

SUMMARY
Gut microbiota profoundly affect gut and systemic diseases, but the mechanism whereby microbiota affect systemic diseases is unclear. It is not known whether specific microbiota regulate T follicular helper (Tfh) cells, whose excessive responses can inflict antibody-mediated autoimmunity. Using the K/BxN autoimmune arthritis model, we demonstrated that Peyer’s patch (PP) Tfh cells were essential for gut commensal segmented filamentous bacteria (SFB)-induced systemic arthritis despite the production of auto-antibodies predominantly occurring in systemic lymphoid tissues, not PPs. We determined that SFB, by driving differentiation and egress of PP Tfh cells into systemic sites, boosted systemic Tfh cell and auto-antibody responses that exacerbated arthritis. SFB induced PP Tfh cell differentiation by limiting the access of interleukin 2 to CD4+ T cells, thereby enhancing Tfh cell master regulator Bcl-6 in a dendritic cell-dependent manner. These findings showed that gut microbiota remotely regulated a systemic disease by driving the induction and egress of gut Tfh cells.
Graphical abstract

INTRODUCTION
Gut microbiota provide essential health benefits to their host, particularly by regulating the immune system (Hooper et al., 2012). Recent studies have shown that alterations in gut microbiota (dysbiosis) can lead to many autoimmune diseases, including diseases with clear association to the gut, notably inflammatory bowel disease (Cerf-Bensussan and Gaboriau-Routhiau, 2010; Wu and Wu, 2012). Dysbiosis has also been implicated in autoimmune diseases that occur outside the gut (gut-distal or systemic), such as autoimmune arthritis, type 1 diabetes, and experimental autoimmune encephalomyelitis (EAE) (Chervonsky, 2013; Scher et al., 2013; Wu et al., 2010; Wu and Wu, 2012). However, the cellular and molecular mechanisms by which microbiota in the gut influence systemic autoimmune diseases such as rheumatoid arthritis (RA) remain largely unknown.
In contrast to the abundant gut-luminal commensals, mucosa-associated commensal species such as segmented filamentous bacteria (SFB) represent a minority among the commensal community, yet they can powerfully modulate host immunity (Hill and Artis, 2010; Ivanov et al., 2009; Palm et al., 2014). We have previously shown that SFB can drive autoimmune arthritis in the K/BxN mouse model of arthritis by inducing gut T helper 17 (Th17) cells to enhance the production of auto-antibodies (Abs) (Wu et al., 2010). However, neutralizing interleukin 17 (IL-17) in vivo still leaves animals with a substantial auto-Ab titer, suggesting that SFB can augment auto-Ab production via other pathways and/or cell types. T follicular helper (Tfh) cells are one likely candidate, because they are a crucial subset of CD4+ T cells that helps B cells produce high-affinity and high-titer Abs (Crotty, 2011, 2014; Ma et al., 2012). Tfh cells co-express high levels of inhibitory co-receptor PD-1 and chemokine receptor CXCR5. The differentiation of Tfh cells requires the master transcription factor Bcl-6. Both dendritic cells (DCs) and B cells are involved in completing the full differentiation program of Tfh cells. Because the function of Tfh cells is to induce germinal center (GC) formation, which helps B cells produce high-titer, high-affinity, isotype-switched Abs and long-lived plasma cells, Tfh cells are known to play a crucial role in generating protective immunity. However, for the exact same reason, an excessive Tfh cell response can lead to many autoimmune conditions including RA (Ueno et al., 2015). It is thus not surprising that specific signaling pathway(s) such as IL-2 and STAT5 signaling pathways have evolved to counter-regulate the Tfh cell response (Ballesteros-Tato et al., 2012; Johnston et al., 2012).
Several specific commensal species have recently been shown to control host immunity by regulating select T cell subtypes including Th1, Th17, and T regulatory (Treg) cells (Hooper et al., 2012; Wu and Wu, 2012). Despite much recent attention on the Tfh cell field, little is known regarding the interaction of commensals and Tfh cells. Most studies have focused on the Tfh cell response induced by infection or immunization with the exception of two pioneer studies showing that impairment of Tfh cells, due to lack of expression of either inhibitory co-receptor PD-1 or ATP-gated ionotropic P2RX7 receptors, can alter the gut commensal community (Kawamoto et al., 2012; Proietti et al., 2014). Here, we looked at the reverse interaction, to determine whether specific microbial species can affect the Tfh cell response and impact host health.
A pressing question that remains largely unanswered is the mechanism by which gut microbiota predispose their host to diseases at gut-distal sites. We addressed this question by using the K/BxN arthritis model to elucidate how autoimmune signals generated in the gut by intestinal commensals are transposed to systemic sites. Our results showed that SFB increased the Tfh cell population not only in Peyer’s patches (PPs), a gut-associated lymphoid tissue (GALT), but also in systemic sites such as the spleen and foot-draining popliteal lymph nodes (PLNs). SFB-induced Tfh cell responses predated arthritis development, and Tfh cells were required for SFB-mediated enhancement of autoimmune arthritis. SFB increased the systemic Tfh cell population by driving the differentiation and egress of PP Tfh cells into systemic sites. This process was crucial for arthritis development because auto-Abs were produced predominantly in systemic sites and much less in PPs. We showed that SFB induced PP Tfh cell differentiation by inhibiting the IL-2 signaling pathway in PPs and identified DCs as the critical cell type required for SFB-mediated Tfh cell induction and IL-2 receptor α (IL-2Rα) suppression in PPs. Our work provides a mechanism by which gut microbiota contribute to gut-distal autoimmune arthritis by triggering the induction and migration of gut Tfh cells into systemic sites.
RESULTS
SFB-Containing Feces Drive K/BxN Arthritis in a Specific-Pathogen-Free Environment
The K/BxN (KRN T cell receptor [TCR] transgenic mice on the C57BL/6 [B6] background x non-obese diabetic [NOD] mice) model is an autoimmune arthritis model in which KRN T cells recognize glucose-6-phosphate isomerase (GPI), the self-antigen (Ag) presented by MHC class II I-Ag7 (from NOD mice) (Monach et al., 2008). The activated T cells can in turn activate B cells to produce anti-GPI auto-Abs. As in many human autoimmune diseases, including RA, auto-Abs play important pathological roles in K/BxN disease development (Ueno et al., 2015). We have previously used germ-free (GF) mice to demonstrate the influence of SFB on the development of arthritis (Wu et al., 2010). However, GF mice are immunocompromised and in physiologic settings, any given commensal species normally co-exists with other commensals. Thus, we established a model by manipulating SFB in specific-pathogen-free (SPF) K/BxN mice. SFB were first introduced to our SFB–SPF mouse colony by gavaging mice with SFB-containing feces from Taconic B6 mice. Later, and in most of the results from this report, SFB were introduced by gavaging with SFB-containing feces collected from the SFB+ mice housed in our SPF colony as described in Figure S1A. Where noted, mice were gavaged with fecal pellets from SFB-monocolonized mice whose feces contain only SFB. Oral gavage of SFB into SPF K/BxN mice that were originally SFB− allowed detection of fecal SFB by quantitative PCR (qPCR), while their littermate controls remained SFB− (Figure S1B). SFB gavage significantly enhanced arthritis in SPF K/BxN mice compared to their non-gavaged littermate controls (Figure 1A). K/BxN mice receiving vertical transfer of maternal SFB also showed a high level of fecal SFB by qPCR and had more severe arthritis than SFB− K/BxN mice (Figures S1C and 1B). We also examined the anti-GPI auto-Ab titer, which was significantly higher in SFB+ than SFB− mice (Figure 1C). We next focused on GC development in the spleen and foot-draining PLNs, because both are the major systemic auto-Ab generating sites in the K/BxN model (Huang et al., 2010). Our results showed that SFB increased GC B cell frequencies and numbers (defined by Fas+PNA+) in the spleen as well as in the PLNs (Figure 1D). Using histological staining, we found significantly larger GCs in SFB+ than SFB− K/BxN mice but no change in the number of splenic GCs between the two groups (Figures 1E and S1D). Thus, SFB increased the disease and systemic GC response in SPF K/BxN mice.
