

Abstract
Approximately one third of all mammalian genes are essential for life. Phenotypes resulting from mouse knockouts of these genes have provided tremendous insight into gene function and congenital disorders. As part of the International Mouse Phenotyping Consortium effort to generate and phenotypically characterize 5000 knockout mouse lines, we have identified 410 lethal genes during the production of the first 1751 unique gene knockouts. Using a standardised phenotyping platform that incorporates high-resolution 3D imaging, we identified novel phenotypes at multiple time points for previously uncharacterized genes and additional phenotypes for genes with previously reported mutant phenotypes. Unexpectedly, our analysis reveals that incomplete penetrance and variable expressivity are common even on a defined genetic background. In addition, we show that human disease genes are enriched for essential genes identified in our screen, thus providing a novel dataset that facilitates prioritization and validation of mutations identified in clinical sequencing efforts.
Keywords: mouse, embryonic lethal, knockout, KOMP, EUCOMM, IMPC
Introduction
Our understanding of the genetic mechanisms required for normal embryonic growth and development has been advanced by the analysis of single mutations generated in individual labs or the identification of mutants through focused mutagenesis screens1–4. Systematic, standardized approaches to mouse phenotypic analysis complement these data, capitalizing on the efficiency provided by scale and reducing the potential for ascertainment bias, ultimately providing a means to achieve genome-wide functional annotation. Moreover, recent challenges in reproducibility of animal model experimentation5,6 emphasize the need for careful standardization of allele design, genetic background, and phenotyping protocols. Building on these principles, the goal of the International Mouse Phenotyping Consortium (IMPC) is to generate a catalogue of gene function through systematic generation and phenotyping of a genome-wide collection of gene knockouts (KO) in the mouse. To date, nearly 5000 new knockout lines have been created by IMPC from the International Knockout Mouse Consortium (IKMC) resource7–12. Here we report the results of the first international, systematic effort to identify and characterize the phenotypes of embryonic lethal mutations using a standardised13, high-throughput pipeline. These findings provide novel insights into gene function, provide new models for inherited disorders, and shed new light on the role of essential genes in a variety of monogenic and complex human disorders.
Results
Intercrosses of 1,751 germ-line transmitted (GLT) heterozygous lines from IMPC production colonies1 identified 410 lines that displayed lethality (Fig. 1a), defined as the absence of homozygous mice after screening of at least 28 pups (p<0.001 Fisher’s exact test) prior to weaning. We also identified 198 “subviable” lines, defined as fewer than 12.5% (half of expected) homozygous preweaning pups (full list of genes available in Supplementary Table 1). The vast majority of the alleles employed in this study were of “tm1.1” or “tm1b” IKMC variants, which disrupt the coding sequence (1704 of 1804 unique alleles; see Extended Data Fig.1 for schematics of each allele and Supplementary Table 2 for all other alleles employed). Centre-to-centre variability in the proportion of essential genes is observed ranging from 4.8%–52.7%, which likely reflects the different biases in gene selection criteria between centres and specific consortium arrangements for lethal gene characterization (TCP and UCD) (Extended Data Fig. 2A,B). No significant bias is observed in the distribution of lethal genes across mouse chromosomes (Extended Data Fig. 2C,D). Overall, however, the lethal proportion (23.4%) is consistent with published observations of null alleles7,9,12,13, particularly when combined with subviable lines (11.3%), resulting in 65.3% viability for IMPC KO lines overall. A main goal of this project is to provide phenotype data for unknown or novel genes, i.e. those with no prior report of a targeted null allele in the mouse (curated in Mouse Genome Informatics). The primary viability data indicated that such unannotated genes displayed an overall viability rate of 66.5%, compared to the 62% viability rate among previously reported null alleles (Extended Data Fig. 2E; novel versus prior gene lists in Supplementary Table 3; list of all first publications or reports of gene knockouts in Supplementary Table 4). These data reveal consistent identification of essential genes in our program, and further support that approximately 35% (24% lethal and 11% subviable) of null mutations across the genome are essential for survival at normal Mendelian ratios.
Figure 1. Large-scale identification of essential genes.

a, Number of viable, subviable and lethal genes identified from intercrosses of IMPC knockout (KO) alleles. b Core essential human genes identified in three studies, 158016, 173917, and 173415 (top row) (see Material and Methods). Pie-charts indicate overlap between core human cell-essential genes and orthologous genes in the mouse: essential (red); non-essential (green) and genes with unknown function in the mouse (blue). c, Numbers and percentages of 242 IMPC lines showing lethality within a particular temporal window. d, Specific LacZ expression for Clcf1, Cgn, Kif26b and widespread expression shown for Psen1.
Functional data from mouse knockouts are highly informative, and thus would be predicted to have a strong impact on Gene Ontology (GO) Consortium14 annotations. For the 1,751 IMPC mouse lines phenotyped to date, IMPC phenotyping provides the only experimental evidence for over 40% of the genes in our dataset. Using the GO Slim tool, which clusters terms associated with each gene into a set of broad categories, we observed enrichment in lethal and subviable genes within several categories (Extended Data Fig. 3). Compared to novel genes, the number of annotations for a majority Process and Function categories was greater for published alleles, highlighting the value of our analysis in assigning function to novel, previously uncharacterized genes.
We used data from three recent publications on genome-wide screens for cell-essential genes in human cells to address the overlap between essential genes in the human and mouse genome15–17. We selected core essential genes from each study and compared to human orthologs of mouse essential genes on the consensus list of curated IMPC-MGI genes. We found that approximately 35% of core essential genes in each study are associated with lethality or subviability in the mouse, with 61–62% of genes currently unknown (Fig. 1b). Of the 19 human essential genes common to all three studies that were nonessential in the mouse, only three (Rbmx, Dkc1, and Sod1) could be reliably confirmed as a targeted knockout of a nonessential gene, highlighting the remarkable concordance between mouse and human in their core essential genes.
To expand the depth of our analysis of essential genes, we developed a comprehensive phenotyping pipeline designed to identify the time of lethality, assign phenotypes, and document LacZ expression patterns at discrete time points (Extended Data Fig. 4)13. A key aspect of the pipeline is the incorporation of optical projection tomography (OPT)18, micro-computed tomography (micro-CT)19,20,21 and high-resolution episcopic microscopy (HREM)22, which provide cost-effective, high-throughput approaches to the collection of phenotype data, including quantitative volumetric analysis (see below). The catalogue of KO lines and all phenotype data are available to the community via the IMPC portal (www.mousephenotype.org), with an embryo phenotyping-specific portal at www.mousephenotype.org/data/embryo (a guide to accessing, viewing and using these data is available on the IMPC portal at http://www.mousephenotype.org/data/documentation/doc-explore)
Using a tiered strategy, we established clear viable vs. lethal (defined if homozygous embryos were absent or lacked a heartbeat) calls at up to four different time points for a total of 283 lethal lines (A comprehensive progress table for all 1861 alleles is provided in Supplementary Table 5), the total number varying by progress through the pipeline. From these data, we established windows of lethality for 242 genes with complete data to more precisely define the timing of embryo death. Figure 1c shows that a majority of lines (147/242; 60.7%) died prior to E12.5 and a majority of these (107/147; 72.8%), development ceased prior to E9.5, the earliest time point examined. Remarkably only 9 total lines die in the E12.5–E15.5 or E15.5–E18.5 windows, while most lines that were viable at E12.5 were also viable at the latest time point examined (E15.5 or E18.5). Although viable, many of these lines show phenotypes at E15.5 and E18.5 (see below), and ultimately succumbed in the perinatal or early postnatal period.
Taking advantage of the LacZ cassette present in most IMPC alleles10,11, gene expression was evaluated in heterozygous embryos at E12.5 in the lethal/subviable lines. Expression patterns fell into three broad categories as shown in Figure 1d (bottom): restricted (e.g., Clcf1, Cgn and Kif26b); ubiquitous (e.g., Psen1); or undetectable expression (not shown). All images and annotations of the expression atlas are available at the IMPC portal, providing a rich and growing in situ expression atlas for the scientific community.
Identification of novel lethal phenotypes
At each time point, gross morphological phenotypes were recorded using a structured set of Mammalian Phenotype (MP) terms (Supplementary Table 6). An analysis of phenotype areas revealed that the most common phenotype overall was growth/developmental delay (Fig. 2a–c) affecting 23.5%, 44.1% and 39.3% of lines at E12.5, E14.5/E15.5 and E18.5, respectively. Abnormalities in cardiovascular development were also common, frequently observed at both E12.5 and E15.5 (Fig. 2a,b), along with craniofacial malformations and defects in development of the limbs and/or tail. At E18.5, a number of mutants exhibited respiratory and/or body wall abnormalities (captured as “other”), in addition to the growth abnormalities seen at other stages.
Figure 2. Systematic identification of novel embryonic phenotypes.

a–c, Frequency of gross morphology phenotypes at E12.5, E15.5, E18.5 (Supplementary Table 6). d–g, Spina bifida in E15.5 Tmem132a knockout embryos (arrow) (n=4 mutants), as well as narrow limbs with fewer digits (n=5 mutants). h–m, Gross morphology of E18.5 Svep1 knockout embryos reveals severe edema (k versus h; white arrowheads). Transverse views reveal an abnormal renal pelvis (i versus l, arrow), hypoplastic lungs, and thin myocardium (arrows in m versus j). Lu=lungs; Li=liver; Sc=spinal cord (n=14 mutants). n–s, Mutant (q) E14.5 Klhdc2 embryos show polydactyly (arrow) and edema (arrowhead) versus controls (n). Sagittal views revealed a thin body wall (arrow in r) and missing adrenals (s) (n=2, E14.5, n=3, E18.5). t–w, E9.5 OPT surface renderings show abnormal allantois development (u), failure to complete turning (u–w) and abnormal heart looping (w) in mutant Gfpt1 embryos (n=4 mutants).
Our pipeline has identified a number of novel phenotypes for previously unreported knockouts. In all cases, 3D imaging revealed additional phenotypes that might have been missed by gross inspection. For example, Tmem132a E15.5 homozygous embryos were smaller than littermates, displayed an obvious spina bifida, and narrow, club-shaped limbs (Fig. 2d,f). Sagittal cross-sections through the micro-CT data showed the abnormal curvature in the spinal column adjacent to the open neural tube, and abnormal head structure in mutants (Fig. 2e,g). Kidney defects were also observed in E15.5 mutant embryos (n=3) and bladder defects were also evident by E18.5 (n=4) (not shown). Svep1 homozygous mutant embryos display multiple defects at both E15.5 and E18.5, severe edema and discolouration (Fig. 2h,k), and die in the perinatal period. Additionally, transverse sections of micro-CT data from E18.5 embryos revealed abnormal development of the kidney pelvis (Fig. 2i, l), severely hypoplastic lungs and a thin myocardium (Fig 2j,m). Homozygous Klhdc2 embryos at E14.5 displayed hindlimb preaxial polydactyly (Fig. 2n,q arrow) and edema (Fig. 2n,q arrowhead). Sections of micro-CT volumes additionally revealed hypoplastic adrenals (Fig. 2s), displaced kidneys, a shorter tongue, and abnormal intestines (Fig 2r).
As stated above, a number of mutants with impaired cardiovascular function were identified (Fig. 2a–c), including Strn3, Atg3, and Slc39a8 (Extended Data Fig. 5). Similarly, cardiovascular defects were common at E9.5, illustrated in detail using OPT (e.g., Tmem100, Extended Data Fig. 6). OPT datasets can be manipulated in three dimensions to reveal additional phenotypes such as abnormal neural tube closure, turning and chorion-allantois fusion seen in homozygous Gfpt mutant embryos (Fig. 2t,u,v,w).
Chtop mutant embryos showed obvious developmental delay, neural tube defects, craniofacial dysmorphology, abnormal eye development, and subcutaneous edema. HREM was used to define further defects at E14.5 revealing major abnormalities in the ribs and vertebrae, the cardiovascular system, and the nervous system at a spatial resolution rivalling standard histological techniques (Fig. 3 a–g).
Figure 3. Deep phenotyping through high-resolution 3D imaging and quantitative volumetric analysis.

