

ABSTRACT
Antimicrobial resistance presents one of the most significant threats to human health, with the emergence of totally drug-resistant organisms. We have combined bioengineering, genetically modified bacteria, longitudinal readouts, and fluidics to develop a transformative platform to address the drug development bottleneck, utilizing Mycobacterium tuberculosis as the model organism. We generated microspheres incorporating virulent reporter bacilli, primary human cells, and an extracellular matrix by using bioelectrospray methodology. Granulomas form within the three-dimensional matrix, and mycobacterial stress genes are upregulated. Pyrazinamide, a vital first-line antibiotic for treating human tuberculosis, kills M. tuberculosis in a three-dimensional culture but not in a standard two-dimensional culture or Middlebrook 7H9 broth, demonstrating that antibiotic sensitivity within microspheres reflects conditions in patients. We then performed pharmacokinetic modeling by combining the microsphere system with a microfluidic plate and demonstrated that we can model the effect of dynamic antibiotic concentrations on mycobacterial killing. The microsphere system is highly tractable, permitting variation of cell content, the extracellular matrix, sphere size, the infectious dose, and the surrounding medium with the potential to address a wide array of human infections and the threat of antimicrobial resistance.
IMPORTANCE
Antimicrobial resistance is a major global threat, and an emerging concept is that infection should be studied in the context of host immune cells. Tuberculosis is a chronic infection that kills over a million people every year and is becoming progressively more resistant to antibiotics. Recent major studies of shorter treatment or new vaccination approaches have not been successful, demonstrating that transformative technologies are required to control tuberculosis. We have developed an entirely new system to study the infection of host cells in a three-dimensional matrix by using bioengineering. We showed that antibiotics that work in patients are effective in this microsphere system but not in standard infection systems. We then combined microspheres with microfluidics to model drug concentration changes in patients and demonstrate the effect of increasing antibiotic concentrations on bacterial survival. This system can be widely applied to address the threat of antimicrobial resistance and develop new treatments.
INTRODUCTION
The progressive emergence of drug-resistant bacteria poses one of the most pressing threats to human health, with the development of totally resistant bacteria potentially leading to a return to the preantibiotic era (1–3). The pipeline of new antibiotics in development is inadequate to combat the rate of evolution of microbial resistance (4, 5). To develop antibiotics, bacteria have traditionally been studied in broth culture, where bacilli are rapidly dividing under optimal growth conditions. However, an emerging concept is that studying pathogens in the context of the host is vital to fully understanding pathogenesis (6, 7). Interaction with host cells modulates multiple facets of bacterial physiology and causes stress-induced changes in bacterial gene expression (8). In parallel, evidence is accumulating that host cell biology is modulated by three-dimensional (3D) extracellular matrix interactions, regulating key processes in the host-pathogen interaction such as cell survival, phagolysosomal fusion, autophagy, and cytokine secretion (9, 10). In patients being treated for infection, the host-pathogen interaction occurs in three dimensions and antibiotic concentrations vary over time according to drug pharmacokinetics (11). Conversely, the vast majority of in vitro studies are done in the absence of human cells, without an extracellular matrix, and at static antibiotic concentrations.
Considering these concepts together, we concluded that a transformative system to address the threat of antimicrobial resistance requires the following elements: primary host cells infected with fully virulent bacteria, culturing within a 3D structure that incorporates a physiological extracellular matrix, and pharmacokinetic modeling of drug concentrations. These criteria represent a significant challenge in the context of virulent organisms because of the high biosafety containment level required and the complexity of bacteria being eluted under flow conditions. We utilized Mycobacterium tuberculosis, a pathogen that is inherently resistant to antibiotics and causes tuberculosis (TB) (12), to develop a system that addresses these technical obstacles and have recently reported on an investigation of the host immune response in this system (13).
TB is the leading cause of death from an infectious disease worldwide (14), and over the last 2 decades, multidrug-resistant, extensively drug-resistant, and totally drug-resistant strains have sequentially emerged, posing the specter of a completely untreatable disease (15). Unfortunately, major recent trials of novel treatment-shortening regimens have not been successful (16), indicating that the model systems that were used to inform these approaches do not sufficiently reflect the disease in humans. Furthermore, pyrazinamide (PZA), one of the most critical antibiotics in human TB treatment, would not have been discovered by current screening approaches. Current models are principally reliant on microbiological broth or solid medium culture, 2D culture, zebrafish, and mice (17). Novel PZA-based regimens show promise (18), and so reliably understanding the action of PZA has become critical, principally focused on mutational analysis of PZA resistance (19). These approaches have limitations, especially in the context of PZA’s complex activation, intracellular activity, and uncertain mode of action. For some other drugs, such as cycloserine, nearly all drug susceptibility systems are unreliable. M. tuberculosis is an obligate pathogen of humans and has a prolonged interaction with host cells, centered on adaption to survival within an intracellular niche (20). In addition, the host-pathogen interaction is spatially organized (21) and the extracellular matrix influences host cell survival (22), suggesting that a fully humanized system structured in three dimensions with an extracellular matrix is needed to identify novel treatments for TB.
Therefore, we developed a platform utilizing M. tuberculosis as the prototype organism. Our system integrated genetically modified virulent reporter bacilli, primary human cells, and a human extracellular matrix by using a bioengineering approach and combined this with a multiparameter longitudinal readout. Within this microsphere system, we demonstrate cellular aggregation and upregulation of mycobacterial stress genes. Critically, PZA is efficacious in the 3D microsphere system but not in standard broth or 2D culture. We then combined microspheres with a microfluidic system to permit pharmacokinetic modeling. We observed more rapid M. tuberculosis killing with higher peak antibiotic concentrations, similar to outcomes in patients with TB (23). Therefore, this system models conditions in patients and can be readily applied to a range of drug-resistant organisms to address the global challenge of antimicrobial resistance.
RESULTS
Granulomas develop within microspheres, and M. tuberculosis stress genes are upregulated.
We incorporated primary human cells, virulent M. tuberculosis, and type I collagen into 3D microspheres by bioelectrospray methodology (see Movie S1 in the supplemental material). Fluorescent staining of monocytes and T cells, followed by infection with mCherry-expressing M. tuberculosis, showed the distribution of cells and bacteria through the microspheres and early granuloma formation from day 4 (Fig. 1A). After 14 days of infection, large cellular aggregates resembling human granulomas developed, while no aggregates formed in uninfected microspheres (Fig. 1B). Granuloma formation was associated with evidence of mycobacterial stress. Multiple stress-related genes were upregulated at day 14 in comparison with M. tuberculosis in 7H9 broth culture analyzed by reverse transcription-quantitative PCR (RT-qPCR) (Fig. 1C), including lipF, the acid stress real-time response gene; recA encoding recombinase A, the key mediator of the SOS response to DNA damage; relA, the nutrient stress-related gene; and sodA, the oxidative-stress response gene. By infecting cells with genetically modified luminescent M. tuberculosis expressing the Lux operon (24), bacterial growth could be monitored longitudinally over time within microspheres in a nondestructive manner (Fig. 1D).
FIG 1 .

Granulomas form within microspheres, and M. tuberculosis stress genes are upregulated. (A) Cellular distribution within microspheres. Primary human PBMCs were separated and fluorescently stained (monocytes blue [i], T cells green [ii]), recombined, and infected with mCherry-expressing M. tuberculosis (red [iii]). The overlay (iv) shows early granuloma development at day 4. (B) Large multicellular granulomas form at day 14 in infected microspheres (ii), which are not observed in uninfected microspheres (i), imaged by inverted microscopy. Scale bar, 50 μm. (C) M. tuberculosis stress genes are upregulated in the microsphere model compared to 7H9 broth culture. The expression of four stress-related mycobacterial genes was analyzed by RT-qPCR in microspheres at day 14 compared to exponentially growing M. tuberculosis (OD600 = 0.25) in 7H9 broth. The ΔΔCT method was used for relative quantification. Data are presented as fold changes normalized to the sigA gene. Data represent the mean results of three independent experiments ± the standard error of the mean. (D) M. tuberculosis growth in microspheres monitored by bacterial luminescence, demonstrating the typical M. tuberculosis luminescence kinetics of infected PBMCs within microspheres (black). Uninfected PBMCs in the microspheres do not luminesce (gray).
Generation of microspheres. Alginate was stained with bromophenol blue to permit visualization during the bioelectrospray process, which is shown in real time. Download MOVIE S1, AVI file, 2.5 MB (2.5MB, avi) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To determine the localization of mycobacteria, we studied microspheres longitudinally. We compared extracellular with cell-associated bacteria by decapsulating microspheres and performing differential centrifugation to separate extracellular mycobacteria from intracellular and cell-adherent mycobacteria. The proportion of cell-associated mycobacteria analyzed by luminescence or CFU counting progressively increased over time (Fig. 2A and B), leading to a 20.7-fold increase in luminescence of cell-associated M. tuberculosis relative to extracellular M. tuberculosis within microspheres at day 15. Gentamicin treatment in a single experiment demonstrated that cell-associated mycobacteria were almost all intracellular, with no significant reduction in CFU counts after the killing of extracellular bacteria. Similarly, the fluorescence of cells infected with green fluorescent protein (GFP)-expressing M. tuberculosis progressively increased over time (Fig. 2C; Fig. S1), demonstrating intracellular proliferation. M. tuberculosis infection did not increase cellular toxicity when measured by the CytoTox Glo 3D assay, with no significant difference in viability between the two conditions (Fig. 2D).
