

Abstract
Ribosome inactivating proteins (RIPs) are among the most toxic agents known. More than a dozen clinical trials against refractory cancers have been initiated using modified RIPs with impressive results. However, dose-limiting toxicity due to vascular leak syndrome limits success of the therapy. We have previously reported some tight-binding transition state analogues of Saporin L3 that mimic small oligonucleotide substrates in which the susceptible adenosine has been replaced by a 9-deazaadenyl hydroxypyrrolidinol derivative. They provide the first step in the development of rescue agents to prevent Saporin L3 toxicity on non-targeted cells. Here we report the synthesis, using solution phase chemistry, of these and a larger group of transition state analogues. They were tested for inhibition against Saporin L3 giving Ki values as low as 3.3 nM and indicating the structural requirements for inhibition.
Keywords: Saporin, Ribosome inactivating protein, Transition state inhibitor, Oligonucleotide, Aza-sugar
1. Introduction
Saporins are ribosome inactivating proteins (RIPs) isolated from the soapwort (Saponaria officinalus) [1]. Like the better known ricin A-chain (RTA), saporins bind to the sarcin – ricin loop of the 28S eukaryotic ribosomal RNA and hydrolytically depurinate adenosine 4234 [2], thereby preventing protein synthesis and causing cell death. Saporins are less specific than RTA, as they also hydrolyze adenine from other sites on RNAs.
Trial cancer therapies have exploited the toxicity of saporin and RTA – antibody constructs targeted to leukemia and lymphoma cells [3]. Off-target toxicity resulting from incomplete uptake or release of RIPs from dying target cells limits the use of these therapies. Inhibitors of RIPs could provide therapy enhancement by inhibition of unwanted RIP following the initial treatment [4]. This paper describes the synthesis and characterization of several potent inhibitors of saporin L3 (SAP) [5,6] a highly active saporin isoform originally isolated from S. officinalus leaves. The inhibitors are characterized with saporin L3 expressed in yeast and mutated to replace alanine 14 with cysteine (SAP A14C), a mutation remote from the catalytic site which provides a chemical attachment site for cell-recognition molecules.
The transition states for RTA and SAP-catalyzed depurination of RNAs have ribocation character from which activated adenine is largely dissociated [7]. The nonhydrolysable adenosine mimic 1 (DIA) captures, in stable form, key features of the transition state geometry. The extended bond length between leaving group adenine and ribose is mimicked by the methylene bridge, the imino-sugar is protonated at physiological pH to mimic the ribocation, and N7 of the 9-deazaadenine ring is protonated at physiological pH. DIA does not significantly inhibit either SAP or RTA, but replacement of the susceptible adenosine in RNA sequences from the reactive part of the sarcin – ricin loop with DIA gives potent inhibitors [8]. The stem loop A-10 (5′-CGCGAGAGCG-3′) along with linear and cyclized GAGA constructs are accepted as substrates by SAP [4], and these molecules provide a template for inhibitor design. SAP accepts truncated RNAs as substrates and is inhibited by the corresponding aza-sugar constructs under physiological conditions whereas RTA requires reduced pH for activity on truncated substrates and for inhibition [4,8].
The crystal structure of a cyclic G(DIA)GA tetramer bound to SAP shows a quadruple π-stack in which deazaadenine is positioned between two tyrosine phenol groups with the 3′- guanosine providing the final layer of the π-stack. These interactions, together with multiple hydrogen bonds, are proposed to provide leaving group activation in catalytic structures [9].
The RNA oligonucleotides containing DIA, 2–4 (Fig. 1), are potent inhibitors of SAP [4]. These oligonucleotides are stabilised against ribonucleases by 2′-O-methylation [10] which, in the context of inhibitor design, is tolerated by SAP [11] and some have phosphate end groups capped with propanediol. They were synthesized in the usual manner for oligonucleotides, on solid support with stepwise addition of each nucleotide phosphoramidite, often requiring multiequivalents of each. We have found that aza -sugar phosphoramidites such as 14 (Scheme 1) give poor and non-reproducible yields under this protocol.
Fig. 1.
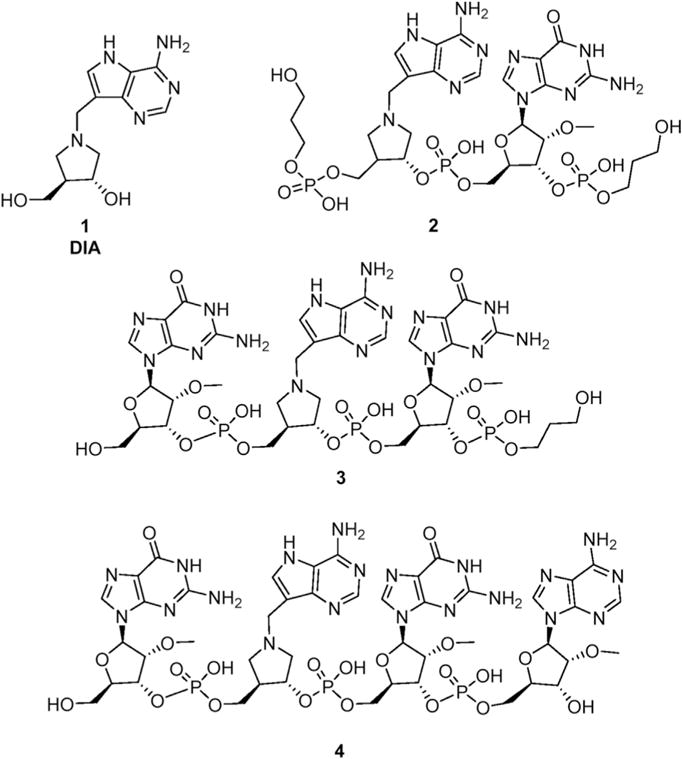
Adenosine mimics 1 and some inhibitors of SAP.
Scheme 1. Synthesis of 4 (GpDIApGpA).
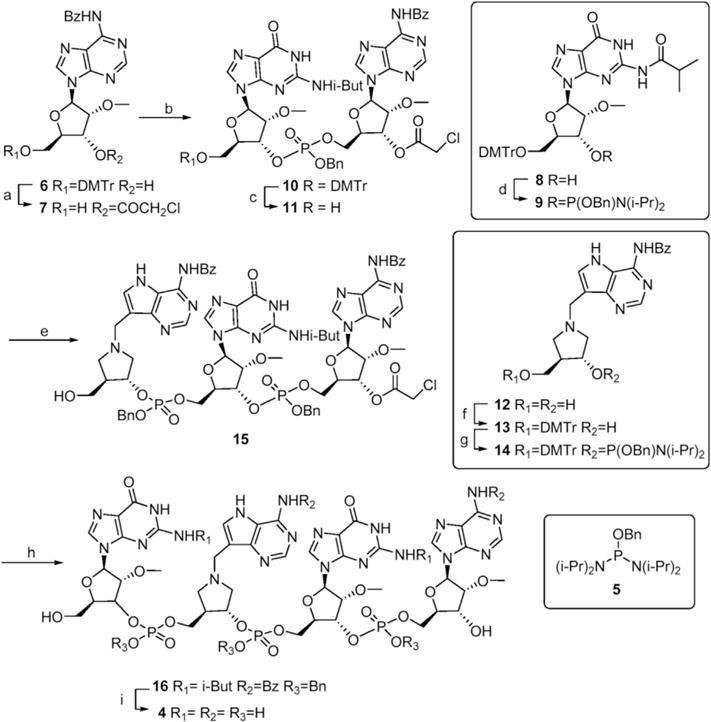
Reagents and conditions a) ClCH2COCl, pyridine, CH2Cl2 then 80% AcOH, 71% b) 9, MTET, CH2Cl2 then t-BuOOH, 85% c) 80% AcOH, 72% d) 5, tetrazole, CH2Cl2, 96% e) 14, MTET, CH2Cl2, then t-BuOOH, then 80% AcOH, 57% f) DMTrCl, pyridine, 84% g) 5, MTET, CH2Cl2 75% h) 9, MTET, t-BuOOH, then 80% AcOH, 6% i) Pd/C, H2, then NH4OH, 40 °C, 39%.
Using conventional solution phase chemistry, which allows for convergent strategies and more efficient use of valuable intermediates, we have synthesized a group of aza-sugar-containing 2′-O-methyl di, tri and tetra RNA nucleotides that probe both aza-sugar structure and the nucleotide context required for inhibition of SAP [12,13]. Compounds 2–4 were resynthesised for comparison and to provide sufficient material for chemical characterization.
2. Results and discussion
2.1. Chemistry
Tetramer 4 (GpDIApGpA) is an inhibitor construct corresponding to the GAGA substrate of SAP. It was synthesized in a linear fashion by stepwise addition to the 3′-terminal adenosine (Scheme 1). Reaction of the 3′-hydroxyl of commercially available 5′-O-dimethoxytrityl-6-N-benzoyl-2′-O-methyladenosine 6 with chloroacetyl chloride followed by hydrolysis of the dimethoxytrityl (DMTr) group with aqueous acetic acid gave 5′-OH adenosine 7 in good yield. The phosphate linkages were protected as benzyl esters as they provide the additional stability over the cyanoethyl group required for solution phase work. Benzyloxydiamidite 5, synthesized from phosphorus trichloride in one pot and purified by partition between hexane and acetonitrile [14], was coupled with guanosine 8 to give the amidite building block 9 which was coupled with adenosine 7 using 5-methyltetrazole (MTET) as the activator. To the best of our knowledge, MTET has not previously been used as an activator in phosphoramidite coupling reactions. It’s predicted pKa of 5.1 [15] is close to that of the commonly used 4,5 – dicyanoimidazole (DCI, pKa 5.0) and it is not subject to the same shipping restrictions as tetrazole. It was effective in many of the reactions reported herein. Oxidation of the intermediate phosphite ester with aqueous 70% t-butylhydroperoxide afforded the benzyl protected phosphate ester 10 in excellent yield as a mixture of diastereomers at phosphorus. After purification by silica gel chromatography the DMTr group was removed with acetic acid to give 11 in good yield.
A Mannich reaction [16] between 6-N-benzoyl-9-deazaadenine, (3R, 4R)-4-(hydroxymethyl)-pyrrolidin-3-ol [17] and formaldehyde gave benzoyl-protected DIA 12 which was selectively tritylated on the primary hydroxyl in good yield to give 13. Preparation of amidite 14 was initially problematic as it was readily hydrolysed (to an H-phosphonate) during purification on silica gel. However, optimization of the chromatography to give rapid elution of 14 as well as resolution from impurities resulted in good yields. Once purified, 14 was stable for at least a week at room temperature and for months at −20 °C. It was coupled with dimer 11 followed by hydrolytic removal of DMTr to give 15 in reasonable yield. Further extension with guanosine phosphoramidite 9 and detritylation gave 16 in poor yield which was fully deprotected, first by hydrogenolysis followed by heating in ammonium hydroxide to give the target 4.
Synthesis of constructs bearing a DIAeguanosine-3′epropanediol diphosphate (DIApGpPD) is illustrated in Scheme 2. Coupling of propanediol amidite 17 and guanosine 8 followed by oxidation of the intermediate phosphite ester gave 18 in good yield. Removal of the DMTr group with methanolic HCl [12] gave 19 in quantitive yield. Instead of using DIA amidite 14 to extend synthon 19 we investigated an alternate approach in which 19 was converted to amidite 20 and then reacted with DIA alcohol 13. Coupling of 20 with 13 catalyzed by tetrazole, gave a moderate yield of 21. Removal of the DMTr group from 21 gave alcohol 22 which was utilized in several ways. Global deprotection gave 23 (DIApGpPD), reaction with propanediol amidite 17 followed by deprotection gave 2 (PDpDIApGpPD), reaction with guanosine amidite 7 and deprotection gave 3 (GpDIApGpPD), and further extension of 24 gave 25 (PDpGpDIApGpPD).
Scheme 2. Synthesis of constructs bearing a DIA – guanosine – propanediol diphosphate (DIApGpPD) at the 3′ end.
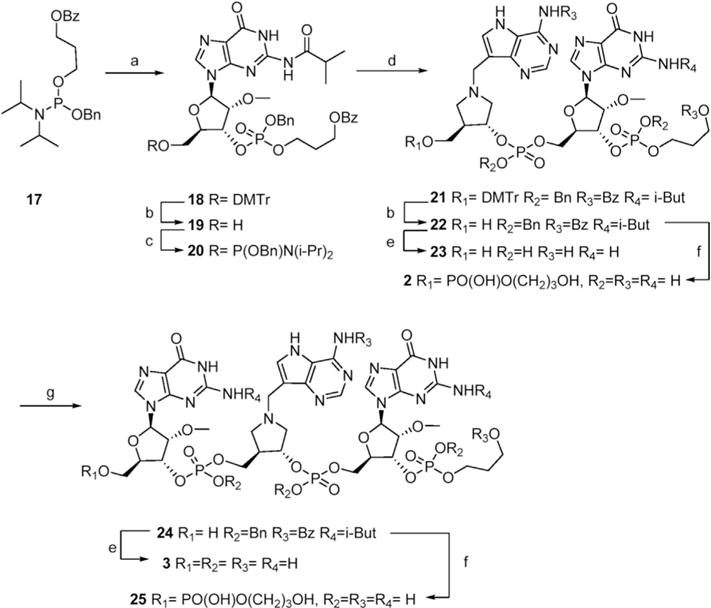
Reagents and conditions a) 8, DCI, CH2Cl2 then t-BuOOH, 74% b) HCl-MeOH, 100% of 19, 75% of 22 c) 5, tetrazole, CH2Cl2, 85% d) 13, tetrazole, CH2Cl2 then t-BuOOH, 42% e) Pd/C, H2, then NH4OH, 50 °C, 24 h, 52% of 23, 29% of 3 f) 17, tetrazole, CH2Cl2 then t-BuOOH then Pd/C, H2, then NH4OH, 50 °C, 24 h, 29% of 2, 7% of 25 g) 9, tetrazole, CH2Cl2 then t-BuOOH then HCl-MeOH, 26%.
Compounds 30 (DIApG), 31 (GpDIApG) and 33 (PDpDIApG) were synthesized in a stepwise fashion from either 3′-O-benzoyl or –elevulinoyl guanosines 26 and 27 (Scheme 3).
Scheme 3. Stepwise synthesis of 30, 31 and 32.
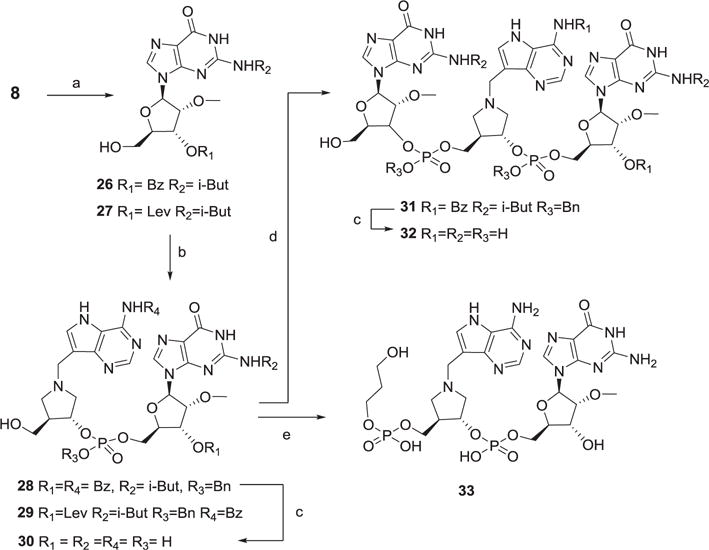
Reagents and conditions a) BzCl, pyridine, then 80% AcOH (for 26, 63%), levulinic acid, EDCI, DMAP, THF, then HCl-MeOH (for 27, 90%) b) 14, tetrazole, CH2Cl2 then t-BuOOH, then for 28, 80% AcOH 95%, for 29 HCl-MeOH, 53% c) Pd(OH)2-C then NH4OH, 50 °C, 24 h, for 30 31%, for 32 23% d) 9, tetrazole, CH2Cl2, then t-BuOOH, then 80% AcOH, 66% e) 17, tetrazole, CH2Cl2, then t-BuOOH, then H2, Pd-C then NH4OH, 50 °C 24 h, 11%.
A convergent synthesis of 39 (PDpGpDIApPD) from protected PDpG and DIApPD synthons is illustrated in Scheme 4. The orthogonal protection afforded by the levulinoyl protective group was used in the preparation of PDpG amidite synthon 36. Thus, 5′-hydroxy levulinoyl guanosine 27 was coupled with propanediol amidite 17 and oxidized to 34. Hydrazine effected selective removal of the levulinoyl group in excellent yield and the resulting alcohol 35 was converted to phosphoramidite 36. Reaction with synthon 37 (prepared from propanediol amidite 17 and DIA alcohol 13) gave a moderate yield of 38 and standard deprotection protocol gave 39. We attempted an analogous convergent strategy to synthesize 41 but coupling of the required diguanosine phosphoramidite with 37 was not successful. Compound 41 was better prepared in a stepwise fashion by two additions of guanosine phosphoramidite 9 followed by deprotection (Scheme 4).
Scheme 4. Convergent synthesis of 39 and synthesis of 41.
