

Summary
When bacterial lineages make the transition from free-living to permanent association with hosts, they can undergo massive gene losses, for which the selective forces within host tissues are unknown. We identified here melanogenic clinical isolates of Pseudomonas aeruginosa with large chromosomal deletions (66 to 270 kbp) and characterized them to investigate how they were selected. When compared with their wild-type parents, melanogenic mutants (i) exhibited a lower fitness in growth conditions found in human tissues, such as hyperosmolarity and presence of aminoglycoside antibiotics, (ii) narrowed their metabolic spectrum with a growth disadvantage with particular carbon sources, including aromatic amino acids and acyclic terpenes, suggesting a reduction of metabolic flexibility. Despite an impaired fitness in rich media, melanogenic mutants can inhibit their wild-type parents and compete with them in coculture. Surprisingly, melanogenic mutants became highly resistant to two intraspecific toxins, the S-pyocins AP41 and S1. Our results suggest that pyocins produced within a population of infecting P. aeruginosa may have selected for bacterial mutants that underwent massive gene losses and that were adapted to the life in diverse bacterial communities in the human host. Intraspecific interactions may therefore be an important factor driving the continuing evolution of pathogens during host infections.
Introduction
When bacterial lineages make the transition from free-living to permanent association with hosts, they often undergo a loss of genes (Moran, 2002). This long-term association with an unchanging environment requires little adaptability of the pathogen, rendering many bacterial genes useless and resulting in greater genetic drift and gene inactivation. Commonly, gene loss and genome reduction improve the ability of pathogens to survive in the host but also restrict the range of hosts available to the bacterium (Bentley and Parkhill, 2004; Ochman and Davalos, 2006).
Pseudomonas aeruginosa is an extremely common opportunistic pathogen. Its large genome (~6.5 Mbp) accounts for its metabolic versatility and affinity for various environmental niches, but also for its infectious capability towards a large set of hosts (Aujoulat et al., 2012). P. aeruginosa is a leading cause of hospital-acquired infections. It also causes a wide range of acute and chronic infections in predisposing situations such as chronic wounds, burn wounds and chronic obstructive pulmonary disease, the latter of which is particularly common among cystic fibrosis patients (Driscoll et al., 2007). Chronic infections with P. aeruginosa are associated with the high-density bacterial assemblages, such as bacterial biofilms, that favour the emergence of variants mostly arising through homologous recombination, recombinatorial DNA repair, and DNA mismatch repair deficiency (Oliver et al., 2000; Stoodley et al., 2002; Boles et al., 2004). The population dynamics within these populations can be affected by bacterial competition through the production of pyocins that kill susceptible subpopulations (Waite and Curtis, 2009).
The evolution and diversification of P. aeruginosa has been extensively studied in the respiratory chronic infection in individuals with cystic fibrosis (CF), during which this pathogen commonly loses genetic information either by gene inactivation or by genome reduction (Ernst et al., 2003; Marvig et al., 2015a, b). For example, mutations of the genes mexZ, mucA and lasR occur very frequently in lineages adapted to CF patients and confer selective advantages in host environments including antibiotic resistance, mucoidy and growth on abundant nitrogen sources, respectively (Marvig et al., 2015a). During in-host evolution, the chromosome of P. aeruginosa can also undergo large deletions of hundreds of genes (Ernst et al., 2003). However, the selective forces for such massive genetic losses in bacteria during chronic infections remain unknown.
In this work, we aimed at exploring the features of chronic infection bacterial isolates with large chromosomal deletions and at identifying the selective forces that could have driven their selection in the host. We focused on pyomelanin-producing mutants in which others have shown large chromosomal deletions (Ernst et al., 2003; Le et al., 2014). Overproduction of the pigment pyomelanin results in an easily recognizable red-brownish pigmented colony. Pyomelanin-producing mutants are regularly retrieved from the cultures of urine, sputum, bile or wounds, and up to 13% of the chronically infected CF patients harbour pyomelanin-producing mutants (Ogunnariwo and Hamilton-Miller, 1975; Mayer-Hamblett et al., 2014). Their pigmentation is due to homogentisic acid, which accumulates due to the inactivation of the gene hmgA (pa2009 in P. aeruginosa strain PAO1), which encodes homogentisate-1,2-dioxygenase (Rodriguez-Rojas et al., 2009). Although the inactivation of hmgA may occur in clinical strains by large chromosomal deletion that included hmgA (Ernst et al., 2003; Le et al., 2014), other authors have described single point mutations in this gene (Murugan et al., 2014; Tielen et al., 2014). First, we compared the genomes and phenotypes of a collection of clinical pyomelanin-producing mutants with those of their wild-type parents. We found that large chromosomal reduction was the main genetic event leading to pyomelanin production. Second, we identified consequent alterations in the fitness, metabolism, production of virulence factors, and resistance to antibiotics and toxins. These results identify the likely selective pressure that drove the emergence of these mutants: intraspecific competition through production of pyocins.
Results and discussion
The genome modifications that occur in P. aeruginosa during chronic infections often include large genomic deletions (>50 kb). These deletions, which tend to occur at specific hotspots distributed throughout the genome, often do not have easily identified phenotypic consequences (Ernst et al., 2003; Spencer et al., 2003; Rau et al., 2012). In contrast, deletions including hmgA lead to an easily recognizable brown colony phenotype. Therefore, pyomelanin-producing mutants are a convenient model to study the causes and consequences of large deletions that occur during chronic infections.
Pyomelanin-producing mutants contain large chromosomal deletions
We first compared the chromosome genetic sequences of one laboratory and four clinical brown mutants of P. aeruginosa with those of their genetically related precursor isolates (Table 1). This comparison revealed large deletions in each case, ranging in size from 66 kpb (isolate Bpm) to 270 kbp (isolate Apm) and representing from 1.05% to 4.32% of the genome. As expected, each deletion eliminated the hmgA gene (Fig. 1A and 1B). Forty-six genes in the vicinity of hmgA (from pa1974 to pa2019, representing 54.6 kbp) were also deleted in all the pyomelanin-producing isolates (Fig. 1C). No other mutations were found in the genome of the brown mutants compared with their wild-type precursors, including in the gene lasR.
Table 1.
Characteristics of the pyomelanin-producing isolates of P. aeruginosa and their precursor wild-type isolates.
| Pair of isolates | Sequence typea | Source | Isolateb | Production of pyomelanin | Isolation location | Type of infection or colonization | Age of the infection/colonization (days)c | Age of the patient (years) | Antibiotic treatmentd | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| PAO1/PAO1 ΔhmgA | ST549 | Laboratory | PAO1 PAO1 ΔhmgA |
No Yes |
–e | – | – | – | – | (Jacobs et al., 2003) |
| PAO1/CIP | ST549 | Laboratory | PAO1 CIP |
No Yes |
– | – | – | – | Ciprofloxacin | This work |
| A | ST253 | Clinical | Awt Apm |
No Yes |
Sinus | Sinusitis | 10 | 72 | Ceftriaxone, fosfomycin, amikacin | This work |
| B | ST298 | Clinical | Bwt Bpm |
No Yes |
Blood culture | Urinary sepsis | 22 | 56 | Cotrimoxazole | This work |
| C | ST253 | Clinical | Cwt Cpm |
No Yes |
Tracheal aspirate | Tracheal colonization | 36 | 49 | Co-amoxiclav | This work |
| D | ST974 | Clinical | Dwt Dpm |
No Yes |
Sputum | Pulmonary infection in cystic-fibrosis patient | 51 | 0.8 | Colimycin | This work |
As determined using the MLST scheme described by Curran et al. (Curran et al., 2004).
