bound Ibu-Gly-Phe on RmoH to form the depsipeptide bond (highlighted in yellow in Figure 3). Mature rimosamide is then released from RmoI by the second Type I thioesterase domain in the system. The taurine dioxygenase (RmoL) is most likely responsible for the hydroxylation of the proline residue at the 3 position; however both the timing of this tailoring and the direct acetyl donor to afford rimosamide A and B require additional studies. It is notable that only acetylated and nonhydroxylated derivatives are observed, with no rimosamides or detoxins containing a free hydroxyl being detected here or reported previously.
Figure 3.

Proposed biosynthetic pathway for the production of rimosamide C from NRPS_502. Production of the rimosamides breaks the normal rules of colinearity in assembly line biosynthesis and is proposed to culminate in formation of an unusual depsipeptide bond formation to generate branching peptide chains. A taurine dioxygenase (RmoL) is likely responsible for the oxidation of the C-3 position of the modified proline core.
In NRPS systems, depsipeptide bonds are formed by nucleophilic attack on thioesterase-bound substrates. Previously, two biosynthetic origins for the hydroxyl nucleophile have been established. The first entails the incorporation of a hydroxy acid monomer into the peptide backbone, such as in cereulide27,28 or arthroamide.29 The second utilizes the side chain of a serine or threonine residue such as in the salinamides.30,31 However, the proposed nucleophilic source in the case of the rimosamides and detoxins does not fit either of these known schemes. The proposed rimosamide biosynthesis represents the first example of the incorporation of a nonpolar amino acid in which the carboxy group is reduced to an alcohol by a ketoreductase before esterification, in a manner similar to ester formation in cyclic and branched polyketides.
Biosynthetic Gene Cluster of the Detoxins
During our MS-based metabolomics experiments of various strains within the S. rimosus clade, we identified the four rimosamides A–D; yet none of the numerous compounds within the detoxin family were observed. This result led us to conclude that GCF NRPS_502 is not responsible for detoxin biosynthesis and that a similar, yet distinct, BGC is present in detoxin-producing strains. S. mobarensis NRRL B-3729 (not previously included in our 179 strains) is a known producer of the detoxins. We obtained S. mobarensis NRRL B-3729 and, during a metabolomics experiment, identified detoxins B1–D2 and E1. Using the rimosamide gene cluster as a guide, we identified a BGC (Figure 4, accession no. NZ_AORZ01000036.1) in the S. mobarensis genome that lacks the biosynthetic machinery to incorporate and load a glycine residue (one of the key differences between the rimosamides and detoxins). S. mobarensis NRRL B-3729 is fermented industrially for the production of transglutaminase (TGase), and the detoxins are common undesired byproducts.32 A knockout of the BGC in this strain should simplify the production process of TGase and purification procedures.
Figure 4.

Analysis of the rimosamide and detoxin BGCs. (A) The rimosamide (top) and detoxin (bottom) BGCs share many of the same ORFs. Homologous ORFs are highlighted, and percentage sequence identity is shown. (B) NRPS and PKS domain predictions are shown for DetF and DetG. (C) Domains responsible for the incorporation of specific monomers as highlighted in the rimosamides (top) and detoxins (bottom).
A representation of the putative detoxin gene cluster and NRPS/PKS domain predictions are shown in Figure 4A and B. Complete annotation of all of the ORFs in the detoxin-producing BGC are shown in Supporting Information Table S3. A comparison of the rimosamides and detoxin BGCs revealed that four ORFs exhibit a high level of sequence similarity (Figure 4A) but that an entire NRPS ORF was absent in the detoxin pathway. RmoH/DetF and RmoI/DetG consist of identical NRPS/PKS domain predictions, but with some functional differences. The starter condensation domain in DetF is likely responsible for the incorporation of the 2-methylbutryl species in the detoxins; in the case of the rimosamides, the starter condensation domain of RmoG (no homologous ORF in the detoxin system) is likely to incorporate the isobutryl tail. A graphical representation depicting the domains that are responsible for the incorporation of corresponding chemical components in the rimosamides (top) and detoxin (bottom) is shown in Figure 4C. The biosynthetic pathway of the detoxins likely follows a similar mechanism as proposed for the rimosamides and is shown in Supporting Information Figure S8. The high level of sequence similarity between rmoI/detG (responsible for depsipeptide bond formation) and rmoL/detJ (responsible for oxidation of the pyrrolidine) further corroborates our assertion that the rimosamides and detoxins likely contain matching stereocenters.