FIG 2 .

M. tuberculosis proliferation within microspheres is intracellular (A, B). PBMCs were infected with luminescent M. tuberculosis and incorporated into microspheres. Cells were released by decapsulation, and extracellular and cell-associated bacteria were separated by differential centrifugation. Open bars; extracellular mycobacteria; checkered bars; cell-associated mycobacteria. Mycobacterial location determined by luminescence and colony counting on 7H11 agar demonstrated that bacterial proliferation was principally cell associated (C). PBMCs were infected with GFP-expressing M. tuberculosis and incorporated into microspheres. Microspheres were decapsulated, and M. tuberculosis localization was analyzed by flow cytometry (i) Uninfected cells. GFP-expressing M. tuberculosis cells at time zero (ii), day 1 (iii), day 4 (iv), day 7 (v), and day 15 (vi) show progressive intracellular proliferation. Data are from a representative experiment performed on two occasions in triplicate (D). M. tuberculosis infection does not reduce cell viability within microspheres. Cellular survival was measured by the CellTiter-Glo 3D Cell Viability Assay. Data are the mean ± the standard error of the mean of an experiment performed in triplicate on two occasions. *, P < 0.05; ***, P < 0.001; ****, P < 0.0001.
Fluorescence-activated cell sorter analysis strategy for GFP-expressing M. tuberculosis associated with cells. Microspheres were generated, and on days 0, 1, 4, 7, and 15, they were decapsulated. Cells were stained for CD14, fixed with 2% paraformaldehyde for 1 h, and then analyzed on a BD Accuri C6 flow cytometer. All events in the high forward and side scatter area gates were then analyzed for CD14 and GFP signals. Columns: i, gating area, cells confirmed to be viable in preliminary experiments; ii, FL4 channel, CD14 antibody; iii, FL1 channel GFP plotted against side scatter; iv, FL1 GFP channel histogram. (A) Unstained, uninfected cells. (B) Uninfected cells stained with an isotype antibody for CD14 antibody, showing no increase in the FL4 channel. (C) Uninfected cells stained with anti-CD14 antibody showing an increase in the FL4 channel. There was only a background signal in the FL1 channel, which detects GFP fluorescence. (D) PBMCs infected with GFP+ M. tuberculosis stained with isotype control antibody. Infection increases the FL1 signal (iii, iv). (E) PBMCs infected with GFP+ M. tuberculosis stained with CD14 antibody. A relative decrease in CD14 expression relative to that in uninfected cells at day 1 is observable (ii). M. tuberculosis infection increases the FL1 signal. (F) Uninfected PBMCs stained for CD14 at day 15. CD14 expression is reduced (ii). (G) Infected PBMCs at day 15 show an increased CD14 signal (ii) and also a progressive increase in the number of GFP-positive M. tuberculosis bacteria associated with cells (iii, iv). Download FIG S1, EPS file, 2.2 MB (2.2MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Standard antibiotics kill M. tuberculosis under all conditions.
Having demonstrated granuloma formation and the M. tuberculosis stress response within microspheres, we first studied standard first-line antibiotics in 2D cell culture and the 3D model to determine the tractability of the model and whether killing efficacy is the same under both conditions. Rifampin, isoniazid (INH), and ethambutol were added to cell culture medium around spheres at physiological concentrations (1, 0.25, and 4 µg/ml, respectively). All three antibiotics inhibited M. tuberculosis growth in both 2D and 3D cell culture systems (Fig. 3A and B, antibiotics added at day 6; Fig. S2, antibiotics added at day 1). Rifampin was the most efficacious at killing M. tuberculosis, and INH was consistently more efficient at controlling M. tuberculosis in the 3D microsphere system than in 2D cell cultures. M. tuberculosis growth analyzed by luminescence correlated closely with CFU counts on Middlebrook 7H11 agar (Fig. 3C).
FIG 3 .
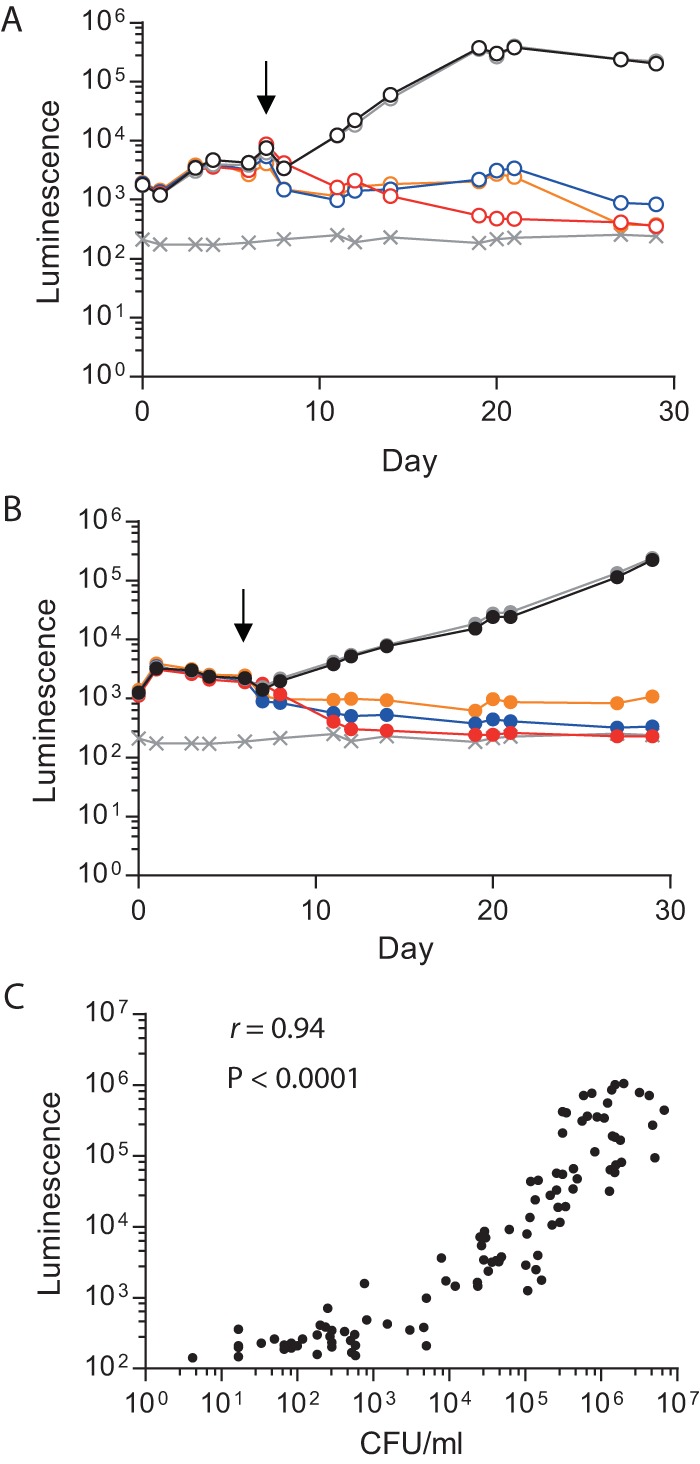
Effects of standard antituberculosis antibiotics on M. tuberculosis growth. Antibiotics (rifampin [red, 1 µg/ml], INH [blue, 0.25 µg/ml], and ethambutol [orange, 4 µg/ml]) were added at day 6 to 2D PBMC cultures or the microsphere system, and M. tuberculosis growth was monitored by measuring luminescence. M. tuberculosis growth was inhibited by all of the antibiotics in both 2D cell cultures (A) and the 3D model (B). M. tuberculosis growth was unaffected in the control sample (black) or by the addition of DMSO (gray), which was used as a solvent for rifampin. Symbols: ×, background level of luminescence; black arrows, antibiotic addition. Data are the mean ± the standard error of the mean of an experiment performed in triplicate and are representative of three separate experiments. (C) M. tuberculosis luminescence closely correlates with CFU counts on Middlebrook 7H11 agar. Spearman r value = 0.94; P < 0.0001.