a–g, Phenotypic features of Chtop null embryos identified by HREM imaging (n = 4 mutants analysed). a, 3D surface model of a mutant embryo b, Sagittal view, revealing abnormal topology of pancreatic tissue (pa). Inlay shows stomach (s) with a defect in.the formation of the diaphragm (dia) (asterisk). c. Coronal plane to reveal double outlet right ventricle (rv), persisting truncus arteriosus (pta), and ventricle septum defects (vs). d, Transverse section showing defects in the right aortic arch (raa) and retroesophageal left subclavian artery (lsa). e, Coronal section showing neural tissue (arrowhead) abnormally protruding into the 3rd brain ventricle (III), and unelevated palatine plates (pp). f, Coronal section showing bicuspid aortic valve (av). g, Sagittal section showing fused spinal ganglia (fsg) and additional ganglion material (ag) caudal to the occipital bone (ob) (2/4 embryos). li, liver; lu, lung; lv, left ventricle; e, esophagus; sc, spinal chord; I, 1st brain ventricle and left telencephalon; II, 2nd brain ventricle and right telencephalon; c, cusp of semilunar valve; sg, spinal ganglion; dr, dorsal roots of spinal nerve; tongue (to); pulmonary valve (pv). h, Coronal and transverse sections through composites of Cbx4−/− embryos registered to an average control dataset. Blue = smaller volumes in the mutant; red = larger volumes. The blue clusters in the top panel correspond to the smaller right and left thymic rudiments, the adrenals in the middle panel, and the trigeminal (V) ganglia in the bottom panel (n = 8 mutants). i, Whole structural volume differences for the left and right thymic rudiment. j, Whole structural volume differences for the adrenal glands. Error bars represent 95% confidence intervals.
In addition to manual annotation, 3D images are amenable to automated computational analyses that can identify mutant anatomical phenotypes that are statistically beyond wildtype variation19,20. As an example, prior studies of Cbx4 knockout mice revealed a clear hypoplastic thymus23. Automated volumetric analysis of E15.5 Cbx4 null mice generated by the IMPC replicated these findings, but also revealed adrenal hypoplasia and smaller trigeminal ganglia using deformation-based morphometry and a novel 3D segmented mouse embryo atlas (Fig. 3h–j). This analysis also identified a smaller cochlea in Eya4 mutant, directing more in depth histopathology analysis to the affected region (Extended Data Fig. 6).
Some centres have expanded the pipeline to include analyses of lines that are lethal between birth and weaning, employing tools such as whole brain MRI. These analyses have identified previously unknown phenotypes for Tox3 at P7, including a smaller cerebellum displaying hypoplasia and dysplasia, and an absent transient external granular layer (Extended Data Fig. 7). Similar analysis of Rsph9, a gene associated with Primary Ciliary Dyskinesia in humans (OMIM #612650), has identified a new mouse model of this disease. All P7 homozygous mice showed enlarged ventricles, while histopathology revealed severe triventricular hydrocephalus with marked rarefaction, cavitation, and loss of periventricular cortical tissue as well as severe sinusitis, typical of ciliary dysfunction (Extended Data Fig. 8).
Subviable genes and incomplete penetrance
Unexpectedly, we observed instances of phenotypes that display incomplete penetrance, including variable lethality (subviability), despite the standard allele structure and defined genetic background. Prior work has shown that lethal genes are much less likely than viable genes to have a paralog, and thus less potential for functional redundancy12. Genes from subviable lines, by contrast, were significantly more likely to have a paralog, similar to viable lines (Fig. 4a). This is consistent with a model where incomplete penetrance and variable expressivity24 are due to cell-autonomous, stochastic variation in gene expression in components of disrupted “buffered” pathways25,26, where paralogs may provide functional redundancy. For example, two alleles of the Acvr2a gene have been generated on a mixed genetic background27,28, and both display variable phenotypes including partial lethality. On a uniform C57BL/6N background we also observe subviability and a wide range of morphological phenotypes at E15.5 including small or missing mandible, cyclopia, and holoprosencephaly (Fig. 4b–i); this is consistent with the normal assembly of ACVR2A into a heteromeric signalling complex with its paralog ACVR2B. Other examples include Rab34, which has three paralogs in the RAB protein family (Rab6a, Rab6b, and Rab36). In addition to the consistent phenotypes of polydactyly and lung hypoplasia, mutants also display highly variable craniofacial malformations, haemorrhage, edema, and exencephaly phenotypes (Fig. 4j–m).
Figure 4. Frequent instances of subviability, incomplete penetrance, and variable expressivity in a uniform genetic context.

a, Comparison of the total percentage of unique genes that lack a paralog between viable, subviable, and lethal. b–i, Surface renderings (b–e) and transverse sections of the heads (f–g) of E15.5 Acvr2a−/− embryos reveal a range of phenotypes (n = 8 mutants). j–m, Variable expressivity of phenotypes is observed in Rab34 mutant embryos at E18.5, (n = 7 mutants).
For all cases of lethal and subviable genes, full cohorts of heterozygous mice are phenotyped as part of the IMPC Adult Phenotyping pipeline, along with surviving subviable homozygous mice in some cases. Viable homozygous animals displayed a greater number of phenotype hits per gene than heterozygous mice from the lethal class, although the average difference was only 1.44 more hits (Extended Data Fig. 9a). However, subviable mice homozygous for a null allele average 5.8 hits per line compared to an average of 4.0 hits per line in homozygote and hemizygote viable lines (Extended Data Fig. 9b).
Novel insights into function of genes associated with human disease
It has been shown that genes causing lethality in the mouse are enriched in disease genes29, 30. We established orthology between genes in the mouse and human, and used the Human Genome Mutation Database (HGMD) to annotate human disease associations31,32. We next compiled an updated list of 3326 essential genes by combining the published data from the Mouse Genome Informatics (MP terms listed in Supplementary Table 7) database and 608 genes identified in the IMPC effort as causing lethality and subviability, along with 4919 nonessential genes. With these updated lists, we report an even stronger enrichment of essential genes relative to nonessential for human disease genes catalogued in the HGMD (odds ratio= 2.00, p-value=6.83e-39, Fig.5a). Consistent with this enrichment, of the 3302 protein-coding HGMD disease genes, 2434 have a reported phenotype and more than half (1253) are essential (Fig. 5b; Supplementary Table 8). Furthermore, we found an enrichment of essential genes in comparison to nonessential genes (odds ratio=1.16, p-value=0.0015) among 6384 genes encompassing or neighbouring the disease- and trait-associated variants in the NHGRI-EBI catalog of published genome-wide association studies (“GWAS hits”)33. (Fig. 5c).
Figure 5. Essential genes are enriched in human disease genes and intolerant to genetic variation.
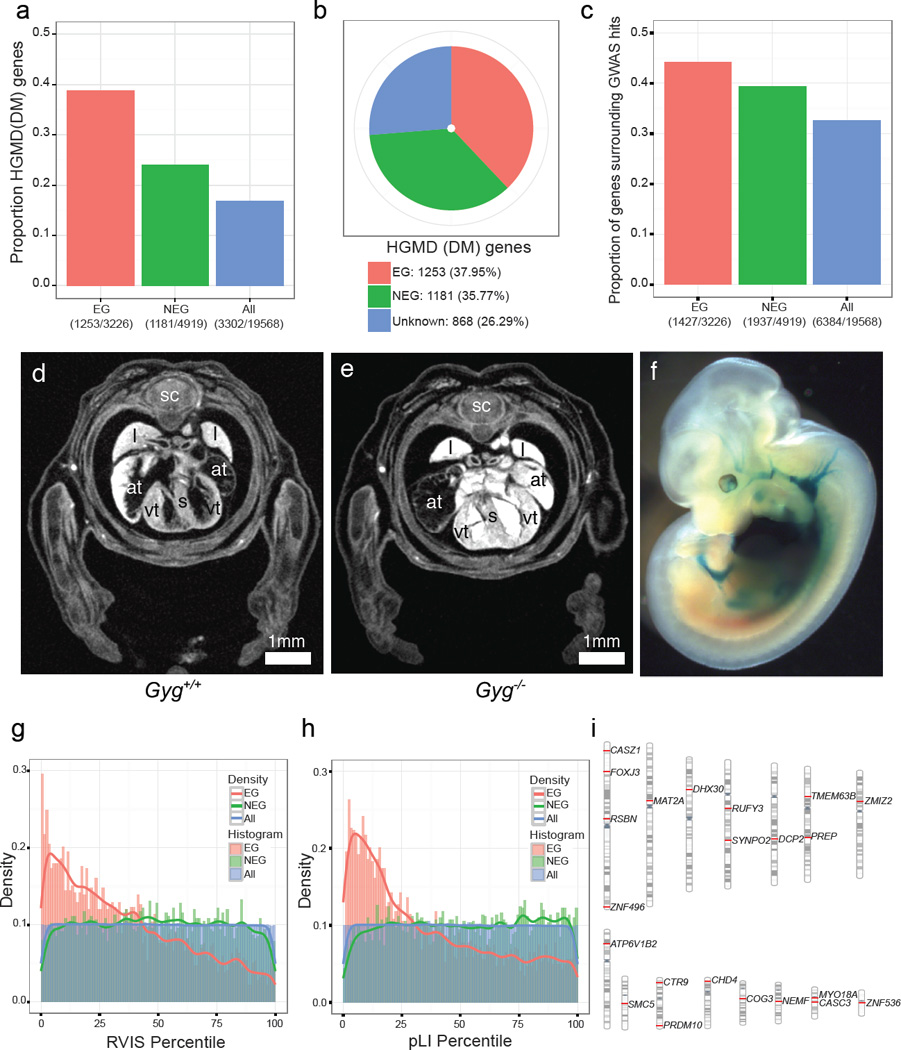
a,. The fractions indicate the number of HGMD disease genes (n=3302) among 3326 Essential genes (EG in red); 4919 Nonessential genes (NEG in green) and 19568 protein-coding genes (ALL in blue). Fisher’s exact test for enrichment: EG vs. NEG (odds ratio=2.00, p=7.80e-46), EG vs. ALL (odds ratio=3.13, p=2.42e-160), NEG vs. ALL (odds ratio=1.56, p=1.83e-29). b, Essentiality status of 3302 HGMD disease genes. c. The fractions indicate the number of genes encompassing or neighbouring GWAS hits33 (n=6384) divided by essentiality status. Fisher’s exact test for enrichment: EG vs. NEG (odds ratio=1.16, p=0.0015), EG vs. ALL (odds ratio=1.56, p=5.80e-31), NEG vs. ALL (odds ratio=1.35, p=1.18e-19). d,e, Gyg null embryos at E15.5 show enlarged atria and a thickened ventricular wall versus controls. f, LacZ expression for Gyg was seen in the heart and vascular system (homozygote shown).. sc=spinal cord; l=lung; s=septum; at=atrium; vt=ventricle. (n = 5 mutants) g, Distribution of percentiles of the Residual Variation Intolerance Score (RVIS) across three classes of genes: EG, NEG and ALL Wilcoxon rank sum test: EG vs. NEG (p<2.2e-16), EG vs. ALL (p<2.2e-16), NEG vs. ALL (p=0.579). h, Distribution of percentiles of the probability of being loss-of-function intolerant (pLI) across three classes of genes: EG, NEG and ALL. Wilcoxon rank sum test: EG vs. NEG (p<2.2e-16), EG vs. ALL (p<2.2e-16), ALL vs. NEG (p= 4.15e-05). i, Chromosomal distribution of 22 human orthologs of RVIS and pLI intolerant essential genes that are not currently included in the catalogues of Mendelian disease genes.