Despite structural similarities, the detoxins and rimosamides were never detected together in a single strain by mass spectrometry. The related but divergent biosynthetic pathways for the rimosamides (from S. rimosus) and detoxins (from S. mobarensis) provide a glimpse of the relationship between these two distantly related strains. This observation, along with the clean loss of an entire NRPS module in the detoxin BGC, leads one to assert that the structural differences between the detoxins and rimosamides are not the result of a promiscuous biosynthetic NRPS or PKS module but rather that each family presents a distinct chemical phenotype, evolved to fill a specific ecological niche. The rimosamide and detoxin gene clusters represent two biosyntheses that likely diverged relatively recently.
Biological Activity of the Rimosamides
The detoxin family of compounds has been reported to antagonize the antibiotic activity of blasticidin S in Bacillus cereus.19 On the basis of their structural and biosynthetic similarity to detoxins, we hypothesized that the rimosamides may exhibit a similar activity. We performed disk diffusion assays and observed that rimosamide A was capable of negating the antibiotic activity of blasticidin S against B. cereus in a dose-dependent manner (Figure 5). It was reported that the detoxins prevent the active uptake of blasticidin S, and we suggest that the rimosamides behave in a similar manner.33 Blasticidin S is produced by S. griseochromogenes, and the corresponding BGC is not present in any of the rimosamide-producing strains.34 This indicates that the rimosamides are potentially produced to serve as a defense mechanism against other Streptomycetes. The rimosamides and detoxins represent examples of the few natural products exhibiting anti-antibiotic activity35 and may provide insight into the complex chemical ecology of these microorganisms.
Figure 5.

Disk diffusion assay of rimosamide A (RA) as an antagonist of the antibiotic blasticidin S (BS) against B. cereus. The assay plate is shown on the top and tabulated results on the bottom. With decreasing concentrations of the rimosamide A, the antagonism of blasticidin S decreases (i.e., inhibition zone increases).
CONCLUSION
Modern approaches to the targeted mining of genomes has provided new small molecules of high interest.36–39 These discoveries generate lead structures for the development of new pharmaceutical agents or provide the genomic information necessary for the heterologous expression of high value compounds. In the case of the rimosamides, their biosynthesis breaks the rules of colinearity in standard NRPS/PKS pathways. This complicates the application of traditional genome mining approaches for natural product discovery and possibly contributed to the orphan status of the detoxins for nearly 50 years after their discovery and structure elucidation. Correlation of metabolites with BGCs based upon their frequency of co-occurrence offers a complementary approach to genome mining for the rapid identification of expressed metabolites but does require implementation on many hundreds of strains to function as a discovery engine. As an untargeted method, it does have the potential for high-throughput discovery of natural products. With a database of hundreds or thousands of new gene cluster/metabolite pairs in hand, the substructures and biosynthetic clues can guide the targeted collection of noncryptic compounds for testing in specific and themed bioassay programs. In time, this approach should provide a new determinism in finding drug leads from the bacterial world, a field traditionally thought of as empirical and stochastic. Here, we described in depth one case aligned with this future regarding natural product discovery.
Gene cluster/metabolite correlation approaches directly explore metabolite expression in multiple strains (unlike indirect methods of studying expression such as transcript-profiling or proteomics).11,40–42 Importantly, metabologenomics can assign the biogenesis of compounds that exhibit unusual or noncanonical biosynthetic steps. Correlation strength between metabolites and their gene clusters will grow substantially by increasing the number of strains interrogated from a few hundred in this study to thousands, suggesting a path forward for high-throughput discovery and greatly accelerated discovery of new compounds from the microbial world.