Figure 1. SFB Exacerbate Arthritis in Specific-Pathogen-Free K/BxN Mice.

(A and B) SFB+ groups were established by one of two methods. Directly gavaging the experimental mice derived from SFB− breeders with SFB (A). The SFB− mice were the ungavaged littermate controls (n = 8–11 mice/group). Alternatively, gavaging the mother first to later give birth to the SFB+ K/BxN litters (B). The SFB− groups were derived from an ungavaged mother (n = 6–8 mice/group). Results are shown as mean ankle thickening ± SEM.
(C) Serum anti-GPI Ab titers were determined by ELISA and are shown as mean + SEM.
(D) Splenocytes or PLN cells from SFB− or SFB+ K/BxN mice were stained with Abs against CD19, CD4, Fas, and PNA (peanut agglutinin). Values in the representative plots indicate the percentage of GC B cells (Fas+PNA+) in total CD19+ B cells. The quantitative data (mean + SEM) are also shown (n = 16/group, data combined from 7 assays).
(E) The area of each GC (PNA+B220+) from splenic cryostat sections is shown. The quantitative results are a combination of 24 GCs measured from four spleens in both SFB− and SFB+ K/BxN mice.
Asterisks indicate statistical significance, *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S1.
Tfh Cells Are Required for SFB-Mediated Enhancement of Autoimmune Arthritis
We next examined whether SFB could facilitate the expansion of Tfh cells that could in turn help GC response. Tfh cells are defined as PD-1+CXCR5+Bcl-6hiCD4+ T cells (Figure 2A). The percentage and absolute number of Tfh cells was significantly increased in both the spleen and PLNs (Figure 2A) of SFB+ versus SFB− K/BxN mice. We also gavaged K/BxN mice with SFB-monocolonized feces and found a similar enhancement of the Tfh cell response compared to mice gavaged with SFB-containing feces from our SPF colony (Figure S2A). Studies have used CD4 and PNA co-staining to evaluate the localization of Tfh cells in the GCs (Johnston et al., 2009). Our data showed a significant boost in the number of GC-located CD4+ T cells in SFB+ K/BxN mice (Figure 2B). SFB did not impact the percentage or number of Treg cells in K/BxN mice, as reported in other strains of mice (Figures S3A and S3B; Goto et al., 2014; Kriegel et al., 2011). Additionally, no significant difference was observed in either the percentage or mean fluorescence intensity (MFI) of CD44 in CD4+ T cells between the SFB− and SFB+ groups, suggesting that SFB did not induce a greater activation status in the general CD4+ T cell population (Figure S3C). We also examined the effect of SFB on Th1 and Th2 cells by analyzing their respective signature cytokines, interferon-γ (IFN-γ) and IL-4. There was no induction of Th1 or Th2 cells by SFB, which again indicated the specificity of SFB-induced immune responses (Figure S3D). To prove the biological significance of this SFB-mediated increase in Tfh cells, we asked whether SFB-induced arthritis required Tfh cells. We addressed this question by using the K/BxN transfer model in which arthritis is generated by transferring KRN T cells into T-cell-deficient hosts, Tcra−/−. BxN mice, on the B6 × NOD background (Monach et al., 2008). Tcra−/−. BxN mice thus bear NOD MHC class II I-Ag7 for self-Ag presentation. In this experiment, Cxcr5−/−. KRN T cells or KRN T cells were used as donor T cells. Cxcr5−/−. KRN T cells are defective in mounting a Tfh cell response because the chemokine receptor CXCR5 is required for Tfh cells to migrate to the GC (Crotty, 2011). When KRN T cells were transferred into SFB- Tcra−/−. BxN recipients, only minimal ankle thickening and auto-Ab titers were observed (Figures 2C and 2D). SFB colonization in Tcra−/−. BxN recipient mice strongly enhanced the development of arthritis and auto-Ab titers in mice receiving KRN T cells but not Cxcr5−/−. KRN T cells. This result suggested that a competent Tfh cell response was required for SFB-mediated arthritis enhancement.
Figure 2. SFB-Mediated Arthritis Augmentation Is Dependent on Tfh Cell Response.

(A) Splenocytes or PLN cells from SFB− or SFB+ K/BxN mice were stained with Abs against CD4, CD19, PD-1, CXCR5, and Bcl-6. Values in the representative plots indicate the percentage of Tfh (PD-1+CXCR5+) cells in total CD4+ T cells. The quantitative data are also shown (n = 16/group, data combined from 7 assays).
(B) Splenic cryostat sections were stained with PNA and anti-CD4 Ab. CD4+ T cells in each GC were counted manually and data are shown as mean + SEM (18 GCs in each group).
(C and D) SFB− KRN or Cxcr5−/−. KRN donor CD4+ T cells were transferred into SFB+ or SFB− Tcra−/−. BxN recipients. Ankle thickening (C) of recipient mice is shown. Day 0 indicates the day of cell transfer. Day 14 sera were collected (D) and anti-GPI Ab titers are shown (n = 6/group, data combined from 3 assays).
See also Figures S2 and 3.
SFB Induce Tfh Cell Differentiation in PPs but Not in the Spleen or PLNs
To determine how SFB expanded the systemic Tfh cell population, we first asked whether SFB increased the survival and/or proliferation of Tfh cells. No difference in Tfh cell apoptosis or proliferation was found in spleen or PLNs between SFB− and SFB+ groups (Figures 3A, 3B, S4A, and S4B), which prompted us to ask whether the increase in the systemic Tfh cell population was from the recruitment of extra-splenic or extra-PLN Tfh cells into the spleen or PLNs. This scenario is conceivable in the K/BxN model because its self-Ag GPI is ubiquitously expressed in tissues, including both the spleen and intestines (as are many self-Ags in other autoimmune diseases) (Monach et al., 2008). SFB tightly adhere to intestinal epithelial cells, especially those covering the PPs (Tannock et al., 1984), which are intestinal lymphoid sites harboring abundant Tfh cells. We thus hypothesized that PPs could have an advantage over systemic immune sites in inducing Tfh cell differentiation, because PPs have access to the adjuvant effect of gut SFB in addition to the self-Ag stimulation that occurs at both sites. Indeed, SFB colonization significantly boosted the percentage and number of Tfh cells in the PPs of K/BxN mice compared to their SFB− counterparts (Figure 3C). K/BxN mice gavaged with SFB-monocolonized feces also displayed a similar Tfh cell boost in PPs (Figure S2B). A larger PP Tfh cell population reflected a higher GC response in the PPs of SFB+ compared to SFB− K/BxN mice (Figure 3D). Because SFB did not decrease apoptosis or increase proliferation (rather, proliferation decreased) of PP Tfh cells (Figures S4C and S4D), we next examined whether SFB increased the PP Tfh cell population by inducing PP Tfh cell differentiation. Bcl-6 is considered a master regulator of Tfh cell differentiation (Crotty, 2011). SFB increased Bcl-6 expression in PP but not splenic or PLN CD4+ T cells, suggesting that SFB preferentially boosted Tfh cell differentiation in PPs but not spleen or PLNs (Figure 3E). Alternatively, because Tfh cells express a higher level of Bcl-6 than non-Tfh cells, increased Bcl-6 protein expression in SFB+ PP CD4+ T cells could simply reflect a higher percentage of Tfh cells in SFB+ than SFB− PP CD4+ T cells that bring up the average value of Bcl-6. To determine whether SFB could truly drive Tfh cell differentiation in PPs, we examined Bcl-6 expression in both non-Tfh and Tfh cells. Our results indeed showed SFB increased Bcl-6 in non-Tfh cells from PPs only but not spleen or PLNs (Figure 3F). Blimp-1 (encoded by Prdm1) is an antagonistic transcriptional factor for Bcl-6, and the balance of Blimp-1 and Bcl-6 is known to control Tfh cell differentiation. In support of the Bcl-6 data, our results showed that SFB downregulated Prdm1 expression in PP non-Tfh cells (Figure S4E). Together, these data suggested that SFB preferentially upregulated Bcl-6 and downregulated Blimp-1 in PP non-Tfh cells, which suggested that SFB truly drove PP Tfh cell differentiation.