The IMPC effort expanded a phenotypic spectrum for over 300 genes associated with known Mendelian diseases. From 194 subviable genes with identified human orthologs, 57 were associated with human disease, of which 34 were previously unreported for their subviable phenotypes (Supplementary Table 9; new reports indicated by ‘N’ in column J). For example, SET binding protein 1 (SETBP1) has been reported as frequently mutated in several types of chronic leukaemia and in Schnizel-Giedion syndrome, a congenital disease characterized by a higher prevalence of tumours, severe mid-face hypoplasia, heart defects, and skeletal anomalies34,35. Among 399 lethal genes, 126 human orthologs have been associated with human diseases, including 52 disease genes for which our data provide the first report of their null phenotype in the mouse (Supplementary Table 10). The human orthologs of these novel lethal genes have been linked to metabolic and storage syndromes (ADSL, DHFR, GYG1, PC), mitochondrial complex deficiencies (ATP5E, NDUFS1, NUBPL, SDHA, SLC25A3, UQCRB), or syndromes caused by disruption of basic processes such as replication or translation initiation (EIF2B3, EIF2B4, ORC1). The severity of clinical manifestation of these human syndromes ranges from neonatal lethality (BBS10, SLC25A3) matching the observed phenotype in the mouse, to neurological disorders and intellectual disability (COQ6, DEPDC5, GOSR2, KDM5C, YARS). These differences in clinical manifestation may be due to differences between underlying biological processes in the mouse and human. Alternatively, a different set of alleles, rather than null, may underlie these dominant or recessive human syndromes. GYG1 mutations have been found in patients with glycogen storage disease XV36 (GSD15; 61350736), and in an additional seven patients with Polyglucosan body myopathy 2 (PGBM2;616199). Both diseases affect skeletal muscle, but PGBM2 is characterized by polyglucosan accumulation in muscle and skeletal myopathy without cardiac involvement37. Homozygous Gyg null embryos die perinatally and show severe heart abnormalities consistent with cardiac hypertrophy evident as early as E15.5 (Fig. 5d,e). At E12.5, LacZ expression was detected specifically in the heart and the carotid and umbilical arteries, correlating strongly with the heart phenotype and heart abnormalities in GSD15 patients (Fig. 5f). Micro-CT images at E18.5 revealed an obvious enlargement of the thymus as well as abnormal morphology of the brain and spinal cord consistent with degeneration (Extended Data Fig. 10a–h). Gyg mutations have not previously been reported in the mouse and this model will be valuable in understanding the distinct roles for Gyg in different organs and potentially the consequences of different alleles in patients. In another example, for a novel human syndrome arising from a chromosomal deletion (16p)38, highlight Kdm8 as a strong candidate amongst a pool of candidate genes (Extended Data Fig. 10i–t).
We also used the updated catalogue of mouse essential and nonessential genes to compare the mutability of their human orthologs in exome sequence of 60,706 subjects in the Exome Aggregation Consortium data (ExAC, Cambridge, MA; http://exac.broadinstitute.org) (Exome Aggregation Consortium et al. submitted). The ExAC data were used to generate intolerance scores for all protein-coding genes by two different complementary methods; a) the Residual Variation Intolerance Score (RVIS) based on intolerance to common missense and truncating single nucleotide variation39 and b) the estimation of probability of being loss-of-function intolerant (pLI score) (Exome Aggregation Consortium et al. submitted). Human orthologs of essential genes are more intolerant to variation (low RVIS and high pLI scores) than orthologs of nonessential genes and all genes in the human genome (p-value<2.2e-16 for lower percentiles in essential genes using the two scoring systems, Fig. 5g and h). Moreover, the IMPC effort identified a set of 22 human orthologs of essential genes, that were not previously associated with human disease (Fig. 5i; Supplementary Table 11), but based on their intolerance to functional variation and lethality of their null alleles in the mouse they represent strong candidates for yet undiagnosed human disease.
Discussion
In this study, we describe the systematic characterization of embryonic lethal phenotypes as part the collaborative effort to generate a genome-wide catalogue of gene function. A unique aspect of our pipeline is the incorporation of high-resolution, high-throughput 3D imaging methods, affording detailed morphological information and automated analysis19. High-resolution datasets are available to the community through a common portal, facilitating additional, in depth analysis by other investigators that will further enrich the phenotype calls reported in the primary screen. These data are provided in real time, without embargo, to create an “open access” environment that allows investigators to rapidly evaluate new models. Importantly, open availability of the mouse models themselves reduces the cost and time lost through duplication of effort.19
Beyond the direct benefit to understanding gene function, this resource has significant relevance to disease-causative genes in humans. We found that the human orthologs of mouse essential genes show evidence of purifying selection in the human population, suggesting a common intolerance to mutation in both mouse and humans. Recent work has identified cases of homozygous loss-of-function in the human population40,41, complementing on-going efforts to discover disease genes in highly consanguineous populations, including mutations that are homozygous lethal42,43, (Salaheen et al. Submitted).
Overall, the data presented here illustrate a rich resource with impact for many scientific communities. The high efficiency and reduced cost of CRISPR/Cas9 technology46 will allow the IMPC to further expand its coverage of the mammalian genome, and additionally provide a means to target genes and sequence features not currently part of the IKMC resource. As current estimates indicate that only a small percentage of genes are studied by the broad research community47, the systematic approach to phenotyping and unrestricted access to data and mouse models provided by the IMPC promises to fill this large gap in our understanding of mammalian gene function.
Online Methods
Standardized, consortium-wide protocols are available at the IMPC portal (www.mousphenotype.org/impress). These procedures define the minimum standards, metadata and protocols for all publically available data. All mouse experiments were conducted in accordance with the governmental and funding regulations of the different member centres. Details of individual centre-specific methods are posted with the IMPReSS procedures. Additional details are provided below.
Mice
All mouse lines in this study are derived from IKMC ES cell resources. All mice are produced and maintained on a C57BL/6N genetic background, with support mice derived from C57BL/6NJ, C57BL/6NTac or C57BL/6NCrl. Husbandry details vary by centre, and can be found at http://www.mousephenotype.org/impress. For timed matings, successful mating and fertilization (0 hour) was calculated to be the midpoint of the dark cycle prior to the appearance of the copulation plug.
Gene list analysis
Gene lists were filtered and analysed using MouseMine at Mouse Genome Informatics (www.mousemine.org). For segmentation of novel and prior reported KO lines, alleles were filtered to include “targeted” and “null” mutations only, as these are comparable to the IKMC alleles in this study. A further filtering step was performed to include only lines for which phenotypic data (normal or abnormal) are reported.
GoSlim Enrichment
Gene lists were analysed using the GOSlim tool hosted at Mouse Genome Informatics: http://www.informatics.jax.org/gotools/MGI_GO_Slim_Chart.html. Both experimental and computational analysis codes were included in the search.
Dissection and Preparation of E9.5 and E15.5 Embryos for OPT and micro-CT Imaging
Embryos were dissected in 37°C phosphate buffered saline (PBS) (minus Ca++/Mg++) containing Heparin (1 unit/1ml PBS). Extra-embryonic membranes were removed and the yolk sac collected for genotyping. The embryos were exsanguinated by severing the umbilical vessels with small scissors and rocking them in warm PBS/Heparin for a maximum of 5 minutes for E9.5 embryos and 15 minutes for E15.5 embryos. Embryos were washed twice with PBS and immersion fixed in 20 – 40 × the volume of 4% paraformaldehyde (PFA) prepared in PBS. E9.5 embryos were fixed for 4 hours at 4°C or 2 hours at room temperature (RT) and E15.5 embryos were fixed overnight at 4°C. After fixation embryos were stored at 4°C in PBS containing 0.02% sodium azide (0.2g/l PBS).
Optical Projection Tomography
Sample Preparation
Each E9.5 embryo was embedded in low-melting point agarose. The agarose plug was then subjected to a dehydration series using methanol (25%, 50%, 75%, 100% × 2) where the methanol solutions are replaced once per day. The agarose plug was then cleared with BABB (1:2 benzyl alcohol/benzyl benzoate) for three days. The BABB solution was replaced once per day during the clearing process.
Imaging
Optical projection imaging was done as previously described1. Briefly, each sample was excited by ultraviolet light filtered by the following excitation filter: Semrock 425/30 BrightLine Bandpass Filter, 25 mm [FF01-425/30-25]. Autofluorescence was captured by a CCD camera, where the emission was filtered using the following emission filter: 473 RazorEdge Long-pass Filter, U-grade, 50.8mm [LP02-473RU-50.8-D]. The sample was rotated 360 degrees at 0.3 degree increments, resulting in 1200 projections. The exposure time varied per sample, but the average was 500 ms. The resultant 3D image file had an isotropic voxel size of [4.45 µm]3.
Micro-Computed Tomography
Sample Preparation
Each E15.5 embryo was subjected to hydrogel stabilization2. Briefly, the embryo was incubated in 20 mL hydrogel solution containing a mixture of ice-cold 4% (wt) PFA, 4% (wt/vol) acrylamide (Bio-Rad, Mississauga, ON, Canada), 0.05% (wt/vol) bis-acrylamide (Bio-Rad, Mississauga, ON, Canada), 0.25% VA044 Initiator (Wako Chemicals USA, Inc., Richmond, VA, USA), 0.05% (wt/vol) saponin (Sigma-Aldrich, St Louis, MO, USA) and PBS at 4°C for 3 days. After incubation, the tube containing the embryo was placed in a dessication chamber where air in the tube was replaced with nitrogen gas. The tube was placed in a 37°C water bath for 3 hours. Lastly, the samples were separated from the encasing gel and place into iodine solution. Each E15.5 mouse embryo was stained with 50 mL of 0.1N iodine solution (Sigma-Aldrich) for 24 hours. The iodine-stained embryo was then embedded in agarose within an 11-mm centrifuge tube and positioned in the micro-CT scanner for imaging.
Imaging
3D datasets were acquired for each mouse embryo using a Skyscan 1172 high-resolution micro-CT scanner (Bruker, Billerica, MA, USA). With the X-ray source at 100 kVp and 100 µA and the use of a 0.5 mm aluminum filter, each specimen was rotated 360° around the vertical axis, generating 1200 views in 5 hours. These image projections were reconstructed into digital cross-sections using the Feldkamp algorithm3 for cone beam CT. The resulting 3D data block contained 2000×1000×1000 voxels of [13.4 µm]3 voxel size.
High-resolution episcopic microscopy (HREM)
Protocols for the preparation and imaging of embryos by HREM are described in detail.4 All analysis was performed on E14.5 embryos.
Automated Image Analysis
The automate image analysis was performed as fully described in5. The segmented 3D atlas of structures used to automate volume measurements was described and presented in6.
Dissection and Preparation of P7 Brains for Whole Brain MRI
Pups were tattooed and genotyped at P3 to determine homozygous viability. At P7, homozygous pups were sedated by intraperitoneal injection of ketamine (150 mg/kg) and xylazine (10 mg/kg) at 0.1ml/10gm body weight. Pups were then trans-cardially flushed with 30mL of PBS (Wisent) containing 1 unit/ml Heparin and 2 mM Gadolinium (Gd) (“ProHance” gadoteridol by Bracco Diagnostics), followed by fixation with 30mL of PBS containing 4% paraformaldehyde (PFA) (Electron Microscopy Sciences) and 2 mM Gd. Flushing and fixation proceeded at a slow flow rate of 1.0 ml/min at room temperature. Following perfusion, the brain was extracted within the skull with the skin, zygomatic bones, eyes, and lower jaw removed. The brain and remaining skull structure were incubated in 35 mls of 4% PFA containing 2 mM Gd overnight at 4°C and then transferred to PBS containing 0.02% sodium azide with 2 mM Gd for at least 3 days prior to imaging.
Image Acquisition
Images were acquired on a 7 Tesla MRI scanner (Varian Inc., Palo Alto, CA)7. The contrast required for registration and assessment of volume is not acceptable with our typical T2-weighted imaging sequence. Therefore, diffusion weighted imaging was performed to enhance the contrast between white and gray matter to aid in the registration and volume measurements.
Diffusion Imaging Sequence
The diffusion sequence uses an in-house custom built 16-coil solenoid array to acquire images from 16 brains in parallel8. The diffusion sequence used was a 3D diffusion-weighted FSE, with TR= 270 ms, echo train length = 6, first TE = 30 ms, TE = 10 ms for the remaining 5 echoes, one average, FOV = 25 mm × 14 mm × 14 mm, and a matrix size of 450 × 250 × 250, which yielded an image with 56 µm isotropic voxels. One b=0 s/mm2 image was acquired and 6 high b-value (b = 2147 s/mm2) images were acquired at the following directions (1,1,0), (1,0,1), (0,1,1), (−1,1,0), (−1,0,1) and (0,1,−1) corresponding to (Gx,Gy,Gz). Total imaging time was ~ 14 hours.