WT, wild-type; PM, pyomelanin-producing isolate.
Duration as first P. aeruginosa isolation.
Antibiotic treatment received by the patients before the emergence of pyomelanin-producing isolates.
Not applicable.
Fig. 1.
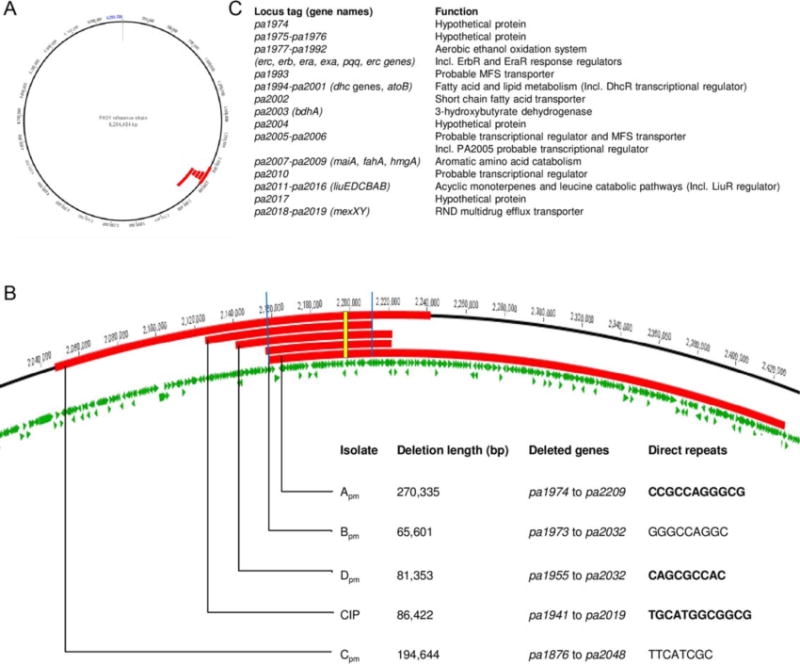
Large chromosomal deletions in pyomelanin-producing P. aeruginosa clinical isolates.
A. Location (in red) relative to sequenced P. aeruginosa strain PAO1 of chromosomal deletions in five brown P. aeruginosa mutants compared with wild-type precursors.
B. Close-up 1A, indicating the deleted region for each pyomelanin-producing mutant in this study. The blue bars surround the genes deleted in all the pyomelanin-producing mutants. The yellow rectangle indicates hmgA gene. The length of the deletion and the deleted genes are indicated. The direct repeats putatively involved in the deletion are detailed (PCR-verified repeats are bold-typed).
C. Name and function of the genes deleted in all the pyomelanin-producing mutants.
We confirmed that deletion of the region pa1974-pa2018, which includes hmgA, occurs commonly during chronic infections by identifying this change in an independent additional collection of 5 other pyomelanin-producing isolates compared with their wild-type parents (Supporting Information table S1). Only one additional brown isolate (3447) had a nucleotide substitution leading to a Leu95Pro change in HmgA when compared with its parent. Because single point mutations in hmgA are fairly uncommon in clinical isolates (i.e., in 1 out of 10 isolates tested), it is unlikely that the in vivo selection of these mutants is only due to properties conferred by hmgA deletion itself. Rather, the brown color of the bacterial colonies due to the hmgA mutation likely represents a surrogate marker of a large chromosomal deletion encompassing this gene that was selected for other reasons.
Comparative sequence analysis of wild-type isolates PAO1, Awt, Bwt, Cwt and Dwt revealed direct repeats (from 8 bp to 12 bp) at the boundaries of the genomic region missing from the brown clinical isolates, among which these repeats occurred in only a single copy at the deletion sites. We confirmed the presence of the direct repeats using PCR and sequencing in the pairs PAO1/CIP, A, and D (Fig. 1B). These data were consistent with a recombination event between these direct repeats, facilitating the excision of the large chromosomal sequences.
Pyomelanin-producing mutants exhibit narrowed metabolic substrate utilization profiles
Screening for phenotypic differences between pyomelanin-producing mutants and their wild-type parents was performed using the Biolog method. This analysis indicated relative growth defects among pyomelanin-producing clinical mutants in high (9.5) pH, in 6% NaCl, and when aromatic amino acids provided the sole sources of either nitrogen or carbon. We first experimentally confirmed that one of these metabolic differences was exhibited by all pyomelanin-producing mutants: a growth defect when tyrosine was the sole C-source (Fig. 2A). As seen with the PAO1 hmgA mutant, this difference was expected due to the fact that HmgA is involved in the catabolic pathway for tyrosine.
Fig. 2.
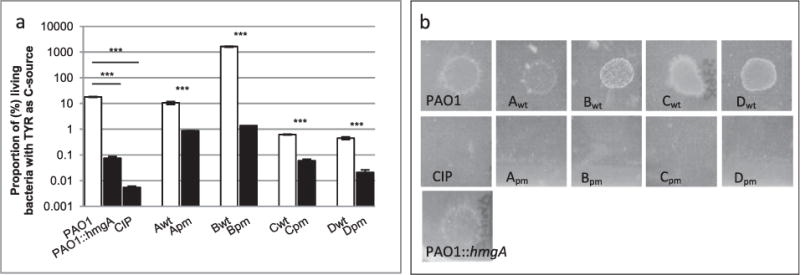
Metabolic changes in pyomelanin-producing mutants of P. aeruginosa.
A. Proportion of living bacteria after 24 h of incubation in minimal media M63 supplemented with 0.05% of tyrosine as the sole C-source as described before (see Supplementary Information). Ratio of means ± SEM from at least 3 independent experiments; two-sided Student’s t-tests; NS, P > 0.05; ***, P < 0.001.
B. Bacterial growth on M63 plates supplemented with 0.1% geraniol as the only C-source and incubated for 48 h at 37°C followed by 10 days at 30°C.
Other metabolic genes were missing from the chromosomes of all tested pyomelanin-producing clinical isolates (Fig. 1C) including the liu genes, necessary for the catabolism of the amino acid leucine and that of acyclic terpenes (e.g., citronellol, geraniol). We confirmed that the brown mutants did not grow with the geraniol as the sole C-source, in contrast with their isogenic parents (Fig. 2B). In addition, deletions of the metabolic genes erbR, eraSR, exaA and pqqC have been shown to impair growth on ethanol, and the deletion of genes dhcRAB are necessary for the use of carnitine (Wargo and Hogan, 2009; Mern et al., 2010; Beaudoin et al., 2012). Taken together, these data suggest a reduction of metabolic versatility, and therefore, of niche range, of the pyomelanin-producing mutants.