Figure 3. SFB Induce Tfh Cell Differentiation in Peyer’s Patches.

(A) Representative plots and quantitative data of splenic Caspase 3/7+ Tfh cells are shown (n = 15/group, data combined from 5 assays).
(B) Representative plots and quantitative data of the percentage of EdU+ splenic Tfh cells are shown (n = 7–8/group, 2 assays).
(C) Representative plots and quantitative data of Tfh cells are shown (n = 16/group, data combined from 7 assays).
(D) Representative plots and quantitative data of GC B cells are shown.
(E and F) Representative histogram plots and quantitative MFI of Bcl-6 expression in splenic, PLN, and PP CD4+ T cells (E) or non-Tfh and Tfh cells (F) are shown (n = 16/group, data combined from 7 assays).
See also Figures S2 and 4.
PP-Derived Tfh Cells Are Detected in the Systemic Lymphoid Tissues where Auto-Abs Are Predominantly Produced
Next, we asked whether SFB-induced Tfh cells in PPs could directly help B cells in PPs and/or exit to systemic sites, helping B cells there. We first asked whether PP Tfh cells provided direct help to PP B cells, leading to in situ production of auto-Abs in PPs that could circulate into the periphery. Immunoglobulin A (IgA) is the predominant isotype of mucosal Abs, but the IgG isotype can also be found in gut or genital mucosal sites (Neutra and Kozlowski, 2006). Because the auto-Ab isotype in the K/BxN model is IgG1, we asked whether SFB-induced PP Tfh cells helped auto-Ab production in PPs by (1) inducing the atypical anti-GPI auto-Abs of the IgA isotype or (2) inducing the typical anti-GPI of the IgG1 isotype despite the PP location. SFB strongly increased the percentage of GPI-specific antibody-secreting cells (ASCs) of the IgG1 isotype in PPs to a similar level observed in the spleen and PLNs (Figure 4A). However, the percentage of anti-GPI auto-Abs of the IgA isotype was minimal in the PPs of both SFB− and SFB+ K/BxN mice. This suggested that SFB-induced PP Tfh cells could help anti-GPI production by the second mechanism mentioned above. Despite the spleen and PPs having similar percentages of anti-GPI-specific B cells, the spleen and even PLNs produced a much higher absolute number of anti-GPI ASCs compared to PPs (Figure 4B). Therefore, we examined whether these increases in the systemic ASCs of SFB+ K/BxN mice were due to the migration of PP Tfh cells to systemic sites where they provided B cell help. The α4β7 integrin subfamily of adhesion molecules are imprinted on gut T cells by PP dendritic cells (Mora et al., 2003). Indeed, we found that SFB increased α4β7+ Tfh cells of gut origin in the spleen and PLNs (Figure 4C).
Figure 4. Detection of PP-Derived Tfh Cells in Systemic Lymphoid Tissue where Auto-Abs Are Predominantly Produced.

(A and B) Data are shown as an average percentage (A) of anti-GPI ASCs of IgG1 and IgA isotypes among total B cells + SEM. The absolute number (B) of total anti-GPI ASCs of the IgG1 isotype per tissue (e.g., the PLN group included two popliteal LNs and the PP group included all PPs from one intestine) is also shown (n = 8–15/group, data combined from 5 assays).
(C) Values in the representative plots indicate the percentage of α4β7-expressing cells in Tfh cells. Quantitative data of the number of integrin α4β7+ Tfh cells are shown (n = 17–22/group, data combined from 8 assays).
(D) The number of PPs per intestine from SFB− or SFB+ KBxN mice is shown (n = 19/group).
(E) The PP-derived CD4+ T cells in spleen are shown as the KikR+ population, which was further gated (indicated by arrows) on Tfh cell markers. Representative plots and quantitative data are shown (n = 7–8/group, data combined from 4 assays).
See also Figure S4.
To ultimately track Tfh cell migration from PPs to systemic lymphoid tissues, we took advantage of the KikGR transgenic mouse line, which ubiquitously expresses the green-to-red photoconvertible fluorescent protein in its cells (Nowotschin and Hadjantonakis, 2009). We first generated KikGR. K/BxN mice and developed a surgical procedure to specifically photoconvert PP cells by treating PPs with violet laser light and subsequently monitored the migration of these photoconverted PP cells to the spleen. The total number of PPs per intestine is similar between SFB− and SFB+ K/BxN mice, with an average of eight to nine PPs per mouse (Figure 4D). To maintain uniformity between the SFB− and SFB+ groups and to perform a time-efficient surgery that ensured a healthy recovery, we consistently treated four PPs per mouse in each group with the violet light. We chose 30 s per PP as the time for violet light exposure because it was the shortest time required to induce >95% photoconverted PP CD4+ T cells (Figure S4F). We subsequently monitored the converted PP cell migration into the spleen 3 days after surgery in our autoimmune model. A significant number of photoconverted, migratory CD4+ T cells from PPs were detected in the spleens of the KikGR. K/BxN mice after PP violet light exposure, regardless of their SFB status (Figure 4E, quantitative data in Figure S4G). However, there was a cell-type-specific contribution to the photoconverted CD4+ T cell pool in the SFB+ spleen: a significant portion of the PP-derived, photoconverted CD4+ T cells in the spleen consisted of Tfh cells, compared to the SFB− group (Figure 4E). Thus, the data from α4β7 expression and photoconversion model independently demonstrated the potential role of SFB-driven and PP-derived Tfh cells in modulating systemic autoimmunity by entering systemic lymphoid tissues to help auto-Ab production.
PP Tfh Cells Are Essential for SFB-Induced Systemic Tfh Cell Response and Autoimmune Arthritis
To ultimately confirm the importance of PPs for an SFB-mediated increase in the systemic Tfh cell population and subsequent worsening of arthritis, PP-null K/BxN mice were generated by administering anti-IL-7 receptor α (IL-7Rα) Abs into pregnant K/BxN breeders on gestation day (gd) 14.5 (Figure 5A; Murai et al., 2003; Yoshida et al., 1999). IL-7 signaling is crucial for the survival of lymphoid tissue inducer cells, and an injection of anti-IL-7Rα blocking Ab is sufficient to prevent PP but not LN development. The absence of PPs was confirmed visually in PP-null K/BxN mice. As reported, this method of depleting PPs did not change the general B and T cell populations in other lymphoid tissues including spleen, thymus, and mesenteric lymph nodes (Figures S5A–S5F; Murai et al., 2003; Yoshida et al., 1999). PP depletion also did not impact the general B and T cell populations in the small intestinal lamina propria (SI-LP) (Figure S5G). Unlike in control mice, SFB induced very limited arthritis development in PP-null K/BxN mice (Figure 5B). The reduced disease severity in the SFB-gavaged PP-null mice was not due to the absence or reduction of SFB colonization after PP removal (Figure 5C). However, we found a significant reduction in SFB-induced systemic Tfh cells in the spleen and PLNs upon PP depletion (Figure 5D). Furthermore, auto-Ab titers in SFB+ PP-null mice were also severely reduced, similar to the titers observed in SFB− control K/BxN mice with intact PPs (Figure 5E).
Figure 5. PP Tfh Cells Are Essential for SFB-Induced Systemic Tfh Cell Response and Arthritis.