Standard antibiotics added on day 1 kill M. tuberculosis in 2D and 3D systems. Rifampin (red), INH (blue), and ethambutol (orange) were added at 1, 0.25, and 4 µg/ml, respectively, at day 1 to either 2D cell cultures (A) or the 3D microsphere system (B). M. tuberculosis growth was inhibited by all of the antibiotics. M. tuberculosis growth was unaffected in the control sample (black) or by DMSO (gray). The × line indicates the background level of luminescence. The black arrow indicates the point of antibiotic administration. Data are the mean ± the standard error of the mean of an experiment performed in triplicate and are representative of three separate experiments. Download FIG S2, EPS file, 1.1 MB (1.1MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PZA is only efficacious in microspheres and not in broth or 2D cell cultures.
Next, we investigated PZA, which is a key antibiotic in treating human disease but has a poorly defined mechanism of action at the concentration described in epithelial cell lining fluid (25). PZA had no effect on M. tuberculosis growth in 7H9 broth without cells at neutral pH (Fig. 4A). In 2D primary cell cultures, PZA had a temporary effect but M. tuberculosis growth rapidly recovered (Fig. 4B). Critically, PZA killed M. tuberculosis in the 3D microsphere system, with luminescence falling to background levels by day 30 when we used the same antibiotic preparation that had no effect in broth and a transient effect in 2D cultures (Fig. 4C). A similar pattern of efficacy was observed when PZA was added to cultures on day 1, with PZA having no effect in 7H9 broth and a temporary effect in 2D cultures but complete control of M. tuberculosis growth in microspheres (Fig. S3). Colony counting on 7H11 agar confirmed that the efficacy of M. tuberculosis killing by PZA was equivalent to that of INH and moxifloxacin (Fig. 4D; Fig. S2D).
FIG 4 .
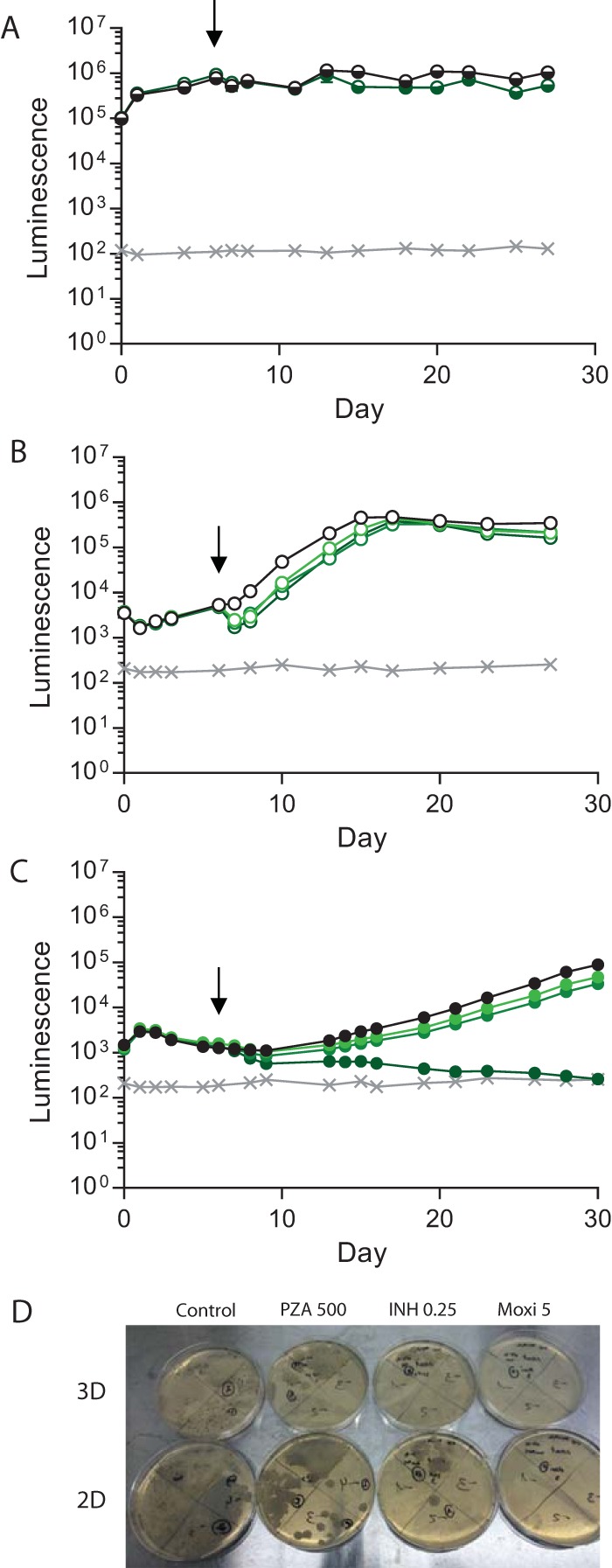
PZA kills M. tuberculosis in the 3D model but not in 7H9 broth or 2D cultures. (A) PZA has no effect on M. tuberculosis growth in 7H9 broth (dark green, 500 µg/ml) compared with an untreated control (black). (B) PZA has a brief effect on M. tuberculosis growth in 2D PBMC cell cultures at 60 µg/ml (light green), 100 µg/ml (medium green), or 500 µg/ml (dark green) in comparison with an untreated control (black), but M. tuberculosis growth rapidly recovers. (C) PZA kills M. tuberculosis in the 3D system at 500 µg/ml (dark green). Minimal killing of M. tuberculosis was observed when 60 µg/ml (light green) or 100 µg/ml (medium green) PZA was added, relative to the control sample (black). Symbols: ×, background level of luminescence; black arrows, antibiotic addition. Data are the mean ± the standard error of the mean of an experiment performed in triplicate and are representative of three separate experiments. (D) Colony counting on 7H11 agar confirms M. tuberculosis killing by PZA. Dilutions of control and 2D PZA plates start from a 1:10 dilution, while all other plates start without dilution. Representative plates are shown.
PZA kills M. tuberculosis more effectively in 3D microspheres than in 2D cultures when added on day 1. (A) PZA at 500 µg/ml (dark green) has no effect on M. tuberculosis growth in 7H9 broth in comparison with that in an untreated control. (B) PZA at a concentration of 60 µg/ml (light green), 100 µg/ml (medium green), or 500 µg/ml (dark green) initially significantly inhibited M. tuberculosis growth in 2D cell cultures compared to that in untreated controls. However, bacterial regrowth occurred, with M. tuberculosis luminescence returning to control levels by day 28. (C) PZA is bactericidal in the 3D system. M. tuberculosis growth was inhibited slightly by a low dose of PZA, while 500 µg/ml (dark green) was bactericidal. Gray lines with × symbols indicate the background levels of luminescence. Black arrows indicate points of antibiotic addition. Data are the mean ± the standard error of the mean of an experiment performed in triplicate and are representative of three separate experiments. (D) Bacteria were plated on 7H11 agar on day 28. CFU counts confirm that the luminescence data reflect bacterial loads. Note that dilutions of control and 2D PZA plates start from a 1:10 dilution, while other plates start without dilution. Representative plates are shown. Download FIG S3, EPS file, 1.6 MB (1.6MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Second-line antibiotics are most efficacious in 3D microspheres.
We then examined the effect of the second-line antibiotics d-cycloserine, moxifloxacin, and linezolid, which are of increasing importance with the emergence of drug-resistant TB. In 7H9 broth, d-cycloserine at a low concentration had a minor inhibitory effect but at a high concentration it was as effective as moxifloxacin (Fig. 5A). In the 2D and 3D systems, d-cycloserine effectively killed M. tuberculosis at low and high concentrations (Fig. 5B and C). Linezolid and moxifloxacin effectively suppressed M. tuberculosis growth under all three conditions. INH, which was included as a control first-line antibiotic, consistently killed M. tuberculosis in 3D microspheres, but bacterial growth resumed in 7H9 broth and the 2D system. A similar pattern was observed when antibiotics were added at day 1, although the inhibition of M. tuberculosis growth by d-cycloserine was more rapid in microspheres than in 2D cultures (Fig. S4). To ensure that the antibiotics were not having a cytotoxic effect on host cells, we analyzed their viability and found no evidence of cytotoxicity after 21 days of culture compared to medium and dimethyl sulfoxide (DMSO) controls (Fig. S5).
FIG 5 .