METHODS
Materials
Streptomyces strains were acquired from U.S. Department of Agriculture, Agricultural Research Service. B. cereus (ATCC 14579) was acquired from ATCC. Stable-isotope labeled amino acids were from Cambridge Isotope Laboratories or CDN Isotopes. Other chemical reagents were from Sigma-Aldrich.
Growth Conditions for Streptomycetes
Strains were streaked out on ISP2 plates and grown for 4 days at 30 °C. Individual colonies were picked and grown in 5 mL of ISP2 medium for 3 days at 30 °C before 100 μL of the culture was transferred to 5 mL of AGS medium, mannitol soy flour medium (MS), or ISP4 medium (media recipes can be found in the Supporting Information) and grown for 10 days at 30 °C. Stable isotope-labeled amino acids were added to the medium to a final concentration of 1 mM when necessary.
Metabolomics Screening
The culture supernatant from strains was separated from cells by centrifugation and separated by solid phase extraction (SPE) using an Oasis Hydrophilic/Lipophilic Balanced (HLB) column from Waters. These samples were separated by a Phenomenex Luna C18 column (250 × 2 mm, 5 μm) eluting with acetonitrile/water with 0.1% (v/v) formic acid from 0 to 100% over 60 min. A Q-Exactive Orbitrap (Thermo Scientific) was used for detection from m/z 250 to 3750. Following each full MS scan, the top five most intense ions were selected for a data dependent MS2 scan.
Targeted Detection of the Rimosamides
Individual colonies of S. rimosus sp. NRRL B-2659 were picked and grown in 5 mL of ISP2 medium for 3 days at 30 °C before 100 μL of the culture was transferred to 5 mL of AGS. The culture supernatant was analyzed by LC–MS. Further information regarding isolation of the rimosamides can be found in the Supporting Information.
Structure Elucidation of Rimosamides
NMR spectroscopy experiments were performed in D2O. 1H, COSY, HSQC, and HMBC experiments were carried out on a Bruker Avance III HD (800 MHz for 1H reference frequency and 201 MHz for 13C) equipped with a 5 mm indirect detection cryoprobe. Chemical shifts were referenced to sodium formate: δ(1H) = 8.44 and δ(13C) = 171.67. 13C chemical shifts were determined via HMBC and HSQC spectra. 1H,1H-J-coupling constants were determined from the acquired 1H or dqfCOSY spectra. ROESY correlations were observed using a mixing time of 400 ms.
Heterologous Expression of the Rimosamides
A fosmid library was constructed using genomic DNA extracted from S. rimosus B2659.26 The library was screened via PCR using primers from rmoB and rmoL. The plasmid from positive fosmids was isolated and recombined in vitro with pAE4 to afford apramycin resistance and integration into the ΦC31 attB site commonly present within the genomes of Streptomyces strains. Recombined plasmid was transformed into E. coli WM6029. Resulting strains were conjugated with S. lividans 66 spores to yield S. lividans 66-pRAM4. Additional information regarding the fosmid library creation and mating with S. lividans 66 is described within the Supporting Information.
Biological Activity Screening of Rimosamide A
Paper disks were treated with 10 μL of 1 mg mL−1 blasticidin S, 10 μL of 2.5 mg mL−1 rimosamide, or 10 μL of mixed stocks. Disks were placed on an ATCC Medium 3 plate seeded with 100 μL of pregrown B. cereus or E. coli and incubated at 37 °C for 16 h. Further information regarding the assays are described in the Supporting Information.
Supplementary Material
Acknowledgments
We thank the Agricultural Research Service of the United States Department of Agriculture for providing us bacterial strains. Research reported in this publication was supported by the Northwestern University Department of Chemistry and the National Institutes of Health under award numbers D012016 (Integrated Molecular Structure Education and Research Center at Northwestern University), GM112739 (F.C.S.), T32-GM008500 (J.A.B.), AT009143 (N.L.K./R.J.T.), and GM077596 (W.W.M.), as well as the NIH supported Chemistry of Life Processes Training Grant T32-GM105538 (R.A.M.).