Registration and Analysis
To visualize and compare the mouse brains for the anatomical volume assessment the 6 high b-value images were averaged together to make a high contrast image necessary for accurate registration. Then these images were linearly (6 parameter followed by a 12 parameter) and nonlinearly registered together. All scans were then resampled with the appropriate transform and averaged to create a population atlas representing the average anatomy of the study sample. All registrations were performed using a combination of the mni_autoreg tools9 and ANTS10. The result of the registration was to have all scans deformed into exact alignment with each other in an unbiased fashion. For the volume measurements, this allowed for the analysis of the deformations needed to take each individual mouse’s anatomy into this final atlas space, the goal being to model how the deformation fields relate to genotype7,11. The Jacobian determinants of the deformation fields are then calculated as measures of volume at each voxel. These measurements were examined on a voxel-wise basis in order to localize the differences found within regions or across the brain. Multiple comparisons were controlled for by using the False Discovery Rate (FDR)12.
LacZ staining
Whole litters of E12.5 embryos were fixed in 4% PFA for 1 hour (range for other centres) in PBS at 4°C with gentle shaking. Embryos were then washed 3× in detergent rinse (2mM MgCl2, 0.02% Igepal, 0.01% sodium deoxycholate and 0.1M phosphate (K2HP04/KH2PO4) buffer, Ph 7.5) at 4°C, then moved to X-gal staining solution (2mM MgCl2, 0.02% Igepal, 0.01% Sodium deoxycholate, 5mM Potassium Ferricyanide, 5mM Potassium Ferrocyanide, 1 mg/mL X-gal in 0.1M phosphate buffer pH, 7.5) for 48 hours at 4°C with gentle shaking in the dark. Stained embryos are rinsed briefly in PBS at room temperature, then postfixed overnight at 4°C in 4% PFA. After three rinses in PBS, embryos are transferred to 50% glycerol/PBS solution for imaging and storage. Images are taken using centre-specific equipment, using standard orientations. Portions of the tail of individual stained embryos were removed for genotyping after imaging and assayed for zygosity and sex.
Identification of human orthologs of essential genes and non-lethal genes
To investigate the relevance of novel developmental phenotypes uncovered in the IMPC project, we combined the IMPC data with phenotype data for targeted loss-of-function mutant lines reported in the Mouse Genome Informatics database (MGI)13. Genes annotated with any of 50 Mouse Phenotype (MP) terms including prenatal, perinatal and postnatal lethal phenotypes (Supplementary table 7)14 were considered to be essential genes (n=3023) (Supplementary table 8). The MGI database was also used to select genes with reported targeted loss-of-function phenotypes that are not embryo or pre-weaning lethal (non-essential genes; n=4995). The IMPC effort expanded these lists with 252 essential genes, 101 genes with sub-viable phenotypes and 701 genes with viable mutant phenotypes. Whenever discrepancy appeared between the lethality status reported in publications (i.e. in MGI) and in the IMPC data, we included phenotypes reported by IMPC as these lines were generated on a defined C57BL/6N background background and phenotyped using a standardized pipeline. We used the MGI mouse-human orthology annotation resulting in 3229 essential and 4757 non-essential human orthologs with unambiguous chromosomal position. Annotations of all human protein-coding genes (Ensembl Genes version 8215), including essential/non-essential status, RVIS16, pLI scores (Exome Aggregation Consortium, submitted) and human disease annotations from HGMD17 and OMIM18, were listed in Supplementary Table 8. Enrichment of HGMD disease genes between our gene sets of interest (i.e. EGs, NEGs and all protein-coding genes) was assessed by two-sided Fisher’s exact test. EG vs. NEG (odds ratio=2.00, p=7.80e-46), EG vs. ALL (odds ratio=3.13, p=2.42e-160), NEG vs. ALL (odds ratio=1.56, p=1.83e-29). Difference in intolerance scores between our gene sets of interest was assessed by one-sided Wilcoxon rank sum test. RVIS: : EG vs. NEG (p<2.2e-16), EG vs. ALL (p<2.2e-16), NEG vs. ALL (p=0.579). pLI: EG vs. NEG (p<2.2e-16), EG vs. ALL (p<2.2e-16), ALL vs. NEG (p= 4.15e-05).
Overlap between cell-essential genes in human haploid cells and human orthologs of essential genes in the mouse
We used data from three recent publications on genome-wide screens for cell-essential genes in human cells to address the overlap between essential genes in the human and mouse genome19,20,21. From these papers, we selected 1580 core EGs (genes above essentiality threshold in at least 3 out 5 cell lines in the study) from Hart et al., 1739 core EGs (genes above essentiality threshold in at least 2 out 4 cell lines in the study) from Wang et al. and 1734 core EGs (genes above essentiality threshold in at least 1 out 2 cell lines in the study) from Blomen et al. We used the combined IMPC-MGI EG list (n=3326, see above) to assess the overlap between human cell-essential genes identified in these three studies and essential genes in the mouse.
Identification of genes encompassing or surrounding disease and trait- associated SNPs (“GWAS hits“)
6384 protein-coding genes encompassing or/and neighboring disease- or trait-associated variants (“GWAS genes”) were obtained from the GWAS Catalog22 (downloaded on April 29, 2016). Specifically, we used the “mapped genes” from the GWAS Catalog which are defined as genes mapped to the strongest SNP from GWAS reports. The mapped genes are defined as the genes encompassing the GWAS SNP(s), (i.e. located in coding or intragenic regions; n=4228) or the two genes that map upstream and downstream of the GWAS SNP(s) (i.e. in intergenic regions; n=3422). Enrichment of GWAS genes between our gene sets of interest was assessed by two-sided Fisher’s exact test. P-values in Fisher’s exact test for enrichment of genes surrounding GWAS hits between: EG vs. NEG (odds ratio=1.16, p=0.0015), EG vs. ALL (odds ratio=1.56, p=5.80e-31), NEG vs. ALL (odds ratio=1.35, p=1.18e-19).
Extended Data
Extended Data Figure 1.
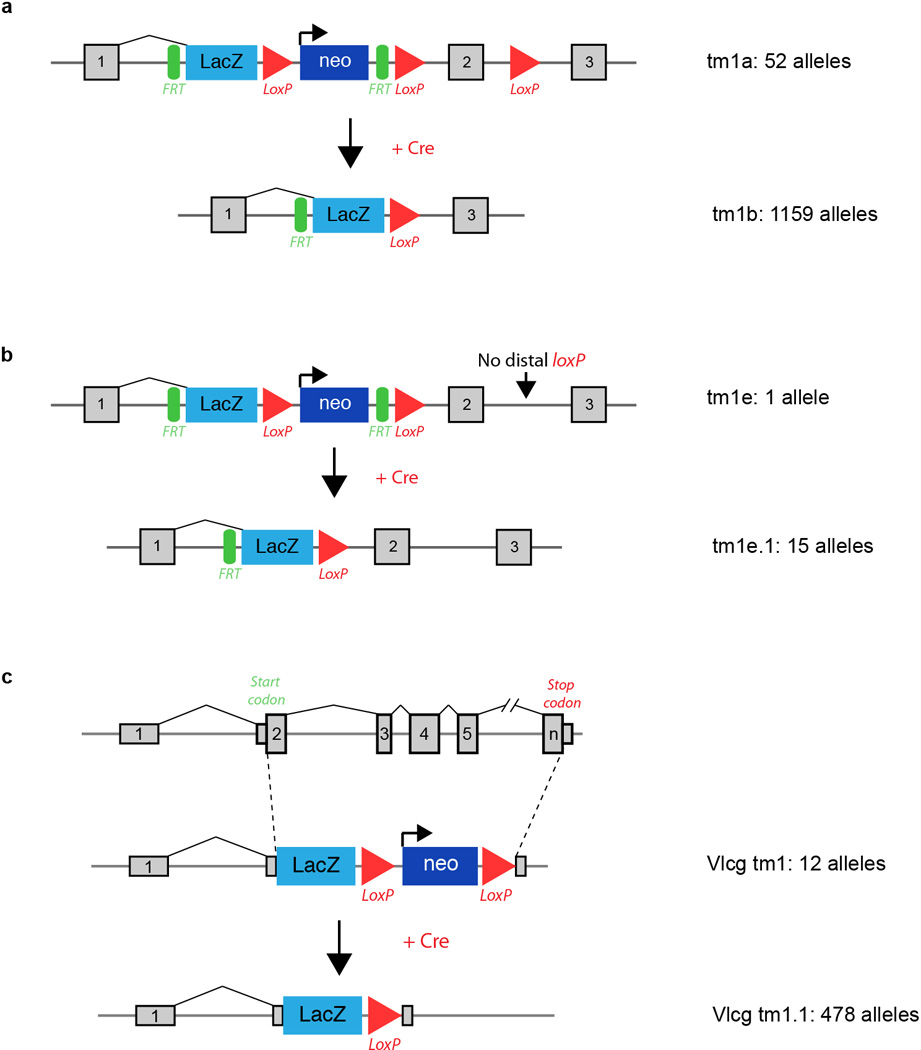
Standard IMPC allele variants and number of alleles for each included in this study. a, Conditional-ready, knockout-first allele (tm1a) design (top) with LacZ reporter, and the Cre converted (tm1b, bottom) version lacking the neo cassette and critical exon. The promoter driven variant is illustrated. b, Schematic of the small number of alleles included where the distal loxP had been lost during targeting (tm1e, top) and the converted (tme.1) variant with the neo cassette removed. c, Velocigene “definitive null” design (top, tm1) where the LacZ cassette replaces the coding sequence of the target gene, and Cre-excised variant (bottom, tm1.1). Details of all alleles used are listed in Supplementary Table 2 and 5. Additional details and schematics of all allele variants are available at: http://www.mousephenotype.org/about-ikmc/targeting-strategies
Extended Data Figure 2.