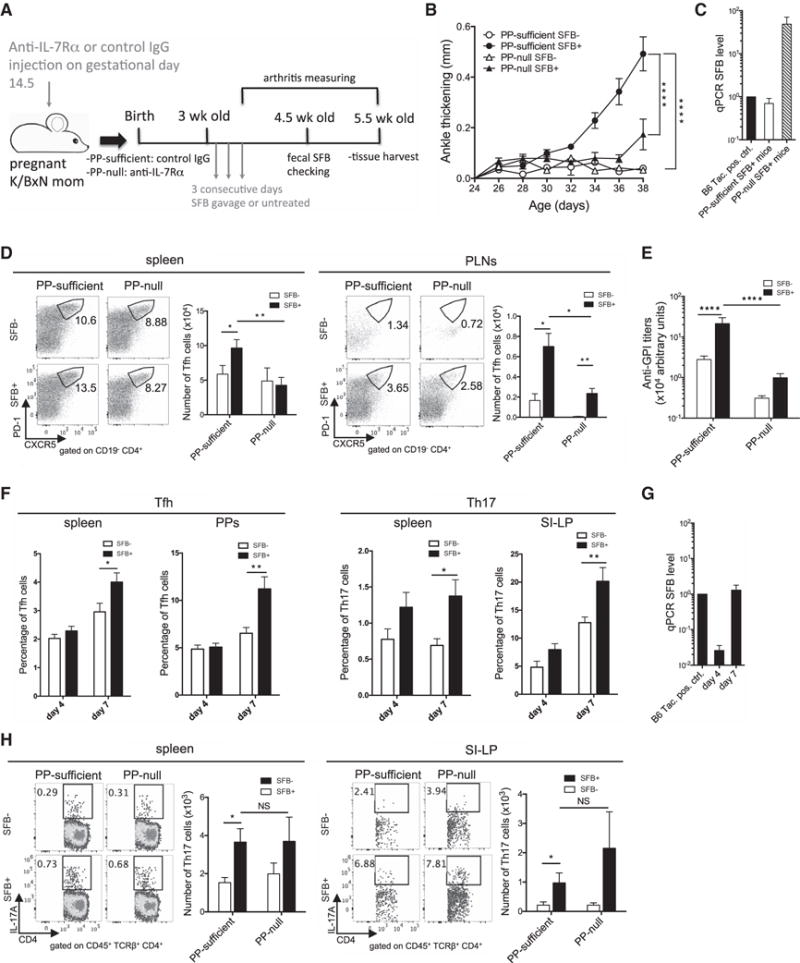
(A) Schematic diagram depicts PP depletion experiment.
(B) PP-null and PP-sufficient K/BxN mice were gavaged with SFB or left untreated. Ankle thickening is shown (6–10 mice/group, data combined from 3 assays).
(C) At 4.5 weeks old, the fecal SFB level of SFB-gavaged PP-null and PP-sufficient K/BxN mice was determined by qPCR. The fecal SFB level of a Taconic B6 mouse (positive control) was used for sample normalization and its SFB level was set as 1 (n = 16–22/group, data combined from 5 assays).
(D) Values in the representative plots indicate the percentage of Tfh cells. Quantitative data of Tfh cell numbers are also shown (n = 8–13/group, data combined from 3 assays).
(E) Anti-GPI Ab titers are shown.
(F) Tfh and Th17 cell analysis on day 4 or day 7 after the first of three consecutive gavages are shown (n = 10/group, data combined from 3 assays at each time point).
(G) Fecal samples from experiments in (F) were collected and their SFB levels are shown.
(H) Values in the representative plots indicate the percentage of Th17 cells. Quantitative data of Th17 cell numbers are also shown (n = 8–12/group, data combined from 3 assays).
See also Figure S5.
Previously, we reported that SFB-induced Th17 cells play a critical role in the development of auto-Abs and arthritis in K/BxN mice (Wu et al., 2010). To delineate the relative contribution of Th17 cells and Tfh cells in SFB-augmented autoimmune arthritis, we performed kinetic experiments measuring systemic and intestinal Tfh and Th17 cell responses (Figure 5F). Earlier we examined the Tfh cell response in age-matched SFB− and SFB+ mice between the ages of 5 and 6.5 weeks old (equivalent to day 14–24 after SFB gavage), by which point mice have already developed arthritis and exhibited strong SFB-induced Tfh cell responses (Figures 1A and 2A). Thus, to catch the onset of Tfh and Th17 cell induction, we picked two earlier time points: day 4 (27 days old) and day 7 (30 days old) after the first of 3 days of consecutive gavage (Figure S5H). We found that the day 7 time point was the earliest that we could detect a significant SFB-mediated induction in both systemic and intestinal Tfh and Th17 cells. The earliest significant SFB-mediated arthritis augmentation occurred at day 10 after SFB gavage (33 days old) (Figures 1A and S5I). Thus, both Tfh and Th17 cell induction in SFB+ mice preceded the onset of SFB-mediated arthritis augmentation. The trend of Tfh and Th17 cell induction corresponded to increasing SFB colonization after gavage (Figure 5G). RORgt (encoded by Rorc) is the master transcriptional regulator for Th17 cells (Korn et al., 2009). We transferred KRN or Rorc−/−. KRN T cells into Tcra−/−. BxN mice to demonstrate that Th17 cells are also critical for the development of arthritis and auto-Abs (Figures S5J and S5K). Together, our data showed that both Tfh and Th17 cells contributed significantly to auto-Ab production, and a lack of either factor strongly ameliorated auto-Ab production and disease development (Figures 2C, 2D, S5J, and S5K). Next, we set out to examine Th17 cells in PP-null mice to determine whether there was a defective Th17 cell response in PP-null mice that could also reduce disease phenotype in SFB+ PP-null mice. The induction of both systemic and SI-LP Th17 cell responses by SFB was intact in SFB+ PP-null mice (Figure 5H). Thus, PP depletion reduced the SFB-mediated Tfh but not Th17 cell response, which resulted in diminished auto-Ab titers and disease development. Together with the data in Figure 4, our results thus far suggested that SFB-induced PP Tfh cells contributed to arthritis development by migrating to systemic sites and augmenting the systemic Tfh cell and auto-Ab response there.
SFB Induce PP Tfh Cell Differentiation by Inhibiting the IL-2 Signaling Pathway in PP CD4+ T Cells
We next investigated the molecular mechanism for SFB-induced Tfh cell differentiation in PPs. IL-6 and IL-21 have been reported to regulate Tfh cell differentiation (Crotty, 2014). We did not find a difference in the percentage or MFI of IL-6- and IL-21-expressing cells in PPs between the SFB− and SFB+ groups (Figures S6A–S6C). There were also no significant differences in IL-6 or IL-21 mRNA expression (Figures S6D and S6E). We then focused on IL-2, because its signaling pathway potently inhibits Tfh cell differentiation by decreasing Bcl-6 expression (Ballesteros-Tato et al., 2012; Johnston et al., 2012). We found that the percentage of IL-2+CD4+ T cells was significantly reduced in the PPs but not spleen of SFB+ versus SFB− K/BxN mice (Figure 6A). Next, given that IL-2Rα (CD25) is a crucial component of the IL-2 high-affinity receptor, and that a 2-fold reduction in IL-2Rα expression (CD25+/−) has been shown to program CD4+ T cells to differentiate into Tfh cells (Johnston et al., 2012), we set out to determine whether SFB colonization affected the expression of CD25 on CD4+ T cells. Foxp3+ Treg cells constitutively express CD25. Thus, CD4+ T cells were gated to exclude Foxp3+ cells to avoid Treg cells skewing the percentage of CD25-expressing cells in the non-Foxp3 CD4+ T cell population. We found that SFB significantly reduced the percentage of IL-2Rα+ non-Foxp3 CD4+ (simplified as IL-2Rα+CD4+ hereafter) T cells in PPs but not spleen (Figure 6B). Additionally, SFB+ mice exhibited a stronger time-dependent reduction of IL-2Rα+CD4+ T cells in PPs compared to SFB− mice after SFB gavage (Figure S6F). Based on these findings, we hypothesized that SFB promoted Tfh cell differentiation in PPs by blocking the inhibition effect of IL-2 during Tfh cell differentiation. To prove the causative effect of our hypothesis, we treated SFB− and SFB+ mice with anti-IL-2 neutralizing Ab or control IgG (Figure S6G). Consistent with other findings suggesting that IL-2 functions as a potent inhibitor of Tfh cell differentiation, systemic administration of anti-IL-2 treatment alone boosted the Tfh cell population in SFB− K/BxN mice in both spleen and PPs (Figure 6C). However, when SFB were added to anti-IL-2-treated mice, there was no further enhancement (NS in Figure 6C). This suggested that the mechanism of SFB-triggered Tfh cell differentiation overlapped with anti-IL-2 treatment, which depended on suppression of the IL-2 signaling pathway. The effect of anti-IL-2 on Tfh cells was also reflected in humoral and disease responses in that anti-IL-2 treatment increased splenic GC B cell response, serum anti-GPI titer, and arthritis development, and SFB did not further boost these responses in the presence of anti-IL-2 (Figures 6D–6F). Together, these data supported the hypothesis that SFB promoted PP Tfh cell differentiation by inhibiting IL-2 signaling, ultimately leading to a boost in systemic Tfh cell response and arthritis development.