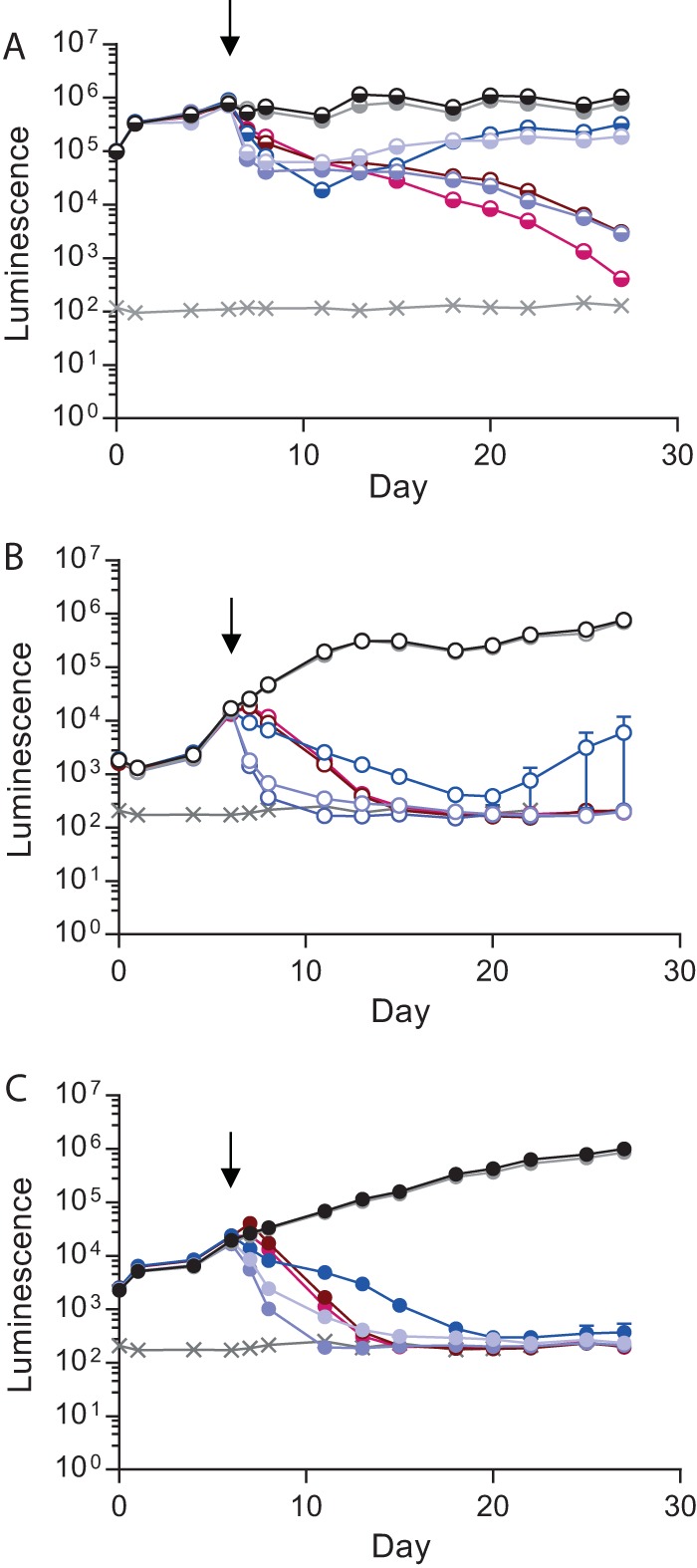
d-Cycloserine has similar effects on M. tuberculosis growth in 2D and 3D cultures. (A) M. tuberculosis in 7H9 broth. d-Cycloserine at a low concentration (20 µg/ml) had a temporary effect on M. tuberculosis growth (light purple) similar to that of INH at 0.25 µg/ml (blue). d-Cycloserine at 200 µg/ml killed M. tuberculosis more rapidly than (dark purple) and as effectively as moxifloxacin (brown, 5 µg/ml). Linezolid was the most effective second-line antibiotic (magenta, 24 µg/ml). The diluent DMSO (gray) did not affect M. tuberculosis growth relative to that in 7H9 broth only (black). (B) M. tuberculosis growth in 2D PBMC cultures. d-Cycloserine at both concentrations inhibited M. tuberculosis growth (purple) more rapidly than the other antibiotics (moxifloxacin [brown, 5 µg/ml], linezolid [magenta, 24 µg/ml], and INH [blue, 0.25 µg/ml]). Shown is M. tuberculosis growth in control samples (black) and with DMSO (gray). (C) M. tuberculosis growth in a 3D cell culture model. d-Cycloserine, linezolid, and moxifloxacin have an efficacy similar to that in a 2D cell culture (purple), while INH (blue) is more consistently bactericidal. Gray lines indicate background levels of luminescence. Black arrows indicate the day antibiotics were added. Data are the mean ± the standard error of the mean of an experiment performed in triplicate and are representative of three separate experiments.
Second-line antibiotics kill M. tuberculosis more rapidly in 3D microspheres than in 2D cell cultures when added on day 1. (A) M. tuberculosis growth in 7H9 broth. d-Cycloserine at low (20 µg/ml) and high (200 µg/ml) concentrations (light and dark purple, respectively) inhibit M. tuberculosis growth. However, bacterial regrowth occurred with the lower dose. INH (blue, 0.25 µg/ml) had a moderate effect on M. tuberculosis growth, and regrowth occurred. Moxifloxacin (brown, 5 µg/ml) and linezolid (magenta, 24 µg/ml) were equally effective but with delayed killing in comparison with high-dose d-cycloserine. M. tuberculosis growth was unaffected in control samples (black; gray with the diluent DMSO). (B) M. tuberculosis growth in 2D cell cultures. d-Cycloserine at a higher dose (dark purple) killed M. tuberculosis with an efficacy equal to that of the other antibiotics tested (5 µg/ml moxifloxacin [brown], 24 µg/ml linezolid [magenta], and 0.25 µg/ml INH [blue]). d-Cycloserine at a lower concentration inhibited M. tuberculosis growth but with a delay in comparison with the other antibiotics. (C) d-Cycloserine kills M. tuberculosis more rapidly in 3D cell cultures. All of the antibiotics investigated were similarly effective against M. tuberculosis, with rapid killing and no bacterial regrowth. Gray lines with × symbols indicate background luminescence. Black arrows indicate points of antibiotic addition. Data are the mean ± the standard error of the mean of an experiment performed in triplicate and are representative of three separate experiments. Download FIG S4, EPS file, 1.5 MB (1.5MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Antibiotics do not cause cytotoxicity within microspheres. Cellular cytotoxicity was investigated at day 21 within the 3D system with the CellTiter-Glo 3D cell viability assay. No antibiotic significantly changed cellular survival either in the absence of infection (A) or after M. tuberculosis infection (B). Similarly, cytotoxicity, analyzed by LDH release, is not different in infected cells treated with antibiotics (C). Download FIG S5, EPS file, 1.2 MB (1.3MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Pharmacokinetic modeling by integration with microfluidics.
In patients, antibiotic concentrations fluctuate over time, as opposed to the static concentrations usually studied in the laboratory. Therefore, we integrated the microsphere system with a microfluidic platform to permit modulation of antibiotic concentrations over time to mimic in vivo pharmacokinetics in patients during treatment (Fig. 6A). We studied rifampin, as its concentrations in plasma correlate with treatment outcomes (23). We manufactured a microfluidic plate from milled poly(methyl methacrylate) (PMMA), providing each well with two inlets and one outlet, permitting a smooth flow of medium through the wells containing microspheres (Fig. 6B). Initially, bacterial luminescence from the 24-well plate was undetectable on a GloMax DISCover plate reader. To overcome this, we used phenol red-free medium, optimized the microsphere density within wells, and placed a custom-made mirror under the plate. These modifications greatly improved the luminescence readout (Fig. 6C and D), and we were able to monitor bacterial growth from experimental time zero.
FIG 6 .

Modeling of antibiotic pharmacokinetics by integrating microspheres with a microfluidic system. (A) Representation of antibiotic pharmacokinetics in human plasma after daily oral administration during treatment. (B) Microfluidic system with two input channels and one exit channel for a 24-well tissue culture plate. (C, D) Placement of a basal mirror doubles the detection of M. tuberculosis luminescence by the GloMax Discover plate reader. Luminescence from infected PBMCs in microspheres in a single well in the absence (no fill) or presence (stripes) of a basal mirror for 24-well (C) and 96-well (D) tissue culture plates. (E) Modeling of antibiotic concentration profiles with a microfluidic system. From day 5 (black arrow), various peak concentrations of antibiotics were introduced for 6 h via the fluidic system and then washed out to approximate pharmacokinetics in vivo. Increasing rifampin concentrations (0.25 µg/ml [salmon], 1 µg/ml [bright red], and 4 µg/ml [dark red]) progressively accelerated M. tuberculosis killing. The black line represents a control sample to which the carrier DMSO was added and identical washes were performed. Three independent experiments were carried out, and the results of a representative experiment are shown.
To model pharmacokinetics in patients, rifampin at a range of concentrations (0.25, 1, and 4 µg/ml) was used to irrigated wells from day 5, incubated for 6 h, and then washed out to leave a minimal antibiotic concentration overnight. A stepwise increase in the rifampin concentration in individual wells produced dose-dependent killing of M. tuberculosis (Fig. 6E). The highest 6-h peak rifampin concentration caused M. tuberculosis killing equal to that of a constant 1 µg/ml antibiotic concentration or that of irrigation with medium with rifampin at 1 μg/ml (Fig. S6A). Luminescence increased overnight in the absence of antibiotics, resulting in a sawtooth pattern of killing that did not occur at a fixed antibiotic concentration and demonstrated rapid recovery of mycobacterial growth once antibiotic pressure was removed. Colony counts on 7H11 agar confirmed that the luminescence data reflected the total bacterial load (Fig. S6B).