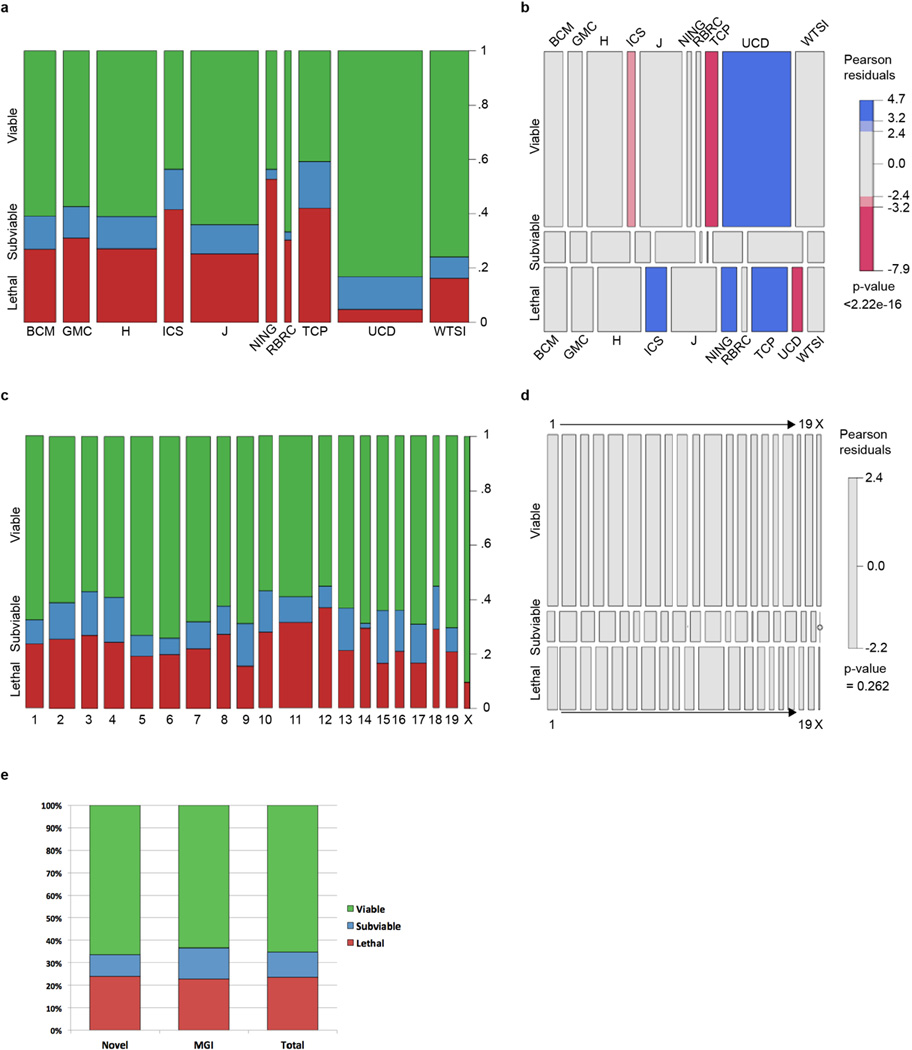
Spine (a) and mosaic (b) plots of progress examining primary viability of IMPC lines for each IMPC Center, segmented by ‘Lethal”, ‘Subviable’ or ‘Viable’ outcome. The mosaic plot shows the significant overrepresentation of viable lines from UCD and lethal lines from ICS, NING, and TCP. c,d, Spine and mosaic plots of primary viability outcome by chromosome, showing no significant deviation from the expected distribution. e, Comparison of the percentage of viable, subviable, and lethal lines between genes for which no targeted KO alleles have been reported (novel) and genes for which one or more KO alleles has been reported. BCM: Baylor College of Medicine; GMC: German Mouse Clinic; H: MRC Harwell; ICS: Institut Clinique del la Souris (PHENOMIN); J: The Jackson Laboratory; NING: Model Animal Research Center, Nanjing University; RBRC: RIKEN BioResource Center; TCP: Toronto Centre for Phenogenomics; UCD: University of California, Davis; WTSI: Wellcome Trust Sanger Institute.
Extended Data Figure 3.
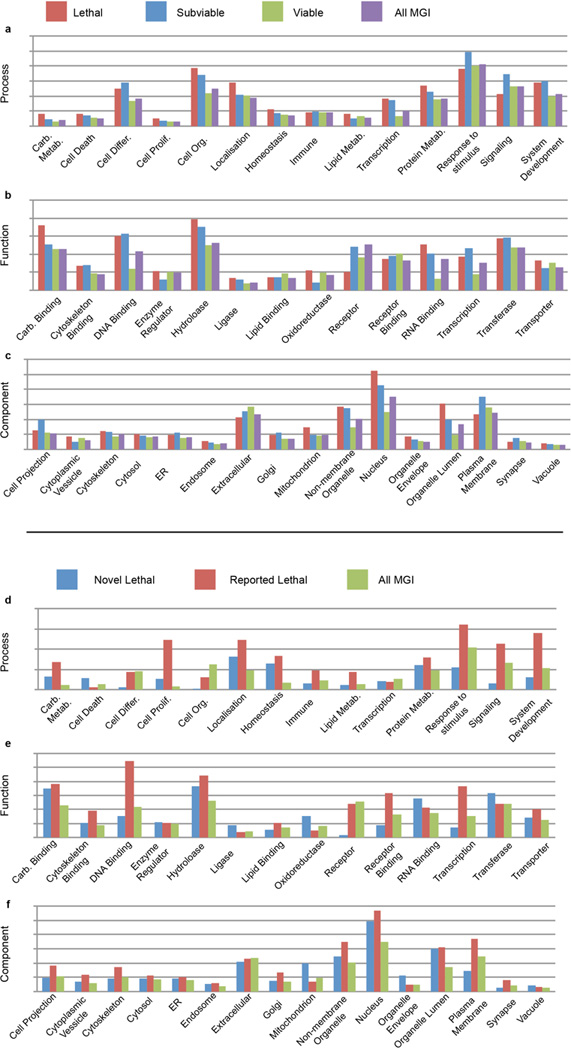
Multiple GOSlim categories show enrichment for lethal and subviable genes versus viable genes from the IMPC dataset. The analysis was performed for GO Process (a), GO Function (b) and GO Component (c) categories. X-axis is the proportion of genes in each class that are annotated for the GOSlim group for each category. d–f, Novel lethal IMPC genes, previously reported IMPC genes and all MGI genes were subject to the same analysis, showing the large effect analysis and characterization of lethal genes has on GO analysis.
Extended Data Figure 4.

Schematic of the IMPC embryonic lethal phenotyping pipeline. Lines are defined as lethal if zero homozygous animals are identified after 28+ animals have been genotyped. The KOMP2/IMPC centres begins with a mid-gestation (E12.5) screen, while the DMDD program initiates screening at the organogenesis phase (E14.5). If no homozygotes are identified (after >=28 embryos screened), centres will examine and characterize embryos at the pattern formation stage (E9.5). Homozygous embryos at this stage will be scored for gross anatomical defects and imaged using OPT. If live homozygotes are identified at E12.5, centres will proceed with the screen at E15.5 or E18.5. This decision is based on the presence of any observable phenotype at E12.5 and is at the discretion of the centre. Embryos collected at E15.5 are imaged via iodine-contrast microCT. Once sufficient numbers are collected, image registration and quantitative volumetric analysis is performed. Each time point should be considered independently, as some included strains have not been completely analysed and progression through each time point is at the discretion of the centre. For each term, two mutants with the same phenotype are required to score a hit.
Extended Data Figure 5.
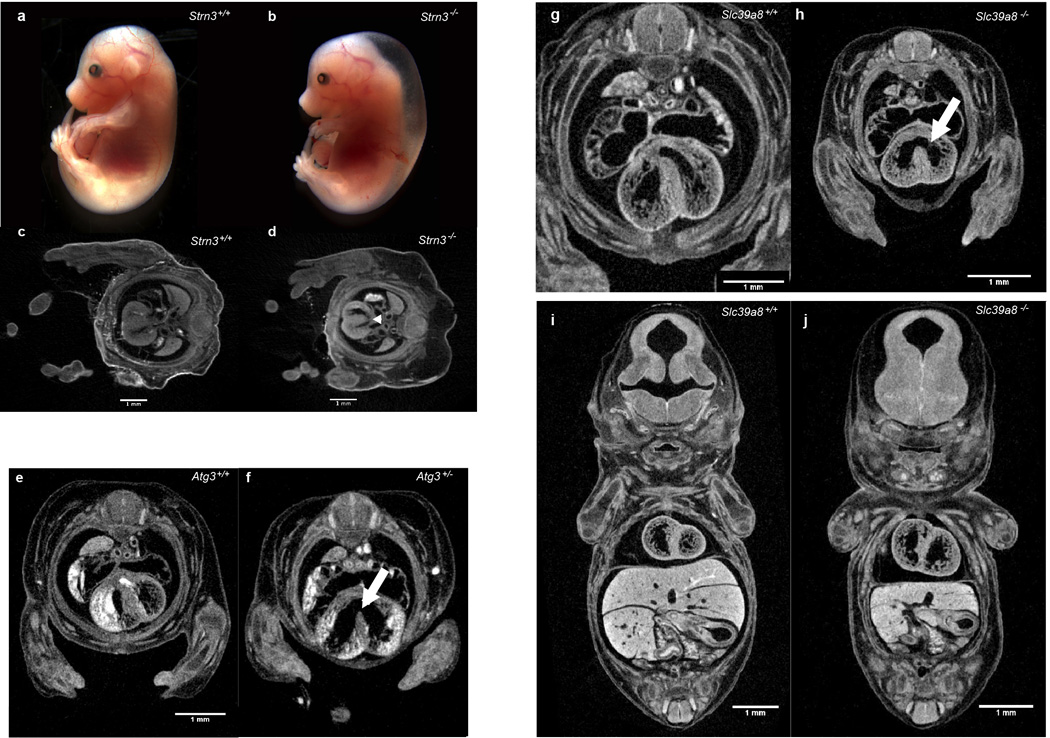
Cardiac defects in Strn3, Atg3, and Slc39a8 mutant embryos a,b severe fetal edema and sporadic hemorrhaging in E15.5 homozygous mutant embryos versus controls (n = 7 mutants analyzed) c,d Subtle, but consistent cardiac septal defects (arrowhead) observed transverse micro-CT volume sections in Strn3−/− embryos (d) versus control (c) (n = 5 mutants analyzed). Atg3+/− (e) and Atg3−/− (f) E14.5 embryos imaged by micro-CT after contrast staining showing evidence of novel heart morphological defects including ventricular septal defects (white arrows in f). Atg3−/− mice also show abnormal atrio-ventricular valves. (n = 4 mutants analyzed). Transverse (g,h) and coronal (i,j) sections through micro-CT volumes of mutant and control Slc39a8 E14.5 embryos reveals novel heart morphological defects including ventricular septal defects (white arrows in h). Slc39a8−/− mice also show the absence of sternum, a small chest cavity and a small liver (j) (n = 4 mutants analyzed).
Extended Data Figure 6.
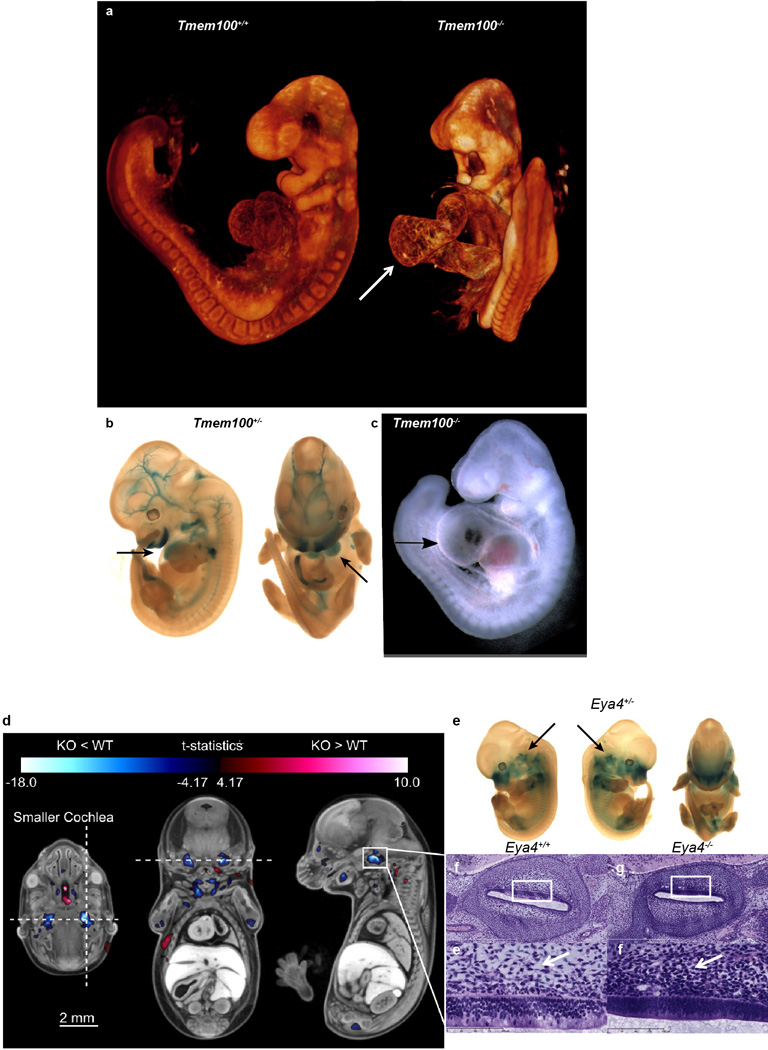
High-resolution 3D imaging reveals phenotypes in Tmem100 and Eya4 mutant embryos. Tmem100−/− embryos have abnormal heart development compared to Tmem100+/+ controls. E9.5 Tmem100−/− embryos had large pericardial effusion and cardiac dysmorphology and enlargement (arrow) when compared to E9.5 Tmem100+/+ (WT) embryos as seen by (a) OPT imaging and (c) bright-field microscopy resulting in lethality. (8 Tmem100+/+ vs. 8 Tmem100−/− with all 8 showing the defect). (b) LacZ expression in the E12.5 Tmem100+/− embryo indicated expression in the heart (arrows), blood vessels and craniofacial regions (blue). d–i, MicroCT imaging revealed a small cochlear volume in E15.5 Eya4−/− embryos. E15.5 Eya4−/− embryos were registered to an average control dataset of the same age followed by automated analysis to show that mutant embryos had a statistically smaller cochlear volume compared to Eya4+/+ (WT) embryos. (d) Transverse, coronal, and sagittal sections through the right cochlea are marked with a horizontal and vertical dashed line in the transverse section to indicate the location of the coronal and sagittal sections, respectively. The color corresponds to areas of larger (red) and smaller (blue) volumes in the KO embryos. The color bar minimum corresponds to a false discovery rate (FDR) threshold of 5%. Hypoplastic bilateral cochlear structures are highlighted in blue. (8 Eya4+/+ (WT) vs. 8 Eya4−/− (KO) with all 8 showing the defect). (e) LacZ imaging in the E12.5 Eya4+/− revealed Eya4 gene expression (blue) in the cochlear region (arrow). (f,g) H&E stained histological sections through the right cochlea of an Eya4+/+ embryo (f) compared to an Eya4−/−embryo (g) confirmed the hypoplastic phenotype. (h,i) Higher magnification of the region (indicated by the white boxes) showed abnormal perilymphatic (periotic) mesenchyme in mutant embryos. In the mutant embryo (i) the perilymphatic mesenchyme did not show rarefaction and had reduced vacuolation versus control (h) (arrows) suggesting the cochlear hypoplasia was due to delayed perilymph development.