Figure 6. SFB Induce PP Tfh Cells by Inhibiting the IL-2 Signaling Pathway in PP CD4+ T Cells.

(A) Representative plots and quantitative data of the percentage of IL-2+ cells in total CD4+ T cells are shown (n = 15–16/group, data combined from 5 assays).
(B) Representative plots and quantitative data of the percentage of CD25+ (IL-2Rα+) cells in Foxp3−CD4+ T cells are shown (n = 10/group, data combined from 3 assays).
(C) Representative plots and quantitative data of the percentage of Tfh cells in total CD4+ T cells are shown (n = 8–10/group, data combined from 5 assays).
(D) Splenocytes from experiments in (C) were stained and analyzed for GC B cells. Data of the percentage of GC B cells are shown.
(E and F) Anti-GPI titers (E) and ankle thickness (F) of the mice from experiments in (C) are shown.
See also Figure S6.
DCs Are Required for SFB-Mediated IL-2Rα Suppression and Bcl-6 Upregulation in PP CD4+ T Cells
Recent studies suggest that both DCs and B cells are involved in promoting Tfh cell differentiation (Crotty, 2011; Ma et al., 2012). We asked whether SFB-mediated Tfh cell differentiation required antigen-presenting cells (APCs) including B cells and/or DCs by using APC-restricted Ag presentation in the K/BxN transfer model. This model, depicted in Figure 7A, was created by transferring KRN T cells and APCs from B6g7 mice (B6 mice congenically expressing NOD MHC class II I-Ag7) into Tcra−/−. B6 mice (T-cell-deficient mice on the B6 background). Unlike the donor APCs, the APCs of recipient Tcra−/−. B6 mice do not have the “correct” MHC class II I-Ag7 molecule to present self-Ag GPI to KRN T cells. We focused our analysis on PPs because these are the sites where SFB-boosted Bcl-6 expression was observed. SFB colonization increased the percentage of PP Tfh cells and Bcl-6 expression in PP CD4+ T cells only when the recipient mice were transferred with both B cells and DCs (Figures 7B and 7C). Although DCs were required to synergize with B cells to induce Tfh cell differentiation in PPs, DCs alone substantially but not significantly increased Tfh cell differentiation in SFB+ K/BxN mice (Figure S7A). No matter whether B cells alone (Figure 7D) or B cells plus DCs (Figure 7E) were used as transferred APCs, no SFB-mediated IL-2Rα+CD4+ T cell suppression was observed in the spleen. In PPs, there was no difference in IL-2Rα+CD4+ T cells between the SFB− and SFB+ groups when B cells were used as APCs (Figure 7D). When DCs were added as additional APCs to the SFB− recipients, there was an increase of PP IL-2Rα+CD4+ population, which was reduced by SFB colonization (Figures 7E). Indeed, DCs themselves were sufficient to suppress IL-2Rα+CD4+ T cell population in SFB+ PPs (Figure S7B), which suggested a crucial role for DCs in mediating SFB-dependent IL-2Rα suppression in PP CD4+ T cells.
Figure 7. DCs Are Required for SFB-Mediated Bcl-6 Upregulation and IL-2Rα Suppression in PP CD4+ T Cells.
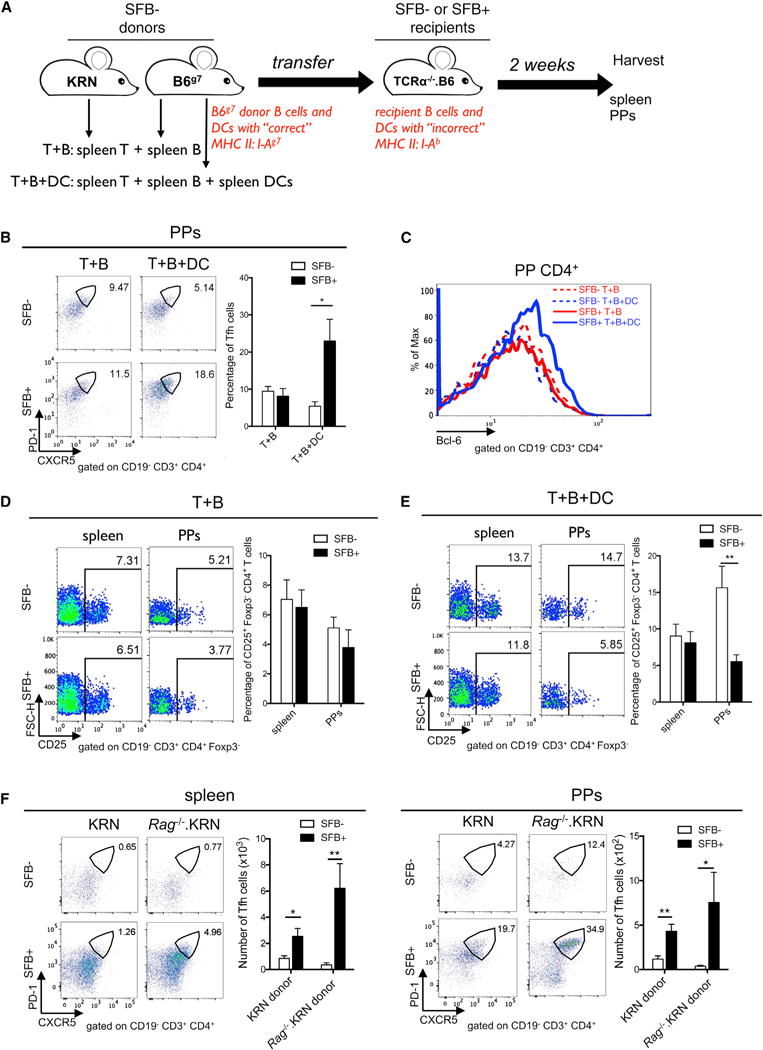
(A) Schematic diagram depicts APC-restricted Ag presentation in the K/BxN transfer model. SFB− donor KRN T cells or APCs were transferred into SFB− or SFB+ Tcra−/−. B6 recipients.
(B) KRN T cells and B6g7 B cells with (T+B+DC) or without (T+B) B6g7 CD11c+ cells were transferred into Tcra−/−. B6 recipients. Representative plots and quantitative data of Tfh cell percentage are shown (n = 9–11/group, data combined from 6 experiments).
(C) Histogram overlay of Bcl-6 expression in PP CD4+ T cells. Data are representative of 6 experiments.
(D and E) Splenocytes and PP cells from the T+B group (D) or T+B+DC group (E) were stained and analyzed for CD25 (IL-2Rα). The percentage of CD25+ cells in Foxp3−CD4+ T cells are shown (n = 9–11/group, data combined from 6 assays).
(F) KRN or Rag−/−. KRN CD4+ T cells were transferred into SFB− or SFB+ Tcra−/−. BxN recipients. Values in the representative plots indicate the percentage of Tfh cells. Quantitative data of Tfh cell numbers are also shown (n = 8–9/group, data combined from 5 assays).
See also Figure S7.