High-dose rifampin for 6 h/day has efficacy equal to that of a constant standard antibiotic concentration. (A) M. tuberculosis growth inhibition in the microfluidic system at different concentrations of rifampin (0.25 [salmon], 1 [bright red], and 4 [dark red] µg/ml). A constant 1 µg/ml rifampin (blue) or addition of rifampin at a concentration of 1 µg/ml without daily washes (checkered box) produced inhibition of M. tuberculosis growth equivalent to that of a 4 µg/ml temporary peak concentration of rifampin, but no overnight regrowth occurred. Controls with DMSO (black) and without DMSO (white squares) were included. (B) Colony counts on 7H11 agar confirm that the luminescence data reflect the total M. tuberculosis loads within microspheres. Download FIG S6, EPS file, 2 MB (2MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Antimicrobial resistance is rapidly emerging as one of the most pressing challenges to global society (2, 3). Innovative approaches to studying the host-pathogen interaction are urgently needed to identify novel treatment approaches that counter the evolution of drug-resistant bacteria. To achieve this, an emerging paradigm is that bacteria should be studied in the context of the host (6). We hypothesized that a transformative platform will require multiple elements that are not currently available in a single system: virulent reporter bacteria, primary human cells, an extracellular matrix, 3D organization of the host-pathogen interaction, and pharmacokinetic modeling. We combined diverse methodologies, including genetically modified bacteria, primary human cell culture, electrostatic microsphere generation, multiparameter readouts, and microfluidics, to develop a platform with all of the requisite elements that could be used in a biosafety level 3 containment environment. In our investigation of the host immune response, we showed that host cell survival is improved in the 3D microsphere model, cellular aggregates form, and host-directed therapies can be studied (13). Here, we investigated the system from the pathogen’s perspective and demonstrated that mycobacterial stress genes are upregulated and key antibiotics used to treat human disease are more efficacious in the microsphere system than in standard culture. The nondestructive readouts permit longitudinal analysis over prolonged periods and real-time pharmacokinetic modeling. This platform has the potential to revolutionize antibiotic discovery and to replace suboptimal animal model systems based on inappropriate host-pathogen combinations.
Miniaturized organoid systems and lab-on-a-chip technologies are rapidly evolving fields for drug discovery, resulting from the widespread belief that studying individual cells or infectious organisms in isolation does not sufficiently reflect conditions in vivo (26, 27). The bioelectrosprayer can produce large numbers of identical microspheres rapidly in a patient-relevant model, fulfilling a principal requirement of such a system (28). 2D systems with flow have been developed to study respiratory epithelial cell infection (29), and while several TB cellular systems have been reported (30–34), none combine 3D culture, collagen, and the potential for high-throughput or longitudinal analysis. The microsphere system is highly tractable, with the ability to modulate infectious organism and doses, host cellular content, the extracellular matrix, microsphere size, and the surrounding medium dynamically.
The need for new antibiotics for TB is particularly acute, given the progressive emergence of drug resistance, recent disappointments in treatment-shortening regimens, and the ongoing global toll of infection (14). PZA is one of the most important antibiotics used to treat human disease, but it was discovered serendipitously in the 1950s and indeed would have not been found by current screening approaches based on the sequential study of MICs in broth culture, murine infections, and human disease (17). PZA has a complex mechanism of action, requiring intracellular acidification. We demonstrated that M. tuberculosis stress genes were upregulated in microspheres and that PZA killed M. tuberculosis in microspheres at neutral pH but not in standard 2D culture systems at a concentration found in epithelial lining fluid (25). This confirms that, within microspheres, bacilli are in a PZA-sensitive compartment and demonstrates the potential to identify other compounds only active against stressed mycobacteria in the correct microenvironment. Similarly, we found that INH was consistently more effective in the 3D system that in the 2D system, with a more rapid fall in luminescence and no failed treatments, supporting the relevance of the model to human TB. Therefore, the system can identify drug resistance in a more clinically relevant fashion and can be used to study novel regimens with variable concentrations in combination rather than single new agents at static concentrations.
The microsphere system has potential for high-throughput use, as >5,000 microspheres can be generated from a single blood donor within an hour and the diameter is compatible with a 384-well format. Mycobacterial killing curves during antibiotic treatment of patients suggest that there are diverse populations within patients’ lungs (35), and therefore, the system can be used to study the separate physiological conditions that drive these within the same experiment, such as hypoxia and nutritional stress. Encapsulation within microspheres permits integration with a microfluidic system without cells and bacteria being lost during irrigation, resolving a significant technical hurdle for pharmacokinetic studies. We were able to show more rapid killing with increased rifampin concentrations, consistent with findings in patients (23, 36). Microfluidics have been used to develop an array of organ-on-a-chip models (37), but we are not aware of development to study virulent containment level 3 pathogens such as M. tuberculosis, which represents additional challenges because of the infection risk. The hollow-fiber model has been used to perform advanced pharmacokinetic modeling, but PZA is only efficacious in this system after acidification to pH 5.8 (38) and so cannot be studied in combination with other agents. Future potential developments for the bioelectrospray platform include dual encapsulation to permit a central lipid-rich caseous core, generating an additional layer of flexibility and modeling drug penetration into necrotic foci (39). Development of the microfluidic plate will permit optimization of combinations of multiple antibiotics in a fully humanized system, with pharmacokinetic modeling of each antibiotic within wells, to identify the best combinations with which to go forward to clinical trials (34).
We developed a bioengineered cell culture platform that replicates key features of human disease and incorporates primary human cells, an extracellular matrix, a 3D structure, virulent bacteria, and pharmacokinetic modeling. The microsphere system is highly tractable, permitting variation of cell content, the extracellular matrix, sphere size, the infectious dose, and the surrounding medium with the potential to address a wide array of human infections. The system can equally be applied to diverse inflammatory and malignant human diseases. Integration with molecular microbiology techniques and clustered regularly interspaced short palindromic repeat gene editing will provide genetically tractable host-pathogen pairings. Therefore, this platform has global applicability to address the threat of antimicrobial resistance and deliver new treatments.
MATERIALS AND METHODS
Bacterial strains, culture conditions, and chemicals.
Bioluminescent M. tuberculosis H37Rv (M. tuberculosis Lux) (24) and mCherry-expressing M. tuberculosis H37Rv (40) were cultured in Middlebrook 7H9 medium (BD Biosciences UK, Oxford, United Kingdom) supplemented with 10% ADC Enrichment (Scientific Laboratory Supplies, Nottingham, UK), 0.2% glycerol, 0.02% Tween 80, and kanamycin (25 µg/ml) or hygromycin (50 µg/ml), respectively. For all experiments, cultures were grown to an optical density at 600 nm (OD600) of 0.6 (approximately 1 × 108 CFU/ml). Bacterial growth in 7H9 broth was monitored by measuring luminescence (GloMax 20/20 Single Tube luminometer; Promega, United Kingdom). Chemicals were purchased from Sigma-Aldrich unless stated otherwise.
Human PBMC isolation and infection.
Ethical approval for these studies was provided by the National Research Ethics Service Committee South Central—Southampton A, ref. 13/SC/0043. Peripheral blood mononuclear cells (PBMCs) were isolated from single-donor buffy coats from the National Health Service Blood and Transplant, Southampton, United Kingdom. Leukocytes were isolated by density gradient centrifugation over Ficoll-Paque (GE Healthcare Life Sciences, United Kingdom). Isolated PBMCs were infected with M. tuberculosis Lux at a multiplicity of infection (MOI) of 0.1 and kept overnight at 37°C in a 5% CO2 incubator in RPMI 1640 medium supplemented with 10 μg/ml ampicillin, 2 mM glutamine, 25 µg/ml kanamycin, and 10% fetal bovine serum (Labtech International Ltd.). The next day, infected PBMCs were transferred from vented flasks to 50-ml Falcon tubes after detachment with Versene solution (Sigma) for 10 min and scraping. After Hanks balanced salt solution (HBSS) without Ca/Mg (Gibco) was added, cells were centrifuged at 320 × g for 8 min at 4°C and the supernatant was decanted. The pelleted cells were resuspended in appropriate volumes of RPMI 1640 medium supplemented with 10 μg/ml ampicillin, 2 mM glutamine, 25 µg/ml kanamycin, and 10% human AB serum (Sigma), referred to as complete RPMI medium.
2D culture.