Extended Data Figure 7.
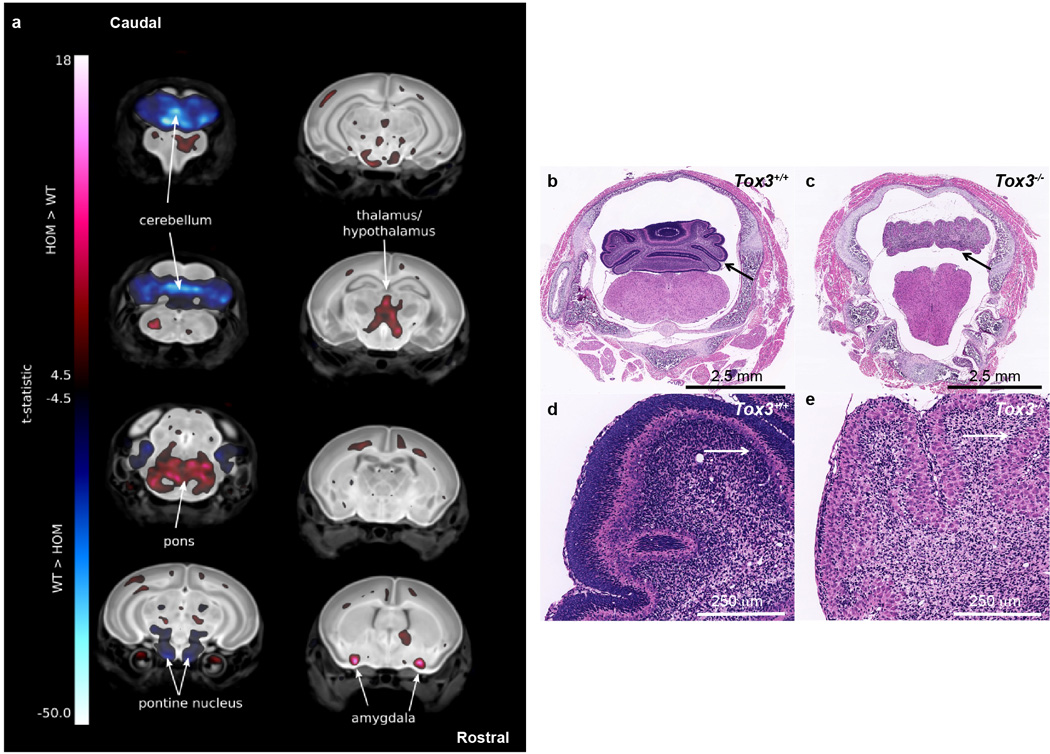
Coronal sections of Whole Brain MRI revealed many volume changes in the P7 Tox3−/− (WT) mice. (a) P7 Tox3−/− (KO) mice brains were registered to an average control dataset of P7 Tox3+/+ (WT) brains. The color corresponds to areas of larger (red) and smaller (blue) relative volumes in the KO embryos. The color bar minimum corresponds to a false discovery rate (FDR) threshold of 5%. KO mice exhibited altered volumes in multiple brain structures including an enlarged pons, amygdala, and thalamus/hypothalamus and a decreased pontine nucleus when compared to the WT brains (arrows). Most striking was the decrease in the size of the cerebellum of the KO mice (arrows). (8 Tox3+/+ (WT) vs. 10 Tox3−/− (KO) with all 10 showing the defects). Histological analysis of Tox3−/− (KO) mice revealed abnormal development of the cerebellum. (b,c) The cerebellum of P7 Tox3−/− (KO) mice is hypoplastic and dysplastic characterized by markedly reduced fissure formation, poor delineation of folia, and disorganized cortical structure and layering (c) when compared to the P7 Tox3+/+ (WT) mice (b) (arrows). In some segments, there was complete absence of folial pattern. (d,e) Higher magnification revealed that the normally transient external granular layer was absent in the Tox3−/− (KO) mice and the subjacent molecular layer was hypotrophic and irregular in thickness and in multiple foci extremely thin or absent; in these foci the Purkinje cells extended to the pial surface (arrows). The Purkinje cell layer was also jumbled with no evidence of cell polarity (e).
Extended Data Figure 8.
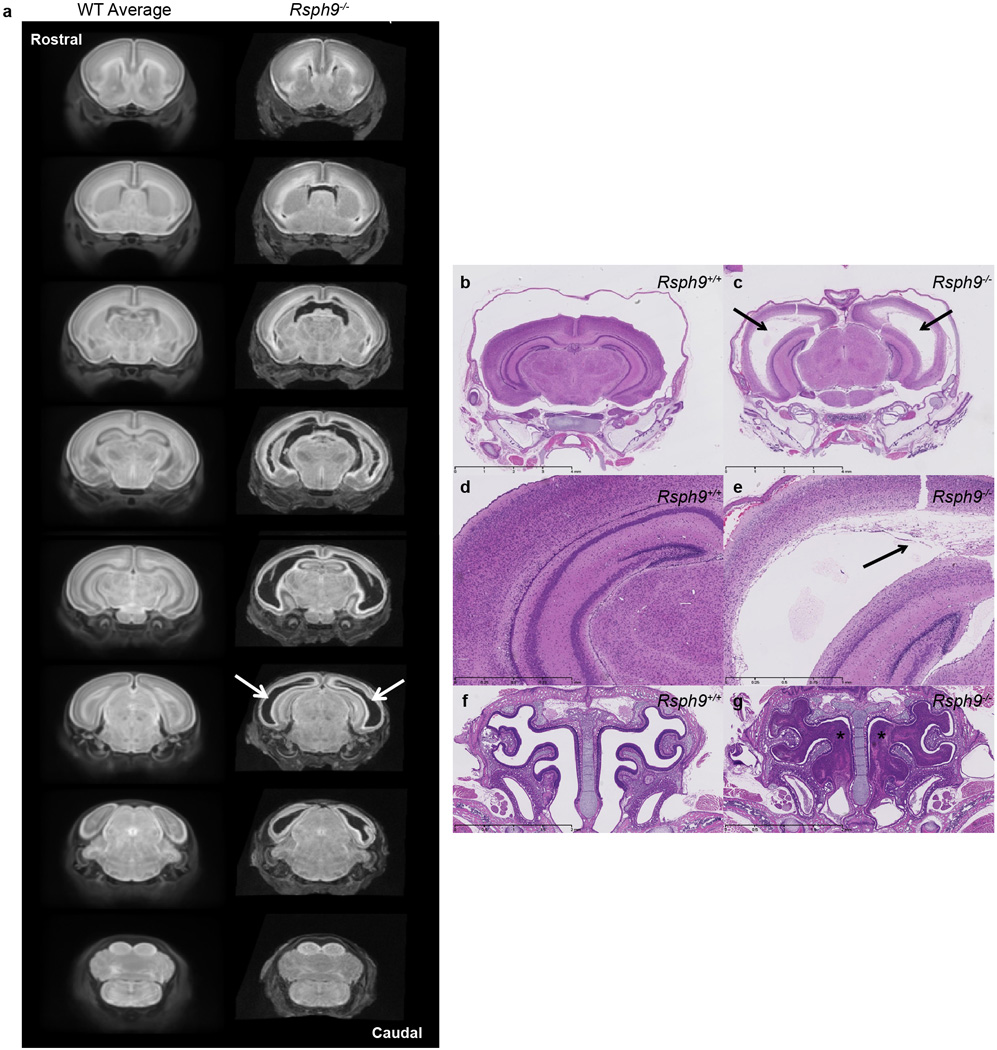
Coronal sections of whole brain MRI revealed enlarged ventricles in the P7 Rsph9−/− (KO) mice. (a) P7 Rsph9−/− mice brains showed enlarged left and right lateral ventricles (arrows) when virtually sectioned from rostral to caudal and compared to a WT average of P7 Rsph9+/+ mice brains. (8 Rsph9+/+ (WT) vs. 10 Rsph9−/− (KO) with all 10 showing the defects). Histological analysis of Rsph9−/− (KO) mice confirmed abnormal brain development. (b,c) Arrows indicate severe hydrocephalus of the left and right lateral ventricles of the Rsph9−/− (KO) P7 mice (c) compared to the Rsph9+/+ (WT) mice (b). The third ventricle was also enlarged but not seen in this section. (d,e) Higher magnification of the cerebrum showed marked rarefaction, cavitation, and loss of periventricular cortical tissue (arrow) in the KO mice (e) compared to WT (d). (f,g) Coronal section through the nasal region revealed that the sinuses of the KO mice were filled with pus (asterisks) (g).
Extended Data Figure 9.
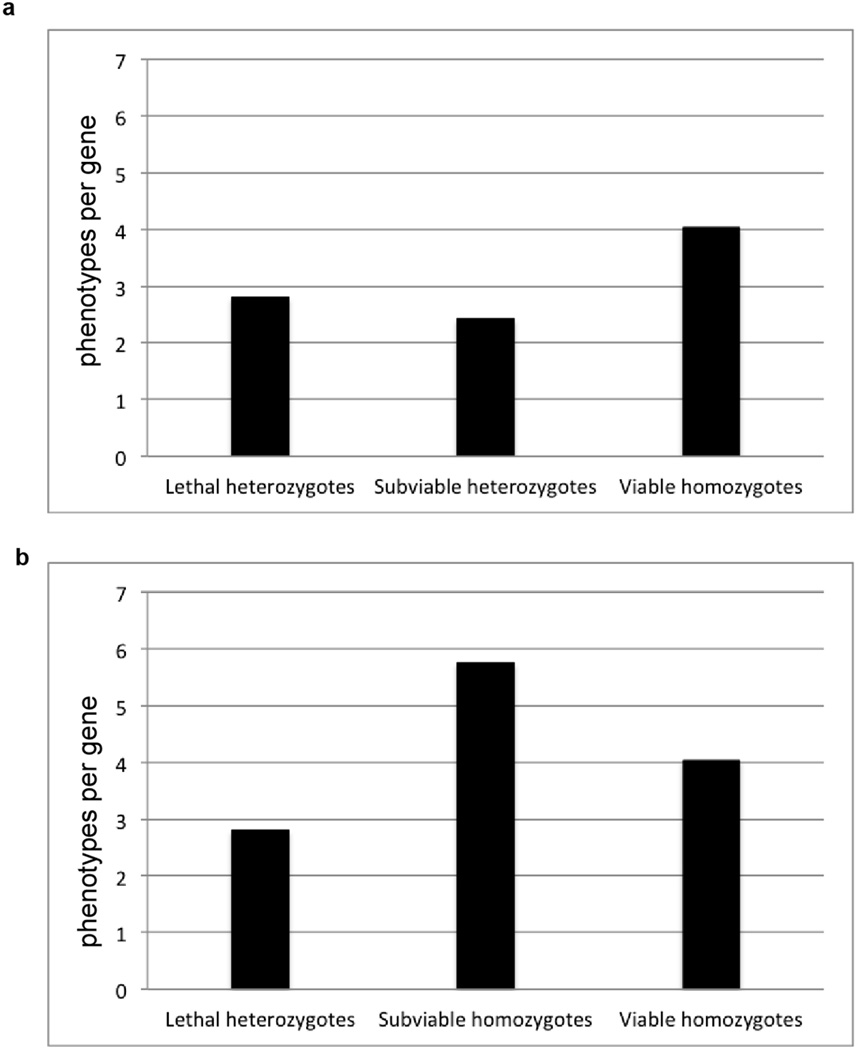
Phenotype hit rates from the adult phenotyping pipeline for lethal, subviable and viable lines. a, Comparison of hit rates between lethal and subviable line heterozygotes versus viable line homozygotes. b, Homozygous subviable cohorts show a dramatically higher hit rate versus lethal line heterozygotes and viable line homozygotes.