Cognate TCR recognition of SFB Ag presented by DCs is crucial for SFB-mediated Th17 cell induction (Goto et al., 2014; Yang et al., 2014). We therefore tested whether SFB-mediated Tfh cell induction also required SFB-specific TCR interaction. It is a well-recognized phenomenon that incomplete allelic exclusion of TCR can lead to dual TCR expression on many strains of TCR transgenic mice. To exclude the possibility of KRN T cells expressing another SFB-specific endogenous TCR in addition to their original KRN TCR, we crossed KRN TCR transgenic mice onto the Rag−/− background. KRN or Rag−/−. KRN CD4+ T cells were transferred into SFB− or SFB+ Tcra−/−. BxN mice. Mice receiving Rag−/−. KRN CD4+ T cells expressed only GPI-specific TCR, as identified by their transgenic TCR Vb6-only expression (Figure S7C). SFB still strongly induced both splenic and PP Tfh cells in mice receiving Rag−/−. KRN CD4+ T cells compared to mice receiving KRN T cells, indicating that TCR recognition of SFB Ag was not required for Tfh cell induction by SFB (Figure 7F). SFB still significantly reduced the percentage of IL-2Rα+CD4+ T cells isolated from mice transferred with Rag−/−. KRN CD4+ T cells (Figure S7D), suggesting that SFB-mediated reduction of IL-2Rα+CD4+ T cells also did not require cognate TCR recognition of SFB Ag. Finally, SFB also enhanced both gut and systemic Tfh cell responses in another systemic autoimmune model, the NZB/NZW F1 nontransgenic mouse model of systemic lupus erythematosus (SLE) (Figure S7E). An increased splenic Tfh cell population also reflected a higher splenic GC response in the SFB+ versus SFB− NZB/NZW F1 mice (Figure S7F). All together, these data suggested that SFB drove Tfh cell response in a TCR-specificity-independent manner, which increases the relevance of our findings because they suggest a broad application of gut microbiota such as SFB in promoting systemic Tfh cell and humoral responses.
DISCUSSION
Despite the sharp rise in dysbiosis-related systemic diseases (Chervonsky, 2013; Wu and Wu, 2012), one of the long-standing questions in the field of host-microbiota interactions is how gut microbiota exert their effects on gut-distal sites. Our study provided evidence that gut commensals, in particular SFB, triggered autoimmune arthritis by driving differentiation and egress of PP Tfh cells into systemic sites. Our data supported a model where exposing DCs to SFB in PPs conditioned PP CD4+ T cells to differentiate into Tfh cells by limiting the access of IL-2 to CD4+ T cells, thereby enhancing their Bcl-6 expression. Subsequently, the PP Tfh cells migrated into systemic sites to help boost auto-Ab production by systemic B cells, ultimately leading to worsening arthritis development. Although numerous commensal species have been identified as inducing other T effector cell types (Hooper et al., 2012; Wu and Wu, 2012), there remains a compelling need to discover specific commensals that impact host health beyond the gut by eliciting robust systemic Tfh cell responses. Our study provided data showing that commensal SFB induced strong gut as well as systemic Tfh cell responses, leading to the exacerbation of auto-Ab production in K/BxN mice.
A central aspect of our finding is that PP Tfh cells could act as the “remote control signal” sent out by the gut microbiota to regulate the immune system at distal sites. Our results showing that PP Tfh cells egressed to systemic sites and drove gut-distal autoimmune responses are supported by a recent study that revises the long-held misconception that Tfh cells are confined to the GC they derive from (Shulman et al., 2013). Shulman et al. (2013) reveal that Tfh cells can readily exit the GC. Consistent with this finding, our results showed that gut Tfh cells with the α4β7 integrin gut-imprinting marker could leave the GC of PPs and be found at systemic sites. Finally, the detection of the newly arrived and PP-derived, photoconverted Tfh cells in the spleen provided the most direct evidence supporting the migration of PP Tfh cells into the spleen. SFB colonization preferentially increased the number of PP Tfh cells arriving in the spleen. In that regard, the presence of circulating CXCR5+ Tfh cells in the blood have been reported in various autoimmune conditions, including RA, SLE, type 1 diabetes, etc. (Ueno et al., 2015). We ultimately identified PPs as the culprit mediating SFB-augmented systemic autoimmune responses by showing a specific reduction in systemic Tfh cell response and arthritis severity in SFB+ PP-null mice. We further tied the importance of PP Tfh cell differentiation to systemic auto-Ab production by dissecting the cellular and molecular mechanisms for SFB-induced PP Tfh cell differentiation. We showed that SFB reduced the IL-2Rα+CD4+ T cell population and boosted Tfh cell differentiation only in PPs but not spleen. Using a DC transfer model, we found that SFB-mediated, DC-dependent IL-2Rα+CD4+ suppression occurred only in PPs. The anti-IL-2 experiments tied our results together and suggested that SFB promoted PP Tfh cell differentiation by inhibiting IL-2 signaling, which ultimately led to a boost in systemic Tfh cell response and arthritis development. To conclude, these data supported our proposed model that SFB unleash the Tfh cell response starting at the PPs by suppressing the IL-2 signaling pathway. This leads to an increase in the PP Tfh cell population that later migrates to systemic lymphoid sites where PP-derived Tfh cells increase the systemic Tfh cell pool and help boost auto-Ab production. The effect of microbiota on the systemic immune response can be mediated by the circulation of microbiota-derived factors, such as peptidoglycan, from the gut into the systemic sites (Clarke et al., 2010). However, our results suggested an alternative mechanism where gut microbiota and/or their products could affect the immune system at distal sites without leaving the gut. Nevertheless, we cannot completely exclude the possibility that SFB-activated PP DCs provide additional help and contribute to the SFB-induced systemic Tfh cell response by migrating to systemic sites and directly activating Tfh cells there. However, this scenario is less likely, because we did not observe SFB-mediated changes in the proliferation, differentiation, and/or apoptosis of systemic Tfh cells.
Previous studies using viral infection models and/or administration of recombinant IL-2 (rIL-2) have identified IL-2 and IL-2-mediated STAT5 signaling as potent negative regulators of Tfh cell differentiation by upregulating Blimp-1 that represses Bcl-6 expression (Ballesteros-Tato et al., 2012; Johnston et al., 2012). However, some fundamental questions remain: Where does IL-2 signaling preferentially occur in vivo, and how do its levels affect Tfh cell response? During a subtle condition such as commensal colonization, can IL-2 contribute to Tfh cell differentiation? Here, we demonstrated that SFB were able to unleash the Tfh cell response by suppressing the IL-2 signaling pathway in PPs. The SFB-mediated increase of Bcl-6 in PP CD4+ T cells was due to an increase of Bcl-6 in the PP non-Tfh cell population, which corresponded with their decreased Blimp-1 expression. These data are in line with previous studies showing that low IL-2 signaling leads to the decrease of Blimp-1, which upregulates Bcl-6 (Ballesteros-Tato et al., 2012; Johnston et al., 2012). We further demonstrated that DC-dependent limitation of IL-2 signaling on PP CD4+ T cells corresponded with Bcl-6 upregulation and Tfh cell induction in PPs. Together, these findings suggested that commensal colonization could modulate IL-2 signaling enough to impact the Tfh cell response in PPs. Because IL-2 plays a pivotal role in the development and function of Treg cells (Liao et al., 2013), we also examined whether the Tfh cell enhancement we observed in anti-IL-2 treatment was due to an anti-IL-2 effect on Treg cells. We found that the systemic administration of anti-IL-2 Abs only slightly reduced the percentage of splenic Treg cells in K/BxN mice, regardless of SFB condition (data not shown). In contrast to the typical Treg cell-suppressing effect on T effector cells, recent studies demonstrated that Treg cells do not suppress, but rather promote, Tfh cell responses (Ballesteros-Tato et al., 2012; León et al., 2014). These studies suggest that our result of anti-IL-2-induced Tfh cells is not likely due to an indirect effect of Treg cells, because we did not see an increase in Treg cells after anti-IL-2 treatment.
One key question is whether intrinsic differences between spleen- and PP-derived CD4+ T cells allow PP CD4+ T cells to downregulate the IL-2 signaling pathway more readily in the presence of SFB. By using the K/BxN transfer model, we demonstrated that SFB could still suppress IL-2Rα and upregulate Bcl-6 expression in splenic CD4+ T cells that repopulated PPs after co-transfer with splenic B cells and DCs. These data suggested that it was the unique environment of PPs, rather than an intrinsic property of the PP-derived CD4+ T cells or APCs themselves, that dictated a preferential generation of Tfh cells in PPs rather than in the spleen. On a related note, two recent studies have demonstrated that Treg and Th17 cells preferentially differentiate into Tfh cells in PPs but not in the spleen (Hirota et al., 2013; Tsuji et al., 2009). It will be of particular interest to investigate whether diminished IL-2 signaling is also the mechanism of re-differentiation of Treg and Th17 cells into Tfh cells in PPs, but not in the spleen or LNs.