Reagents and Conditions a) 17, DCI, CH2Cl2 then t-BuOOH, 50% b) N2H4-H2O, MeOH, 5 h, 70% c) 5, DCI, CH2Cl2, 86% d) 17, tetrazole, CH2Cl2 then t-BuOOH then HCl-MeOH,, 66% e) 36, tetrazole, CH2Cl2 then t-BuOOH, 32% f) Pd-C, H2 then NH4OH, 50 °C, 24 h, 43% g) 9, tetrazole, CH2Cl2 then t-BuOOH then HCl-MeOH, 50% h) 9, tetrazole, CH2Cl2 then t-BuOOH then HCl-MeOH, then Pd-C, H2 then NH4OH, 50°C 24 h.
The methylene bridge between the pyrrolidine and deazaadenine in DIA models the dissociative nature of the transition state for hydrolysis by SAP. In compound 42 (DEIA) we have increased the pyrrolidine – deaazaadenine distance by incorporation of an ethylene bridge as a comparison. Mesylation of 9-hydroxyethyl-9-deazaadenine [18] and nucleophilic displacement with (3R, 4R)-4-(hydroxymethyl)-pyrrolidin-3-ol [17] gave 42 (Scheme 5) which was selectively tritylated to give 43. The potentially troublesome conversion of 43 to a phosphoramidite was circumvented by treating 43 with the PDpG amidite 20 followed by hydrolysis of the DMTr group to give 44. Compound 45 (PDpDEIApGpPD) was then accessed by the deprotection methods described above.
Scheme 5. Synthesis of DIEA compound 45.
Reagents and conditions a) DMTrCl, pyridine, 33% b) 20, tetrazole, CH2Cl2 then t-BuOOH, then HCl-MeOH, 40% c) 17, tetrazole, CH2Cl2 then t-BuOOH, then H2, Pd-C, then NH4OH, 50°C, 24 h, 9%.
Constructs with serinol as the aza-sugar component are potent inhibitors of SAP [19]. The use of homoserinol (2-(aminomethyl) propane-1,3-diol) in this context offers the possibility for additional flexibility enabling stronger interactions with SAP. The Mannich reaction between protected homoserinol 46, formadehyde and 9-deazaadenine gave 47 (Scheme 6) [20,21] Standard protecting group manipulations gave racemic monotrityl compound 49 which was treated with phosphorodiamidite 5 and then the product was coupled with guanosine derivative 19 to give 50. Acid hydrolysis gave 51 and then coupling with amidite 17 followed by global deprotection gave racemic 52. NMR peaks of Fmoc protected compounds 49–51 were extremely broad, presumably due to NMR-timescale inversion or rotation whereas 52 had well resolved NMR spectra consistent with the desired structure.
Scheme 6. Synthesis of Homoserinol construct 52.
Reagents and conditions a) 9-deazaadenine, CH2O, EtOH, 60 °C, 52% b) H2, Pd(OH)2–C, 93% c) FmocCl, NaHCO3, MeOH, then BzCl, pyridine then THF – AcOH, 2:1 then DMTrCl, pyridine, 35%. d) 5, tetrazole, CH2Cl2 then 19, tetrazole, CH2Cl2 then, t-BuOOH, 58% e) HCl-MeOH, 61% f) 17, tetrazole, CH2Cl2 then t-BuOOH, 19% then H2, Pd – C then NH4OH, 50 °C, 19%.
2.2. Inhibitory studies
Inhibition of SAP was studied in a competitive assay using stem-loop RNA A10 (5′CGCGAGAGCG3′) as the substrate. Product formation was quantitated in continuous assays by linking formation of adenine from RNA substrates to the production of light from luciferase [4].
The DIA (1) inhibitor constructs show strong relationships between structure and activity. All constructs with dissociation constants (Ki* values, including slow-onset properties) < 40 nM contain the DIApG substructure. When complexed with SAP this moiety forms a quadruple π-stack with the phenolic rings of SAP tyrosines 73 and 123 [9]. Our data shows this stack is essential but not sufficient for binding. Comparison of compounds 23 and 30 with 2, 3, 4 and 25 indicates that tight binding requires the DIApG motif to be flanked by phosphate esters, i.e. pDIApGp. Hydrogen bonds to these terminal phosphates are evident in inhibitor – SAP crystal structures and are important for stabilization of the SAP – nucleotide complex [9]. Compounds 39 and 41 which have three phosphate groups but not the DIApG substructure are weak inhibitors. Propanediol can replace the terminal G and A of the inhibitor 6; either separately (3) or together (2) with little effect on inhibition. Adding an additional phosphate ester to 3, as in 25, does not increase inhibitor affinity. A crystal structure of a circular construct of pGpDIApGpAp complexed with SAP shows interactions with SAP only through the central pDIApGp motif [9] and the correlation between structure and inhibition presented here are fully consistent with this binding model.
The PDpDEIApGpPD construct 45 with an additional methylene separating the aza-sugar and deazaadenine is almost as good an inhibitor as the equivalent DIA analogue 2, indicating that SAP can accommodate both increased distance from the phosphodiester backbone and additional flexibility. Constructs containing acylic ribocation mimics homoserinol (52) and serinol [19] are also good inhibitors, but less potent than those containing the cyclic amines.
The molecular electrostatic potential surfaces (MEPs) of compounds 2, 45 and 52 reveal similarity to the MEP of the transition state (Fig. 2) [22] while the MEP of the catalytically inactive substrate analogue PDpApGPD differs significantly, which is also consistent with the weaker binding of this compound (Ki* = 16.1 μM; Fig. 2 and Table 1).
Fig. 2.
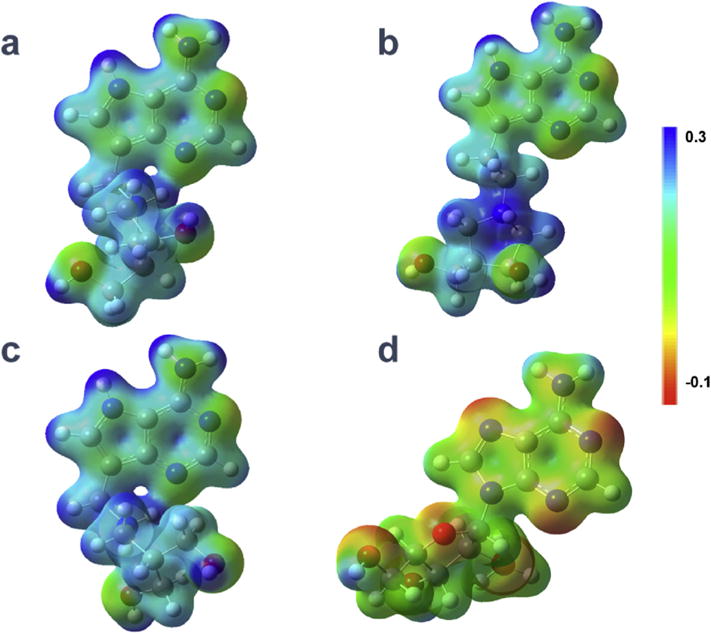
Comparison of MEPs for the transition state mimic residues of saporin inhibitors. (a) Compound 2 (PDpDIApGpPD, 3.3 nM, from Yuan et al. [22].); (b) compound 45 (PDpDEIApGppd, 12.4 nM); (c) compound 54 (PDpHomoSerpGpPD, 26.8 nM); and (d) catalytically inactive substrate analogue PDpApGpPD ( 16.1 μM).
Table 1.
Inhibition constants for SAP inhibitors.
| Compound | Construct | Inhibition constant (nM)a |
|---|---|---|
| 2 | PDpDIApGpPD | 3.3 ± 0.2 |
| 3 | GpDIApGpPD | 10.5 ± 0.9 |
| 4 | GpDIApGpA | 6.1 ± 0.3 |
| 23 | DIApGpPD | 10,700 ± 800 |
| 25 | PDpGpDIApGpPD | 7.4 ± 0.4 |
| 30 | DIApG | 21,300 ± 800 |
| 32 | GpDIApG | 33.0 ± 1.7 |
| 33 | PDpDIApG | 19.3 ± 1.7 |
| 39 | PDpGpDIApPD | 188 ± 12 |
| 41 | GpGpDIApPD | 212 ± 17 |
| 45 | PDpDEIApGpPD | 12.4 ± 0.9 |
| 52 | PDpHomoSerpGpPD | 26.8 ± 0.3 |
| PDpSerpGpPD | 43.2 ± 1.1b | |
| PDpApGpPD (substrate analogue) | 16,000 ± 1500 |
is the dissociation constant Kd for the inhibitor interactions with SAP following slow–onset inhibition.
Values taken from Schramm et al. [19].
Compound 2 also inhibits the hydrolysis of yeast tRNA and rabbit rRNA by both SAP and SAP-A14C (Fig. 3). In rabbit reticulocyte cell-free protein translation studies, SAP A14C inhibited translation of luciferase mRNA with an IC50 of 330 pM. Addition of inhibitor 2 to translation mixtures rescued the translation of luciferase with an IC50 of 13 nM in the presence of 10 nM SAP A14C (Fig. 4). Thus, a stoichiometric excess of 3 nM of compound 2 relative to SAP is required for rescue of translation, fully consistent with the Ki* value of 3.3 nM (Table 1).
Fig. 3.
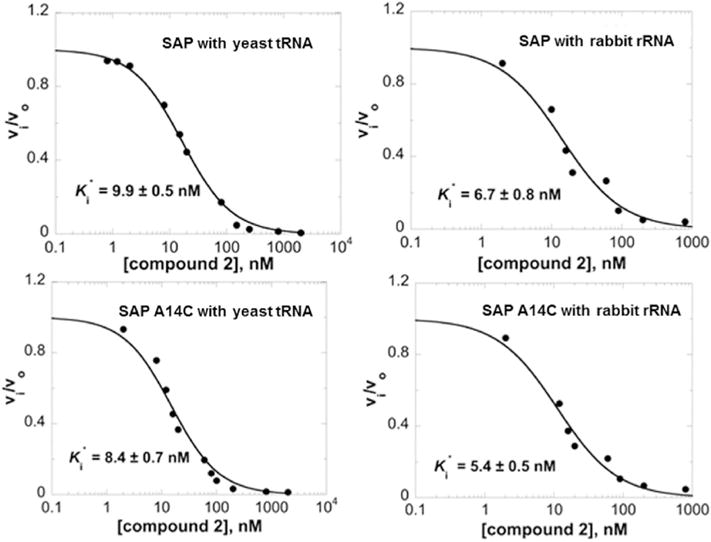
Competitive inhibition of SAP as a function of compound 2 with a fixed concentration of isolated rabbit 80S ribosome (100 nM) or yeast tRNA (1.0 mg/mL). Kinetics were measured after a 10 min enzyme-inhibitor preincubation for slow onset binding ( ).
Fig. 4. Inhibition of translation of luciferase mRNA in rabbit reticulocyte extract assays.
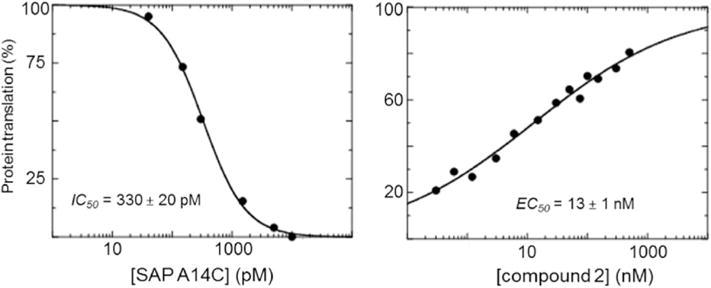
Left panel, SAP A14C inhibition of luciferase translation in rabbit reticulocyte lysate assay. Right panel, Luciferase translation was rescued from 10 nM SAP A14C by compound 2 with an apparent IC50 of 13 nM, a 3 nM stoichiometric excess relative to saporin concentration.
3. Conclusion
We have synthesized a suite of oligonucleotide inhibitors of SAP using solution phase techniques. These compounds define the structural parameters required for inhibition of SAP that is consistent with x-ray crystal structure studies. The low nanomolar dissociation constants for the best compounds encourage physiological applications as SAP rescue agents.
4. Experimental section
4.1. General
Air sensitive reactions were performed under argon. Organic solutions were dried over anhydrous MgSO4 and the solvents were evaporated under reduced pressure. Anhydrous and chromatography solvents were obtained commercially and used without any further purification. Thin layer chromatography (tlc) was performed on aluminium sheets coated with 60 F254 silica gel. Organic compounds were visualized under uv light or using a dip of N,N-dimethylaminobenzaldehyde (0.6%) in methanol – conc. HCl, 6:1, or KMnO4 (0.5% in water). Column chromatography was performed on silica gel (40–63 μm) or on prepacked columns using an automated system with continuous gradient facility. 1H NMR spectra were measured in CDCl3, CD3OD (internal Me4Si, δ 0) or D2O (HOD, δ 4.79). 1H-decoupled 13C NMR spectra were measured in CDCl3 (centre line, δ 77.0), CD3OD (centre line, δ 49.0) or D2O (no internal reference). 1H-decoupled 31P NMR spectra were measured with no internal reference. Assignments of 1H and 13C resonances were based on 2D (1H-1H DQF-COSY, 1H-13C HSQC, HMBC, 31P-1H COSY) and DEPT experiments. HPLC used either a Kinetex C-18 column eluted with a gradient of MeCN in water containing 0.1% TFA or a Poroshell 120 EC-C-18 column eluted with a gradient of MeOH in water containing 0.01% formic acid or Et3NHOAc (10 mM, pH 6.5). Detection was at 254 nM. LCMS utilized a quadrupole mass spectrometer with electrospray ionization. Preparative HPLC utilized a Luna C-18 column eluted with ca 20% MeOH in Et3NHOAc (50 μM, pH 6.5) High resolution mass spectra (HRMS) were recorded with electrospray ionization on a Q-TOF tandem mass spectrometer.
5. Methods
5.1. Method 1 – coupling and oxidation
Alcohol (1 equiv) and phosphoramidite (1.5–4 equiv) were dissolved in a little dry DCM and diluted with dry acetonitrile. The solution was concentrated and held under oil pump vacuum for 1 h. Dry DCM (approx. 20 mL/g) and activator (tetrazole, MTET or 4, 5-dicyanoimidazole (DCI), 2–5 equiv) were added and the solution stirred for 1–2 h when tlc or HPLC showed conversion of the alcohol to a new material. t-Butylhydroperoxide (20% solution in toluene or 70% solution in water, 3–6 equiv) was added and the solution stirred for a further 3 h. The solution was diluted with CHCl3 and extracted with Na2S2O3 (10%, aq), NaHCO3 (10%, aq) and brine. Drying, concentration and chromatography on silica, eluting with a gradient (0–15%) of MeOH in CHCl3 – EtOAc, 2:1 (unless otherwise stated), gave the phosphate ester product as a mixture of diastereomers.
5.2. Method 2 – hydrolytic cleavage of the dimethoxytrityl group
Methanolic HCl (5 μM) was freshly prepared from MeOH and AcCl. DMTr nucleotide (10–20 mg/mL) was stirred in this for 10–20 min, when tlc showed complete reaction. The solution was bought to pH 7 by the addition of NaHCO3 (aq, 10%) and concentrated to dryness. The residue was taken up in CHCl3, MgSO4 was added and the solution filtered through diatomaceous earth. The detritylated nucleotide was isolated by chromatography, eluting with a gradient (0–15%) of MeOH in CHCl3 – EtOAc, 2:1.
5.3. Method 3 – global deprotection of oligonucleotides
The protected oligonucleotide (5–100 mg) was dissolved in THF (5–20 mL) and stirred with Pd(OH)2 on carbon (50 wt%) or Pd on carbon (5%) or Pd black under a balloon of hydrogen. After 24 h, or when LCMS showed hydrogenolysis of all the benzyl groups, water (5 mL) was added and the catalyst removed by filtration. Solvents were removed and the residue taken up in ammonium hydroxide (27%). The solution was stirred at 50 °C for 24 h and then concentrated and purified as described.
5.4. Compound 7
To a solution of 6 (1.18 g, 1.72 mmol) in dry CH2Cl2 (10 mL) was added pyridine (0.421 mL 5.15 mmol) and then chloroacetyl chloride (0.195 mL, 2.40 mmol) and the resulting solution was stirred at rt for 1 h, then washed with water, HCl (1 M), NaHCO3 (10%, aq), dried and concentrated to dryness. Chromatography (40% and 50% EtOAc in CHCl3 gave a foam (1.07 g, HRMS – found 764.2476, calc for C41H39N5O8Cl [M+H]+, 764.2487) which was dissolved in AcOH (80%, aq, 20 mL) and stirred at rt for 2 h. Concentration and chromatography (EtOAc) gave 7 (0.54 g, 1.1 mmol, 71%) as a colourless foam. 1H NMR (CDCl3, 500 MHz) δ 9.26 (s, 1H), 8.77 (s, 1H), 8.10 (s, 1H), 8.06–7.99 (m, 2H), 7.65–7.58 (m, 1H), 7.52 (dd, J = 10.6, 4.8 Hz, 2H), 6.18 (d, J = 10.9 Hz, 1H), 5.90 (d, J = 7.9 Hz, 1H), 5.75 (d, J = 4.9 Hz, 1H), 4.82 (dd, J = 7.9, 4.9 Hz, 1H), 4.40 (s, 1H), 4.24–4.17 (m, 2H), 4.00 (d, J = 13.0 Hz, 1H), 3.83 (t, J = 11.8 Hz, 1H), 3.29 (s, 3H). 13C NMR (CDCl3, 126 MHz) δ 166.5, 164.6, 152.2, 150.6, 150.5, 143.2, 133.4, 133.0, 128.9, 128.0, 124.7, 89.4, 85.8, 81.2, 74.0, 62.8, 59.6, 40.6. HRMS – found 462.1176, calc for C20H21N5O6Cl [M+H]+ 462.1180.