Extended Data Figure 10.
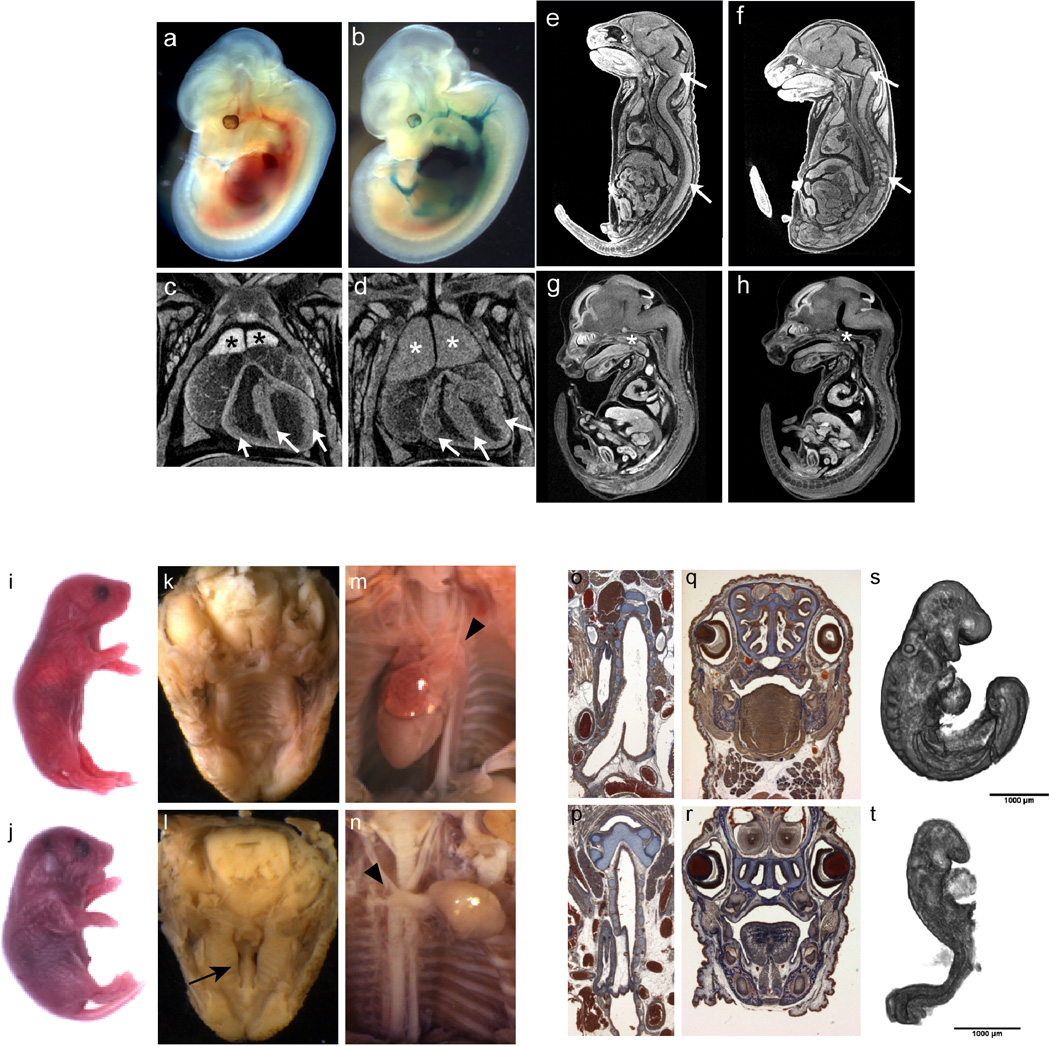
LacZ expression in Gyg heterozygous and homozygous embryos at E12.5 showed specific, strong expression in the heart and surrounding major vessels (i.e, the dorsal aorta, the carotid artery and umbilical artery) (a,b), consistent with smooth muscle cells at this stage. Homozygous embryos were recovered at expected proportions at E12.5, E15.5 and E18.5 and could not be distinguished from wild type and heterozygous embryos by outward appearance. However, inspection of cross-sections through the whole embryo microCT images of E18.5 and E15.5 embryos showed abnormalities in several areas of the developing embryo. Thickened myocardium was evident in the hearts of 2 of 3 homozygotes examined at E15.5 as shown in Figure 5. Coronal cross-sections also confirmed thickened myocardium in E18.5 mutant hearts (arrows; n=5 mutants), compare wt (c) to Gygtm1b/tm1b(d). From the E18.5 sections, it was also obvious that the thymus was enlarged in mutants (n=5 mutants) compared with controls (*), but the thymus appeared normal in E15.5 mutant embryos (data not shown). E18.5 mutant embryos also exhibited abnormal gaps in the brain and spinal cord that we interpret as neural degeneration; compare wt littermates (e) to Gygtm1b/tm1b mutants (n=5 mutants) (f). Abnormalities in the nervous system, similar to abnormalities in the heart, were obvious at E15.5. Representative images are shown from sagittal cross-sections through a wild type (g) and a homozygous Gyg mutant E15.5 embryo (h) (n=3). E15.5 Gygtm1b/tm1b mutant embryos have a flattened forebrain with reduced lateral ventricles, as well as excess space within the cephalic and cervical flexures. i–t, tm1a and tm1b alleles can lead to phenotypes of differing strength in Kdm8 mutants. Abnormal phenotype of Kmd8tm1a/tm1a mice at E18.5: i, k, m, o, q: WT fetuses; j, l, n, p, r: mutant fetuses. i, j, gross morphological appearance of E18.5 fetuses. k–n, photomicrographs of the palate and heart taken during necropsy. g– j, histological sections at similar levels of the trachea and the nasal cavities. (n = 4 mutants analyzed at E18.5). Morphology of control (s) and mutants (t) Kmd8 embryos at E9.5 captured by OPT showing developmental delay at that stage, including small size and lack of turning. Arrows, unfused palatal shelves; arrowheads, arch of the aorta. (n = 7 mutants analyzed at E9.5) Bar = 1 mm
Supplementary Material
Acknowledgments
The authors would like to thank all of the IMPC members and partners for their contribution to the consortium effort, including this study, and acknowledge the contributions of Janet Rossant, S. Lee Adamson, and Tania Bubela. This work was supported by NIH grants U42 OD011185 (S.A.M), U54 HG006332 (R.E.B, K.S), U54 HG006348-S1 and OD011174 (A.L.B.), HG006364-03S1 and U42 OD011175 (K.C.K.L.), U54 HG006370 (P.F., A-M.M., H.E.P., S.D.M.B.) and additional support provided by the The Wellcome Trust, Medical Research Council Strategic Award (L.T., S.W., S.D.M.B.), Government of Canada through Genome Canada and Ontario Genomics (OGI-051)(C.M., S.D.M.B.), Wellcome Trust Strategic Award “Deciphering the Mechanisms of Developmental Disorders (DMDD)” (WT100160) (D.A., T.M.), National Centre for Scientific Research (CNRS), the French National Institute of Health and Medical Research (INSERM), the University of Strasbourg (UDS), the “Centre Européen de Recherche en Biologie et en Médecine”, the “Agence Nationale de la Recherche” under the frame programme “Investissements d’Avenir” labelled ANR-10-IDEX-0002-02, ANR-10-INBS- 07 PHENOMIN to (Y.H.), The German Federal Ministry of Education and Research by Infrafrontier grant 01KX1012 (S.M., V.G.D., H.F., M.HdA.)
Footnotes
Code accessibility
Analysis code for bioinformatics data presented in Figures 1 and 5 can be accessed at https://github.com/IMPC2015/code.
Additional Contributors
19The International Mouse Phenotyping Consortium
The Jackson Laboratory: Matthew McKay, Barbara Urban, Caroline Lund, Erin Froeter, Taylor LaCasse, Adrienne Mehalow, Emily Gordon, Leah Rae Donahue, Robert Taft, Peter Kutney, Stephanie Dion, Leslie Goodwin, Susan Kales, Rachel Urban, Kristina Palmer
Infrastructure Nationale PHENOMIN, Institut Clinique de la Souris (ICS): Fabien Pertuy, Deborah Bitz, Bruno Weber, Patrice Goetz-Reiner, Hughes Jacobs, Elise Le Marchand, Amal El Amri, Leila El Fertak, Hamid Ennah, Dalila Ali-Hadji, Abdel Ayadi, Marie Wattenhofer-Donze, Sylvie Jacquot, Philippe André, Marie-Christine Birling, Guillaume Pavlovic, Tania Sorg.
Charles River Laboratories: Iva Morse, Frank Benso
MRC Harwell: Michelle E Stewart, Carol Copley, Jackie Harrison, Samantha Joynson
The Toronto Centre for Phenogenomics: Ruolin Guo, Dawei Qu, Shoshana Spring, Lisa Yu, Jacob Ellegood, Lily Morikawa, Xueyuan Shang, Pat Feugas, Amie Creighton, Patricia Castellanos Penton, Ozge Danisment
The Wellcome Trust Sanger Institute: Nicola Griggs, Catherine L. Tudor, Angela L. Green, Cecilia Icoresi Mazzeo, Emma Siragher, Charlotte Lillistone, Elizabeth Tuck, Diane Gleeson, Debarati Sethi, Tanya Bayzetinova, Jonathan Burvill, Bishoy Habib, Lauren Weavers, Ryea Maswood, Evelina Miklejewska, Michael Woods, Evelyn Grau, Stuart Newman, Caroline Sinclair, Ellen Brown
RIKEN BioResource Center: Shinya Ayabe, Mizuho Iwama, Ayumi Murakami
Contributions
M.E.D, A.M.F., X.J, L.T., M.D.W., J.K.W, T.F.M, W.J.W., H.W., D.J.A., M.B., and S.A.M. contributed to the data analysis and writing of the paper, A.Y., A.B., L.B., L.B.C., F.C., B.D., H.F., A. Galli, A.G., V. G-D., S.G., S.M., S.A.M., L.M.J.N., E.R., J.R.S., M.S., W.C.S., R.R.S., L.T., S.W., J.K.W., generated animal models and identified lethal genes, M.E.D, A.M.F., X.J., H.W., L.T., J.M.B., N.R.H., T.F.M., M.E.Dolan, S.A.M. contributed to gene list analysis, H.A., L.B, L.B.C., C.N.B., J.C., J.M.D., M.E.D, S.M.E., A.M.F. A. Galli, C-W.H., S.J.J., S.K., L.C.K., L.L., M.M., M.L.M., T.M., S.A.M., S.N., L.M.J.N., K.A.P., D.R., E.R., Z. S-K., M.T., L.T., A.T., O.W., W.J.W., J.K.W., L.W., contributed to the secondary lethal screen and data analysis, J.M.B., D.C., J.G., N.R.H, T.N.L., J.M., I.T., and J.W. provided informatics support, M.D.W. and R.M.H. performed the automated 3D analysis, J.M.B, N.R.H, I.T., J.W., and H.W. developed and implemented the IMPC portal, X.J, M.J.D., S.A.M., M.L., K.E.S., D.G.M., D.J.A., and M.B. contributed to the essential gene and human disease analysis, M.E.D, A.M.F., X.J, L.T., M.D.W., J.K.W, T.F.M, W.J.W., H.W., S.W., R.R-S., J.M.D., D.G.M., D.B.W., G.P.T-V, X.G., P.F., W.C.S., A.B, M.J.J., H.E.P., M.M, S.W., R.E.B., K.S., M.H.d.A, Y.H., T.M., A.-M.M., R.M.H., S.D.M.B., D.J.A., K.C.K.L., C.M., A.L.B., M.B., and S.A.M. contributed to the design, management, execution of the work and review of the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
Data accessibility
All data is freely available from the IMPC database hosted at EMBL-EBI via a web portal (mousephenotype.org), ftp (ftp://ftp.ebi.ac.uk/pub/databases/impc) and automatic programmatic interfaces. An archived version of the database will be maintained after cessation of funding (exp. 2021) for an additional 5 years. Allele and phenotype summaries are additionally archived with Mouse Genome Informatics at the Jackson Laboratory via direct data submissions (J:136110, J:148605, J:157064, J:157065, J:188991, J:211773).