We would like to emphasize that, despite the fact that SFB have strong immunomodulation effects, their impact on T effector cells is rather specific. SFB do not activate the general CD4+ T cell population. Moreover, SFB induce Th17 and Tfh, but not Th1 and Th2, cells. Thus, our data clearly demonstrated that commensal SFB induce a unique immune profile with multiple players, Tfh and Th17 cells, that simultaneously affect disease development. Induction of Th17 cells is one of the best-known immunostimulatory effects of SFB. In that regard, IL-2 signaling has been reported to inhibit Th17 cell differentiation. It will be interesting to examine whether SFB-mediated inhibition of IL-2 signaling also contributes to the induction of Th17 cells (Ivanov et al., 2009). However, we would like to emphasize that there are some major differences between the induction mechanism of Th17 and Tfh cells by SFB. First, we and others have shown that the induction of Th17 cells by SFB is a completely PP- or GALT-independent process (Goto et al., 2014; Lécuyer et al., 2014). However, PPs, a component of GALT, are indispensable for SFB-mediated Tfh cell induction. This is most likely due to the fact that Tfh cell development requires a well-structured GC microenvironment, and PPs can provide that exact requirement. Second, induction of Th17 cell differentiation by SFB requires SFB-specific TCR recognition (Goto et al., 2014; Yang et al., 2014). In contrast, our results suggest that, with unlimited access to self-Ag ubiquitously expressed in tissues, including intestine, T cells can differentiate into Tfh cells upon receiving an SFB-mediated, DC-derived bystander signal that restrains IL-2 signaling, regardless of TCR Ag specificity. This was demonstrated by using the K/BxN transfer model with Rag−/−. KRN CD4+ T cells as donor cells, which are devoid of endogenous TCR that can potentially recognize SFB. It is very unlikely that SFB induce differentiation of Rag−/−. KRN T cells into Tfh cells by molecularly mimicking the GPI antigen, because our data showed that SFB also induced Tfh cell differentiation in NZB/NZW F1 mice, a non-transgenic murine SLE model. SFB are one of the most potent commensal types known to induce GC formation and IgA production (Lécuyer et al., 2014). Our finding that SFB mediated bystander activation of GPI-specific Tfh cells is supported by a recent report showing that SFB induced vigorous GC formation but a very low percentage of SFB-specific IgA in PPs compared to a commensal strain of E. coli (Lécuyer et al., 2014).
Most gut commensals reside in the gut lumen, spatially separated from the host mucosal immune system. In contrast, mucosa-associated commensal species, though representing a minority within the commensal community, can powerfully modulate host immunity. A consortium of gut bacteria highly coated with IgA induces strong intestinal inflammation in both humans and mice (Palm et al., 2014). SFB, which are highly IgA-coated and mucosa-associated gut microbiota, have been shown to impact many disease models. Notably, in contrast to SFB’s detrimental role in the K/BxN model and EAE model (Lee et al., 2011; Wu et al., 2010), SFB can actually protect NOD mice from developing type 1 diabetes (Kriegel et al., 2011). These data provide valuable insights indicating that the same commensal can cause different disease outcomes, probably depending on whether the commensal-mediated immunomodulation is enhancing or inhibiting the pathogenesis of each disease. For example, auto-Abs are a key pathogenic factor and diseases can be induced by passive transfer of auto-Abs in the K/BxN and EAE models (Monach et al., 2008; Urich et al., 2006). In contrast, type 1 diabetes in NOD mice is a T-cell-mediated autoimmune disease, and although B cells of NOD mice produce autoantibodies, these are not thought to play a diabetogenic role (Serreze et al., 1998). Thus, SFB, with their Tfh cell- and auto-Ab-boosting effect, are more likely to play pathogenic roles in K/BxN than NOD mice.
Without infection, PP Tfh cells are best known for their role in maintaining gut homeostasis by inducing the generation of PP GCs and mucosal IgA. However, we elucidated an unexpected role of PP Tfh cells wherein they functioned as a messenger of SFB and transposed the initial autoimmune signal from the gut into systemic sites. This report provided a mechanistic confirmation of the long-standing hypothesis that several systemic autoimmune diseases in human patients could be initiated at mucosal sites (Deane and El-Gabalawy, 2014). Overall, we demonstrated that gut microbiota trigger the host autoimmune response by tipping the balance of Tfh cells. Serious attention needs to be paid to this field, because dysbiosis-related immune disorders have been rising at an alarming rate in the industrialized world (Chervonsky, 2013; Wu and Wu, 2012).
EXPERIMENTAL PROCEDURES
Mice
K/BxN mice were generated by crossing KRN TCR transgenic mice on the B6 background with NOD mice. KRN, Tcra−/−. B6, Tcra−/−. NOD, Rag−/−, and B6g7 mice were originally obtained from the mouse colony of Drs. Diane Mathis and Christophe Benoist at the Jackson Laboratory. Other strains are listed in Supplemental Experimental Procedures. All experiments were conducted according to the guidelines of the University of Arizona Institutional Animal Care and Use Committee.
Antibodies and Flow Cytometry
Cells were run on an LSRII (BD Biosciences) and analyzed by FlowJo. Abs used are listed in the Supplemental Experimental Procedures.
Microbiota Reconstitution and Quantification
SFB were initially introduced to our mouse colony by gavaging mice with SFB-containing feces from Taconic B6 mice. Later, SFB were passed by gavaging mice with SFB-containing feces collected from the SFB+ mice housed in our colony. Where noted, fecal pellets collected from GF mice monocolonized with SFB were used as gavaging materials. The details of SFB gavaging and quantification are described in the Supplemental Experimental Procedures.
In Vivo Blocking Abs Assay
K/BxN mice were intraperitoneally (i.p.) injected with anti-IL-2 (JES6-1A12, BioXCell, 0.5 mg per mouse), or control Rat IgG (Jackson ImmunoResearch) 1 day after weaning. The mice treated with blocking Abs or control IgG were treated 3 more times, every 3 days.
Generation of PP-Null K/BxN Mice
On gd 14.5, pregnant K/BxN breeders were i.p. injected with 0.6 mg anti-IL-7Rα (A7R34, BioXCell) or control rat IgG (Jackson ImmunoResearch) to produce the pups that were PP null (from anti-IL-7Rα-treated mothers) or PP sufficient (from IgG-treated mothers).
Adoptive Transfer
Splenic CD4+ T cells (5 × 105) were enriched by CD4-conjugated MACS beads (Miltenyi) from SFB− KRN, Rorc−/−. KRN, or Cxcr5−/−. KRN mice, and adoptively transferred by intravenous (i.v.) injection into SFB− or SFB+ Tcra−/−. BxN recipients. SFB status was manipulated by gavaging recipient mice with SFB prior to cell transfer. For other adoptive transfer experiments, please see the Supplemental Experimental Procedures.
Photoconversion Procedures
Survival surgeries were performed under sterile conditions. Anesthetized mice were shaved at the abdominal area with electric clippers. The shaved area was wiped with alcohol prep pads. Mice were positioned on their back and a small incision (~1.5 cm) was made at the midline, below the costal margin. The PPs in small intestine were identified and then covered with sterile foil with a ~4 mm hole to leave only one PP exposed to a violet laser at a time (405 nm; peak power < 30 mW). A total of four PPs per intestine were exposed to violet laser. Each PP was exposed to violet light for 30 s and PBS was dripped on the intestine after each PP exposure during surgery to avoid drying. The abdominal muscles were closed with a 4-0 synthetic absorbable suture. The skin incision was closed with clips using a Reflex 9mm Wound Clip Applier (Robot Surgical Instrument Co.). After surgery, the mice were placed on a heating pad for recovery.
Statistical Analysis
Differences were considered significant when p < 0.05 by Student’s t test (two-tailed, unpaired) or two-way analysis of variance (ANOVA). To compare ankle thickening, the area under the curve (AUC) was calculated for each mouse within an experimental set followed by Student’s t test between groups (Prism 6, Graph-Pad Software). *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Supplementary Material
Highlights.