Footnotes
Accession Codes
The assembled genome of S. rimosus NRRL B-2659 is available from the National Center for Biotechnology Information under the accession number ASM33118v1. All sequences used for metabologenomics can be accessed through NCBI BioProject PRJNA238534
ORCID
Ryan A. McClure: 0000-0002-0163-9290
Neil. L Kelleher: 0000-0002-8815-3372
Author Contributions
R.A.M. collected and analyzed metabolomics data and performed the isolation of the rimosamides. A.W.G. performed in silico analysis of the biosynthetic gene cluster and performed the metabologenomics correlation of the rimosamides. R.A.M., J.A.B., and F.C.S. performed the structure elucidation of the rimosamides. R.A.M. and K.S.J. generated the fosmid library and heterologous expression. R.A.M. performed the biological activity assays. W.W.M. and N.L.K. designed the correlative methodology. R.A.M., A.W.G., W.W.M., R.J.T., and N.L.K. prepared and revised the manuscript.
The authors declare no competing financial interest.
- Strain list, plasmids, primers, phylogenetic analysis, isotope labeling, NMR spectra and interpretation, bioinformatics analysis, and additional methods (PDF)
References
- 1.Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 2.Gross H. Strategies to unravel the function of orphan biosynthesis pathways: recent examples and future prospects. Appl Microbiol Biotechnol. 2007;75:267–277. doi: 10.1007/s00253-007-0900-5. [DOI] [PubMed] [Google Scholar]
- 3.Scherlach K, Hertweck C. Triggering cryptic natural product biosynthesis in microorganisms. Org Biomol Chem. 2009;7:1753–1760. doi: 10.1039/b821578b. [DOI] [PubMed] [Google Scholar]
- 4.Chiang YM, Chang SL, Oakley BR, Wang CC. Recent advances in awakening silent biosynthetic gene clusters and linking orphan clusters to natural products in microorganisms. Curr Opin Chem Biol. 2011;15:137–143. doi: 10.1016/j.cbpa.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mann J. Natural products in cancer chemotherapy: past, present and future. Nat Rev Cancer. 2002;2:143–148. doi: 10.1038/nrc723. [DOI] [PubMed] [Google Scholar]
- 6.Li JWH, Vederas JC. Drug discovery and natural products: End of an era or an endless frontier? Science. 2009;325:161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 7.Demain AL, Vaishnav P. Natural products for cancer chemotherapy. Microb Biotechnol. 2011;4:687–699. doi: 10.1111/j.1751-7915.2010.00221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cragg GM, Newman DJ. Natural products: a continuing source of novel drug leads. Biochim Biophys Acta, Gen Subj. 2013;1830:3670–3695. doi: 10.1016/j.bbagen.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li C, Hazzard C, Florova G, Reynolds KA. High titer production of tetracenomycins by heterologous expression of the pathway in a Streptomyces cinnamonensis industrial monensin producer strain. Metab Eng. 2009;11:319–327. doi: 10.1016/j.ymben.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 10.Duncan KR, Crusemann M, Lechner A, Sarkar A, Li J, Ziemert N, Wang M, Bandeira N, Moore BS, Dorrestein PC, Jensen PR. Molecular networking and pattern-based genome mining improves discovery of biosynthetic gene clusters and their products from Salinispora species. Chem Biol. 2015;22:460–471. doi: 10.1016/j.chembiol.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meier JL, Niessen S, Hoover HS, Foley TL, Cravatt BF, Burkart MD. An orthogonal active site identification system (OASIS) for proteomic profiling of natural product biosynthesis. ACS Chem Biol. 2009;4:948–957. doi: 10.1021/cb9002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, McClure RA, Zheng Y, Thomson RJ, Kelleher NL. Proteomics guided discovery of flavopeptins: anti-proliferative aldehydes synthesized by a reductase domain-containing non-ribosomal peptide synthetase. J Am Chem Soc. 2013;135:10449–10456. doi: 10.1021/ja4031193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doroghazi JR, Albright JC, Goering AW, Ju K, Haines RR, Tchalukov KA, Labeda DP, Kelleher NL, Metcalf WW. A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol. 2014;10:963–968. doi: 10.1038/nchembio.