References
- 1.Cox BJ, et al. Phenotypic annotation of the mouse X chromosome. Genome Res. 2010;20:1154–1164. doi: 10.1101/gr.105106.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Justice MJ. Capitalizing on large-scale mouse mutagenesis screens. Nat Rev Genet. 2000;1:109–115. doi: 10.1038/35038549. [DOI] [PubMed] [Google Scholar]
- 3.Kasarskis A, Manova K, Anderson KV. A phenotype-based screen for embryonic lethal mutations in the mouse. Proc Natl Acad Sci U S A. 1998;95:7485–7490. doi: 10.1073/pnas.95.13.7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kile BT, Hilton DJ. The art and design of genetic screens: mouse. Nat Rev Genet. 2005;6:557–567. doi: 10.1038/nrg1636. [DOI] [PubMed] [Google Scholar]
- 5.Perrin S. Preclinical research: Make mouse studies work. Nature. 2014;507:423–425. doi: 10.1038/507423a. [DOI] [PubMed] [Google Scholar]
- 6.Prinz F, Schlange T, Asadullah K. Believe it or not: how much can we rely on published data on potential drug targets? Nat Rev Drug Discov. 2011;10:712. doi: 10.1038/nrd3439-c1. [DOI] [PubMed] [Google Scholar]
- 7.Ayadi A, et al. Mouse large-scale phenotyping initiatives: overview of the European Mouse Disease Clinic (EUMODIC) and of the Wellcome Trust Sanger Institute Mouse Genetics Project. Mamm Genome. 2012;23:600–610. doi: 10.1007/s00335-012-9418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradley A, et al. The mammalian gene function resource: the International Knockout Mouse Consortium. Mamm Genome. 2012;23:580–586. doi: 10.1007/s00335-012-9422-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Angelis MH, et al. Analysis of mammalian gene function through broad-based phenotypic screens across a consortium of mouse clinics. Nat Genet. 2015;47:969–978. doi: 10.1038/ng.3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Skarnes WC, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474:337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valenzuela DM, et al. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol. 2003;21:652–659. doi: 10.1038/nbt822. [DOI] [PubMed] [Google Scholar]
- 12.White JK, et al. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell. 2013;154:452–464. doi: 10.1016/j.cell.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adams D, et al. Bloomsbury report on mouse embryo phenotyping: recommendations from the IMPC workshop on embryonic lethal screening. Dis Model Mech. 2013;6:571–579. doi: 10.1242/dmm.011833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blomen VA, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350:1092–1096. doi: 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- 16.Hart T, et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163:1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 17.Wang T, et al. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharpe J, et al. Optical projection tomography as a tool for 3D microscopy and gene expression studies. Science. 2002;296:541–545. doi: 10.1126/science.1068206. [DOI] [PubMed] [Google Scholar]
- 19.Wong MD, Dorr AE, Walls JR, Lerch JP, Henkelman RM. A novel 3D mouse embryo atlas based on micro-CT. Development. 2012;139:3248–3256. doi: 10.1242/dev.082016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong MD, Maezawa Y, Lerch JP, Henkelman RM. Automated pipeline for anatomical phenotyping of mouse embryos using micro-CT. Development. 2014;141:2533–2541. doi: 10.1242/dev.107722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong MD, et al. 4D atlas of the mouse embryo for precise morphological staging. Development. 2015;142:3583–3591. doi: 10.1242/dev.125872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weninger WJ, et al. Phenotyping structural abnormalities in mouse embryos using high-resolution episcopic microscopy. Dis Model Mech. 2014;7:1143–1152. doi: 10.1242/dmm.016337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu B, et al. Cbx4 regulates the proliferation of thymic epithelial cells and thymus function. Development. 2013;140:780–788. doi: 10.1242/dev.085035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horvitz HR, Sulston JE. Isolation and genetic characterization of cell-lineage mutants of the nematode Caenorhabditis elegans. Genetics. 1980;96:435–454. doi: 10.1093/genetics/96.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burga A, Casanueva MO, Lehner B. Predicting mutation outcome from early stochastic variation in genetic interaction partners. Nature. 2011;480:250–253. doi: 10.1038/nature10665. [DOI] [PubMed] [Google Scholar]
- 26.Raj A, Rifkin SA, Andersen E, van Oudenaarden A. Variability in gene expression underlies incomplete penetrance. Nature. 2010;463:913–918. doi: 10.1038/nature08781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matzuk MM, Kumar TR, Bradley A. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature. 1995;374:356–360. doi: 10.1038/374356a0. [DOI] [PubMed] [Google Scholar]
- 28.Song J, et al. The type II activin receptors are essential for egg cylinder growth, gastrulation, and rostral head development in mice. Dev Biol. 1999;213:157–169. doi: 10.1006/dbio.1999.9370. [DOI] [PubMed] [Google Scholar]
- 29.Georgi B, Voight BF, Bucan M. From mouse to human: evolutionary genomics analysis of human orthologs of essential genes. PLoS Genet. 2013;9:e1003484. doi: 10.1371/journal.pgen.1003484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dickerson JE, Zhu A, Robertson DL, Hentges KE. Defining the role of essential genes in human disease. PLoS One. 2011;6:e27368. doi: 10.1371/journal.pone.0027368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stenson PD, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 32.Stenson PD, et al. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet. 2014;133:1–9. doi: 10.1007/s00439-013-1358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Welter D, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schinzel A, Giedion A. A syndrome of severe midface retraction, multiple skull anomalies, clubfeet, and cardiac and renal malformations in sibs. Am J Med Genet. 1978;1:361–375. doi: 10.1002/ajmg.1320010402. [DOI] [PubMed] [Google Scholar]
- 35.Piazza R, et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat Genet. 2013;45:18–24. doi: 10.1038/ng.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moslemi AR, et al. Glycogenin-1 deficiency and inactivated priming of glycogen synthesis. N Engl J Med. 2010;362:1203–1210. doi: 10.1056/NEJMoa0900661. [DOI] [PubMed] [Google Scholar]
- 37.Malfatti E, et al. A new muscle glycogen storage disease associated with glycogenin-1 deficiency. Ann Neurol. 2014;76:891–898. doi: 10.1002/ana.24284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ballif BC, et al. Discovery of a previously unrecognized microdeletion syndrome of 16p11.2–p12.2. Nat Genet. 2007;39:1071–1073. doi: 10.1038/ng2107. [DOI] [PubMed] [Google Scholar]
- 39.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sulem P, et al. Identification of a large set of rare complete human knockouts. Nat Genet. 2015 doi: 10.1038/ng.3243. [DOI] [PubMed] [Google Scholar]
- 41.MacArthur DG, et al. A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alkuraya FS. Human knockout research: new horizons and opportunities. Trends Genet. 2015;31:108–115. doi: 10.1016/j.tig.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 43.Narasimhan VM, et al. Health and population effects of rare gene knockouts in adult humans with related parents. Science. 2016;352:474–477. doi: 10.1126/science.aac8624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Threadgill DW, et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–234. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- 45.Simon MM, et al. A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol. 2013;14:R82. doi: 10.1186/gb-2013-14-7-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang H, et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edwards AM, et al. Too many roads not taken. Nature. 2011;470:163–165. doi: 10.1038/470163a. [DOI] [PubMed] [Google Scholar]
Methods Supplementary References
- 1.Wong MD, Dazai J, Walls JR, Gale NW, Henkelman RM. Design and implementation of a custom built optical projection tomography system. PLoS One. 2013;8:e73491. doi: 10.1371/journal.pone.0073491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong MD, Spring S, Henkelman RM. Structural stabilization of tissue for embryo phenotyping using micro-CT with iodine staining. PLoS One. 2013;8:e84321. doi: 10.1371/journal.pone.0084321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feldkamp L, Davis L. Practical cone-beam tomography. J Opt Soc Am A. 1984 [Google Scholar]
- 4.Weninger WJ, et al. Phenotyping structural abnormalities in mouse embryos using high-resolution episcopic microscopy. Dis Model Mech. 2014;7:1143–1152. doi: 10.1242/dmm.016337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wong MD, Maezawa Y, Lerch JP, Henkelman RM. Automated pipeline for anatomical phenotyping of mouse embryos using micro-CT. Development. 2014;141:2533–2541. doi: 10.1242/dev.107722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wong MD, Dorr AE, Walls JR, Lerch JP, Henkelman RM. A novel 3D mouse embryo atlas based on micro-CT. Development. 2012;139:3248–3256. doi: 10.1242/dev.082016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nieman BJ, Flenniken AM, Adamson SL, Henkelman RM, Sled JG. Anatomical phenotyping in the brain and skull of a mutant mouse by magnetic resonance imaging and computed tomography. Physiol Genomics. 2006;24:154–162. doi: 10.1152/physiolgenomics.00217.2005. [DOI] [PubMed] [Google Scholar]
- 8.Nieman BJ, et al. Mouse behavioral mutants have neuroimaging abnormalities. Hum Brain Mapp. 2007;28:567–575. doi: 10.1002/hbm.20408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collins DL, Neelin P, Peters TM, Evans AC. Automatic 3D intersubject registration of MR volumetric data in standardized Talairach space. J Comput Assist Tomogr. 1994;18:192–205. [PubMed] [Google Scholar]
- 10.Avants BB, et al. A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage. 2011;54:2033–2044. doi: 10.1016/j.neuroimage.2010.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lau JC, et al. Longitudinal neuroanatomical changes determined by deformation-based morphometry in a mouse model of Alzheimer's disease. Neuroimage. 2008;42:19–27. doi: 10.1016/j.neuroimage.2008.04.252. [DOI] [PubMed] [Google Scholar]
- 12.Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage. 2002;15:870–878. doi: 10.1006/nimg.2001.1037. [DOI] [PubMed] [Google Scholar]
- 13.Eppig JT, et al. The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res. 2015;43:D726–D736. doi: 10.1093/nar/gku967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith CL, Eppig JT. Expanding the mammalian phenotype ontology to support automated exchange of high throughput mouse phenotyping data generated by large-scale mouse knockout screens. J Biomed Semantics. 2015;6:11. doi: 10.1186/s13326-015-0009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cunningham F, et al. Ensembl 2015. Nucleic Acids Res. 2015;43:D662–D669. doi: 10.1093/nar/gku1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stenson PD, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–581. doi: 10.1002/humu.10212. [DOI] [PubMed] [Google Scholar]
- 18.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hart T, et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163 doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 20.Wang T, et al. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blomen VA, et al. Gene essentiality and synthetic lethality in haploid human cells. Science. 2015;350:1092–1096. doi: 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- 22.Welter D, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic acids research. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.