5.5. Compound 9
A solution of 8 (3.0 g, 4.5 mmol) and MTET (0.33 g, 3.9 mmol, 0.88 equiv) in dry CH2Cl2 (100 mL) was added to a solution of 5 (3.0 g, 9.0 mmol, 2.0 equiv) in dry CH2Cl2 (30 mL). After stirring for 2 h, Et3N (1.3 mL) was added and the solution was concentrated to a small volume. Chromatography eluting with hexanes – CHCl3 1:1, CHCl3 and then 3% MeOH in CHCl3, all containing 1% Et3N gave 9 (3.92 g, 4.3 mmol, 96%) as a 3:2 mix of stereoisomer. 1H NMR (CDCl3, 500 MHz) δ 7.78 (m, 1H), 7.54 (m, 2H), 7.40 (m, 4H), 7.34–7.08 (m, 10H), 6.78 (m, 4H), 5.84 (t, J = 7.4 Hz, 1H), 4.76 (m, 2H), 4.64 (m, 1H), 4.49 (m, 1H), 4.31 (m, 1H), 3.74 (m, 6H), 3.62 (m, 2H), 3.51 (m, 1H), 3.43 (m, 3H), 3.11 (m, 1H), 2.54 (q, J = 7.3 Hz, 1H), 1.22–0.71 (m, 18H). 13C NMR (CDCl3, 126 MHz) δ 178.3, 158.8, 155.7, 148.3, 147.1, 145.1, 144.9, 138.8, 136.1, 135.7, 130.0, 128.2, 128.1, 128.0, 127.3, 127.1, 127.0, 126.9, 122.5, 113.3, 86.6, 86.4, 84.5, 84.1, 81.8, 81.2, (71.0, 70.1) both d, JC,P = 17 Hz), (65.8, 65.2, both d, JC,P = 18 Hz), 63.3, 58.7, 58.2, 55.2, 46.2, (43.3, 43.0, both d, JC,P = 13 Hz), 36.0, 24.7, 24.6, 24.5, 24.4, 18.5. 31P NMR (CDCl3, 202 MHz)δ 150.7, 149.9.
5.6. Compound 10
7 (1.24 g, 2.68 mmol) and phosphoramidite 9 (3.70 g, 4.08 mmol) were coupled according to the Method 1 using MTET (1.5 equiv) as the activator. Chromatography (0–10% MeOH in EtOAc) gave 10 (2.95 g, 2.30 mmol, 85%), a foam, as a mixture of stereoisomers. 1H NMR (CDCl3, 500 MHz) δ 11.98 and 11.93 (s, 1H), 9.13 (m, 2H), 8.80 and 8.75 (s, 1H), 8.33 and 8.18 (s, 1H), 8.05–7.93 (m, 2H), 7.80 and 7.76 (s, 1H), 7.63–7.44 (m, 3H), 7.43–7.10 (m, 15H), 6.80–6.70 (m, 4H), 6.15 and 6.06 (d, J = 5.6 Hz, 1H), 5.83 and 5.78 (d, J = 6 Hz, 1H), 5.69–5.48 (m, 2H), 5.17–5.08 (m, 1H), 4.99–4.74 (m, 2H), 4.48–4.07 (m, 4H), 3.74, 3.73, 3.73 and 3.72 (s, 2 × 3H), 3.48, 3.39, 3.35 and 3.32 (s, 2 × 3H), 3.47–3.28 (m, 1H), 3.19 and 3.07 (dd, J = 11.0, 3.0 Hz, 1H), 2.16–2.01 (m, 1H), 1.03, 0.97, 0.92 and 0.86 (d, J = 6.9 Hz, 6H). 13C NMR (CDCl3, 126 MHz) δ 179.0,178.8, 166.6, 166.5, 164.8, 164.7, 158.7, 155.5, 155.4, 152.9, 152.8, 151.8, 151.6, 149.8, 148.1, 147.7, 147.6, 144.5, 142.1, 141.8, 138.7, 138.4, 135.6, 135.3, 135.0, 133.2, 133.0, 130.0, 129.2, 128.9, 128.8, 128.7, 128.0, 128.0, 127.8, 127.2, 127.0, 123.9, 122.0, 113.3, 113.2, 87.2, 87.1, 86.6, 85.4, 82.1, 81.9, 81.2, 81.1, 80.8, 80.7, 80.6, 75.1, 72.3, 72.2, 70.2, 70.0, 66.3, 66.1, 62.5, 60.4, 59.4, 58.8, 58.7, 55.2, 40.4, 36.13, 36.08, 18.72, 18.66, 18.57. HRMS – found 1283.3995, calc for C63H65ClN10O16P [M+H]+, 1283.4006.
5.7. Compound 11
Compound 10 (2.80 g, 2.18 mmol) in AcOH (80% aq, 30 mL) was stirred at rt. After 3 h, CHCl3 was added and the mixture was washed with water (×2) and then NaHCO3 (10%, aq), dried and evaporated. Chromatography (0–20% MeOH in EtOAc) gave two stereoisomers of 11 as colourless foams, first isomer A, (0.67 g) and then isomer B (0.89 g, total 1.56 g, 1.58 mmol, 72%). For Isomer A: 1H NMR (CDCl3, 500 MHz) δ 12.21 (s, 1H),10.19 (s, 1H), 9.50 (s, 1H), 8.80 (s, 1H), 8.35 (s, 1H), 8.11–7.91 (m, 3H), 7.64–7.51 (m, 1H), 7.51–7.41 (m, 2H), 7.40–7.22 (m, 5H), 6.14 (d, J = 5.7 Hz, 1H), 5.78 (t, J = 7.2 Hz, 1H), 5.60 (dd, J = 5.1, 3.6 Hz, 1H), 5.23–5.04 (m, 4H), 4.88 (t, J = 5.5 Hz, 1H), 4.54 (t, J = 5.3 Hz, 1H), 4.46–4.36 (m, 3H), 4.18 (s, 2H), 3.88–3.80 (m, 1H), 3.58 (d, J = 11.9 Hz, 1H), 3.37 (s, 3H), 3.28 (s, 3H), 2.88 (s, 1H), 2.81–2.71 (m, 1H), 1.19–1.14 (m, 6H); 13C NMR (CDCl3, 126 MHz) δ 179.8, 166.7, 165.1, 155.4, 152.8, 151.8, 149.9, 148.2, 148.0, 142.2, 138.9, 135.2, 135.1, 133.2, 132.9, 129.0, 128.84, 128.78,128.1, 128.0, 123.9, 121.7, 87.1, 86.8, 84.9, 81.7, 80.9, 80.8, 76.1, 72.3, 70.2, 70.1, 66.4, 61.5, 59.4, 58.7, 40.5, 36.1, 19.1, 18.8. HRMS-found 981.2706, calc for C42H47ClN10O14P [M+H]+, 981.2699. For isomer B: 1H NMR (CDCl3, 500 MHz) δ 12.13 (s, 1H), 10.19 (s, 1H), 9.51 (s, 1H), 8.75 (s, 1H), 8.33 (s, 1H), 8.10–7.86 (m, 3H), 7.54 (t, J = 7.4 Hz, 1H), 7.50–7.22 (m, 7H), 6.11 (d, J = 5.6 Hz, 1H), 5.74 (d, J = 6.1 Hz, 1H), 5.60 (dd, J = 4.9, 3.9 Hz, 1H), 5.33 – 5.04 (m, 4H), 4.87 (t, J = 5.4 Hz, 1H), 4.55 (t, J = 5.1 Hz, 1H), 4.45–4.19 (m, 3H), 4.18 (s, 2H), 3.88 (d, J = 10.1 Hz, 1H), 3.67 (d, J = 11.8 Hz, 1H), 3.36 (s, 3H), 3.27 (s, 3H), 2.94 (s, 1H), 2.78–2.69 (m, 1H), 1.18 (m, 6H); 13C NMR (CDCl3, 126 MHz) δ 179.6, 166.7, 165.1, 155.5, 152.7, 151.8, 149.9, 148.1, 147.9, 142.3, 138.8, 135.23, 135.18, 133.2, 132.9, 129.0, 128.8, 128.1, 127.9, 124.0, 121.5, 87.1, 86.8, 84.4, 81.5, 80.7, 75.6, 72.2, 70.3, 70.2, 66.2, 61.1, 59.4, 58.6, 40.5, 36.1, 18.9. HRMS Found 981.2704, calc for C42H47ClN10O14P [M+H]+, 981.2699.
5.8. Compound 12
Aqueous formaldehyde (1.25 mL, 16.6 mmol, 37 wt %) was added to a suspension of (3R, 4R)-4-(hydroxymethyl)-pyrrolidin-3-ol (1.95 g, 16.6 mmol) [17] and 6-N-benzoyl-9-deazaadenine (3.6 g, 15.1 mmol) in H2O (40 mL) and EtOH (20 mL) and the mixture warmed to 60°C. After 4 h tlc analysis of the homogeneous reaction mixture showed none of the starting amine. Silica gel (ca 20 g) was added and the resulting suspension concentrated to afford a solid residue which was chased consecutively with MeOH and CHCl3. The resulting residue was purified by chromatography on silica gel (20%–40% MeOH in CHCl3 then 1:1 dioxane:H2O) to afford 12 (5.55 g, 52%) as a pale yellow solid. 1H NMR (CD3OD, 500 MHz) δ 8.57 (s, 1H), 8.10 (m, 2H), 7.75 (s, 1H), 7.64 (tt, J = 7.4, 1.3 Hz, 1H), 7.57–7.53 (m, 2H), 4.00 (dt, J = 6.2, 4.1 Hz, 1H), 3.93 (d, J = 13.4 Hz, 1H), 3.89 (d, J = 13.4, 1H), 3.62 (dd, J = 10.7, 5.9 Hz, 1H), 3.50 (dd, J = 10.8, 7.7 Hz, 1H), 3.00 (dd, J = 9.8, 8.3 Hz, 1H), 2.84 (dd, J = 10.1, 6.3 Hz, 1H), 2.67 (dd, J = 10.1, 4.2 Hz, 1H), 2.44 (dd, J = 9.8, 6.7 Hz, 1H), 2.21–2.14 (m, 1H). 13C NMR (CD3OD, 126 MHz) δ 168.8, 151.7, 150.1, 144.7, 134.5, 133.9, 133.1, 129.8, 129.4, 117.9, 112.4, 74.2, 64.2, 62.6, 56.8, 51.2, 48.9. HRMS found, 368.1724 calc for C19H22N5O3 [M+H]+, 368.1723.
5.9. Compound 13
12 (2.90 g, 7.893 mmol) was dissolved in pyridine (50 mL), concentrated under vacuum and then redissolved in pyridine (65 mL). 4,4′-DMTrCl (3.03 g, 8.68 mmol) was added and the resulting mixture stirred for 1 h at rt under an argon atmosphere. The reaction mixture was diluted with CHCl3 (250 mL), washed with H2O (100 mL) and then NaHCO3 (10%, aq.), and the organic layer was dried, filtered and concentrated. The residue was purified by chromatography on silica gel (CHCl3 then EtOAc, then 10%–20% MeOH in CHCl3) to afford 13 (4.45 g, 84%) as a syrup.1H NMR (CDCl3, 500 MHz) δ 10.93 (s, 1H), 9.40 (s, 1H), 8.49 (s, 1H), 8.01–7.99 (m, 2H), 7.61 (tt, J = 7.4, 1.8 Hz, 1H), 7.52–7.49 (m, 3H), 7.40–7.38 (m, 2H), 7.29–7.23 (m, 7H), 7.18 (tt, J = 7.3, 2.0 Hz, 1H), 6.81–6.78 (m, 4H), 4.07 (quintet, J = 2.8 Hz, 1H), 3.93 (d, J = 13.4 Hz, 1H), 3.84 (d, J = 13.4, 1H), 3.76 (s, 6H), 3.15–3.09 (m, 4H), 2.85 (dd, J = 10.2, 2.4 Hz, 1H), 2.60 (dd, J = 10.2, 5.6 Hz, 1H), 2.41 (d quintet, J = 7.3, 3.3 Hz, 1H), 2.20 (t, J = 8.6 Hz, 1H) 13C NMR (CDCl3, 126 MHz) δ 166.6, 158.4, 151.2, 149.2, 145.0, 142.2, 136.2, 136.2, 133.2, 132.8, 130.4, 130.1, 129.0, 128.2, 127.8, 126.7, 115.8, 113.1, 86.0, 74.8, 64.6, 61.6, 56.2, 55.2, 48.8, 48.2. HRMS – found, 670.3026 calc for C40H40N5O5 [M+H]+, 670.3029.
5.10. Compound 14
Compound 13 (1.00 g, 1.49 mmol) and MTET (0.259 g, 2.99 mmol) were dissolved, with warming, in dry MeCN (40 mL), evaporated to dryness and held under oil pump vacuum. 5 (1.01 g, 2.99 mmol) in dry MeCN (20 mL), was concentrated to dryness, held under oil pump vacuum and redissolved in dry CH2Cl2 (5.0 mL). This solution was added dropwise over 30 min to a solution of the 13-MTET mixture in dry CH2Cl2 (5.0 mL). After a further1 h stirring at rt the reaction was quenched with Et3N (0.63 mL, 4.48 mmol) and then hexanes (15 mL) was added and the mixture was applied directly to a silica gel column (25 × 4 cm), which had been washed with CHCl3 – hexanes 1:1 containing 2% Et3N. The column was eluted with this solvent and then with CHCl3 followed by 70% EtOAc in CHCl3 (both containing 2% Et3N) to give 14 (1.02 g, 1.12 mmol, 75%) as a foam and a mixture of stereoisomers. 1H NMR (CDCl3, 500 MHz) δ 10.88 (s, 1H), 9.15 (bs, 1H), 8.51 (s, 1H), 8.00 (m, 2H), 7.61 (m, 1H), 7.50 (m, 3H), 7.40 (m, 2H), 7.31–7.13 (m, 12H), 6.76 (m, 4H), 4.60 (m, 2H), 4.21 (m, 0.5H), 4.13 (m, 0.5H), 3.98 (dd, J = 3.1, 13.7 Hz, 1H) 3.87 (dd, J = 2.4, 13.7 Hz, 1H), 3.74 (m, 6H), 3.57 (m, 2H), 3.28 (dd, J = 5.3, 8.9 Hz, 0.5H), 3.23 (dd, J = 4.8, 8.6 Hz, 0.5H), 3.03 (m, 2H), 2.86 (dd, J = 6.6, 9.8 Hz, 0.5H), 2.82 (m, 1H), 2.75 (dd, J = 4.5, 9.8 Hz, 0.5H), 2.56 (m, 2H) 1.08 (m, 12H). 13C NMR (CDCl3, 126 MHz) δ 166.5, 158.3, 151.2, 149.2, 145.2, 142.1, 139.7, 136.4, 133.2, 132.8, 130.1, 128.5, 128.3, 128.2, 127.7, 127.0, 126.6, 115.8, 113.5, 113.0, 85.8, 75.6, 75.1, 65.3, 65.1, 64.3, 61.0, 56.4, 55.2, 48.5, 47.5, 43.0, 24.5. 31P NMR (CDCl3, 202 MHz) δ 147.3, 147.1.
5.11. Compound 15
Alcohol 11 (0.53 g, 0.540 mmol) and MTET (0.14 g, 1.62 mmol) were dissolved, with warming, in dry MeCN (40 mL) and evaporated to dryness. 14 (1.23 g, 1.35 mmol) in dry MeCN (10 mL), was concentrated to dryness and redissolved in dry CH2Cl2 (5.0 mL). This solution was added dropwise to a solution of the 11-tetrazole mixture in dry CH2Cl2 (10.0 mL) held at −15°C under argon. The solution was stirred and allowed to warm to 0°C over 45 min t-BuOOH (70 mass% in H2O, 0.37 mL, 2.70 mmol) was added and the solution was allowed to warm to rt. After 30 min the solution was washed with NaHCO3 (10%, aq) dried and evaporated. Partial purification by flash chromatography gave a foam (1.04 g). This material was dissolved in 80% aq AcOH (20 mL), stirred at rt for 16 h and then evaporated. The residue was dissolved in CHCl3 and washed with NaHCO3 (10%, aq), dried and evaporated. Chromatography of the residue (0–20% MeOH in CHCl3) gave 15 (0.465 g, 0.31 mmol, 57%) as a foam and a mixture of four stereoisomers. 1H NMR (CDCl3, 500 MHz) δ 12.19 (br s, 1H), 11.05–10.66 (m, 2H), 9.74–9.05 (m, 2H), 8.81–8.23 (m, 2H), 8.09–7.70 (m, 3H), 7.66–7.19 (m, 20H), 6.20–6.10 (m, 1H), 5.74–5.63 (m, 1H), 5.56 (m, 1H), 5.18–4.67 (m, 7H), 4.52–4.03 (m, 5H), 4.01–3.16 (m, 15H), 3.12–2.15 (m, 5H), 1.24–1.09 (m, 6H). 13C NMR (CDCl3, 126 MHz) δ 166.6, 166.5, 165.2, 158.6, 155.6, 152.8, 151.7, 151.0, 150.0, 149.4, 149.2, 148.1, 147.5, 142.3, 141.8, 139.6, 135.1, 133.2, 133.1, 132.8, 132.7, 130.3, 129.2, 129.1, 128.9, 128.8, 128.7, 128.6, 128.5, 128.3, 128.2, 128.1, 128.0, 127.81, 127.75, 127.5, 127.0, 127.0, 123.8, 122.8, 116.0, 115.8, 113.2, 87.4, 87.0, 86.9, 81.5, 81.0, 80.7, 79.1, 78.5, 75.5, 72.2, 70.3, 69.8, 67.5, 66.3, 65.2, 63.7, 62.8, 60.3, 59.4, 58.9, 58.7, 55.5, 55.2, 48.7, 48.3, 47.8, 40.4, 35.8, 29.5, 22.8, 22.2, 19.2, 18.8. HRMS Found 1500.4386, calc for C68H73ClN15O19P2 [M+H]+, 1500.4371.