Pyomelanin-producing mutants are more susceptible to harsh environments
The gene mexY was missing on the chromosome of all the melanogenic-producing mutant isolates. The deletion of this protein transporter, involved in the intrinsic resistance to aminoglycosides, resulted in a twofold to fourfold higher susceptibility of the brown mutants to tobramycin, an aminoglycoside antibiotic used widely to treat P. aeruginosa infection (Fig. 3A; Ramos Aires et al., 1999; Vogne et al., 2004). In contrast, we did not observe any clear trend in the susceptibility of brown mutants to β-lactams, to ciprofloxacine, and to colistin (Supporting Information table 2).
Fig. 3.
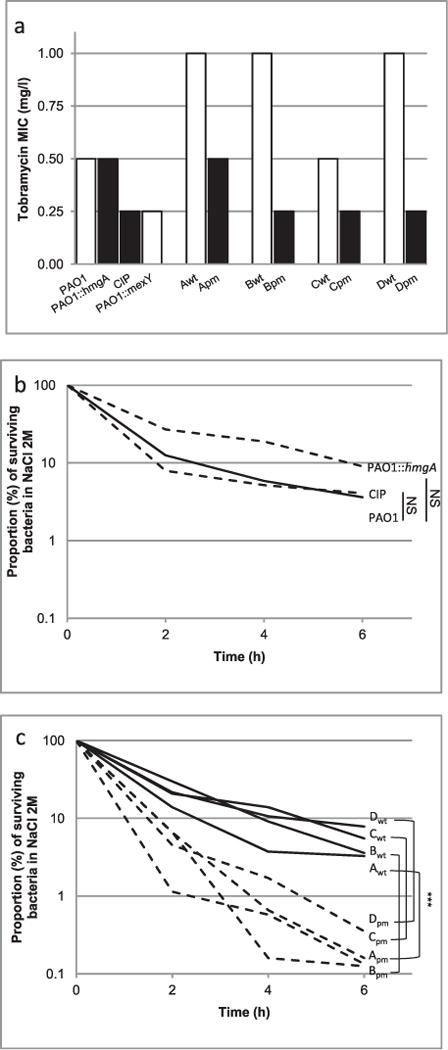
Survival of the pyomelanin-producing mutants of P. aeruginosa in harsh environments.
A. The minimal inhibitory concentrations of tobramycin were determined by Etest.
B and C. The resistance to osmotic shock was determined in Luria-Bertani broth supplemented with 2M NaCl. Ratio of means ± SEM from at least 3 independent experiments; two-sided Student’s t-tests; NS, P > 0.05; ***P < 0.001.
We experimentally confirmed the higher susceptibility to basic pH (pH9.5) of three out of four pyomelanin-producing clinical mutants. This feature was independent of the deletion of hmgA (Supporting Information fig. S1a). Although PAO1 and its two melanogenic derivatives behaved similarly in a hyperosmotic media containing 2 M of NaCl (Fig. 3B), all the clinical mutants producing pyomelanin were more susceptible to this osmotic shock than were their isogenic wild-type parents (Fig. 3C). This finding was particularly unexpected for the isolate Dpm retrieved from the lung of a CF-patient, as the osmolarity of CF airway secretions are thought to be high in osmolarity (Henderson et al., 2014).
It has been shown that the production of pyomelanin after the inactivation of hmgA increases the resistance of P. aeruginosa to oxidative stress (Rodriguez-Rojas et al., 2009). Here, although we confirmed the higher resistance of an engineered hmgA mutant to challenge with 50 mM of H2O2 for 45 min compared with its parent PAO1, only one clinical mutant (Apm) out of the four that underwent a large deletion was more resistant to H2O2 than its isogenic counterparts (Supporting Information fig. S1b). This finding makes it unlikely that pyomelanin-producing mutants are selected in the host based on a higher resistance to reactive oxygen species. Therefore, loss of other genes in the vicinity of hmgA presumably counterbalanced the protective effect of pyomelanin. We found that all the pyomelanin-producing clinical mutants were more susceptible to hyperosmolarity and the presence of aminoglycosides, and that three out of four were more susceptible to high pH. These stress conditions are found in the human tissues. However, these findings must be interpreted with caution. For example, oxidative stress in vivo presumably occurs primarily due to superoxide anions produced by macrophages and monocytes in response to engulfed bacteria or bacterial products and that help to kill phagocytosed bacteria. However, exposure to these products may not occur in vivo as they do in in vitro cultures due either to biofilm formation, physicochemical variability and/or gradients, or other unknown mechanisms (Costerton et al., 1999). Therefore, the relationships between in vitro and in vivo susceptibilities to specific conditions are difficult to predict.
Pyomelanin-producing mutants produced less virulence factors in vitro
We compared the in vitro production of virulence factors (elastase, rhamnolipids and pyocyanin), biofilm formation, haemolytic activities, and swarming, swimming and twitching motilities among these isolates and strains. Production of rhamnolipids, swimming, swarming and twitching motilities were not affected in the pyomelanin-producing mutants (data not shown). In contrast, the production levels of pyocyanin, elastase and the haemolytic activity were significantly reduced in three, one and three clinical mutants, respectively (Supporting Information fig. S2). The haemolytic activity is presumably attributed to PlcH, which gene (pa0844) is present in the genome of all the mutants and parental strains. In contrast, the production of none of the virulence factors tested was affected in the PAO1 hmgA mutant (Supporting Information fig. S2). The gene galU (pa2023) encodes a UDP-glucose pyrophosphorylase that catalyzes an essential step for the synthesis of a complete lipopolysaccharide (LPS) outer core. GalU mutants produce truncated (rough) LPS molecules and display attenuated virulence in part due to higher serum sensitivity (Dean and Goldberg, 2002; Priebe et al., 2004). As determined by the O-serotyping by specific antisera, the wild-type isolates PAO1, Awt, Bwt and Cwt harboured the O:5, O:16, O:11, and O:16 antigens, respectively. Consistent with the deletion of the galU gene from their genomes (Fig. 1C), the pyomelanin-producing clinical isolates Apm, Bpm, and Cpm autoaggregated, presumably due to the lack of the parental O-antigen (Dean and Goldberg, 2002). We measured the kinetics of biofilm formation of all the isolates and found that three out of the four pyomelanin-producing clinical isolates (Bpm, Cpm, Dpm) significantly produced less early biofilm than their wild-type parents (Fig. 4). This impairment was not clearly related to the size and the gene content of the chromosomal deletions underwent by the brown mutants.
Fig. 4.
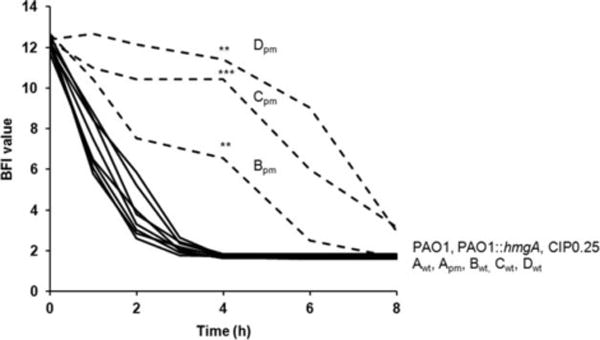
Kinetics of the early phase of biofilm formation of pyomelanin-producing mutants of P. aeruginosa and their wild-type parents. Measures were done by the BioFilm Ring Test® (BioFilm Control). Means of BFI (BioFilm Indice) for four replicates from two independent experiments. Standard deviations were not displayed for ease of reading. Statistical differences at 4 h were calculated with two-sided Student’s t-tests; **P < 0.01; ***P < 0.001.