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goering AW, McClure RA, Doroghazi JR, Albright JC, Haverland NA, Zhang YB, Ju KS, Thomson RJ, Metcalf WW, Kelleher NL. Metabologenomics: Correlation of microbial gene clusters with metabolites drives discovery of a nonribosomal peptide with an unusual amino acid monomer. ACS Cent Sci. 2016;2:99–108. doi: 10.1021/acscentsci.5b00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doroghazi JR, Metcalf WW. Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genomics. 2013;14:611. doi: 10.1186/1471-2164-14-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baltz RH. Renaissance in antibacterial discovery from actinomycetes. Curr Opin Pharmacol. 2008;8:557–563. doi: 10.1016/j.coph.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 17.Subramani R, Aalbersberg W. Marine actinomycetes: an ongoing source of novel bioactive metabolites. Microbiol Res. 2012;167:571–580. doi: 10.1016/j.micres.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 18.Otake N, Kakinuma K, Yonehara H. Separation of detoxin complex and characterization of two active principles, detoxin C1 and D1. J Antibiot. 1968;21:371–373. doi: 10.7164/antibiotics.21.371. [DOI] [PubMed] [Google Scholar]
- 19.Yonehara H, Seto H, Aizawa S, Hidaka T, Shimazu A, Otake N. The detoxin complex, selective antagonists of blasticidin S. J Antibiot. 1968;21:369–370. doi: 10.7164/antibiotics.21.369. [DOI] [PubMed] [Google Scholar]
- 20.Bachmann BO, Ravel J. Chapter 8. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009;458:181–217. doi: 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- 21.Rottig M, Medema MH, Blin K, Weber T, Rausch C, Kohlbacher O. NRPSpredictor2–a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res. 2011;39:W362–367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T. antiSMASH 2.0–a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41:W204–212. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng YQ, Tang GL, Shen B. Type I polyketide synthase requiring a discrete acyltransferase for polyketide biosynthesis. Proc Natl Acad Sci U S A. 2003;100:3149–3154. doi: 10.1073/pnas.0537286100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neidig ML, Brown CD, Light KM, Fujimori DG, Nolan EM, Price JC, Barr EW, Bollinger JM, Jr, Krebs C, Walsh CT, Solomon EI. CD and MCD of CytC3 and taurine dioxygenase: role of the facial triad in alpha-KG-dependent oxygenases. J Am Chem Soc. 2007;129:14224–14231. doi: 10.1021/ja074557r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pohle S, Appelt C, Roux M, Fiedler HP, Sussmuth RD. Biosynthetic gene cluster of the non-ribosomally synthesized cyclodepsipeptide skyllamycin: deciphering unprecedented ways of unusual hydroxylation reactions. J Am Chem Soc. 2011;133:6194–6205. doi: 10.1021/ja108971p. [DOI] [PubMed] [Google Scholar]
- 26.Eliot AC, Griffin BM, Thomas PM, Johannes TW, Kelleher NL, Zhao H, Metcalf WW. Cloning, expression, and biochemical characterization of Streptomyces rubellomurinus genes required for biosynthesis of antimalarial compound FR900098. Chem Biol. 2008;15:765–770. doi: 10.1016/j.chembiol.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agata N, Ohta M, Mori M, Isobe M. A novel dodecadepsipeptide, cereulide, is an emetic toxin of Bacillus cereus. FEMS Microbiol Lett. 1995;129:17–20. doi: 10.1016/0378-1097(95)00119-P. [DOI] [PubMed] [Google Scholar]
- 28.Marxen S, Stark TD, Rutschle A, Lucking G, Frenzel E, Scherer S, Ehling-Schulz M, Hofmann T. Depsipeptide intermediates interrogate proposed biosynthesis of cereulide, the emetic toxin of Bacillus cereus. Sci Rep. 2015;5:10637. doi: 10.1038/srep10637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Igarashi Y, Yamamoto K, Fukuda T, Shojima A, Nakayama J, Carro L, Trujillo ME. Arthroamide, a cyclic depsipeptide with quorum sensing inhibitory activity from Arthrobacter sp. J Nat Prod. 2015;78:2827–2831. doi: 10.1021/acs.jnatprod.5b00540. [DOI] [PubMed] [Google Scholar]