Infected cells were resuspended in 50 ml of complete RPMI medium, and 1 ml was equally distributed into 2-ml Eppendorf tubes at a final concentration of 3 × 106/ml. Cultures were incubated at 37°C in 5% CO2. M. tuberculosis luminescence was monitored with a GloMax 20/20 luminometer. Antibiotics were added at predetermined time points. For colony counts, cultures were treated with 1% saponin in HBSS and bacteria were plated onto 7H11 agar at serial dilutions. For RT-qPCR analysis, infected cells were plated into six-well plates at a final concentration of 2.5 × 106/ml.
3D culture.
Infected cells were resuspended in complete RPMI medium, mixed with sterile alginate-collagen at 1 × 106 cells/ml, and injected into an Electrostatic Bead Generator (Nisco, Zurich, Switzerland) to form microspheres via a Harvard syringe driver as described previously (41). After generation, microspheres were equally distributed into 2-ml Eppendorf tubes (microsphere volume, 0.4 ml), immersed in 1 ml of complete RPMI medium, and incubated at 37°C in 5% CO2. M. tuberculosis luminescence was monitored with a GloMax 20/20 luminometer. For CFU counts, microspheres were dissolved in 55 mM sodium citrate–10 mM EDTA with 1% saponin in HBSS and bacteria were plated onto 7H11 agar. For RT-qPCR analysis, microspheres were cultured in 50-ml Falcon tubes (microsphere volume, 10 ml) in complete RPMI medium.
Immunofluorescence and confocal imaging.
PBMCs were separated into monocytes and lymphocytes with MACS Cell Separation Columns (Miltenyi Biotec, Inc., Surrey, United Kingdom). Cells were then labeled with CellTracker Blue or CellTrace CFSE (Thermo, Fisher Scientific, United Kingdom) separately in accordance with the manufacturer’s recommendations before infection with mCherry-expressing M. tuberculosis H37Rv (40) at an MOI of 0.1. Microspheres were generated and fixed in 4% paraformaldehyde after 4 days. Confocal images were acquired on a Leica TCS SP5 confocal microscope and processed with ImageJ 1.5 0d (NIH, Bethesda, MD).
Transcription analysis by RT-qPCR.
For bacteria grown in 7H9 broth (OD600 of 0.25) and 2D cultures, total RNA was extracted by centrifugation at 13,000 rpm for 10 min and the addition of 500 µl of RNAprotect Bacteria Reagent (Qiagen). The resuspended pellet was left for 10 min at room temperature prior to repeat centrifugation and resuspension of the pellet in 1 ml of TRIzol (Life Technologies, Inc.) and stored at −80°C. For 3D cultures, RNAlater solution (Ambion) was used to preserve RNA overnight at 4°C. Cells were decapsulated with 100 mM sodium citrate and centrifuged at 3,000 × g for 30 min, and the pellet was resuspended in 1 ml of TRIzol (Life Technologies, Inc.) and stored at −80°C until use. Thawed samples were transferred to Lysis Matrix B tubes containing 0.1-mm silica beads (Q-Biogene) and homogenized in a MagnaLyser instrument (Roche) at 4,000 rpm for 5 × 45 s with incubation on ice for 1 min between homogenizations. Samples were centrifuged for 1 min at 16,100 × g at 4°C, and the supernatant was transferred to a new Eppendorf tube. After phenol-chloroform extraction, the nucleic acids were precipitated with isopropanol, washed with 75% ethanol, air dried for 10 to 15 min, and finally resuspended in nuclease-free water (Fisher Scientific). Genomic DNA was removed with a DNA-free kit (AM1906; Ambion) in accordance with the manufacturer’s instructions. RNA was further purified with the Qiagen RNeasy minikit (Qiagen), subjected with on-column DNase digestion with the RNase-free DNase set (79254; Qiagen), repurified with an RNeasy minikit, and eluted in 50 µl of RNase- and DNase-free water (Fisher Scientific). The first-strand cDNA was synthesized in 10-µl reaction volumes with the High Capacity cDNA RT kit (Applied Biosystems). The cDNA samples were diluted 1:3 in nuclease-free water, and real-time qPCR was performed with 10-µl reaction volumes containing FastStart Universal Probe Master with Rox (Roche), LNA-based probe (designed with the Universal ProbeLibrary System Technology [Roche]) (Table 1), oligonucleotides (Sigma) (Table 1), and 1 µl of a cDNA preparation. Reactions were run on a 7900HT Fast real-time PCR system (Applied Biosystems) with the following program: 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C and 1 min at 60°C. All samples were amplified in triplicate, and threshold cycle (CT) values of ≥40 were considered negative. Expression data were normalized to the M. tuberculosis housekeeping gene sigA, and relative quantifications were carried out by the ΔΔCT method.
TABLE 1.
Primers and probes used in this study
| Primer or probe | 5′–3′ sequence |
|---|---|
| lipF-FOR | ATGAGCCGCTCGACCATA |
| lipF-REV | GAGCCGGAAACGTGAATAAG |
| Roche UPL LNA-probe 160 | FAMa-TGCCGCCG-dark quencher dye |
| recA-FOR | AGGAGAATGCCCGCAACT |
| recA-REV | CTTCTTCTCGATCTCGTCAGC |
| Roche UPL LNA-probe 22 | FAM-TGGTGGAG-dark quencher dye |
| relA-FOR | CGCATCATCGAGGTGCTAT |
| relA-REV | CCTGGATTGCCACCAGAA |
| Roche UPL LNA-probe 152 | FAM-TCGCCGTC-dark quencher dye |
| sodA-FOR | TGGCCGAATACACCTTGC |
| sodA-REV | GAGATGTGCGGTTCCAGTG |
| Roche UPL LNA-probe 85 | FAM-GACCTGGA-dark quencher dye |
| sigA-FOR | AGCTGGCCAAAGAGATGGA |
| sigA-REV | GGGCGTATTGCTGGATTTC |
| Roche UPL LNA-probe 133 | FAM-GGAGAAGG-dark quencher dye |
FAM, 6-carboxyfluorescein.
Eukaryotic cell viability assay.
Microspheres containing PBMCs alone or M. tuberculosis-infected PBMCs were incubated in 96-well plates for 21 days. Cell viability was analyzed at day 21 with the CellTiter-Glo 3D Cell Viability Assay (Promega) in accordance with the manufacturer’s instructions. Luminescence was analyzed by a GloMax Discover 96-well plate reader (Promega, United Kingdom). To measure cell toxicity, lactate dehydrogenase (LDH) release was analyzed by a colorimetric activity assay (Roche, Burgess Hill, United Kingdom).
Analysis of M. tuberculosis location.
PBMCs were infected with luminescent M. tuberculosis and incorporated into microspheres. At predefined time points, microspheres were decapsulated and cell-associated M. tuberculosis was pelleted by centrifugation at 380 × g for 8 min as previously described (42–44). At days 7 and 15, additional samples of cell-associated M. tuberculosis were treated with 100 µg/ml gentamicin for 90 min at 37°C in a 5% CO2 incubator to remove noninternalized bacteria and then washed with phosphate-buffered saline (PBS). Mycobacterial location was analyzed by measuring luminescence in the supernatant and pellet and also by colony counting on Middlebrook 7H11 agar. For flow cytometry, PBMCs were infected with GFP-expressing M. tuberculosis at an MOI of 0.1. Microspheres were made as described above, and on days 0, 1, 4, 7, and 15, microspheres were decapsulated and stained with allophycocyanin-conjugated anti-human CD14 antibody (ImmunoTools, Friesoythe, Germany). Cells were fixed with 2% paraformaldehyde and analyzed on a BD Accuri C6 flow cytometer. All events in the high forward and side scatter areas stained with CD14 were included in the analysis. Flow cytometry data were analyzed with BD Accuri C6 software (ver. 1.0.264.21). Experiments were done at least two times in triplicate.
Microfluidic system manufacture.
The lid template was based on the original plate lid (Berthold Technologies, United Kingdom). The lid was manufactured from a 5-mm-thick PMMA sheet (Weatherall Equipment & Instruments Ltd.) by micromilling on a ProtoMat 100 micromill (LPKF Laser & Electronics AG, Garbsen, Germany). The tools used for fabrication were a 3.00-mm end mill and a 1.59-mm drill (ACS Industries United Kingdom). The 3.00-mm cutting tool was used to cut out the holding sockets for the Iso-Disc syringe filters (PTFE-4-4, 4 mm [diameter] by 0.45 μm; Supelco, USA) and to cut out the exact 127.90- by 85.85-mm outline of the lid. The inlets for each well were created by drilling pairs of holes through with the 1.59-mm drill and then inserting 30-mm (length) by 0.87-mm (inner diameter) by 1.59-mm (outer diameter) stainless steel tubing (Swagelok, United Kingdom). The stainless steel tubing was terminated with polytetrafluoroethylene tubing (0.75-mm inner diameter) to luer lock syringe connectors. The outlet port was designed to accommodate the Iso-Disc syringe filter. Three 0.15-mm holes were drilled through each Iso-Disc syringe filter to allow withdrawal of the liquid from each well during experiments with a 1-ml syringe (via the outlet port).