5.12. Compound 16
15 (0.44 g, 0.29 mmol) and 9 (0.665 g, 0.73 mmol) were coupled according to Method 1 using MTET (2 equiv) as the activator. After partial purification by chromatography (0–10% MeOH in CHCl3) the residue was dissolved in AcOH (80%, 10 mL), stirred at rt for 16 h then evaporated, dissolved in CHCl3, washed with aq NaHCO3 dried and evaporated. Chromatography (0–25% MeOH in CHCl3) gave 16 (35 mg, 0.018 mmol, 6%). 1H NMR (CDCl3, 500 MHz) δ 12.24 (m, 2H), 10.75 (m, 2H), 9.58 (m, 2H), 8.73 (m, 1H), 8.57–8.18 (m, 2H), 8.14–7.76 (m, 6H),, 7.68–7.13 (m, 21H), 6.23–5.53 (m, 3H), 5.32–4.83 (m, 8H), 4.75–3.50 (m, 17H), 3.43–3.13 (m, 9H), 3.05–2.13 (m, 8H), 1.30–1.08 (m, 16H). 13C NMR (CDCl3, 126 MHz) δ 180.1, 179.9, 179.6, 166.8, 166.6, 165.6, 165.3, 161.8, 155.9, 155.6, 152.5, 151.3, 151.0, 149.8, 149.3, 149.1, 148.5, 148.4, 148.1, 142.4, 141.8, 141.7, 140.6, 140.3, 140.0, 139.0, 138.2, 135.4, 135.2, 135.1, 133.5, 133.2, 132.8, 132.6, 130.6, 128.9, 128.7, 128.2, 128.1, 128.0, 127.3, 123.7, 122.2, 121.9, 121.4, 116.1, 115.9, 87.3, 87.2, 87.0, 86.8, 86.6, 86.1, 84.9, 84.7, 83.2, 83.0, 82.4, 82.2, 81.7, 81.5, 81.3, 79.9, 79.7, 79.5, 79.3, 75.6, 75.4, 75.0, 70.1, 69.9, 69.7, 69.1, 68.7, 67.8, 67.5, 67.1, 66.7, 61.5, 61.3, 61.1, 59.6, 59.2, 59.1, 59.0, 58.9, 58.8, 58.7, 58.5, 55.2, 54.3, 47.8, 47.6, 47.4, 47.3, 46.5, 46.2, 45.6, 36.0, 35.8, 19.2, 19.1, 19.0, 18.8. HRMS Found 1943.6171, calc for C88H98N20O26P3 [M+H]+, 1943.6174.
5.13. Compound 4
A solution of 16 (0.035 g, 0.018 mmol) in THF (3 mL), and NH3-MeOH (7 N, ~1 mL) was strried with Pd/C (10%) under H2. After 40 h the solution was filtered through glass fibre paper and the solids were washed with THF – H2O, (1:1) and then H2O and all were evaporated. This material was dissolved in water, filtered through a 3 μm filter and lyophilized. A solution of this material in NH4OH (27%, 5 mL) was stirred and heated in a sealed vessel at 40 °C for 18 h and then evaporated to dryness. The residue was purified by preparative HPLC and lyophilized (×4) to give 4 (5.3 mg, 0.004 mmol, 39%) as a non – stoichiometric Et3 N salt 1H NMR (500 MHz, D2O) δ 8.38 (s, 1H), 8.34 (s, 1H), 8.20 (s, 1H), 8.01 (s, 1H), 7.94 (m, 2H), 6.15 (d, J = 4.9 Hz, 1H), 5.89 (d, J = 6.4 Hz, 1H), 5.84 (d, J = 4.5 Hz, 1H), 5.01 (m, 1H), 4.93 (m, 2H), 4.69 (m, 1H), 4.58 (m, 3H), 4.48–4.34 (m, 5H), 4.34–4.02 (m, 7H), 3.97–3.82 (m, 2H), 3.75 (m, 1H), 3.64 (m, 1H), 3.54–3.48 (m, 10 H), 2.99 (s, 1H); 13C NMR (126 MHz, D2O) δ 158.6, 158.2, 154.5, 153.6, 153.5, 152.2, 151.8, 151.2, 150.4, 148.4, 145.1, 139.7, 137.9, 137.1, 134.4, 118.3, 116.4, 116.0, 112.5, 101.9, 85.8, 85.6, 84.9, 83.74, 83.68, 83.0, 82.2, 81.2, 76.2, 73.0, 72.0, 68.7, 64.8, 64.7, 64.6, 61.2, 59.4, 58.3, 58.1, 57.9, 53.8, 46.8, 46.0. HRMS – found 1323.3246, (M−H)− calc for C45H58N20O22P3 [M−H]− 1323.3247.
5.14. Phosphoramidite 17
A solution of 3-hydroxypropyl benzoate (1.5 g, 8.3 mmol, dried by evaporation from MeCN) in dry CH2Cl2 (30 mL) was added to an ice cooled solution of 5 (4.2 g, 12 mmol, 1.5 equiv) in dry CH2Cl2 containing suspended tetrazole (0.58 g, 8.3 mmol, 1 equiv). The suspension was stirred in the ice bath for 10 min and then allowed to warm to rt. After 20 min Et3N (5 mL) was added and the solution was concentrated to approximately 15 mL. The resulting slurry was filtered through a silica gel plug that had been washed with 2% Et3N in hexanes. The plug was washed with hexanes containing 2% of each of Et3N and EtOAc. The resulting crude was further purified by chromatography using hexanes containing 2% Et3N as eluent to give 17 as a colourless oil. (2.35 g, 5.8 mmol, 67%) 1H NMR (CDCl3, 500 MHz) δ 7.96 (d, J = 8.3 Hz, 2H), 7.46 (t, J = 7.5 Hz, 1H), 7.34 (t, J = 7.8 Hz, 2H), 7.29–7.21 (m, 4H), 7.16 (m, 1H), 4.68 (dd, JH,P = 8.3, JH,H = 12.6 Hz, 1H), 4.59 (dd, JH,P = 8.3, JH,H = 12.6 Hz, 1H), 4.36 (t, J = 6.3 Hz, 2H), 3.77 (m, 1H), 3.72 (m, 1H) 3.58 (m, 2H), 2.00 (q, J = 6.3 Hz, 2H), 1.12 (s, 3H), 1.12 (s, 3H), 1.11 s, (3H), 1.10 (s, 3H)· 13CNMR (CDCl3, 126 MHz) δ 166.6 (q), 139.5 (q), 132.8 (CH), 130.4 (q) 129.6, 128.3, 127.2, 127.0 (CH), 65.3 (CH2, d, JC,P = 18 Hz), 62.0 (CH2), 60.1 (CH2, d, JC,P = 18 Hz), 43.0 (CH2, d, JC,P = 13 Hz), 30.6 (CH2), 24.6 (CH3). 31P NMR (CDCl3, 202 MHz) δ 147.2. HRMS Found 418.2140, calc for C23H33NO4P [M+H]+, 418.2147.
5.15. Compound 18
Alcohol 8 (2.0 g, 2.99 mmol) and phosphoramidite 15 (2.0 g, 2.2 mmol, 1.6 equiv) were coupled according to Method 1 using DCI (1.1 g, 9.0 mmol, 3 equiv) as the activator to give a pale coloured foam of 18 as a 1:1 mixture of diastereomers (2.2 g, 2.2 mmol, 74%). 1H NMR (CDCl3, 500 MHz) δ 11.92 (s, 1H), 8.92 (s, 0.5H), 8.91 (s, 0.5H), 7.97 (d, J = 8.0 Hz, 2H), 7.74 (s, 0.5H), 7.73 (s, 0.5H), 7.55 (m, 1H), 7.42–7.14 (m, 16H), 6.74 (m, 4H), 5.85 (dt, J = 7.7, 5.1 Hz, 0.5H), 5.80 (m, 1H), 5.76 (dt, J = 8.1, 4.5 Hz, 0.5H), 5.11 (d, J = 8.2 Hz, 2 × 0.5H), 4.95 (dd, J = 8.7, 11.8 Hz, 0.5H), 4.85 (m, 1.5H), 4.43 (m, 2× 0.5H), 4.32 (m, 1.5H), 4.24 (m, 2× 0.5H), 4.19 (m, 0.5H), 4.04 (m, 2× 0.5H), 3.75 (s, 1.5H), 3.74 (s, 1.5H), 3.73 (s, 1.5H), 3.72 (s, 1.5H),3.51 (m, 0.5H), 3.50 (s, 1.5H), 3.47 (dd, J = 2.3, 11.1 Hz, 0.5H), 3.40 (s, 1.5H), 3.15 (dd, J = 3.1, 11.1 Hz, 0.5H), 3.09 (dd, J = 3.1, 11.1 Hz, 0.5H), 2.10 (m, 2H), 1.98 (m, 1H), 1.06 (m, 3H), 0.93 (m, 3H). 13C NMR (CDCl3, 126 MHz) δ 178.7, 166.6, 166.4, 158.7, 155.5, 147.9, 147.7, 147.5, 138.9, 138.7, 135.7, 135.4, 133.2, 129.9, 129.5, 128.8, 128.7, 128.4, 128.0, 127.9, 127.8, 127.1, 122.2, 113.2, 86.5, 86.4, 86.0, 85.6, 81.3, 81.1, 74.6, 74.4, 69.8, 69.6, 65.1,64.8, 62.3, 62.2, 61.0, 60.8, 58.7, 58.6, 55.5, 36.2, 29.5, 18.6. 31P NMR (CDCl3, 202 MHz) δ −1.2, −1.3. HRMS – found 1002.3688, calc for C53H57N5O13P [M+H]+, 1002.3691.
5.16. Compound 19
18 (2.2 g, 2.2 mmol) was hydrolysed according to Method 2 to give 19 as a colourless foam and a mixture of stereoisomers (1.5 g, 2.2 mmol, 100%). 1H NMR (CDCl3, 500 MHz) δ 12.15 (s, 1H), 9.41 (s, 0.5H), 9.32 (s, 0.5H), 7.99 (d, J = 8.0 Hz. 2H), 7.82 (s, 0.5H), 7.78 (s, 0.5H), 7.56 (m, 1H), 7.45–7.29 (m, 7H), 5.75 (m, 1H), 5.35 (m, 1H), 5.14 (m, 2H), 4.86 (bm, 1H) 4.54 (m, 0.5H), 4.51 (m, 0.5H), 4.44 (m, 2H), 4.37 (s, 0.5H), 4.27 (m, 2H), 4.20 (s, 0.5H), 3.94 (d, J = 12.3 Hz, 0.5H), 3.87 (d, J = 12.3 Hz, 0.5H), 3.74 (m, 0.5H), 3.62 (m, 0.5H), 3.35 (s, 1.5H), 3.28 (s, 1.5H), 2.73 (quin, J = 7.0 Hz, 1H), 2.15 (m, 2H), 1.26 (m, 6H). 13C NMR (CDCl3, 126 MHz) δ 179.3, 166.7, 166.5, 155.5, 148.0, 147.8, 139.0, 135.5, 135.4, 133.2, 130.0, 129.5, 128.8, 128.7, 128.5, 128.0, 127.9, 121.9, 121.8, 87.2, 86.9, 84.9, 84.8, 81.8, 81.5, 75.8, 75.4, 69.9, 69.8, 65.1, 65.0, 61.5, 61.0, 60.9, 58.7, 58.5, 36.2, 29.6, 18.9. 31P NMR (CDCl3, 202 MHz) δ −1.5, −1.6. HRMS- found 722.2197, calc for C32H39N5O11PNa [M+Na]+, 722.2203.
5.17. Compound 20
19 (0.70 g, 1.0 mmol) and tetrazole (70 mg, 1.0 mmol, 1.0 equiv) were dried by evaporation from MeCN. After redissolution in CH2Cl2 (10 mL) the mixture was added to a solution of 5 (0.68 g, 2 mmol, 2 equiv) in CH2Cl2 (5 mL). After stirring for 40 min Et3N (1 mL) was added and the mixture concentrated to a small volume. Chromatography eluting with 20–40% EtOAc and 2% Et3N in CHCl3 gave 20 (0.8 g, 0.85 mmol, 85%) as a mixture of stereoisomers. 31P NMR (CDCl3, 202 MHz) δ 149.2, 149.1, 147.9, 147.7, −1.4, −1.5, −1.6.
5.18. Compound 21
20 (0.8 g, 0.85 mmol) was coupled with 13 (0.38 g, 0.56 mmol, 0.67 equiv) according to Method 1 using tetrazole (0.18 g, 3 equiv) as the activator to give 21 as a colourless foam (0.38 g, 0.24 mmol, 42% based on 13). 1H NMR (CDCl3, 500 MHz) δ 12.24–12.06 (m, 1H), 10.92 (m,1H), 10.78 (m, 1H), 8.98 (m, 1H), 8.56 (m, 1H), 8.02 (m, 4H), 7.61 (m, 2H), 7.54 (m, 3H), 7.45–7.10 (m, 22H), 6.77 (m, 4H), 5.69–5.59 (m, 1H), 5.17–4.92 (m, 5H), 4.84 (m, 1H), 4.68 (m, 1H), 4.45–4.33 (m, 3H), 4.30–4.10 (m, 4H), 3.99 (m, 1H), 3.90 (m, 1H), 3.74 (m, 7H), 3.60 (m, 1H), 3.27–2.98 (m, 5H), 2.85 (m, 1H), 2.68 (m, 1H), 2.56 (m, 1H), 2.21 (m, 1H), 2.07 (m, 2H), 1.20–0.91 (m, 6H). 13C NMR (CDCl3, 126 MHz) δ 179.9, 166.4, 158.5, 155.7, 151.1, 149.3, 148.1, 147.9, 144.8, 142.1, 140.1, 136.0, 133.2, 132.6, 130.1, 129.6, 129.1, 128.7, 128.4, 128.1, 127.8, 126.9, 123.0, 116.0, 113.1, 87.3, 86.2, 82.1, 81.4, 78.3, 75.2, 69.7, 66.6, 65.1, 63.3, 60.9, 59.3, 58.6, 55.9, 55.2, 48.0, 47.2, 35.7, 29.6, 19.2, 18.8. 31P NMR (CDCl3, 202 MHz) δ −1.2, −1.5, −3.5. HRMS – found 1521.5349, calc for C79H83N10O18P2 [M+H]+, 1521.5362.
5.19. Compound 22
21 (0.38 g, 0.24 mmol) was hydrolysed according to Method 2 to give 22 (0.21 g, 0.18 mmol, 75%) as a colourless foam. 1H NMR (CDCl3, 500 MHz) δ 12.24 (bs, 1H), 10.92 (bm, 1H), 10.73 (bm, 1H), 9.00 (bs, 1H), 8.56 (m, 1H), 8.02 (m, 4H), 7.61 (m, 2H), 7.54 (m, 3H), 7.45–7.10 (m, 13H), 5.60 (m, 1H), 5.33–4.94 (m, 6H), 4.94–4.67 (m, 2H), 4.49–4.04 (m, 6H), 4.01–3.65 (m, 2H), 3.55 (m, 1H), 3.46 (m, 1H) 3.31–3.16 (m, 3H), 3.09–2.86 (m, 1H), 2.73 (m, 1H), 2.69–2.48 (m, 2H), 2.33 (m, 2H), 2.11 (m, 2H), 1.24–1.08 (m, 6H). 13C NMR (CDCl3, 126 MHz) δ 180.0, 166.5, 155.7, 151.0, 149.3, 148.1, 142,2, 139.7, 135.4, 135.2, 133.2, 132.7, 130.2, 129.5, 129.1, 128.9, 128.8, 128.7, 128.5, 128.1, 127.9, 127.8, 122.9, 116.0, 87.6, 87.5, 81.8, 80.9, 79.2, 74.8, 69.8, 66.3, 65.0, 63.3, 60.9, 60.4, 59.7, 58.6, 55.2, 48.2, 47.8, 35.8, 29.5, 19.2, 18.8. 31P NMR (CDCl3, 202 MHz) δ −1.2, −1.5, −2.7 HRMS – found 1219.4042 calc for C58H65N10O16P2 [M+H]+, 1219.4055. HPLC (Poroshell, 60–100% MeOH over 6 min) Rt 3.92, 3.97, 4.06, >90%.