Fitness and coculture
The persistence of the pyomelanin-producing mutants in the source patients led us to suspect that the chromosome deletions observed in the pyomelanin-producing mutants conferred a growth advantage in vivo (Mayer-Hamblett et al., 2014). Therefore, we searched for in vitro conditions under which these mutants had a growth advantage. Surprisingly, we found a lower relative fitness of pyomelanin-producing strains under both aerobic and anaerobic conditions in rich liquid media in comparison with their wild-type parents (Fig. 5A and 5B). We then hypothesized that the brown mutants had an advantage over their wild-type parents in biofilm conditions (Costerton et al., 1999). To test this hypothesis, we cocultured each pair of isolates in growth conditions that favor the formation of a biofilm (Waite and Curtis, 2009). While the CIP laboratory mutant outgrew its wild-type parent PAO1 after 24 h of biofilm growth, the isolates Cpm and Dpm stayed in similar proportion with their wild-type parent after 24 h of coculture despite their lower fitness in anaerobiosis (Fig. 5C). In contrast, the PAO1-derivated hmgA mutant and the clinical isolates Apm and Bpm were outcompeted by their wild-type parents after 24 h of coculture. To determine how some pyomelanin-producing mutants could compete with their parents in biofilms, we tested the ability each strain’s supernatant to inhibit growth of its isogenic counterpart. Others have shown that a general stress response known as the SOS reponse is triggered in biofilms (Beloin et al., 2004; Waite and Curtis, 2009). To stimulate the SOS response and to mimic this biofilm stress, each strain was cultured in this experiment in media containing 1 mg/l of the SOS-inducing mitomycin C (Hocquet et al., 2012). Consistent with the biofilm competition results, the supernatants of the three pyomelanin-producing mutants that effectively competed with their wild-type parents (CIP, Cpm and Dpm) also inhibited growth of those parent strains on agar media (Fig. 5D). The role of the 19 genes (pa1955-pa1973; Fig. 1b) that are deleted only in CIP, Cpm and Dpm while present in the other brown mutants still remains to be explored.
Fig. 5.
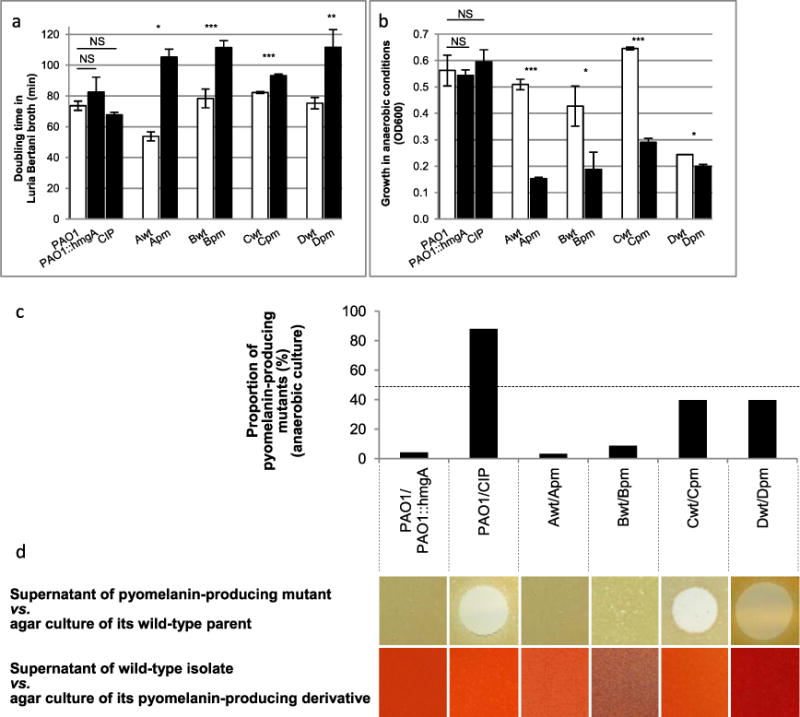
Brown mutants can compete with their wild-type parents in coculture despite lower fitness in monoculture. Means 6 SEM from at least 3 independent experiments; two-sided Student’s t-tests; NS, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001.
A. Growth in Luria-Bertani broth under aerobic conditions at 37°C under shaking at 150 r.p.m.
B. Growth in Luria-Bertani broth supplemented with 0.4 mM KNO3 under anaerobic conditions without shaking.
C. Relative proportion of pyomelanin-producing mutants after 24 h of coculture with their isogenic parents in biofilm under anaerobic conditions as described elsewhere (Waite and Curtis, 2009).
D. Inhibitory activity of 30 μl of the supernatant of an overnight culture of pyomelanin-producing mutants in MH broth containing 1 mg/l of the SOS response inducer mitomycin C against a culture on MH agar of their wild-type parent (up). Inhibitory activity of 30 μl of the supernatant of a 12-h culture of the wild-type parent in MH broth containing 1 mg/l of mitomycin C against a culture on MH agar of their pyomelanin-producing derivative (down). As a control, 30 μl of MH broth containing 1 mg/l of mitomycin C incubated overnight had no inhibitory activity against any of the strains and isolates shown on agar culture (data not shown). The 2 variant colonies growing in the inhibition spot of the Cwt were found – after an overnight subculture on MH agar – as susceptible to the Cpm supernatant as their parent (data not shown).
Pyomelanin-producing mutants are resistant to S-pyocins
Bacteria are known to diversify both in vivo during chronic infections and in vitro during growth in biofilms, and biofilms are thought to underlie many chronic infections (Boles et al., 2004; Marvig et al., 2015b). It has been shown that the dynamics of variants that arise in biofilms can be shaped by the production of pyocins, which are produced at increased levels during anaerobic biofilm growth (Waite and Curtis, 2009). P. aeruginosa is capable of producing at least three different pyocins (of S-, R- and F-types) that kill other strains from the same species. More specifically, S-pyocins kill bacteria through DNase, tRNase, or pore-forming activity (Michel-Briand and Baysse, 2002). Interestingly, Holloway et al. described in 1973 two mutants tolerant to the S-pyocin AP41 that ‘produced copious brown pigment in the media’, suggesting a potential relationship between pyocin resistance and brown mutant selection (Holloway et al., 1973). Therefore, we tested the inhibitory effect of the S-pyocins AP41, S1, S2, S3, and the R-pyocins R1, R2, R3, R5 on our collection and found that, unlike their wild-type parents, the mutants CIP, Apm, Bpm and Cpm were all resistant to AP41, and the mutants Apm, Bpm and Dpm were resistant to S1 (Table 2; Additional file 4: Supporting Information). Therefore, each brown clinical mutant had acquired new resistance to at least one tested S-pyocin. These resistances were not conferred by deletion of hmgA, since the PAO1 hmgA mutant remained as susceptible to AP41 as its parent strain was. The resistance to some pyocins has been attributed to modification of the lipopolysaccharides which production depends (?) on galU gene, deleted in all the brown mutants (Kohler et al., 2010). We then tested the resistance to the S-pyocins AP41 and S1 of the PAO1galU mutant (El Garch et al., 2007) and found that it was as susceptible to the S-pyocins tested as its wild-type parent PAO1. No change in the suceptibility to pyocins S2 and S3 and to the R-pyocins R1, R2, R3, R5 occurred in pyomelanin-producing isolates (data not shown). Unlike the killing mechanism of pyocins S2 and S3, which depends on the FpvA ferripyoverdine receptors, the resistance mechanisms to pyocins S1 and AP41 are still unknown (Denayer et al., 2007). Our data confirm that the resistance to pyocins mostly occurs via mechanisms independent from specific immunity genes (Ghoul et al., 2015). The resistances to S1 and AP41 pyocins could be due to the loss of a membrane receptor that allows the entry of these toxins into the bacterial cell. As we did not find any obvious receptor-encoding genes within the chromosomal region missing from the brown mutants, we speculated the presence within this region of a gene encoding a regulator which deletion abolishes or downregulates the production of a pyocin receptor. These results could indicate that the relative resistance to AP41 and/or S1-pyocins of the pyomelanin-producing mutants plays a key role in the survival of these adapted mutants in patients; in support of this concept, nearly all (98%) the clinical isolates of P. aeruginosa isolated from CF-patients have been observed to produce at least one S-pyocin (Ghoul et al., 2015). However, we are aware that the pyocins that we did not test (S-pyocins S4, S5, S6, R-pyocin R4 and F-pyocins) could also play a role in bacterial competition.