Microfluidic experiments.
For microfluidic experiments, microspheres were placed in 24-well plates (Berthold Technologies, United Kingdom) with RPMI without phenol red (Gibco) supplemented with 10 μg/ml ampicillin, 2 mM glutamine, 25 µg/ml kanamycin, and 10% human AB serum (Sigma). M. tuberculosis Lux luminescence was monitored with a GloMax Discover plate reader (Promega, United Kingdom). Rifampin was added to cultures at either day 4 or 5. At 9 a.m. each day, wells were treated with different doses of antibiotic, and after 6 h, wells were irrigated five times with RPMI. A custom-made mirror was placed under the 24-well clear-bottom plate to maximize luminescence collection for detection.
Statistical analyses.
Statistical analyses were preformed with GraphPad Prism. Differences were considered significant at P < 0.05.
ACKNOWLEDGMENTS
We thank Jennifer Russell and Regina Teo, University of Southampton, for excellent technical assistance. We thank Nuria Andreu and Siouxsie Wiles for providing the Lux-expressing M. tuberculosis strain and Tanya Parish for the mCherry-expressing M. tuberculosis strain.
This work was supported by The UK Antimicrobial Resistance Cross Council Initiative funded by the Biotechnology and Biological Sciences Research Council and the Medical Research Council (MR/N006631/1), the United States National Institutes of Health (R33AI102239), and the UK National Centre for the 3Rs (NC/L001039/1). We are grateful to NAMRIP (Network for antimicrobial Resistance and Infection Prevention) for support, including pump-priming funding via NAMRIP’s EPSRC grant NAMRA (EP/M027260/1), part of the EPSRC, Network for Antimicrobial Action, Bridging the Gap program.
Funding Statement
This work was supported by the UK Antimicrobial Resistance Cross Council Initiative funded by the Biotechnology and Biological Sciences Research Council and the Medical Research Council MR/N006631/1, the US National Institute for Health R33AI102239, the UK National Centre for the 3Rs NC/L001039/1, and the University of Southampton Network for Antimicrobial Resistance and Prevention (NAMRIP).
Footnotes
Citation Bielecka MK, Tezera LB, Zmijan R, Drobniewski F, Zhang X, Jayasinghe S, Elkington P. 2017. A bioengineered three-dimensional cell culture platform integrated with microfluidics to address antimicrobial resistance in tuberculosis. mBio 8:e02073-16. https://doi.org/10.1128/mBio.02073-16.
REFERENCES
- 1.Cantón R, Morosini MI. 2011. Emergence and spread of antibiotic resistance following exposure to antibiotics. FEMS Microbiol Rev 35:977–991. doi: 10.1111/j.1574-6976.2011.00295.x. [DOI] [PubMed] [Google Scholar]
- 2.Davies J, Davies D. 2010. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev 74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.WHO 2014. Antimicrobial resistance: global report on surveillance 2014. World Health Organization, Geneva, Switzerland: http://www.who.int/drugresistance/documents/surveillancereport/en/. Accessed 6 January 2016. [Google Scholar]
- 4.Bush K, Courvalin P, Dantas G, Davies J, Eisenstein B, Huovinen P, Jacoby GA, Kishony R, Kreiswirth BN, Kutter E, Lerner SA, Levy S, Lewis K, Lomovskaya O, Miller JH, Mobashery S, Piddock LJ, Projan S, Thomas CM, Tomasz A, Tulkens PM, Walsh TR, Watson JD, Witkowski J, Witte W, Wright G, Yeh P, Zgurskaya HI. 2011. Tackling antibiotic resistance. Nat Rev Microbiol 9:894–896. doi: 10.1038/nrmicro2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spellberg B, Bartlett JG, Gilbert DN. 2013. The future of antibiotics and resistance. N Engl J Med 368:299–302. doi: 10.1056/NEJMp1215093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhavsar AP, Guttman JA, Finlay BB. 2007. Manipulation of host-cell pathways by bacterial pathogens. Nature 449:827–834. doi: 10.1038/nature06247. [DOI] [PubMed] [Google Scholar]
- 7.Scortti M, Lacharme-Lora L, Wagner M, Chico-Calero I, Losito P, Vázquez-Boland JA. 2006. Coexpression of virulence and fosfomycin susceptibility in Listeria: molecular basis of an antimicrobial in vitro-in vivo paradox. Nat Med 12:515–517. doi: 10.1038/nm1396. [DOI] [PubMed] [Google Scholar]
- 8.Westermann AJ, Gorski SA, Vogel J. 2012. Dual RNA-seq of pathogen and host. Nat Rev Microbiol 10:618–630. doi: 10.1038/nrmicro2852. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz MA, Chen CS. 2013. Cell biology. Deconstructing dimensionality. Science 339:402–404. doi: 10.1126/science.1233814. [DOI] [PubMed] [Google Scholar]
- 10.Bonnans C, Chou J, Werb Z. 2014. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 15:786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nielsen EI, Friberg LE. 2013. Pharmacokinetic-pharmacodynamic modeling of antibacterial drugs. Pharmacol Rev 65:1053–1090. doi: 10.1124/pr.111.005769. [DOI] [PubMed] [Google Scholar]
- 12.Eldholm V, Balloux F. 2016. Antimicrobial resistance in Mycobacterium tuberculosis: the odd one out. Trends Microbiol 24:637–648. doi: 10.1016/j.tim.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 13.Tezera LB, Bielecka MK, Chancellor A, Reichmann MT, Al Shammari B, Brace P, Batty A, Tocheva A, Jogai S, Marshall BG, Tebruegge M, Jayasinghe SN, Mansour S, Elkington PT. 2017. Dissection of the host-pathogen interaction in human tuberculosis using a bioengineered 3-dimensional model. Elife 6:e21283. doi: 10.7554/eLife.21283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallis RS, Maeurer M, Mwaba P, Chakaya J, Rustomjee R, Migliori GB, Marais B, Schito M, Churchyard G, Swaminathan S, Hoelscher M, Zumla A. 2016. Tuberculosis—advances in development of new drugs, treatment regimens, host-directed therapies, and biomarkers. Lancet Infect Dis 16:e34–e46. doi: 10.1016/S1473-3099(16)00070-0. [DOI] [PubMed] [Google Scholar]
- 15.Udwadia ZF. 2012. MDR, XDR, TDR tuberculosis: ominous progression. Thorax 67:286–288. doi: 10.1136/thoraxjnl-2012-201663. [DOI] [PubMed] [Google Scholar]
- 16.Warner DF, Mizrahi V. 2014. Shortening treatment for tuberculosis—to basics. N Engl J Med 371:1642–1643. doi: 10.1056/NEJMe1410977. [DOI] [PubMed] [Google Scholar]
- 17.Dartois V, Barry CE III. 2013. A medicinal chemists’ guide to the unique difficulties of lead optimization for tuberculosis. Bioorg Med Chem Lett 23:4741–4750. doi: 10.1016/j.bmcl.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diacon AH, Dawson R, von Groote-Bidlingmaier F, Symons G, Venter A, Donald PR, van Niekerk C, Everitt D, Hutchings J, Burger DA, Schall R, Mendel CM. 2015. Bactericidal activity of pyrazinamide and clofazimine alone and in combinations with pretomanid and bedaquiline. Am J Respir Crit Care Med 191:943–953. doi: 10.1164/rccm.201410-1801OC. [DOI] [PubMed] [Google Scholar]
- 19.Casali N, Nikolayevskyy V, Balabanova Y, Harris SR, Ignatyeva O, Kontsevaya I, Corander J, Bryant J, Parkhill J, Nejentsev S, Horstmann RD, Brown T, Drobniewski F. 2014. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat Genet 46:279–286. doi: 10.1038/ng.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Russell DG. 2011. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol Rev 240:252–268. doi: 10.1111/j.1600-065X.2010.00984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, Daudelin IB, Chen PY, Booty MG, Kim JH, Eum SY, Via LE, Behar SM, Barry CE III, Mann M, Dartois V, Rubin EJ. 2016. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med 22:531–538. doi: 10.1038/nm.4073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Al Shammari B, Shiomi T, Tezera L, Bielecka MK, Workman V, Sathyamoorthy T, Mauri F, Jayasinghe SN, Robertson BD, D’Armiento J, Friedland JS, Elkington PT. 2015. The extracellular matrix regulates granuloma necrosis in tuberculosis. J Infect Dis 212:463–473. doi: 10.1093/infdis/jiv076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasipanodya JG, McIlleron H, Burger A, Wash PA, Smith P, Gumbo T. 2013. Serum drug concentrations predictive of pulmonary tuberculosis outcomes. J Infect Dis 208:1464–1473. doi: 10.1093/infdis/jit352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andreu N, Zelmer A, Fletcher T, Elkington PT, Ward TH, Ripoll J, Parish T, Bancroft GJ, Schaible U, Robertson BD, Wiles S. 2010. Optimisation of bioluminescent reporters for use with mycobacteria. PLoS One 5:e10777. doi: 10.1371/journal.pone.0010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conte JE Jr., Golden JA, Duncan S, McKenna E, Zurlinden E. 1999. Intrapulmonary concentrations of pyrazinamide. Antimicrob Agents Chemother 43:1329–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrila J, Radtke AL, Crabbé A, Sarker SF, Herbst-Kralovetz MM, Ott CM, Nickerson CA. 2010. Organotypic 3D cell culture models: using the rotating wall vessel to study host-pathogen interactions. Nat Rev Microbiol 8:791–801. doi: 10.1038/nrmicro2423. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Tan S, Huang L, Abramovitch RB, Rohde KH, Zimmerman MD, Chen C, Dartois V, VanderVen BC, Russell DG. 2016. Immune activation of the host cell induces drug tolerance in Mycobacterium tuberculosis both in vitro and in vivo. J Exp Med 213:809–825. doi: 10.1084/jem.20151248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neuži P, Giselbrecht S, Länge K, Huang TJ, Manz A. 2012. Revisiting lab-on-a-chip technology for drug discovery. Nat Rev Drug Discov 11:620–632. doi: 10.1038/nrd3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benam KH, Villenave R, Lucchesi C, Varone A, Hubeau C, Lee HH, Alves SE, Salmon M, Ferrante TC, Weaver JC, Bahinski A, Hamilton GA, Ingber DE. 2016. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat Methods 13:151–157. doi: 10.1038/nmeth.3697. [DOI] [PubMed] [Google Scholar]