5.20. Compound 23
22 (170 mg, 0.16 mmol) was deprotected according to Method 3. Chromatography of the crude on silica gel using 30–50% NH4OH (7 M) in dioxane as the eluant followed by lyophilization gave the ammonium salt of 23 (42 mg, 52%) as a white solid. 1H NMR (D2O) δ 7.96 (s, 1H), 7.81 (s, 1H), 7.52 (s, 1H), 5.74 (d, J = 6.1 Hz, 1H), 4.84 (m, 1H), 4.64–4.61 (m, 1H), 4.40–4.36 (m, 1H), 4.38 – 4.34 (m, 1H), 4.31 (br quintet, J = 2.6 Hz, 1H), 4.25 (d, J = 13.9 Hz, 1H), 4.03 (dt, J = 12.0, 3.5 Hz, 1H), 3.97 (q, J = 6.4 Hz, 2H), 3.95–3.91 (m, 1H), 3.74 (dd, J = 12.0, 8.5 Hz, 1H), 3.67 (t, J = 6.4 Hz, 3H), 3.57 (brd, J = 5.5 Hz, 1H), 3.41 (s, 3H), 3.35 (brd, J = 12.4 Hz, 1H), 3.18–3.12 (m, 2H), 2.68 (br quintet, J = 6.4 Hz, 1H), 1.85 (quintet, J = 6.4 Hz, 2H). 13C NMR (D2O) δ 158.5, 153.7, 151.4, 150.3, 149.8, 143.8, 136.9, 131.5, 115.9, 113.4, 103.1, 85.1, 83.0, 81.4, 75.5, 72.3, 64.7, 63.2, 60.2, 58.3, 58.2, 58.0, 53.8, 47.8, 47.4, 32.5 (d, J = 6.6 Hz). 31P NMR (D2O): δ = −0.11, −1.37. HRMS – found 759.2011, calc for C26H37N10O13P2 [M−H]− 759.2017. HPLC (Kinetex, 0–30% MeCN over 15 min) Rt 8.18 min, 97%.
5.21. Compound 2
Alcohol 22 (0.090 g, 0.074 mmol) and amidite 17 (0.123 g, 0.3 mmol, 4 equiv) were coupled according to Method 1 using tetrazole (0.0063 g, 1.2 equiv) as the activator. The syrup (61 mg) obtained by chromatography was deprotected as described in Method 3. Chromatography of the crude on silica gel using 30–50% NH4OH (7 M) in dioxane as the eluant followed by lyophilization gave the ammonium salt of 2 (19 mg, 44%) as a white solid that was further purified by preparative HPLC. 1H NMR (D2O) δ 8.30 (s, 1H), 7.99 (s, 1H), 7.80 (s, 1H), 5.91 (d, J = 6.4 Hz, 1H), 4.97 (m, 2H), 4.59 (m, 1H), 4.47 (m, 2H), 4.18 (dt, J =11.9, 4.0 Hz, 1H), 4.14–3.87 (m, 9H), 3.78 (t, J = 6.4 Hz, 2H), 3.74 (t, J = 6.4 Hz, 2H), 3.72 (dd, J = 4.2, 11.9 Hz, 0.5 H), 3.63 (dd, J = 6.5, 11.9 Hz, 0.5 H), 3.55 (bd, J = 13.2 Hz, 1H), 3.51 (s, 3H), 3.43 (d, J = 5.9 Hz, 1H), 3.41 (d, J = 5.4 Hz, 1H), 2.96 (brs, 1H), 1.96 (q, J = 6.3 Hz, 2H), 1.91 (q, J = 6.3 Hz, 2H). 13C NMR (D2O) δ 158.6, 153.7, 151.6, 150.6, 149.4, 143.2, 137.4, 132.2, 116.2, 113.6, 103.1, 85.3, 83.1, 81.1, 76.0, 72.4, 66.6, 65.0, 64.1, 63.1, 63.1, 58.6, 58.3, 58.2, 53.6, 48.3, 46.0, 32.5 (d, J = 7.0 Hz), 32.4 (d, J = 7.0 Hz). 31P NMR (D2O) δ 0.42, −0.07, −1.34. HRMS – found 897.2017, calc for C29H44N10O17P3 [M – H]−, 897.2099. HPLC (Poroshell, 0–350% MeOH over 5 min) Rt 2.01 min, >97%.
5.22. Compound 24
22 (0.21 g, 0.18 mmol) and phosphoramidite 9 (0.36 g, 0.2 mmol, 1.3 equiv) were coupled according to the Method 1 using tetrazole (0.020 g, 3 equiv) as the activator. The resulting colourless foam (0.18 g) was hydrolysed according to Method 3 to give 24 (0.085 g, 26%) as a pale foam. 1H NMR (CDCl3, 500 MHz) δ 12.24 (bm, 2H), 10.87 (bm, 3H), 9.30 (bm, 1H), 8.56–7.05 (m, 29H), 5.87–5.60 (m, 2H), 5.45–3.50 (m, 25H), 3.50–3.12 (m, 8H), 2.90–2.35 (m, 6H), 2.09 (m, 2H), 1.33–1.00 (m, 12H). 13C NMR (CDCl3, 126 MHz) δ 180.1, 166.5, 155.7, 150.9, 149.4, 148.3, 142.5, 140.0, 138.9, 135.4, 135.1, 133.2, 132.6, 131.2, 129.5, 129.0, 128.7, 128.4, 128.1, 127.9, 122.3, 121.4, 115.9, 87.2, 86.5, 84.9, 81.7, 81.3, 79.2, 75.8, 74.6, 69.9, 66.8, 65.1, 61.3, 60.9, 58.6, 53.9, 47.5, 46.4, 35.8, 29.6, 19.2, 19.1. 31P NMR (CDCl3, 202 MHz) δ −1.2, −2.1, −2.5, −3.5. HRMS – found 1738.5563, calc for C80H91N15O24P3 [M+H]+, 1738.5574. HPLC (Poroshell, 50–100% MeOH over 6 min) RT 4.86–4.97, >88%.
5.23. Compound 3
24 (0.030 mg, 0.017 mmol) was hydrogenolysed and hydrolysed according to Method 3 and the resulting crude was purified on a plug of C-18 silica and then by preparative HPLC. The eluate was lyophilized to give 3 as a non – stoichiometric Et3N salt (6.5 mg, 4.9 mmol, 29%). 1H NMR (D2O, 500 MHz) δ 8.20 (s, 1H), 8.02 (s, 1H), 7.98 (s, 1H), 7.77 (s, 1H), 5.92 (d, J = 6.2 Hz, 1H) 5.89 (d, J = 6.2 Hz, 1H), 5.00 (m, 1H), 4.95 (m, 1H), 4.91 (m, 1H), 4.58 (t, J = 5.8 Hz, 1H), 4.53 (m, 2H), 4.47 (m, 2H), 4.39 (m, 1H), 4.21–4.03 (m, 6H), 3.93 (m, 1H), 3.75 (t, J = 6.4 Hz, 2H), 3.71 (dd, J = 4.4, 11.8 Hz, 1H), 3.62 (m, 2H), 3.55–3.44 (m, 8H), 3.26 (q, J = 7.4 Hz, 10H, Et3N), 2.99 (bs, 1H), 1.93 (m, 2H), 1.34 (t, J = 7.4 Hz, 15H, Et3N). 13C NMR (D2O, 126 MHz) δ 158.7, 158.5, 153.7, 153.6, 151.6, 151.3, 150.4, 148.3, 137.8, 137.3, 132.7, 116.5, 116.1, 113.3, 103.1, 85.8, 85.3, 84.8, 83.1, 81.2, 76.0, 72.9, 72.4, 65.1, 64.6, 63.1, 62.6, 59.0, 58.3, 58.2, 58.1, 53.8, 48.5, 46.7, 46.0, 32.5, 8.3. 31P NMR (CDCl3, 202 MHz) δ −0.1, −0.8, −1.3. HRMS-found 1118.2651 calc for C37H51N15O20P3 [M−H]−, 1118.2648. HPLC (Poroshell,-50% MeOH over 5 min) RT 2.72, >97%.
5.24. Compound 25
24 (0.055 g, 0.032 mmol) and phosphoramidite 17 (0.053 g, 0.013 mmol, 4 equiv) were coupled according to Method 1 using tetrazole (0.011 g, 0.16 mmol, 5 equiv) as the activator. The resulting pale coloured foam (0.28 g) was hydrogenolysed and hydrolysed according Method 3 and the resulting crude was purified on a plug of C-18 silica and then by preparative HPLC. The eluate was lyophilized to give 25 (4.9 mg, 3.1 μmol, 6.4%) as a non – stoichiometric Et3N salt contaminated with 5 mass% benzamide. 1H NMR (D2O, 500 MHz) δ 8.31 (s, 1H), 8.12 (s, 1H), 8.02 (s, 1H), 7.89 (s, 1H), 5.93 (d, J = 6.4 Hz, 1H) 5.91 (d, J = 6.6 Hz, 1H), 5.01 (m, 1H), 4.98 (m, 1H), 4.92 (m, 1H), 4.68 (m, 1H), 4.57 (m, 3H), 4.51 (m, 2H), 4.25–4.10 (m, 6H), 4.07 (q, J = 6.5 Hz, 2H), 3.92 (m, 3H), 3.77 (t, J = 6.4 Hz, 2H), 3.68 (bd, J = 13 Hz, 1H), 3.64 (t, J = 6.4 Hz, 2H), 3.57 (m, 1H), 3.50 (m, 7H), 3.27 (q, J = 7.4 Hz, 12H, Et3N), 3.01 (bs, 1H), 1.94 (q, J = 6.4 Hz, 2H), 1.81 (q, J = 6.4 Hz, 2H), 1.35 (t, J = 7.4 Hz, 18H, Et3N) 13C NMR (D2O, 126 MHz) δ 158.8, 158.6, 153.8, 151.9, 151.6, 150.5, 148.6, 138.1, 137.5, 132.7, 116.2, 113.0, 85.3, 84.9, 83.2, 81.1, 76.1, 73.2, 72.3, 65.1, 64.6, 63.1, 62.9, 59.1, 58.3, 58.2, 58.1, 53.9, 48.5, 46.8, 46.0, 32.5, 32.48.3. 31P NMR (CDCl3, 202 MHz) δ 0.3, 0.1, −0.9. HRMS- found 1256.273, calc for C40H58N15O24P4 [M – H]−,1256.2730, found 1256.2738. HPLC (Poroshell, 5–50% MeOH over 5 min) Rt 2.08, 96%.
5.25. Compound 26
BzCl (51 μL, 0.44 mmol) was added to a solution of 8 (0.24 g, 0.36 mmol) in pyridine (5 mL) and the resulting mixture stirred for 1 h under an atmosphere of argon. The reaction mixture was concentrated and dissolved in EtOAc (25 mL), washed with H2O (10 mL) and NaHCO3 (10%, aq, 10 mL), dried and concentrated. The residue was purified by chromatography (50–100% EtOAc in hexanes) to afford a foam (0.28 g). which was dissolved in AcOH (80%, 5 mL) and stirred at rt for 15 min. Solvents were evaporated and the residue purified by chromatography on silica gel (EtOAc, then 10% MeOH in CHCl3) to afford 26 (0.12 g, 63%) as a foam. 1H NMR (CDCl3, 500 MHz) δ 8.09–8.06 (m, 2H), 7.98 (s, 1H), 7.60 (tt, J = 7.5, 1.2 Hz, 1H), 7.47 (t, J = 7.8 Hz, 2H), 5.90 (d, J = 6.9 Hz, 1H), 5.75 (dd, J = 5.2, 2.2 Hz, 1H), 4.70 (dd, J = 6.9, 5.2 Hz, 1H), 4.41 (q, J = 2.2 Hz, 1H), 4.03 (dd, J = 12.7, 2.6 Hz, 1H), 3.90 (brd, J =12.4 Hz, 1H), 3.29 (s, 3H), 2.78 (septet, J = 6.9 Hz, 1H), 1.28 (d, J = 4.7 Hz, 3H), 1.27 (d, J = 4.7 Hz, 3H). 13C NMR (CDCl3, 126 MHz) δ 179.1, 165.7, 155.3, 147.9, 147.5, 139.1, 133.6, 129.8, 129.3, 128.6, 122.3, 83.4, 85.0, 81.5, 72.2, 62.3, 59.1, 36.3, 19.0, 18.9.
5.26. Compound 27
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide-HCl (1.2 g, 6.0 mmol, 2.0 equiv.) and 4-dimethylaminopyridine (0.018 g, 0.15 mmol, 0.05 equiv) were added to a vigorously stirred solution of 8 (2.0 g, 3.0 mmol) and levulinic acid (0.71 g, 6.0 mmol, 2 equiv) in dry THF (50 mL). After 14 h the mixture was diluted with EtOAc and extracted with water, KHSO4 (10%, aqueous), Na2CO3 (10%, aqueous) and brine. The organic phase was dried and concentrated to give crude levulinyl ester. This material was hydrolysed according to Method 2 to give 27 (1.2 g, 2.6 mmol, 90%) as a pale coloured foam. 1H NMR (CDCl3, 500 MHz) δ 8.78 (s, 1H), 7.82 (s, 1H), 7.27 (s, 1H), 5.77 (d, J = 7.0 Hz, 1H), 5.55 (m, 1H), 5.27 (bs, 1H), 4.53 (dd, J = 5.3, 7.3 Hz, 1H), 4.28 (m, 1H), 3.95 (dd, J = 2.2, 12.6 Hz, 1H) 3.80 (d, J = 12.6 Hz, 1H), 3.28 (s, 3H), 287–2.67 (m, 5H), 2.20 (s, 3H), 1.28 (m, 6H). 13C NMR (CDCl3, 126 MHz) δ 206.4, 180.0, 172.1, 155.2, 147.8, 147.3, 139.0, 122.4, 88.2, 84.9, 81.4, 72.0, 62.4, 59.1, 37.9, 36.4, 29.8, 27.9, 18.9. HRMS – found 466.1936 calc for C20H28N5O8 [M+H]+, 466.1938.
5.27. Compound 28
Alcohol 26 (0.078 g, 0.17 mmol) and phosphoramidite 14 (0.21 g, 0.23 mmol) were coupled according to Method 1 using tetrazole (0.023 g, 2 equiv) as the activator. The crude was purified by silica gel chromatography (4% Et3N in EtOAc then 5% MeOH and 1% Et3N in CHCl3) and the residue stirred in AcOH (80%) for 4 h. Concentration and silica chromatography (2% Et3N in MeCN – CHCl3, 1:1 then 5% MeOH and 1% Et3N in CHCl3) gave 28 (0.16 g, 95%). 1H NMR (CDCl3, 500 MHz) δ 8.49 (s, 1H), 8.06–8.00 (m, 4H), 7.73 (s, 1H), 7.61 (dt, J = 7.4, 1.1 Hz, 2H), 7.52 (t, J = 7.7 Hz, 2H), 7.42 (brt, J = 7.7 Hz, 2H), 7.39–7.32 (m, 6H), 5.75 (d, J = 7.6 Hz, 1H), 5.61 (dd, J = 5.3, 1.6 Hz, 1H), 5.10 (d, J = 9.0 Hz, 2H), 4.97 (dd, J = 7.5, 5.3 Hz, 1H), 4.77 (br septet, J = 3.0 Hz, 1H), 4.39–4.30 (m, 2H), 4.28–4.25 (m, 1H), 3.87 (d, J = 13.3 Hz, 1H), 3.69 (d, J = 13.3 Hz, 1H), 3.57 (dd, J = 10.7, 4.9 Hz, 1H), 3.52 (dd, J = 10.7, 5.7 Hz, 1H), 3.21 (s, 3H), 2.94 (dd, J = 8.8, 7.4 Hz, 1H), 2.77 (quintet, J = 6.8 Hz, 1H), 2.70 (dd, J = 10.9, 3.2 Hz, 1H), 2.63 (dd, J = 10.9, 5.6 Hz, 1H), 2.40–2.37 (m, 1H) 2.36–2.32 (m, 1H), 1.23 (d, J = 6.9 Hz, 3H), 1.19 (d, J = 6.9 Hz, 3H). 13C NMR (CDCl3, 126 MHz) δ 180.1, 166.5, 165.7, 155.7, 1511, 149.3, 148.2, 148.1, 142.2, 139.8, 133.6, 133.2, 132.7, 130.1, 129.8, 129.1, 129.0, 128.9, 128.7, 128.6, 128.1, 127.8, 127.7, 123.0, 116.0, 112.4, 88.4, 81.6, 81.5, 81.0, 81.0, 79.1, 71.1, 69.9, 69.8, 66.8, 66.7, 63.4, 59.7, 59.0, 55.4, 48.3, 48.2, 47.9, 46.1, 35.8, 19.2, 18.8. HRMS – found 991.3508 calc for C48H52N10O12P [M+H]+, 991.3504.