Table 2.
Susceptibility to S-pyocins of the pyomelanin-producing isolates of P. aeruginosa and their wild-type parents.
| Isolate | Lysis by S-type pyocins
|
|
|---|---|---|
| AP41 | S1 | |
| PAO1 | +a | − |
| PAO1::hmgA | + | − |
| CIP | − | − |
| Awt | + | + |
| Apm | − | − |
| Bwt | + | + |
| Bpm | − | − |
| Cwt | + | − |
| Cpm | − | − |
| Dwt | − | + |
| Dpm | − | − |
+, lysis; −, no lysis.
Emergence of pyomelanin-producing mutants
The four patients from whom pyomelanin-producing mutants were isolated differed in terms of the anatomic location of their infections, which included sinusitis, endocarditis, pulmonary infection and endotracheal colonization, all of which have been proposed to involve bacterial bio-films (Costerton et al., 1999). In addition, the source patients were treated with unrelated antibiotics (Table 1). Elsewhere, Cabot et al. have shown that meropenem, another antibiotic, can select pyomelanin-producing mutants (Cabot et al., 2016). Biofilms are structured multicellular communities of microorganisms associated with surfaces that are believed to play a role in at least 65% of all human infections, in particular device-related infections, body surface infections, and other chronic infections (Costerton et al., 1999). Therefore, the brown mutants tested here could all have occurred in biofilms in vivo.
The resistance of pyomelanin-producing mutants to S-pyocins was the only unifying characteristic that could have played a selective role. We then hypothesized that pyocin resistance was selected during biofilm growth in these mutants. Pyocin-resistant mutants have been readily selected in vitro by the S-pyocin AP41, and Boles et al. further showed that these mutants mostly emerge in biofilm, depending on RecA for recombination (Holloway et al., 1973; Boles et al., 2004). To test whether pyomelanin-producing mutants could have been specifically selected from bacterial biofilms of wild-type isolates, we grew PAO1, Awt, Bwt and Cwt both in planktonic and biofilm cultures before selection on MH agar plates supplemented with AP41 and quantified the proportion of pyocin AP41-resistant mutants that produced pyomelanin. Dwt was deliberately not tested since already resistant to AP41. While no pyomelanin-producing mutants were retrieved from planktonic cultures, such mutants were easily selected from biofilms, as previously shown (Boles et al., 2004). The average frequency of emergence of AP41-resistant mutants was 1.9 × 10−3 from biofilms, with pyomelanin-producing mutants representing a large part of pyocin-resistant mutants (from 31% to 64%, depending on the strain tested, PAO1, Awt, Bwt, or Cwt), with an average frequency of pyomelanin-producing mutants of 1.1 × 10−3. The emergence of pyomelanin-producing mutants depends on the presence of the DNA recombinase RecA, which is activated in biofilm, while independent from the error-prone DNA polymerase DinB (Boles et al., 2004). Given our observation of direct repeats at the boundaries of the chromosomal deletions (Fig. 1B) and the relative rarity of point mutation in hmgA in our pyomelanin-producing mutant collection, these results indicate that recombination is the primary mechanism of selection of these mutants.
We found here that all the tested pyomelanin-producing clinical isolates contained large chromosomal deletions (≥65.6 kbp). The diversity of the genetic background of the strains (Table 1; Supporting Information fig. S3) and the size change of the chromosomal deletion in mutants (Fig. 1B) certainly account for the variability of their phenotypes. However, all the brown mutants tested displayed a lower fitness, a narrowed substrate utilization profiles and an increased in vitro susceptibility to hyperosmolarity and antibiotics. While these isolates also exhibited reduced growth in monoculture compared with their wild-type parents, the mutants could inhibit their parents in coculture. Importantly, the pyomelanin-producing mutants all displayed increased resistance to S-pyocins, likely favoring their survival in mixed population of P. aeruginosa in patients. Our results suggest that the production of pyomelanin resulting from the deletion of hmgA is a phenotypic marker of a large chromosomal deletion in P. aeruginosa mutants adapted to the life in diverse bacterial communities in the human host. These pyomelanin-producing mutants lost much of the metabolic flexibility exhibited by free-living bacteria and produced less virulence factors in vitro.
The pathogen chromosome reductions that occur in many chronic infections often result in the loss of unnecessary regulatory elements. The relatively static environment of host tissues eliminates the extreme environmental fluctuations encountered by free-living bacteria (Moran, 2002). Interactions between the host and the bacteria can drive the microbe evolution (Marvig et al., 2015a). We found that pyomelanin-producing mutants emerged in diverse clinical situations and from various genetic backgrounds. This suggests that the selection of pyomelanin-producing mutants is unlikely due to host interactions or antibiotic treatments. Here, we propose that the bacterial adaption to the host can be shaped by pyocin-mediated interbacterial interactions that are facilitated by growth in biofilms.
Experimental procedures
Bacterial isolates
We used six pyomelanin-producing mutants of P. aeruginosa (suffixed with ‘pm’) and their non-pyomelanin-producing parents (suffixed with wt, for wild-type). Each clinical pair (A, B, C and D) was isolated the same day in the same specimen from the same patient. The genetic similarity of P. aeruginosa clinical isolates was investigated by pulsed field gel electrophoresis (PFGE; CHEF-DR III; Bio-Rad, Hercules, California) with the use of DraI enzyme, as described elsewhere (Supporting Information fig. S3; Talon et al., 1996). We also used two derivatives from the reference strain PAO1: the mutant PAO1::hmgA with hmgA gene inactivated with the transposon ISphoA/hah (Jacobs et al., 2003) and the mutant CIP fortuitously isolated from PAO1 culture on agar containing 0.25 mg/l of ciprofloxacin. Table 1 details the characteristics of the clinical isolates and the patient source.