SFB enhance autoimmune arthritis, reflected by elevated auto-Ab, GC, and Tfh cell responses
SFB-driven differentiation and egress of PP Tfh cells to systemic sites cause disease
SFB induce PP Tfh cell differentiation by limiting the access of IL-2 to PP CD4+ T cells
DCs are required for SFB-mediated IL-2Rα suppression and Bcl-6 upregulation in PPs
Acknowledgments
We thank Dr. Haochu Huang for the initial supply of rGPI. We thank Drs. Dominik Schenten, Vesna Pulko, and Taras Kreslavsky for comments about the manuscript and Ms. Kelly O’Callaghan for editing. This work was supported by grants from the NIH (R56AI107117 and R01AI107117) and by the Southwest Clinic and Research Institute Fund to H.-J.J.W. This work was also supported by funds from the Microbial Pathogenesis Program at the University of Arizona to H.-J.J.W.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2016.03.013.
AUTHOR CONTRIBUTIONS
F.T. performed the experiments and analyzed the data. H.-J.J.W. designed the experiments, analyzed the data, and wrote the manuscript. C.N.K. and K.M.F. performed ELISA and ELISPOT analysis. C.P.B. contributed to some FACS analysis. E.W. set up the SFB colonization and quantification protocol. N.L.T. performed SFB quantification. Y.U. provided the SFB-monocolonized fecal pellets.
References
- Ballesteros-Tato A, León B, Graf BA, Moquin A, Adams PS, Lund FE, Randall TD. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. 2012;36:847–856. doi: 10.1016/j.immuni.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerf-Bensussan N, Gaboriau-Routhiau V. The immune system and the gut microbiota: friends or foes? Nat Rev Immunol. 2010;10:735–744. doi: 10.1038/nri2850. [DOI] [PubMed] [Google Scholar]
- Chervonsky AV. Microbiota and autoimmunity. Cold Spring Harb Perspect Biol. 2013;5:a007294. doi: 10.1101/cshperspect.a007294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke TB, Davis KM, Lysenko ES, Zhou AY, Yu Y, Weiser JN. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat Med. 2010;16:228–231. doi: 10.1038/nm.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S. Follicular helper CD4 T cells (TFH) Annu Rev Immunol. 2011;29:621–663. doi: 10.1146/annurev-immunol-031210-101400. [DOI] [PubMed] [Google Scholar]
- Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane KD, El-Gabalawy H. Pathogenesis and prevention of rheumatic disease: focus on preclinical RA and SLE. Nat Rev Rheumatol. 2014;10:212–228. doi: 10.1038/nrrheum.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, Laufer TM, Ignatowicz L, Ivanov II. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 2014;40:594–607. doi: 10.1016/j.immuni.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Artis D. Intestinal bacteria and the regulation of immune cell homeostasis. Annu Rev Immunol. 2010;28:623–667. doi: 10.1146/annurev-immunol-030409-101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Turner JE, Villa M, Duarte JH, Demengeot J, Steinmetz OM, Stockinger B. Plasticity of Th17 cells in Peyer’s patches is responsible for the induction of T cell-dependent IgA responses. Nat Immunol. 2013;14:372–379. doi: 10.1038/ni.2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Littman DR, Macpherson AJ. Interactions between the microbiota and the immune system. Science. 2012;336:1268–1273. doi: 10.1126/science.1223490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Benoist C, Mathis D. Rituximab specifically depletes short-lived autoreactive plasma cells in a mouse model of inflammatory arthritis. Proc Natl Acad Sci USA. 2010;107:4658–4663. doi: 10.1073/pnas.1001074107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J, Crotty S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1010. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. 2012;209:243–250. doi: 10.1084/jem.20111174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto S, Tran TH, Maruya M, Suzuki K, Doi Y, Tsutsui Y, Kato LM, Fagarasan S. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science. 2012;336:485–489. doi: 10.1126/science.1217718. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci USA. 2011;108:11548–11553. doi: 10.1073/pnas.1108924108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lécuyer E, Rakotobe S, Lengliné-Garnier H, Lebreton C, Picard M, Juste C, Fritzen R, Eberl G, McCoy KD, Macpherson AJ, et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity. 2014;40:608–620. doi: 10.1016/j.immuni.2014.03.009. [DOI] [PubMed] [Google Scholar]
- Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- León B, Bradley JE, Lund FE, Randall TD, Ballesteros-Tato A. FoxP3+ regulatory T cells promote influenza-specific Tfh responses by controlling IL-2 availability. Nat Commun. 2014;5:3495. doi: 10.1038/ncomms4495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity. 2013;38:13–25. doi: 10.1016/j.immuni.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med. 2012;209:1241–1253. doi: 10.1084/jem.20120994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monach PA, Mathis D, Benoist C. The K/BxN arthritis model. Curr Protoc Immunol 15 Unit. 2008;15:22. doi: 10.1002/0471142735.im1522s81. [DOI] [PubMed] [Google Scholar]
- Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH. Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature. 2003;424:88–93. doi: 10.1038/nature01726. [DOI] [PubMed] [Google Scholar]
- Murai M, Yoneyama H, Ezaki T, Suematsu M, Terashima Y, Harada A, Hamada H, Asakura H, Ishikawa H, Matsushima K. Peyer’s patch is the essential site in initiating murine acute and lethal graft-versus-host reaction. Nat Immunol. 2003;4:154–160. doi: 10.1038/ni879. [DOI] [PubMed] [Google Scholar]
- Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006;6:148–158. doi: 10.1038/nri1777. [DOI] [PubMed] [Google Scholar]
- Nowotschin S, Hadjantonakis AK. Use of KikGR a photoconvertible green-to-red fluorescent protein for cell labeling and lineage analysis in ES cells and mouse embryos. BMC Dev Biol. 2009;9:49. doi: 10.1186/1471-213X-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158:1000–1010. doi: 10.1016/j.cell.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proietti M, Cornacchione V, Rezzonico Jost T, Romagnani A, Faliti CE, Perruzza L, Rigoni R, Radaelli E, Caprioli F, Preziuso S, et al. ATP-gated ionotropic P2×7 receptor controls follicular T helper cell numbers in Peyer’s patches to promote host-microbiota mutualism. Immunity. 2014;41:789–801. doi: 10.1016/j.immuni.2014.10.010. [DOI] [PubMed] [Google Scholar]
- Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife. 2013;2:e01202. doi: 10.7554/eLife.01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH, Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol. 1998;161:3912–3918. [PubMed] [Google Scholar]
- Shulman Z, Gitlin AD, Targ S, Jankovic M, Pasqual G, Nussenzweig MC, Victora GD. T follicular helper cell dynamics in germinal centers. Science. 2013;341:673–677. doi: 10.1126/science.1241680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock GW, Miller JR, Savage DC. Host specificity of filamentous, segmented microorganisms adherent to the small bowel epithelium in mice and rats. Appl Environ Microbiol. 1984;47:441–442. doi: 10.1128/aem.47.2.441-442.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji M, Komatsu N, Kawamoto S, Suzuki K, Kanagawa O, Honjo T, Hori S, Fagarasan S. Preferential generation of follicular B helper T cells from Foxp3+ T cells in gut Peyer’s patches. Science. 2009;323:1488–1492. doi: 10.1126/science.1169152. [DOI] [PubMed] [Google Scholar]
- Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol. 2015;16:142–152. doi: 10.1038/ni.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urich E, Gutcher I, Prinz M, Becher B. Autoantibody-mediated demyelination depends on complement activation but not activatory Fc-receptors. Proc Natl Acad Sci USA. 2006;103:18697–18702. doi: 10.1073/pnas.0607283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HJ, Wu E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes. 2012;3:4–14. doi: 10.4161/gmic.19320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Torchinsky MB, Gobert M, Xiong H, Xu M, Linehan JL, Alonzo F, Ng C, Chen A, Lin X, et al. Focused specificity of intestinal TH17 cells towards commensal bacterial antigens. Nature. 2014;510:152–156. doi: 10.1038/nature13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Honda K, Shinkura R, Adachi S, Nishikawa S, Maki K, Ikuta K, Nishikawa SI. IL-7 receptor alpha+ CD3(−) cells in the embryonic intestine induces the organizing center of Peyer’s patches. Int Immunol. 1999;11:643–655. doi: 10.1093/intimm/11.5.643. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.