5.28. Compound 29
A solution of 27 (0.16 g, 0.35 mmol) and phosphoramidite 14 (0.53 g, 0.58 mmol, 1.5 equiv) were coupled according to Method 1 using tetrazole (0.12 g, 1.7 mmol, 3 equiv) as the activator to give a pale coloured foam (0.38 g) which was hydrolysed according to Method 2 to give 29 (0.19 g, 0.19 mmol, 53%). 1H NMR (CDCl3, 500 MHz) δ 12.22 (bs, 1H), 11.00 (bs, 1H), 10.67 (bm, 1H), 9.03 (bs, 1H), 8.53 (s, 1H), 8.05 (m, 2H), 7.60 (m, 5H), 7.38 (m, 5H), 5.63 (dd, J = 8.2 Hz, 1H), 5.35 (m, J = 5.2 Hz, 1H), 5.17–4.77 (m, 4H), 4.39–3.82 (m, 5H), 3.53 (m, 2H), 3.19 (m, 4H), 2.93–2.55 (m, 7H), 2.40 (m, 1H), 2.19 (m, 4H), 1.29–1.10 (m, 6H). 13C NMR (CDCl3, 126 MHz) δ 206.4, 180.2, 172.2, 166.6, 156.0, 151.1, 149.7, 148.1, 142.6, 139.7, 133.4, 132.8, 129.1, 128.8, 128.2, 127.8, 127.7, 122.9, 115.9, 113.2, 88.1, 81.5, 78.8, 71.0, 70.1, 66.9, 59.0, 47.8, 37.8, 35.8, 29.8, 27.8, 19.2, 18.8. 31P NMR (CDCl3, 202 MHz) δ −3.0. HRMS – found 985.3609 calc for C46H54N10O13P [M+H]+, 985.3609.
5.29. Compound 30
28 (0.16 g, 0.16 mmol) was deprotected according to Method 3. The crude product was washed with MeOH (20 mL) to give 30 (10 mg, 31%) as a white solid. 1H NMR (CDCl3, 500 MHz) δ 7.96 (s, 1H), 7.77 (s, 1H), 7.44 (s, 1H), 5.75 (d, J = 5.6 Hz, 1H), 4.55–4.51 (m, 1H), 4.71 (t, J = 4.5 Hz, 1H), 4.20 (t, J = 5.4 Hz, 1H), 4.15 (quintet, J = 3.0 Hz, 1H), 4.08 (d, J = 13.8 Hz, 1H), 3.99 (dt, J = 11.8, 3.5 Hz, 1H), 3.91 (dt, J = 11.8, 3.0 Hz, 1H), 3.85 (d, J = 13.8 Hz, 1H), 3.64 (dd, J = 11.4, 6.0 Hz, 1H), 3.57 (dd, J = 11.4, 6.5 Hz, 1H), 3.40 (s, 3H), 3.06 (d, J = 11.9 Hz, 1H), 2.73 (dd, J = 11.9, 5.2 Hz, 1H), 2.66 (t, J = 9.2 Hz, 1H), 2.59–2.53 (m, 1H). 13C NMR (CDCl3, 126 MHz) δ 158.9, 154.0, 151.3, 150.3, 149.9, 144.7, 136.8, 130.3, 116.0, 113.4, 107.1, 85.2, 83.7 (d, J = 9.1 Hz), 82.8, 76.6 (d, J = 4.6 Hz), 68.8, 64.5 (d, J = 4.6 Hz), 61.1, 58.3, 58.2, 54.7, 48.0 (d, J = 5.3 Hz), 47.7. HRMS – found 623.2087 calc for C23H31N10O9P [M+H]+, 623.2091. HPLC (Kinetex, 0–50% MeCN, Rt 8.27 min, 84%).
5.30. Compound 31
Alcohol 28 (0.13 g, 0.13 mmol) and phosphoramidite 9 (0.26 g, 0.29 mmol, 2.2 equiv) were coupled according to Method A using tetrazole (0.023 g, 2.5 equiv) as the activator. Chromatography gave a white solid (0.11 g, HRMS – found 1812.6326, calc for C91H95N15O22P2 [M+H]+, 1812.6330) which was dissolved in AcOH (5 mL, 80%) and stirred at rt for 3 h. The solution was concentrated to dryness and the residue taken up in CHCl3, washed with NaHCO3 (10%, aq), dried and evaporated. Chromatography (0–20% MeOH in EtOAc) gave 31 as a white solid (58 mg, 0.0384 mmol, 66%). 1H NMR (CDCl3, 500 MHz) δ 12.27 (s, 1H), 11.13–10.56 (m, 3H), 9.26 (s, 1H), 8.67–8.38 (m, 1H), 8.16–7.67 (m, 5H), 7.69–7.03 (m, 18H), 5.98–5.64 (m, 2H), 5.64–5.47 (m, 1H), 5.37–3.56 (m, 18H), 3.38–3.10 (m, 7H), 3.06–2.60 (m, 5H), 2.56–2.34 (m, 2H), 2.32–2.15 (m, 1H), 1.33–1.08 (m, 12H). 13C NMR (CDCl3, 126 MHz) δ 180.2, 180.1, 166.6, 165.6, 155.8, 155.7, 155.6, 151.1, 151.0, 149.2, 148.4, 148.33, 148.28, 148.1, 142.4, 142.3, 142.2, 140.24, 140.17, 139.2, 138.7, 138.4, 135.5, 135.4, 135.1, 133.7, 133.2, 132.64, 132.60, 130.6, 130.4, 130.3, 129.8, 129.7, 129.0, 128.9, 128.8, 128.7, 128.6, 128.2, 128.10, 128.06, 128.06, 128.03, 127.8, 127.4, 127.3, 122.6, 122.5, 121.8, 121.6, 121.5, 116.1, 116.0, 115.9, 112.3, 112.1, 88.4, 88.3, 86.6, 86.3, 85.2, 85.0, 81.5, 81.4, 80.1, 79.8, 79.6, 79.5, 79.3, 75.8, 71.4, 71.3, 69.9, 69.81, 69.76, 67.0, 61.5, 61.4, 60.4, 59.7, 59.3, 59.2, 59.0, 58.5, 54.5, 54.4, 54.3, 54.1, 47.7, 47.6, 46.6, 36.0, 35.9, 19.3, 19.11, 19.05, 18.9, 18.8. HRMS – found 1510.5031, calc for C70H78N15O20P2 [M+H]+, 1510.5023.
5.31. Compound 32
31 (0.055 g, 0.036 mmol) was deprotected according to Method 3. Silica chromatography (dioxane – NH4OH, 1:1) gave clean 32 (8.0 mg, 8.1 μmol, 23%) as well as some slightly impure material (20 mg). 1H NMR (D2O, 500 MHz) δ 8.12 (s, 1H), 7.99 (s, 1H), 7.94 (s, 1H), 7.69 (s, 1H), 5.89–5.85 (m, 2H), 4.97 (s, 1H), 4.88 (s, 1H), 4.59–4.23 (m, 7H), 4.18–4.03 (m, 4H), 3.97–3.79 (m, 3H), 3.69–3.34 (m, 9H), 2.97 (s, 1H); 13C NMR (D2O, 126 MHz) δ 158.6, 158.4, 153.6, 151.2, 150.4, 149.8, 137.7, 137.0, 131.9, 116.5, 116.0, 113.5, 103.3, 85.7, 85.5, 84.9, 83.5, 83.4, 82.7, 81.3, 75.7, 73.0, 68.5, 64.8, 64.4, 61.2, 58.8, 58.2, 58.1, 53.7, 48.5, 45.9. HRMS – found 980.2565, calc for C34H44N15O16P2 [M – H]−, 980.2566, HPLC (Kinetex, 3–30% MeOH) Rt 7.42 min, 96%.
5.32. Compound 33
29 (0.09 g, 0.091 mmol) and 17 (0.095 g, 0.22 mmol, 2.5 equiv) were coupled according to Method 1 using tetrazole (0.026 g, 0.37 mmol, 4 equiv) as the activator to give a pale coloured foam (0.060 g) which was hydrolysed according to Method 3. The crude was purified on a plug of C-18 silica and then by preparative HPLC. The eluate was lyophilized to give 33 (8.0 mg, 10 mmol, 11%) as a pale foam. 1H NMR (D2O, 500 MHz) δ 8.10 (s, 1H), 7.86 (s, 1H), 7.63 (s, 1H), 5.80 (d, J = 5.7 Hz, 1H), 4.79 (s, 1H), 4.48 (t, J = 4.6 Hz, 1H), 4.44 (d, J = 13.0 Hz, 1H), 4.35 (d, J = 13.0 Hz, 1H), 4.28 (t, J = 5.3 Hz, 1H), 4.18 (m, 1H), 4.03 (m, 2H), 3.92 (m, 1H), 3.83 (m, 4H), 3.63 (t, J = 6.4 Hz, 2H), 3.46 (d, J = 13.1 Hz, 1H), 3.41 (s, 3H), 3.36 (m, 1H), 3.29 (dd, J = 12.5, 6.2 Hz, 1H), 3.17 (q, J = 7.4 Hz, Et3N), 2.84 (m, 1H), 1.79 (quin, J = 6.4 Hz, 2H), 1.25 (t, J = 7.4 Hz, Et3N). 13C NMR (D2O, 126 MHz) δ 158.6, 153.7, 150.5, 149.8, 143.6, 137.1, 132.1, 116.1, 113.6, 103.1, 85.5, 83.6, 82.5, 75.8, 68.8, 63.9, 63.0, 58.5, 58.2, 53.5, 48.3, 46.7, 45.8, 32.4, 8.3. 31P NMR (D2O, 202 MHz) δ 0.5, −1.1. HRMS - found 759.2020 calc for C26H37N10O13P2 [M − H]−, 759.2017. HPLC (5–50% MeOH over 6 min) Rt 1.84 min, >99%.
5.33. Compound 34
27 (0.77 g, 1.65 mmol) and phosphoramidite 17 (1.0 g, 2.48 mmol, 1.5 equiv) were coupled according to Method 1 using DCI (0.59 g, 5 mmol, 3 equiv) as the activator to give 34 (0.65 g, 0.82 mmol, 50%) as a colourless foam and a mixture of stereoisomers. 1H NMR (CDCl3, 500 MHz) δ 12.20 (bs, 1H), 11.01 (bs, 0.5H), 10.90 (bs, 0.5H), 8.01 (d, J = 7.6 Hz, 1H), 7.95 (d, J = 7.6 Hz, 1H), 7.71 (s, 0.5H), 7.67 (s, 0.5H), 7.56 (m, 1H), 7.41 (m, 4H), 7.25 (m, 2H), 7.16 (m, 1H), 5.74 (d, J = 7.7 Hz, 0.5H), 5.68 (d, J = 7.7 Hz, 0.5H), 5.51 (d, J = 5.4 Hz, 0.5H), 5.39 (d, J = 5.4 Hz, 0.5H), 5.14 (d, J = 9.3 Hz, 1H), 5.01 (m, 1H), 4.94 (dd, J = 5.4, 7.7 Hz, 0.5H), 4.84 (dd, J = 5.4, 7.3 Hz, 0.5H), 4.46 (t, J = 6.0 Hz, 1H), 4.41–4.28 (m, 4H), 4.13 (m, 1H), 3.25 (s, 1.5H), 3.21 (s, 1.5H), 2.89–2.62 (m, 5H), 2.20 (m, 5H), 2.04 (m, 1H), 1.19 (m, 6H). 13C NMR (CDCl3, 126 MHz) δ 206.1, 179.8, 171.9, 171.8, 166.0, 155.5, 155.4, 147.9, 139.4, 135.0, 134.7, 132.9, 129.7, 129.3, 129.2, 128.7, 128.6, 128.4, 128.2, 127.9, 127.0, 122.7, 88.0, 81.3, 78.7, 70.7, 68.8, 69.3, 66.8, 66.6, 64.8, 64.5, 60.5, 60.4, 58.8, 37.6, 35.4, 29.5, 29.4, 29.2, 27.86, 18.9, 18.8, 18.6. 31P NMR (CDCl3, 202 MHz) δ −2.5, −2.6. HRMS – found 798.2749 calc for C37H45N5O13P [M+H]+, 798.2752.
5.34. Compound 35
Hydrazine hydrate (0.1 mL, 50%) was added to a solution of 34 (0.57 g, 0.72 mmol) in MeOH (5 mL). After 5 h the reaction was quenched with excess acetone and concentrated. Chromatography of the residue, eluting with a gradient of methanol (0–10%) in CHCl3 – EtOAc, 2:1, gave 35 (0.35 g, 0.50 mmol, 70%). as a pale coloured foam and a mixture of stereoisomers. 1H NMR (CDCl3, 500 MHz) δ 12.30 (bs, 0.5H), 12.24 (bs, 0.5H), 11.06 (bs, 0.5H), 10.94 (bs, 0.5H), 8.01 (m, 1H), 7.95 (m, 1H), 7.79 (s, 0.5H), 7.76 (s, 0.5H), 7.55 (m, 1H), 7.39 (m, 4H), 7.25 (m, 2H), 7.18 (m, 1H), 5.88 (d, J = 6.3 Hz, 0.5H), 5.82 (d, J = 6.1 Hz, 0.5H), 5.14 (m, 1H), 5.00 (m, 1H), 4.57 (m, 2H), 4.45 (t, J = 6.0 Hz, 1H), 4.33 (m, 4H), 4.13 (m, 1H), 3.97 (bs, 1H), 3.34 (s, 1.5H), 3.32 (s, 1.5H), 2.86 (m, 0.5H), 2.79 (m, 0.5H), 2.18 (m, 1H), 2.04 (m, 1H), 1.22 (m, 6H). 13C NMR (CDCl3, 126 MHz) δ 180.2, 180.1, 166.3, 166.2, 155.6, 148.1, 139.2, 135.2, 135.0, 133.0, 129.8, 129.7, 129.4, 128.8, 128.6, 128.5, 128.3, 128.0, 127.2, 122.4, 88.1, 88.0, 83.4, 83.4, 81.3, 69.8, 69.5, 69.4, 67.4, 67.2, 64.9, 64.6, 60.7, 60.5, 60.3, 58.5, 35.6, 29.5, 29.4, 19.0, 18.9, 18.8.31P NMR (CDCl3, 202 MHz) δ −1.9. HRMS – found 700.2377 calc for C32H39N5O11P [M+H]+, 700.2384.
5.35. Compound 36
A solution of 35 (0.18 g, 0.26 mmol) and DCI (0.036 g. 0.31 mmol, 1.2 equiv) in dry CH2Cl2 (5 mL) was added dropwise to a solution of 5 (0.13 g, 0.31 mmol, 1.2 equiv) in dry CH2Cl2 (2 mL). After 1 h the reaction was quenched with Et3N (1 mL) and concentrated. Column chromatography of the residue, (hexanes – CHCl3. 1:1 then CHCl3, both with 1% Et3N) gave 36 (0.25 g, 0.26 mmol, 86%), 31P NMR (CDCl3, 202 MHz) δ 151.6, 149.6, −2.0, −1.7.
5.36. Compound 37
13 (0.10 g, 0.15 mmol) and phosphoramidite 17 (0.076 g, 0.18 mmol, 1.2 equiv) were coupled together according to Method 1 using tetrazole (0.021 g, 0.30 mmol, 2 equiv) as the activator. The resulting pale foam (0.11 g) was hydrolysed according to Method 2 to give 37 (0.33 g, 0.48 mmol, 66%) as a colourless foam and a mixture of diastereomers. 1H NMR (CDCl3, 500 MHz) δ 10.95 (bs, 1H), 9.33 (bs, 1H), 8.53 (s, 1H), 7.99 (m, 4H), 7.62 (m, 1H), 7.53 (m, 4H), 7.41 (m, 2H), 7.32 (m, 5H), 5.04 (m, 2H), 4.81 (m, 0.5H), 4.77 (m, 0.5H) 4.34 (m, 2H), 4.16 (m, 2H), 3.93 (dd, J = 13.6, 6.3 Hz, 1H), 3.86 (dd, J = 13.7, 8.1 Hz, 1H), 3.61 (m, 2H), 3.18 (bs, 1H), 3.01 (dd, J = 10.7, 6.5 Hz, 0.5H), 2.93 (m, 1.5H), 2.76 (dd, J = 10.7, 3.6 Hz, 0.5H), 2.72 (dd, J = 10.7, 3.6 Hz, 0.5H), 2.45 (m, 2H), 2.07 (m, 2H). 13C NMR (CDCl3, 126 MHz) δ 166.6, 166.2, 150.9, 149.1, 142.3, 135.6, 135.7, 133.0, 132.9,132.7130.1, 129.9, 129.4, 128.8, 128.4, 128.2, 127.8, 127.7, 115.8, 112.5, 80.3, 69.3, 64.3, 63.6, 60.8, 60.1, 55.4, 48.4, 47.7, 29.4. 31P NMR (CDCl3, 202 MHz) δ −1.2, for −1.3. HRMS- found 700.2534 calc C36H39N5O8P [M+H]+, 700.2536.