Genome comparison
The reads from the chromosomes of the pyomelanin-producing isolates and their wild-type parents were generated with Illumina GAIIx sequencer (Illumina, San Diego, CA). The reads were mapped onto the genomic sequence of the reference strain P. aeruginosa PAO1 using BWA and Samtools (Stover et al., 2000; Li and Durbin, 2009; Li et al., 2009). For each pair, we looked for regions of the PAO1 genome that were covered by reads from the wild-type parent but not by reads from its pyomelanin-producing derivative. Only regions with a minimum coverage of 10 and a minimum quality score of 15 were considered. The position of the deleted region was validated by the graphical view of reads distribution. To confirm the position of the deletions in pyomelanin-producing isolates, we designed four primers for each pair of strains to perform three PCR reactions (Supporting Information table S3). A first PCR with primers 1 and 2 overlapped the 3′-edge of the deletion in the chromosome of the wild-type parent. A second PCR with primers 3 and 4 overlapped the 5′-edge of the deletion. A third PCR with primers 1 and 4 overlapped the ‘scar’ of the deletion in the chromosome of the pyomelanin-producing isolates. Direct repeats potentially involved in the deletion were identified with Repfind (http://genometools.org). We predicted the sequence type (ST) of the isolates from their whole genome sequence data with the MLST 1.7 tool (Larsen et al., 2012).
Confirmation of deletion by PCR
To confirm the chromosomal deletion in other pyomelanin-producing clinical mutants, we PCR amplified internal fragments of the genes pa1974, pa1989, pa2009 and pa2019 in an independent collection of six pyomelanin-producing clinical mutants and in their wild-type parents (Supporting Information table S1).
Phenotype determination
We screened the metabolic changes in the pyomelanin-producing mutants with the Phenotype MicroArray analysis (Biolog, Hayward, CA) in duplicate according to the manufacturer’s instructions. The bacterial growth of the pyomelanin-producing mutants was assessed in 950 conditions (i.e., with various carbon, nitrogen, phosphorus, sulfur and peptide nitrogen sources, various nutrient supplements, with various osmolytes) and compared with that of their isogenic parent. The conditions are available at http://www.biolog.com/pdf/pm_lit/PM1-PM10.pdf. The supplemental materials detail the conditions under which we assessed the bacterial fitness, the changes in utilization of aromatic amino acids and terpene compounds, the survival in harsh environment, and the production of virulence factors. We evaluated the kinetics of early biofilm formation of all the isolates of the collection using the Biofilm Ring Test (BioFilm Control, Saint Beauzire, France) as previously described (Olivares et al., 2016).
Coculture
Cocultures of pyomelanin-producing mutants with their wild-type parents were performed in anaerobic biofilms which favor the production of pyocins (Waite and Curtis, 2009). Briefly, 105 CFU of an overnight culture of the pyomelanin-producing mutants and 105 CFU of their wild-type parents were mixed and grown on 0.45-μm nitrocellulose filters (Millipore) placed on reduced-strength LB agar (20%) containing 1% KNO3 to permit anaerobic respiration. Dishes were incubated 24 h at 37°C in an anaerobic cabinet (MACS – MG 500 Work Station; DW Scientific). The proportion of each cocultured isolate was assessed by plating an appropriate dilution of the culture on MHA incubated overnight at 37°C. Randomly-chosen colonies (n > 100) were streaked on MHA incubated overnight at 37°C before observation of the colony pigmentation.
Pyocin resistance determination
E. coli DH5α strains harboring plasmids pYMSS11, pYMPS1, pPYS3-3pY or pYK211 (encoding for the pyocins S1, S2, S3 or AP41, respectively) were cultured 6 h at 37°C in LB broth containing 100 mg/l of ampicillin and then centrifuged (Sano and Kageyama, 1993; Sano et al., 1993; Duport et al., 1995). The pellet was resuspended in PBS buffer and sonicated on ice. The lysate was cleared by centrifugation 20 min, 1200 g at 4°C. The supernatant was sterilized through a 0.22 μm pore size filter and stored at −80°C upon utilization. MHA plates were inoculated with a lawn of P. aeruginosa isolates, according to CLSI disk diffusion method, before dropping 30 μl of the pyocin solution and overnight incubation at 37°C.
Emergence of pyomelanin-producing mutants
To assess the conditions under which the pyomelanin-producing mutants emerge, we cultured the wild-type isolates PAO1, Awt, Bwt, Cwt and Dwt in 5 ml of MH broth at 37°C under shaking either 18 h or 5 days with daily media changes. Dilutions of the resulting cultures were then plated on MHA plates containing 1.5% of the pyocin AP41 and incubated 24 h at 37°C. Fifty randomly-chosen AP41-resistant mutants were streaked on MHA for the observation of colony pigmentation.
Statistical analysis
Student’s t-tests were used to determine statistical significance for comparisons of survival in tyrosine and in harsh environment, of biofilm and growth parameters, and of production of virulence factors. Graphical examination supports the assumption of normality and homogeneous variation across experiments. The chosen significance threshold was 0.05 for all tests.
Supplementary Material
Figure S1. Survival of the pyomelanin-producing mutants of P. aeruginosa in high pH and under oxidative stress. a, proportion of surviving bacteria after 24h of incubation at 37°C under shaking in LB broth pH9.5. Ratio of means ± SEM from at least 3 independent experiments; two-sided Student’s t-tests; NS, P>0.05; **, P<0.01; ***, P<0.001. b, proportion of surviving bacteria after 45 min of exposure to 50 mM H2O2 (Rodriguez-Rojas et al., 2009). Ratio of means ± SEM from at least 3 independent experiments; two-sided Student’s t-tests; NS, P>0.05; *, P<0.05, **, P<0.01; ***, P<0.001.
Figure S2. Production of virulence factors by pyomelanin-producing P. aeruginosa. Means or ratio of means ± SEM from at least 3 independent experiments; two-sided Student’s t-tests; NS, P>0.05; *, P<0.05; **, P<0.01; ***, P<0.001. a, elastase activity was quantitated with Elastin Congo Red assay. b, proportion of sheep red blood cells that were hemolyzed after incubation with the supernatant of an overnight bacterial culture. c, the production of pyocyanin was quantified as previously described (see Suppl. materials).
Figure S3. Pulsed-field gel electrophoresis profiles of DraI-digested DNA from P. aeruginosa isolates. a, first series with A, B, C, and D pairs. b, complementary series. The brackets indicate two isogenic isolates isolated from the same patient – a wild-type isolate and its pyomelanin-producing mutant.
Table S1. PCR detection of the genes pa1974, pa1989, pa2009 and pa2018 among a collection of pyomelanin-producing clinical isolates of P. aeruginosa and in their isogenic parents. + and − signs indicate a positive and a negative PCR reaction with specific primers, respectively.
Table S2. Susceptibility of the pyomelanin-producing mutants and their isogenic counterparts to the major anti-pseudomonal compounds.
Table S3. Primers used in the study.
Acknowledgments
We thank Beth Ramage, Marie-Pierre Blanc, and Marion Manzoni for their technical support and also Yumico Sano for the gift of pYMPS1 and pYK211. We thank Emmanuelle Dé, Thierry Jouenne and Christine Baysse for helpful discussion.