- 30.Moore BS, Trischman JA, Seng D, Kho D, Jensen PR, Fenical W. Salinamides, antiinflammatory depsipeptides from a marine streptomycete. J Org Chem. 1999;64:1145–1150. [Google Scholar]
- 31.Ray L, Yamanaka K, Moore BS. A peptidyl-transesterifying type I thioesterase in salinamide biosynthesis. Angew Chem, Int Ed. 2016;55:364–367. doi: 10.1002/anie.201508576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagy V, Szakacs G. Production of transglutaminase by Streptomyces isolates in solid-state fermentation. Lett Appl Microbiol. 2008;47:122–127. doi: 10.1111/j.1472-765X.2008.02395.x. [DOI] [PubMed] [Google Scholar]
- 33.Shimazu A, Yamaki H, Furihata K, Endo T, Otake N, Yonehara H. Effect of detoxin D on blasticidin S uptake in Bacillus cereus. Experientia. 1981;37:365–366. doi: 10.1007/BF01959864. [DOI] [PubMed] [Google Scholar]
- 34.Cone MC, Yin X, Grochowski LL, Parker MR, Zabriskie TM. The blasticidin S biosynthesis gene cluster from Streptomyces griseochromogenes: sequence analysis, organization, and initial characterization. ChemBioChem. 2003;4:821–828. doi: 10.1002/cbic.200300583. [DOI] [PubMed] [Google Scholar]
- 35.Ocampo PS, Lazar V, Papp B, Arnoldini M, Abel zur Wiesch P, Busa-Fekete R, Fekete G, Pal C, Ackermann M, Bonhoeffer S. Antagonism between bacteriostatic and bactericidal antibiotics is prevalent. Antimicrob Agents Chemother. 2014;58:4573–4582. doi: 10.1128/AAC.02463-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwan JC, Donia MS, Han AW, Hirose E, Haygood MG, Schmidt EW. Genome streamlining and chemical defense in a coral reef symbiosis. Proc Natl Acad Sci U S A. 2012;109:20655–20660. doi: 10.1073/pnas.1213820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Udwary DW, Zeigler L, Asolkar RN, Singan V, Lapidus A, Fenical W, Jensen PR, Moore BS. Genome sequencing reveals complex secondary metabolome in the marine actinomycete Salinispora tropica. Proc Natl Acad Sci U S A. 2007;104:10376–10381. doi: 10.1073/pnas.0700962104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laureti L, Song L, Huang S, Corre C, Leblond P, Challis GL, Aigle B. Identification of a bioactive 51-membered macrolide complex by activation of a silent polyketide synthase in Streptomyces ambofaciens. Proc Natl Acad Sci U S A. 2011;108:6258–6263. doi: 10.1073/pnas.1019077108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juguet M, Lautru S, Francou FX, Nezbedova S, Leblond P, Gondry M, Pernodet JL. An iterative nonribosomal peptide synthetase assembles the pyrrole-amide antibiotic congocidine in Streptomyces ambofaciens. Chem Biol. 2009;16:421–431. doi: 10.1016/j.chembiol.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 40.Bumpus SB, Evans BS, Thomas PM, Ntai I, Kelleher NL. A proteomics approach to discovering natural products and their biosynthetic pathways. Nat Biotechnol. 2009;27:951–956. doi: 10.1038/nbt.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Albright JC, Goering AW, Doroghazi JR, Metcalf WW, Kelleher NL. Strain-specific proteogenomics accelerates the discovery of natural products via their biosynthetic pathways. J Ind Microbiol Biotechnol. 2014;41:451–459. doi: 10.1007/s10295-013-1373-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gubbens J, Zhu H, Girard G, Song L, Florea BI, Aston P, Ichinose K, Filippov DV, Choi YH, Overkleeft HS, Challis GL, van Wezel GP. Natural product proteomining, a quantitative proteomics platform, allows rapid discovery of bio-synthetic gene clusters for different classes of natural products. Chem Biol. 2014;21:707–718. doi: 10.1016/j.chembiol.2014.03.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.