5.37. Compound 38
36 (0.18 g, 0.19 mmol) was coupled with 37 (0.094 g, 0.14 mmol, 0.7 equiv) according to Method 1 using tetrazole (0.04 g, 0.58 mmol, 3 equiv) as the activator to give 38 (0.050 g, 0.033 mmol, 32%) as a pale coloured foam and a mixture of stereoisomers.1H NMR (CDCl3, 500 MHz) δ 12.17 (m, 1H), 10.90 (m, 2H), 9.12 (bs, 1H), 8.57 (s, 1H), 7.98 (m, 6H), 7.72 (m, 1H), 7.54 (m, 5H), 7.45–7.17 (m, 18H), 7.11 (m, 1H), 5.73 (m, 1H), 5.10 (m, 7H), 4.82 (m, 1H), 4.69 (m, 1H), 4.51–4.02 (m, 13H), 3.90 (m, 2H), 3.22 (m, 3H), 3.00–2.57 (m, 5H), 2.39 (m, 2H), 2.14 (m, 2H), 1.98 (m, 2H), 1.56 (m, 6H). 13C NMR (CDCl3, 126 MHz) δ 179.9, 166.4, 155.6, 151.2, 149.3, 148.1, 142.2, 139.5, 135.6, 135.2, 135.0, 133.3, 133.2, 133.1, 132.6, 130.3, 129.9, 129.8, 129.5, 129.1, 128.9, 128.8, 128.7, 128.6, 128.4, 128.1, 127.9, 127.7, 127.2, 122.7, 115.8, 112.4, 87.1, 82.2, 82.0, 79.4, 79.0, 78.7, 75.1, 69.9, 69.8, 69.5, 67.7, 66.8, 65.0, 64.6, 60.9, 60.7, 59.8, 58.7, 58.6, 54.6, 47.6, 46.6, 35.8, 29.5, 19.1, 18.9, 18.8. 31P NMR (CDCl3, 202 MHz) δ −1.1, −1.3, −1.5, −1.6, −1.7, −1.8, −1.9, −2.0, −2.1.
5.38. Compound 39
38 (0.045 g, 0.029 mmol) was deprotected according to Method 3. Chromatography on C-18 silica (10% MeOH in aqueous Et3NHOAc (0.05 M) gave 39 (0.015 g, 0.013 mmol, 43%) as a non – stoichiometric Et3N salt contaminated with 10 mass% BzNH2. 1H NMR (D2O, 500 MHz) δ 8.13 (s, 1H), 8.08 (s, 1H), 7.58 (s, 1H), 5.88 (d, J = 6.7 Hz, 1H), 4.93 (m, 1H), 4.50 (m, 3H), 4.09 (m, 3H), 3.94 (m. 7H), 3.69 (dt, J = 6.4, 3.0 Hz, 2H), 3.62 (t, J = 6.4 Hz, 2H), 3.35 (s, 3H), 3.18 (q, J = 7.3 Hz, Et3N), 3.04 (m, 1H), 2.93 (dd, J = 6.0, 10.8 Hz, 1H), 2.62 (m, 1H), 2.51 (dd, J = 7.8, 9.7 Hz, 1H), 1.84 (quin, J = 6.4 Hz, 2H), 1.80 (quin, J = 6.4 Hz, 2H), 1.29 (t, J = 7.3 Hz, Et3N). 13C NMR (D2O, 126 MHz) δ 163.3, 157.4, 151.9, 150.3, 149.8, 145.3, 136.2, 132.5, 130.0, 128.7, 127.4, 117.0, 113.3, 109.3, 84.5, 83.4, 81.3, 76.3, 73.1, 65.9, 65.2, 62.9, 62.7, 59.3, 58.2, 57.9, 54.0, 46.6, 32.4, 8.3. 31P NMR (CDCl3, 202 MHz) δ 0.4, −0.1, −0.6. HRMS – found 897.2098 calc for C29H44N10O17P3 [M+H]+, calculated 897.2099. HPLC (Poroshell, 0–30% MeCN over 12 min) Rt 4.25 min, >98%.
5.39. Compound 40
37 (0.26 g, 0.37 mmol) and phosphoramidite 9 (0.67 g, 0.74 mmol 2 equiv) were coupled according to Method 1 using tetrazole (0.078 g, 1.1 mmol, 3 equiv) as activator. The resulting pale foam (0.34 g) was hydrolysed according to Method 2 to give 40 (0.22 g, 0.18 mmol, 50%) as a colourless foam and a mixture of stereoisomers 1H NMR (CDCl3, 500 MHz) δ 12.16 (bs, 1H), 10.92 (bs, 1H), 9.86 (m, 1H), 8.92 (bs, 1H), 8.57 (m, 1H), 8.08–7.86 (m, 5H), 7.67–7.25 (m, 17H), 5.84 (m, 1H), 5.41–5.17 (m, 1H), 5.07 (m, 4H), 4.69 (m, 1H), 4.52 (m, 1H), 4.33 (m, 3H), 4.12 (m, 4H), 3.96–3.58 (m, 4H), 3.30 (m, 4H), 2.92 (m, 2H), 2.81 (m, 1H), 2.64 (m, 2H), 2.44 (m, 1H), 2.04 (m, 2H), 1.20 (m, 6H). 13C NMR (CDCl3, 126 MHz) δ 179.6, 166.3, 155.4, 151.1, 149.4, 148.2, 142.2, 138.5, 135.6, 133.3, 133.1, 132.6, 130.2, 129.5, 129.1, 128.6, 128.4, 128.1, 127.9, 127.6, 115.8, 86.0, 84.9, 84.1, 81.7, 78.7, 76.5, 75.4, 69.6, 67.4, 64.7, 61.5, 60.9, 59.7, 58.8, 54.4, 47.6, 46.3, 35.9, 29.6, 18.9. 31P NMR (CDCl3, 202 MHz) δ −1.5, −1.6, −1.7, −2.1, −2.4. HRMS – found 1219.4041 calc for C58H65N10O16P2 [M+H]+, 1219.4055.
5.40. Compound 41
40 (0.22 g, 0.18 mmol) and phosphoramidite 9 (0.34 g, 0.37 mmol 2 equiv) were coupled according to Method 1 using tetrazole (0.039 g, 0.56 mmol, 3 equiv) as activator to give the protected triphosphate (0.23 g, 0.12 mmol, 66%). HRMS – found 2040.6893 calc for C101H109N15O26P3 [M+H]+, 2040.6881. Acidic hydrolysis (Method 2) and hydrogenolysis and basic hydrolysis (Method 3) gave a crude material that was purified first on a plug of C-18 silica and then by preparative HPLC. The eluate was lyophilized to give 41 (1.5 mg, 1.1 μmol, 0.6%) a non – stoichiometric Et3N salt. 1H NMR (D2O, 500 MHz) δ 8.10 (s, 1H), 8.00 (s, 1H), 7.90 (s, 1H), 7.65 (s, 1H), 5.81 (m, 2H), 4.92 (m, 1H), 4.81 (m, 1H), 4.67 (m, 1H), 4.57 (m, 1H), 4.45 (m, 1H), 4.39 (m, 3H), 4.25 (m, 1H), 4.16 (m, 2H), 4.02 (m, 2H), 3.87 (m, 2H), 3.69 (m, 3H), 3.60 (m, 2H), 3.50 (m, 1H), 3.45 (m, 1H), 3.39 (s, 3H), 3.33 (s, 3H) 3.18 (q, J = 7.3 Hz, Et), 3.15 (m, 1H), 2.82 (m, 1H), 1.76 (quin, J = 6.4 Hz, 2H), 1.26 (t, J = 7.3 Hz, Et3N). 13C NMR (D2O, 126 MHz, excluding quaternary carbons) δ 150.9, 138.2, 132.0, 86.4, 84.9, 83.7, 81.7, 76.4, 73.3, 65.5, 63.5, 61.5, 59.7, 58.5, 54.3, 48.5, 47.3, 32.9, 8.8. 31P NMR (D2O, 202 MHz) δ −0.5, −0.8, −1.0. HRMS – found 1120.2810, calc for C37H53N15O20P3 [M+H]+, 1120.2804. HPLC (Poroshell, 50–100% MeOH over 6 min) Rt 2.76 min, 97%.
5.41. Compound 42
MsCl (0.40 mL, 1.9 equiv) and then Et3N (0.80 mL, 2.1 equiv) were added to a stirred solution of 6-N-benzoyl-9-deaza-9-(2-hydroxyethyl)adenine (0.75 g, 2.7 mmol) [18] in CH2Cl2 (100 mL). After 4 h the solution was washed with H2O and NaHCO3 (10%, aq). The organic phase was dried and evaporated to a white solid (0.90 g) which was taken up in DMF (30 mL). (3R, 4R)-4-Hydroxymethylpyrrolidin-3-ol [17] (0.55 g, 4.7 mmol) and Na2CO3 (0.75 g) were added and the solution stirred at 90 °C. The solvent was evaporated under reduced pressure and the residue suspended in CHCl3 – MeOH, 9:1 and filtered. Concentration of the solvent and chromatography of the residue (MeOH with 1% NH4OH (0–60%) in CHCl3) gave 42 (0.78 g, 2.1 mmol, 76%). 1H (NMR CDCl3-CD3OD, 2:1, 500 MHz) δ 8.56 (s, 1H), 8.08 (d, J = 7.1 Hz, 2H), 7.66 (m, 1H), 7.58 (m, 3H), 4.13 (dt, J = 3.7, 5.9 Hz, 1H), 3.66 (dd, J = 6.3, 10.8 Hz, 1H), 3.61 (dd, J = 6.5, 10.9 Hz, 1H) 3.15 (dd, J = 8.2, 10.0 Hz, 1H), 3.06 (t, J = 7.6 Hz, 2H), 2.92 (m, 3H), 2.81 (dd, J = 3.6, 10.3 Hz, 1H), 2.51 (dd, J = 6.5, 9.7 Hz, 1H), 2.27 (m, 1H). 13C NMR CDCl3-CD3OD, 2:1, 126 MHz) δ 166.7, 149.9, 148.1, 142.6, 132.7, 132.3, 129.1, 128.5, 127.6, 115.9, 113.0, 72.8, 62.8, 61.6, 56.0, 55.5, 49.2, 22.1. HRMS – found 382.1878 calc for C20H24N5O3 [M+H]+, 382.1879.
5.42. Compound 43
A solution of alcohol 42 (0.77 g, 2.0 mmol) in dry pyridine was concentrated to dryness, held under oil pump vacuum for 0.5 h and then redissolved in dry pyridine (5 mL) to which DMTrCl (1.0 g, 1.5 equiv) was added. The solution was stirred for 16 h and then partitioned between CHCl3 and H2O. The organic phase was dried, concentrated and the residue purified by chromatography (MeOH (0–10%) in CHCl3) to give 43 (0.45 g, 33%). A quantity of ditritylated material was also recovered that was converted to the desired monotrityl product by stirring for 40 min in MeOH- HCl. 1H NMR (CDCl3, 500 MHz) δ 10.8 (bs, 1H), 9.03 (bs, 1H), 8.56 (s, 1H), 8.00 (d, J = 7.1 Hz, 2H), 7.64 (tt, J = 7.4, 1.8, 1H), 7.54 (m, 2H), 7.43 (m, 3H), 7.33–7.25 (m, 6H), 7.23 (m, 1H), 6.82 (m, 4H), 4.06 (m, 1H), 3.78 (s, 6H), 3.13 (m, 2H), 3.02 (t, J = 7.6 Hz, 2H), 2.82 (m, 3H), 2.57 (dd, J = 5.4, 9.8 Hz, 1H), 2.39 (m, 1H) 2.15 (dd, J =7.9, 9.6, 1H), 2.11 (bs, 1H). 13C NMR (CDCl3, 126 MHz) δ 166.3, 158.5, 151.2, 148.9, 142.0, 136.3, 133.2, 132.8, 130.1, 129.1, 128.7, 128.2, 127.8, 127.7, 126.7, 115.9, 114.8, 113.1, 86.0, 74.8, 64.7, 61.8, 56.2, 56.0, 55.2, 48.7, 23.1. HRMS -found 684.3176 calc for C41H42N5O5 [M+H]+, 684.3186.
5.43. Compound 44
43 (0.17 g, 0.24 mmol) and phosphoramidite 20 (0.33 g, 0.35 mmol, 1.5 equiv) were coupled according to the Method 1 using tetrazole (0.074 g, 3 equiv) as the activator. Chromatography gave of a glassy solid (0.28 g. HRMS – found 1535.5515 calc for C80H85N10O18P2 [M+H]+, 1535.5519) which was hydrolysed according to Method 2 to give 44 (0.12 g, 0.10 mmol, 40%) as a mixture of diastereomers. 1H NMR (CDCl3, 500 MHz) δ 12.2 (bs, 1H), 10.8 (bs, 2H), 9.21 (bs, 1H), 8.48 (s, 1H), 8.00 (m, 4H), 7.63 (m, 2H), 7.54 (m, 3H), 7.45–7.28 (m, 15H), 5.68 (m, 1H), 5.11 (m, 5H), 4.81 (m, 2H), 4.41 (m, 3H), 4.25 (m, 4H), 3.53 (m, 2H), 3.31–3.20 (m, 4H), 3.13–2.60 (m, 7H), 2.42 (m, 1H), 2.12 (m, 2H) 1.2 (m, 6H). 13C NMR (CDCl3, 126 MHz) δ 180.1, 166.4, 155.6, 150.7, 148.8, 148.1, 142.3, 139.7, 135.3, 133.2, 132.7, 129.9, 129.5, 129.0, 128.8, 128.5, 128.2, 128.1, 127.9, 127.8, 122.8, 115.9, 87.6, 81.8, 80.6, 79.2, 74.6, 70.0, 66.6, 65.1, 62.4, 60.9, 60.1, 58.6, 55.6, 55.0, 47.9, 35.8, 29.6, 22.5, 19.2, 18.9. 31P NMR (CDCl3, 202 MHz) δ −1.2, calc −1.4, −2.8. HRMS – found 1233.4215 calc for C59H67N10O16P2 [M+H]+, 1233.4212.
5.44. Compound 45
44 (0.11 g, 0.089 mmol) and 17 (0.074 g, 0.52 mmol, 2.0 equiv) were coupled according to Method 1 using tetrazole (0.025 g, 4 equiv) as the activator. Chromatography gave a glassy solid (0.040 g, (HRMS – found 1565.5018 calc for C76H84N10O21P3 [M + H)]+, 1565.5025). This material was deprotected according to Method 3 and purified on a column of C-18 silica (MeOH (15%) in Et3NHOAc (50 mM, pH 6). Lyophilization gave 45 (0.011 g, 7.7 μM, 8.6%) as a non-stoichiometric Et3NH+ salt contaminated with 15 mol% BzNH2. 1H NMR (D2O, 500 MHz) δ 8.13 (s, 1H), 8.03 (s, 1H), 7.40 (s, 1H), 5.88 (d, J = 5.8, 1H), 5.00 (dt, J = 4.6, 8.6, 4.96 (m, 1H), 4.53 (m, 2H), 4.28 (dm, J = 11.6, 1H), 4.21 (dt, J = 11.6, 3.7, 1H), 4.09 (q, J = 6.35, 3H), 4.03 (q, J = 5.8, 3H), 3.92 (dd, J = 9.0, 12.2, 3.77 (m, 3.52 (s, 3H), 3.44 (m, 3H), 3.25 (m, Et3N + 1H), 2.96 (m, 3H), 1.95 (m, 4H), 1.33 (m, Et3N). 13C NMR (D2O, 126 MHz) δ 158.1, 153.4, 151.2, 150.2, 148.4, 137.2, 128.8, 128.7, 127.4, 115.8, 113.1, 109.5, 85.2, 82.8, 81.4, 76.2, 72.3, 65.3, 64.2, 63.2, 63.1, 62.5, 59.5, 58.3, 58.2, 55.4, 54.0, 46.7, 45.7, 32.5, 20.7, 8.3. 31P NMR (D2O, 202 MHz) δ −0.5, −0.1, −1.1. HRMS – found 935.2196 calc for C30H47N10O17P3Na [M+Na]+, 935.2197. HPLC (Poroshell, 0–50% MeOH over 5 min) Rt 2.49 min, > 95%.
5.45. Compound 47
9-Deazaadenine [20] (1.49 g, 1.1 equiv), and then formaldehyde (aq, 37 mass%, 1.56 mL, 2 equiv) were added to a solution of amine 46 [21] (2.38 g, 10.1 mmol) in EtOH (20 mL) and H2O (10 mL). The resulting suspension was warmed to 60 °C and stirred for 2 h. Silica gel (10 g) was added, solvents evaporated and the resulting solid purified by chromatography (10% 7 N NH3 in MeOH – CHCl3) to afford the title compound 47 (2.00 g, 52%) as a white solid. 1H NMR (CD3OD, 500 MHz) δ 8.17 (s, 1H), 7.46 (s, 1H), 7.33–7.17 (m, 5H), 3.90 (dd, J = 11.8, 4.3 Hz, 2H), 3.80 (s, 2H), 3.60 (s, 2H), 3.53 (dd, J = 11.8, 7.3 Hz, 2H), 2.42 (d, J = 7.5 Hz, 2H), 2.02–1.95 (m, 1H), 1.32 (s, 3H), 1.20 (s, 3H). 13C NMR (d4-MeOH, 126 MHz) δ 152.0, 150.8, 147.2, 141.0, 130.0, 129.7, 129.2, 127.9, 115.2, 113.8, 99.2, 64.1, 60.2, 53.6, 48.6, 33.8, 25.4, 23.3.