Footnotes
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site:
Conflict of interests
The authors have declared that no competing interests exist.
Ethics statement
Approval and written informed consent from all subjects or their legally authorized representatives were obtained before study initiation. The study was approved by the ethical committee ‘Comité d’Etude Clinique’ of the Besançon hospital, Besançon, France.
References
- Aujoulat F, Roger F, Bourdier A, Lotthe A, Lamy B, Marchandin H, Jumas-Bilak E. From environment to man: genome evolution and adaptation of human opportunistic bacterial pathogens. Genes (Basel) 2012;3:191–232. doi: 10.3390/genes3020191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoin T, Zhang L, Hinz AJ, Parr CJ, Mah TF. The biofilm-specific antibiotic resistance gene ndvB is important for expression of ethanol oxidation genes in Pseudomonas aeruginosa biofilms. J Bacteriol. 2012;194:3128–3136. doi: 10.1128/JB.06178-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloin C, Valle J, Latour-Lambert P, Faure P, Kzreminski M, Balestrino D, et al. Global impact of mature biofilm lifestyle on Escherichia coli K-12 gene expression. Mol Microbiol. 2004;51:659–674. doi: 10.1046/j.1365-2958.2003.03865.x. [DOI] [PubMed] [Google Scholar]
- Bentley SD, Parkhill J. Comparative genomic structure of prokaryotes. Annu Rev Genet. 2004;38:771–791. doi: 10.1146/annurev.genet.38.072902.094318. [DOI] [PubMed] [Google Scholar]
- Boles BR, Thoendel M, Singh PK. Self-generated diversity produces “insurance effects” in biofilm communities. Proc Natl Acad Sci USA. 2004;101:16630–16635. doi: 10.1073/pnas.0407460101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabot G, Zamorano L, Moyà B, Juan C, Navas A, Blázquez J, Oliver A. Evolution of Pseudomonas aeruginosa antimicrobial resistance and fitness under low and high mutation rates. Antimicrobial Agents and Chemotherapy. 2016;60:1767–1778. doi: 10.1128/AAC.02676-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- Dean CR, Goldberg JB. Pseudomonas aeruginosa galU is required for a complete lipopolysaccharide core and repairs a secondary mutation in a PA103 (serogroup O11) wbpM mutant. FEMS Microbiol Lett. 2002;210:277–283. doi: 10.1111/j.1574-6968.2002.tb11193.x. [DOI] [PubMed] [Google Scholar]
- Denayer S, Matthijs S, Cornelis P. Pyocin S2 (Sa) kills Pseudomonas aeruginosa strains via the FpvA Type I ferripyoverdine receptor. J Bacteriol. 2007;189:7663–7668. doi: 10.1128/JB.00992-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll J, Brody S, Kollef M. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs. 2007;67:351–368. doi: 10.2165/00003495-200767030-00003. [DOI] [PubMed] [Google Scholar]
- Duport C, Baysse C, Michel-Briand Y. Molecular characterization of pyocin S3, a novel S-type pyocin from Pseudomonas aeruginosa. J Biol Chem. 1995;270:8920–8927. doi: 10.1074/jbc.270.15.8920. [DOI] [PubMed] [Google Scholar]
- El Garch F, Jeannot K, Hocquet D, Llanes-Barakat C, Plésiat P. Cumulative effects of several nonenzymatic mechanisms on the resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob Agents Chemother. 2007;51:1016–1021. doi: 10.1128/AAC.00704-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst RK, D’Argenio DA, Ichikawa JK, Bangera MG, Selgrade S, Burns JL, et al. Genome mosaicism is conserved but not unique in Pseudomonas aeruginosa isolates from the airways of young children with cystic fibrosis. Environ Microbiol. 2003;5:1341–1349. doi: 10.1111/j.1462-2920.2003.00518.x. [DOI] [PubMed] [Google Scholar]
- Ghoul M, West SA, Johansen HK, Molin S, Harrison OB, Maiden MCJ, Jelsbak L, Bruce JB, Griffin AS. Bacteriocinmediated competition in cystic fibrosis lung infections. Proc R Soc B. 2015;282:20150972. doi: 10.1098/rspb.2015.0972. http://dx.doi.org/10.1098/rspb.2015.0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson AG, Ehre C, Button B, Abdullah LH, Cai LH, Leigh MW, et al. Cystic fibrosis airway secretions exhibit mucin hyperconcentration and increased osmotic pressure. J Clin Invest. 2014;124:3047–3060. doi: 10.1172/JCI73469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocquet D, Llanes C, Thouverez M, Kulasekara HD, Bertrand X, Plésiat P, et al. Evidence for induction of integron-based antibiotic resistance by the SOS response in a clinical setting. PLoS Pathog. 2012;8:e1002778. doi: 10.1371/journal.ppat.1002778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway BW, Rossiter H, Burgess D, Dodge J. Aeruginocin tolerant mutants of Pseudomonas aeruginosa. Genet Res. 1973;22:239–253. doi: 10.1017/s0016672300013069. [DOI] [PubMed] [Google Scholar]
- Jacobs MA, Alwood A, Thaipisuttikul I, Spencer D, Haugen E, Ernst S, et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2003;100:14339–14344. doi: 10.1073/pnas.2036282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler T, Donner V, van Delden C. Lipopolysaccharide as shield and receptor for R-pyocin-mediated killing in Pseudomonas aeruginosa. J Bacteriol. 2010;192:1921–1928. doi: 10.1128/JB.01459-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen MV, Cosentino S, Rasmussen S, Friis C, Hasman H, Marvig RL, et al. Multilocus sequence typing of total-genome-sequenced bacteria. J Clin Microbiol. 2012;50:1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le S, Yao X, Lu S, Tan Y, Rao X, Li M, et al. Chromosomal DNA deletion confers phage resistance to Pseudomonas aeruginosa. Sci Rep. 2014;4:4738. doi: 10.1038/srep04738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvig RL, Sommer LM, Molin S, Johansen HK. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat Genet. 2015a;47:57–64. doi: 10.1038/ng.3148. [DOI] [PubMed] [Google Scholar]
- Marvig RL, Dolce D, Sommer LM, Petersen B, Ciofu O, Campana S, et al. Within-host microevolution of Pseudomonas aeruginosa in Italian cystic fibrosis patients. BMC Microbiol. 2015b;15:1–13. doi: 10.1186/s12866-015-0563-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Hamblett N, Rosenfeld M, Gibson RL, Ramsey BW, Kulasekara HD, Retsch-Bogart GZ, et al. Pseudomonas aeruginosa in vitro phenotypes distinguish cystic fibrosis infection stages and outcomes. Am J Respir Crit Care Med. 2014;190:289–297. doi: 10.1164/rccm.201404-0681OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mern DS, Ha SW, Khodaverdi V, Gliese N, Görisch H. A complex regulatory network controls aerobic ethanol oxidation in Pseudomonas aeruginosa: indication of four levels of sensor kinases and response regulators. Microbiology. 2010;156:1505–1516. doi: 10.1099/mic.0.032847-0. [DOI] [PubMed] [Google Scholar]
- Michel-Briand Y, Baysse C. The pyocins of Pseudomonas aeruginosa. Biochimie. 2002;84:499–510. doi: 10.1016/s0300-9084(02)01422-0. [DOI] [PubMed] [Google Scholar]
- Moran NA. Microbial minimalism: genome reduction in bacterial pathogens. Cell. 2002;108:583–586. doi: 10.1016/s0092-8674(02)00665-7. [DOI] [PubMed] [Google Scholar]
- Murugan N, Malathi J, Umashankar V, Madhavan HNR. Comparative genomic analysis of multidrug-resistant Pseudomonas aeruginosa clinical Isolates VRFPA06 and VRFPA08 with VRFPA07. Genome Announc. 2014;2:e00140–00114. doi: 10.1128/genomeA.00140-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Davalos LM. The nature and dynamics of bacterial genomes. Science. 2006;311:1730–1733. doi: 10.1126/science.1119966. [DOI] [PubMed] [Google Scholar]
- Ogunnariwo J, Hamilton-Miller JMT. Brown- and red-pigmented Pseudomonas aeruginosa: differentiation between melanin and pyorubrin. J Med Microbiol. 1975;8:199–203. doi: 10.1099/00222615-8-1-199. [DOI] [PubMed] [Google Scholar]
- Olivares E, Badel-Berchoux S, Provot C, Jaulhac B, Prévost G, Bernardi T, Jehl F. The BioFilm Ring Test: a rapid method for routine analysis of Pseudomonas aeruginosa biofilm formation kinetics. J Clin Microbiol. 2016;54:657–661. doi: 10.1128/JCM.02938-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- Priebe GP, Dean CR, Zaidi T, Meluleni GJ, Coleman FT, Coutinho YS, et al. The galU Gene of Pseudomonas aeruginosa is required for corneal infection and efficient systemic spread following pneumonia but not for infection confined to the lung. Infect Immun. 2004;72:4224–4232. doi: 10.1128/IAI.72.7.4224-4232.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos AJ, Köhler T, Nikaido H, Plésiat P. Involvement of an efflux system in the natural resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob Agents Chemother. 1999;43:2624–2628. doi: 10.1128/aac.43.11.2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau MH, Marvig RL, Ehrlich GD, Molin S, Jelsbak L. Deletion and acquisition of genomic content during early stage adaptation of Pseudomonas aeruginosa to a human host environment. Environ Microbiol. 2012;14:2200–2211. doi: 10.1111/j.1462-2920.2012.02795.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Rojas A, Mena A, Martin S, Borrell N, Oliver A, Blazquez J. Inactivation of the hmgA gene of Pseudomonas aeruginosa leads to pyomelanin hyperproduction, stress resistance and increased persistence in chronic lung infection. Microbiology. 2009;155:1050–1057. doi: 10.1099/mic.0.024745-0. [DOI] [PubMed] [Google Scholar]
- Sano Y, Kageyama M. A novel transposon-like structure carries the genes for pyocin AP41, a Pseudomonas aeruginosa bacteriocin with a DNase domain homology to E2 group colicins. Mol Gen Genet. 1993;237:161–170. doi: 10.1007/BF00282797. [DOI] [PubMed] [Google Scholar]
- Sano Y, Matsui H, Kobayashi M, Kageyama M. Molecular structures and functions of pyocins S1 and S2 in Pseudomonas aeruginosa. J Bacteriol. 1993;175:2907–2916. doi: 10.1128/jb.175.10.2907-2916.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer DH, Kas A, Smith EE, Raymond CK, Sims EH, Hastings M, et al. Whole-genome sequence variation among multiple isolates of Pseudomonas aeruginosa. J Bacteriol. 2003;185:1316–1325. doi: 10.1128/JB.185.4.1316-1325.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoodley P, Sauer K, Davies DG, Costerton JW. Biofilms as complex differentiated communities. Annu Rev Microbiol. 2002;56:187–209. doi: 10.1146/annurev.micro.56.012302.160705. [DOI] [PubMed] [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Talon D, Cailleaux V, Thouverez M, Michel-Briand Y. Discriminatory power and usefulness of pulsed-field gel electrophoresis in epidemiological studies of Pseudomonas aeruginosa. J Hosp Infect. 1996;32:135–145. doi: 10.1016/s0195-6701(96)90055-9. [DOI] [PubMed] [Google Scholar]
- Tielen P, Wibberg D, Blom J, Rosin N, Meyer AK, Bunk B, et al. Genome sequence of the small-colony variant Pseudomonas aeruginosa MH27, isolated from a chronic urethral catheter infection. Genome Announc. 2014;2:e01174–13. doi: 10.1128/genomeA.01174-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogne C, Aires JR, Bailly C, Hocquet D, Plésiat P. Role of the multidrug efflux system MexXY in the emergence of moderate resistance to aminoglycosides among Pseudomonas aeruginosa isolates from patients with cystic fibrosis. Antimicrob Agents Chemother. 2004;48:1676–1680. doi: 10.1128/AAC.48.5.1676-1680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite RD, Curtis MA. Pseudomonas aeruginosa PAO1 pyocin production affects population dynamics within mixed-culture biofilms. J Bacteriol. 2009;191:1349–1354. doi: 10.1128/JB.01458-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wargo MJ, Hogan DA. Identification of genes required for Pseudomonas aeruginosa carnitine catabolism. Microbiology. 2009;155:2411–2419. doi: 10.1099/mic.0.028787-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Survival of the pyomelanin-producing mutants of P. aeruginosa in high pH and under oxidative stress. a, proportion of surviving bacteria after 24h of incubation at 37°C under shaking in LB broth pH9.5. Ratio of means ± SEM from at least 3 independent experiments; two-sided Student’s t-tests; NS, P>0.05; **, P<0.01; ***, P<0.001. b, proportion of surviving bacteria after 45 min of exposure to 50 mM H2O2 (Rodriguez-Rojas et al., 2009). Ratio of means ± SEM from at least 3 independent experiments; two-sided Student’s t-tests; NS, P>0.05; *, P<0.05, **, P<0.01; ***, P<0.001.
Figure S2. Production of virulence factors by pyomelanin-producing P. aeruginosa. Means or ratio of means ± SEM from at least 3 independent experiments; two-sided Student’s t-tests; NS, P>0.05; *, P<0.05; **, P<0.01; ***, P<0.001. a, elastase activity was quantitated with Elastin Congo Red assay. b, proportion of sheep red blood cells that were hemolyzed after incubation with the supernatant of an overnight bacterial culture. c, the production of pyocyanin was quantified as previously described (see Suppl. materials).
Figure S3. Pulsed-field gel electrophoresis profiles of DraI-digested DNA from P. aeruginosa isolates. a, first series with A, B, C, and D pairs. b, complementary series. The brackets indicate two isogenic isolates isolated from the same patient – a wild-type isolate and its pyomelanin-producing mutant.
Table S1. PCR detection of the genes pa1974, pa1989, pa2009 and pa2018 among a collection of pyomelanin-producing clinical isolates of P. aeruginosa and in their isogenic parents. + and − signs indicate a positive and a negative PCR reaction with specific primers, respectively.
Table S2. Susceptibility of the pyomelanin-producing mutants and their isogenic counterparts to the major anti-pseudomonal compounds.
Table S3. Primers used in the study.