5.46. Compound 48
A suspension of 47 (2.0 g, 5.2 mmol) and Pd(OH)2 – C (0.5 g) in a mixture of MeOH (20 mL) and NH4OH (2 mL) was stirred under a balloon of hydrogen for 48 h. Filtration through diatomaceous earth and evaporation of solvent gave a residue that was absorbed onto silica gel (10 g) and purified by chromatography (20% 7 N NH3 in MeOH – CHCl3) to afford 48 (1.39 g, 93%) as a white solid. 1H NMR (CD3OD, 500 MHz) δ 8.15 (s, 1H), 7.47 (s, 1H), 3.94 (dd, J = 1.8, 4.3 Hz, 2H), 3.91 (s, 2H), 3.66 (dd, J = 11.8, 7.3 Hz, 2H), 2.62 (d, J = 6.9 Hz, 2H), 1.94–1.87 (m, 1H), 1.35 (s, 6H). 13C NMR (d4-MeOH, 126 MHz) δ 152.1, 150.9, 146.6, 129.1, 115.4, 114.4, 99.3, 64.1, 49.3, 49.0, 44.0, 35.8, 25.6, 23.0. HRMS – found 292.1776 calc for C14H22N5O2 [M+H]+, 292.1773.
5.47. Compound 49
Fmoc chloride (3.40 g, 13 mmol) was added to a rapidly stirred suspension of 48 (2.5 g, 8.6 mmol) in MeOH (250 mL) and NaHCO3 (10% aq, 25 mL). After stirring overnight silica gel (250 g) was added, solvents evaporated and the resulting solid purified by chromatography (20% MeOH in CHCl3) to afford a gum (2.8 g) which was committed to the next step without characterization. BzCl (1.9 mL, 16 mmol) was added dropwise to a solution of this gum in pyridine (50 mL) and the resulting mixture stirred for 48 h. Solvent was evaporated and the residue partitioned between CHCl3 and NaHCO3 (10% aq). The organic layer was separated and washed with water, dried and concentrated onto silica (20 g) and purified by chromatography (CHCl3 then EtOAc then 5% MeOH in CHCl3) to afford a gum (2.24 g) which was committed to the next step without characterization. The gum was dissolved in THF (40 mL), AcOH (20 mL) and H2O (20 mL) and stirred for 48 h at rt and then concentrated. The resulting syrup was dissolved in CHCl3 (200 mL) and washed with NaHCO3 (10% aq) and brine. The organic layer was dried and concentrated and the resulting residue purified by chromatography. (EtOAc then 10% MeOH in CHCl3) to give a white foam (2.1 g) which was committed to the next step without characterization. DMTrCl (1.26 g, 3.6 mmol) was added to this foam dissolved in pyridine (25 mL) and the solution stirred for 24 h. The mixture was concentrated and the residue dissolved in CHCl3, washed with water, HCl (10% aq) and NaHCO3 (10% aq), The organic phase was dried and concentrated and the resulting residue purified by chromatography (50–100% EtOAc in hexanes) to afford 49 (2.61 g, 3.0 mmol, 35% over 4 steps) as a foam. 1H NMR (CDCl3, 500 MHz) δ 10.80 (m, 1H), 9.24 (s, 1H), 8.44 (m, 1H), 7.99 (m, 2H), 7.84–7.07 (m, 20H), 6.76 (m, 4H), 6.55 (s, 1H), 4.79–4.33 (m, 3H), 4.24 (m, 2H), 3.73 (s, 6H), 3.64–3.22 (m, 5H), 3.21–2.99 (m, 2H) 2.03 (s, 1H). 13C NMR (CDCl3, 126 MHz) δ 166.6, 158.4, 157.4, 156.5, 150.8, 149.9, 149.4, 145.1, 144.0, 142.0, 141.5, 136.2, 136.1, 135.9, 133.3, 132.8, 131.3, 130.1, 129.1, 128.2, 127.7, 127.7, 127.2, 126.6, 124.9, 124.7, 123.7, 120.1, 119.9, 115.4, 113.0, 112.7, 86.1, 66.7, 63.6, 63.2, 62.8, 61.0, 55.2, 47.5, 45.9, 41.7, 41.0, 40.8, 40.4. HRMS – found, 880.3700, calc for C54H50N5O7 [M+H]+, 880.3710.
5.48. Compound 50
Alcohol 49 (0.16 g, 0.18 mmol) was dissolved in dry MeCN (3 mL), evaporated to dryness and redissolved in MeCN (1 mL) and dry DCM (1 mL). Tetrazole (0.013 g, 1 equiv) and then 5 (0.12 g, 2 equiv) in DCM (1 mL) were added. After 0.5 h the reaction was quenched with Et3N (0.2 mL), concentrated and chromatographed (hexanes – EtOAc – Et3N, 3:2:0.02) to give a phosphoramidite (0.16 g, 31P NMR δ 146.5 ppm). This material was reacted with 19 (0.080 g, 0.12 mmol 0.8 equiv) using tetrazole (0.020 g, 2 equiv) as the activator according to Method 1 to give 50 (0.12 g, 0.069 mmol, 58% based on 19) The 1H and 13C NMR spectra were of little value for characterization of this complex mixture of diastereomers and rotamers. 31P NMR (CDCl3, 202 MHz) δ −1.2, −1.3, −1.5, −2.3. HRMS – found 1731.6045, calc for C93H93N10O20P2 [M+H]+, 1731.6043.
5.49. Compound 51
Hydrolysis according to Method 2 gave alcohol 51 (0.060 g, 0.41 mmol, 61%). 31P NMR δ −1.2, −1.3, −1.4, −2.1. HPLC and LCMS (Kinetex, 60–90% MeCN over 15 min) Rt 5.3 and 5.6 min, (each with base peak 715.2 ([M+H]2+), together > 95%).
5.50. Compound 52
Alcohol 51 (0.034 g, 0.024 mmol) and phosphoramidite 17 (0.030 g, 3 equiv) were coupled according to Method 1 using tetrazole (0.005 g, 3 equiv) as the activator. This material was deprotected according to Method 3 and purified on a column of C-18 silica eluted with 10 and 15% MeOH in Et3NHOAc. Lyophilization gave 54 (4.0 mg, 4.5 mmol, 19%) as a non-stoichiometric Et3NH+ salt. 1H NMR (D2O, 500 MHz) δ 8.19 (s, 1H), 7.99 (m, 1H), 7.45 (s, 1H), 5.94 (m, 1H), 4.97 (m, 1H), 4.51 (m, 2H), 4.09 (m, 4H), 3.89 (m, 8H), 3.78 (m, 2H), 3.66 (m, 2H), 3.50 (s, 3H), 2.71 (m, 2H), 2.23 (m, 1H), 1.97 (m, 2H), 1.80 (m, 2H). 13C NMR (D2O, 126 MHz) δ 166.1, 159.7, 151.7, 150.4, 149.9, 147.1, 144.8, 135.4, 129.0, 117.3, 113.7, 111.4, 84.5, 83.3, 81.7, 72.8, 64.9, 64.6, 63.2, 62.8, 58.3, 46.3, 41.5, 39.6, 32.5. 31P NMR (D2O, 202 MHz) δ 0.8, 0.2, −0.1. HRMS – found 887.2252, calc for C28H46N10O17P3 [M+H]+, 887.2255. HPLC (Kinetex, 0–30% MeCN over 15 min) Rt 8.57 min, >95%.
5.51. Saporin L3 kinetic studies
Saporin L3 and saporin L3 A14C were expressed as excreted proteins in yeast cultures and purified to near-homogeneity as previously reported [5,22].
Inhibition of SAP was studied in a competitive assay using RNA A10 (5′CGCGAGAGCG3′) as the substrate. Product formation was quantitated in a continuous assay by linking adenine formation to the production of ATP and the action of luciferase to produce light.4 Inhibitors at varying concentrations were preincubated with enzyme for 10 min at 20 °C in a buffer containing 200 μL of ATPlite (Perkin-Elmer) added into 1 mL of 100 mM Tris-acetate pH 7.7, 2 mM phosphoenolpyruvic acid, 2 mM sodium pyrophosphate, 2 mM 5-phospho-D-ribose 1-diphosphate pentasodium salt, 15 mM (NH4)2SO4, 15 mM (NH4)2MoO4, 10 mM Mg2SO4, 4 units of adenine phosphoribosyl transferase and 8 units of pyruvate phosphate dikinase). The reaction was initiated by adding 100 μM RNA A10. The equilibrium dissociation constants were calculated by fitting the final, equilibrium reaction rates to equation (1) for competitive inhibition, where v and vo are steady-state rates in the presence and absence of inhibitor, respectively; KA10 is the A10 RNA Michaelis constant[4] and [s] and [I] are the concentrations of the A10 RNA substrate and inhibitor, respectively.
| (1) |
In cases where the concentration of inhibitor is < 10-fold the concentration of enzyme, the following correction was applied:
| (2) |
where I′ is the effective inhibitor concentration; I is the concentration of inhibitor used in the assay; v and vo are initial rates in the presence and absence of inhibitor, respectively; and Et is the total saporin enzyme concentration used in the assay.
5.52. RNA assays
Yeast tRNA used as substrate was purchased from Invitrogen. Rabbit reticulocyte lysate (untreated) was purchased from Promega (Madison, WI). The rRNA was purified as previously described [5]. The concentration of ribosomal RNA was calculated by the absorbance at 260 nm using the extinction coefficient of 5 × 10−7 cm−1 M−1.
6. Protein translation assays
The protein translation inhibited by saporin L3 was studied using a flexi rabbit reticulocyte lysate translation system following the supplier’s instructions. The reaction mixtures (50 μL) containing 35 μL of rabbit reticulocyte lysate, 0.5 μL of amino acid mixture, minus leucine, 0.5 μL of amino acid mixture minus methionine, 1.4 μL of 2.5 M KCl, 1 μL of RNasin ribonuclease inhibitor, 1 μL of luciferase-encoding mRNA and different concentrations of saporin L3 A14C ranging from 0.3 to 500 nM were incubated at 30 °C for 90 min. A 10 μL aliquot was measured with a luciferase detection kit following the supplier’s protocol in a 96-well plate on a luminometer. The percent translation relative to control was plotted versus the log of SAP A14C concentration and fit to a dose-response curve for the calculation of IC50.
Rescue of translation by inhibitor 2
10 nM SAP A14C was pre-incubated with increasing concentrations of compound 2 for 10 min in 5 μL pH 7.4 buffer. The preincubated samples were incubated with translation reaction (45 μL) at 30 °C for 90 min. Then the luminescence was measured with a luciferase detection kit as described above. A sample with the maximum inhibitor concentration without SAP A14C was used as a control. The percent translation relative to control was plotted versus the log of inhibitor concentration and fit to a dose-response curve for the calculation of EC50.
All molecular structure optimizations and energy and frequency calculations were performed using density functional theory in B3LYP and using a 6–31 g* basis set as implemented in Gaussian 09.[23] The MEPs were calculated with the CUBE program from Gaussian and the files were visualized with GaussView 3.0.
Supplementary Material
Acknowledgments
This work was supported in part by the National Institutes of Health, USA (Grants CA072444 and GM41916) and by funds provided by the Albert Einstein College of Medicine. Dr. Herbert Wong (Callaghan Innovation) assisted with NMR spectroscopy. Dr. Yinrong Lu (Callaghan Innovation) obtained the high resolution mass spectra.
Abbreviations
- RTA
Ricin A-chain
- SAP
saporin L3
- MTET
methyltetrazole
Appendix A. Supplementary data
Supplementary data associated with this article can be found in the online version, at http://dx.doi.org/10.1016/j.ejmech.2016.10.059. These data include MOL files and InChiKeys of the most important compounds described in this article.
Footnotes
Supporting information
NMR spectra of all compounds. HPLC traces for compounds 2, 3, 23, 25, 30, 32, 33, 39, 41, 45 and 52.
References
- 1.Ferreras J, Barbieri L, Girbes T, Battelli MG, Rojo MA, Arias FJ, Rocher MA, Soriano F, Mendez E, Stirpe F. Distribution and properties of major ribosome-inactivating proteins (28 S rRNA N-glycosidases) of the plant Saponaria officinalis L. (Caryophyllaceae) BBA – Gene Struct Expr. 1993;1216:31–42. doi: 10.1016/0167-4781(93)90034-b. [DOI] [PubMed] [Google Scholar]
- 2.Endo Y, Mitsui K, Motizuki M, Tsurugi K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 S ribosomal RNA caused by the toxins. J Biol Chem. 1987;262:5908–5912. [PubMed] [Google Scholar]
- 3.Gilabert-Oriol R, Weng A, von Mallinckrodt B, Melzig MF, Fuchs H, Thakur M. Immunotoxins constructed with ribosome-inactivating proteins and their enhancers: a lethal cocktail with tumor specific efficacy. Curr Pharm Des. 2014;20:6584–6643. doi: 10.2174/1381612820666140826153913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sturm MB, Tyler PC, Evans GB, Schramm VL. Transition state analogues rescue ribosomes from saporin-L1 ribosome inactivating protein. Biochemistry. 2009;48:9941–9948. doi: 10.1021/bi901425h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan H, Du Q, Sturm MB, Schramm VL. Soapwort saporin L3 expression in yeast, mutagenesis, and RNA substrate specificity. Biochemistry. 2015;54:4565–4574. doi: 10.1021/acs.biochem.5b00405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tartarini A, Pittaluga E, Marcozzi G, Testone G, Rodrigues-Pousada RA, Giannino D, Spano L. Differential expression of saporin genes upon wounding, ABA treatment and leaf development, Physiol. Plant. 2010;140:141–152. doi: 10.1111/j.1399-3054.2010.01388.x. [DOI] [PubMed] [Google Scholar]
- 7.Chen XY, Berti PJ, Schramm VL. Ricin A-chain: kinetic isotope effects and transition state structure with stem-loop RNA. J Am Chem Soc. 2000;122:1609–1617. [Google Scholar]
- 8.Sturm MB, Roday S, Schramm VL. Circular DNA and DNA/RNA hybrid molecules as scaffolds for ricin inhibitor design. J Am Chem Soc. 2007;129:5544–5550. doi: 10.1021/ja068054h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho MC, Sturm MB, Almo SC, Schramm VL. Transition state analogues in structures of ricin and saporin ribosome-inactivating proteins. Proc Natl Acad Sci. 2009;106:20276–20281. doi: 10.1073/pnas.0911606106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Manoharan M. 2′-Carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochim Biophys Acta (BBA) 1999;1489:117–130. doi: 10.1016/s0167-4781(99)00138-4. [DOI] [PubMed] [Google Scholar]
- 11.Amukele TK, Schramm VL. Ricin a-chain substrate specificity in RNA, DNA, and hybrid Stem Loop structuresy. Biochemistry. 2004;43:4913–4922. doi: 10.1021/bi0498508. [DOI] [PubMed] [Google Scholar]
- 12.de Koning MC, Ghisaidoobe ABT, Duynstee HI, Ten Kortenaar PBW, Filippov DV, van der Marel GA. Simple and efficient solution-phase synthesis of oligonucleotides using extractive work-up. Org Proc Res Devel. 2006;10:1238–1245. [Google Scholar]
- 13.Chen C, Chen W, Chen Y, Lee M, Huang C, Chanda K, Sun C. Convergent solution phase synthesis of chimeric oligonucleotides by a 2+2 and 3+3 phosphoramidite strategy. Aust J Chem. 2010;63:227–235. [Google Scholar]
- 14.Guanti G, Banfi L, Basso A, Bondanza L, Guglieri G, Powles K, Riva R. Optimized synthesis of phosphatidylserine. Amino Acids. 2010;39:367–373. doi: 10.1007/s00726-009-0447-0. [DOI] [PubMed] [Google Scholar]
- 15.Calculated using Advanced Chemistry Development (ACD/Labs) Software V11.02 (© 1994–2015 ACD/Labs) accessed through SciFinder.
- 16.Evans GB, Furneaux RH, Tyler PC, Schramm VL. Synthesis of a transition state analogue inhibitor of purine nucleoside phosphorylase via the mannich reaction. Org Lett. 2003;5:3639–3640. doi: 10.1021/ol035293q. [DOI] [PubMed] [Google Scholar]
- 17.Mason JM, Clinch K, Evans GB, Furneaux RH, Lenz DH, Mee SPH, Tyler PC, Wilcox SJ. A practical synthesis of (3R,4R)-N-tert-butoxycarbonyl-4-hydroxymethylpyrrolidin-3-ol. Org Biomol Chem. 2007;5:2800–2802. doi: 10.1039/b708796a. [DOI] [PubMed] [Google Scholar]
- 18.Evans GB, Furneaux RH, Schramm VL, Tyler PC, Mee SPH, Zubkova OV. Preparation of Inhibitors of Nucleoside Phosphorylases and Nucleosidases. US 20090233948 United States Patent.
- 19.Schramm VL, Mason JM, Evans GB, Tyler PC. Saporin-L1 Inhibitors and Uses Thereof. US2011/201674 United States Patent.
- 20.Evans GB, Cameron SA, Luxenburger A, Guan R, Suarez J, Thomas K, Schramm VL, Tyler PC. Tight binding enantiomers of pre-clinical drug candidates. Bioorg Med Chem. 2015;23:5326–5333. doi: 10.1016/j.bmc.2015.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clinch K, Evans GB, Fröhlich RFG, Furneaux RH, Kelly PM, Legentil L, Murkin AS, Li L, Schramm VL, Tyler PC, Woolhouse AD. Third-generation immucillins: syntheses and bioactivities of acyclic immucillin inhibitors of human purine nucleoside phosphorylase. J Med Chem. 2009;52:1126–1143. doi: 10.1021/jm801421q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan H, Stratton CF, Schramm VL. Transition state structure of RNA depu-rination by saporin L3. ACS Chem Biol. 2016;11:1383–1390. doi: 10.1021/acschembio.5b01069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd J, Brothers EN, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc; Wallingford, CT, USA: 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.