- 30.Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, Daffé M, Emile JF, Marchou B, Cardona PJ, de Chastellier C, Altare F. 2008. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog 4:e1000204. doi: 10.1371/journal.ppat.1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guirado E, Mbawuike U, Keiser TL, Arcos J, Azad AK, Wang SH, Schlesinger LS. 2015. Characterization of host and microbial determinants in individuals with latent tuberculosis infection using a human granuloma model. mBio 6:e02537-14. doi: 10.1128/mBio.02537-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Braian C, Svensson M, Brighenti S, Lerm M, Parasa VR. 2015. A 3D human lung tissue model for functional studies on Mycobacterium tuberculosis infection. J Vis Exp doi: 10.3791/53084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kapoor N, Pawar S, Sirakova TD, Deb C, Warren WL, Kolattukudy PE. 2013. Human granuloma in vitro model, for TB dormancy and resuscitation. PLoS One 8:e53657. doi: 10.1371/journal.pone.0053657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silva A, Lee BY, Clemens DL, Kee T, Ding X, Ho CM, Horwitz MA. 2016. Output-driven feedback system control platform optimizes combinatorial therapy of tuberculosis using a macrophage cell culture model. Proc Natl Acad Sci U S A 113:E2172–E2179. doi: 10.1073/pnas.1600812113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horsburgh CR Jr., Barry CE III, Lange C. 2015. Treatment of tuberculosis. N Engl J Med 373:2149–2160. doi: 10.1056/NEJMra1413919. [DOI] [PubMed] [Google Scholar]
- 36.Boeree MJ, Diacon AH, Dawson R, Narunsky K, du Bois J, Venter A, Phillips PP, Gillespie SH, McHugh TD, Hoelscher M, Heinrich N, Rehal S, van Soolingen D, van Ingen J, Magis-Escurra C, Burger D, Plemper van Balen G, Aarnoutse RE, PanACEA Consortium . 2015. A dose-ranging trial to optimize the dose of rifampin in the treatment of tuberculosis. Am J Respir Crit Care Med 191:1058–1065. doi: 10.1164/rccm.201407-1264OC. [DOI] [PubMed] [Google Scholar]
- 37.Sackmann EK, Fulton AL, Beebe DJ. 2014. The present and future role of microfluidics in biomedical research. Nature 507:181–189. doi: 10.1038/nature13118. [DOI] [PubMed] [Google Scholar]
- 38.Gumbo T, Dona CS, Meek C, Leff R. 2009. Pharmacokinetics-pharmacodynamics of pyrazinamide in a novel in vitro model of tuberculosis for sterilizing effect: a paradigm for faster assessment of new antituberculosis drugs. Antimicrob Agents Chemother 53:3197–3204. doi: 10.1128/AAC.01681-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dartois V. 2014. The path of anti-tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat Rev Microbiol 12:159–167. doi: 10.1038/nrmicro3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carroll P, Schreuder LJ, Muwanguzi-Karugaba J, Wiles S, Robertson BD, Ripoll J, Ward TH, Bancroft GJ, Schaible UE, Parish T. 2010. Sensitive detection of gene expression in mycobacteria under replicating and non-replicating conditions using optimized far-red reporters. PLoS One 5:e9823. doi: 10.1371/journal.pone.0009823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Workman VL, Tezera LB, Elkington PT, Jayasinghe SN. 2014. Controlled generation of microspheres incorporating extracellular matrix fibrils for three-dimensional cell culture. Adv Funct Mater 24:2648–2657. doi: 10.1002/adfm.201303891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bange FC, Brown AM, Jacobs WR Jr.. 1996. Leucine auxotrophy restricts growth of Mycobacterium bovis BCG in macrophages. Infect Immun 64:1794–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho S, Mehra V, Thoma-Uszynski S, Stenger S, Serbina N, Mazzaccaro RJ, Flynn JL, Barnes PF, Southwood S, Celis E, Bloom BR, Modlin RL, Sette A. 2000. Antimicrobial activity of MHC class I-restricted CD8+ T cells in human tuberculosis. Proc Natl Acad Sci U S A 97:12210–12215. doi: 10.1073/pnas.210391497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stegelmann F, Bastian M, Swoboda K, Bhat R, Kiessler V, Krensky AM, Roellinghoff M, Modlin RL, Stenger S. 2005. Coordinate expression of CC chemokine ligand 5, granulysin, and perforin in CD8+ T cells provides a host defense mechanism against Mycobacterium tuberculosis. J Immunol 175:7474–7483. doi: 10.4049/jimmunol.175.11.7474. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Generation of microspheres. Alginate was stained with bromophenol blue to permit visualization during the bioelectrospray process, which is shown in real time. Download MOVIE S1, AVI file, 2.5 MB (2.5MB, avi) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Fluorescence-activated cell sorter analysis strategy for GFP-expressing M. tuberculosis associated with cells. Microspheres were generated, and on days 0, 1, 4, 7, and 15, they were decapsulated. Cells were stained for CD14, fixed with 2% paraformaldehyde for 1 h, and then analyzed on a BD Accuri C6 flow cytometer. All events in the high forward and side scatter area gates were then analyzed for CD14 and GFP signals. Columns: i, gating area, cells confirmed to be viable in preliminary experiments; ii, FL4 channel, CD14 antibody; iii, FL1 channel GFP plotted against side scatter; iv, FL1 GFP channel histogram. (A) Unstained, uninfected cells. (B) Uninfected cells stained with an isotype antibody for CD14 antibody, showing no increase in the FL4 channel. (C) Uninfected cells stained with anti-CD14 antibody showing an increase in the FL4 channel. There was only a background signal in the FL1 channel, which detects GFP fluorescence. (D) PBMCs infected with GFP+ M. tuberculosis stained with isotype control antibody. Infection increases the FL1 signal (iii, iv). (E) PBMCs infected with GFP+ M. tuberculosis stained with CD14 antibody. A relative decrease in CD14 expression relative to that in uninfected cells at day 1 is observable (ii). M. tuberculosis infection increases the FL1 signal. (F) Uninfected PBMCs stained for CD14 at day 15. CD14 expression is reduced (ii). (G) Infected PBMCs at day 15 show an increased CD14 signal (ii) and also a progressive increase in the number of GFP-positive M. tuberculosis bacteria associated with cells (iii, iv). Download FIG S1, EPS file, 2.2 MB (2.2MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Standard antibiotics added on day 1 kill M. tuberculosis in 2D and 3D systems. Rifampin (red), INH (blue), and ethambutol (orange) were added at 1, 0.25, and 4 µg/ml, respectively, at day 1 to either 2D cell cultures (A) or the 3D microsphere system (B). M. tuberculosis growth was inhibited by all of the antibiotics. M. tuberculosis growth was unaffected in the control sample (black) or by DMSO (gray). The × line indicates the background level of luminescence. The black arrow indicates the point of antibiotic administration. Data are the mean ± the standard error of the mean of an experiment performed in triplicate and are representative of three separate experiments. Download FIG S2, EPS file, 1.1 MB (1.1MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
PZA kills M. tuberculosis more effectively in 3D microspheres than in 2D cultures when added on day 1. (A) PZA at 500 µg/ml (dark green) has no effect on M. tuberculosis growth in 7H9 broth in comparison with that in an untreated control. (B) PZA at a concentration of 60 µg/ml (light green), 100 µg/ml (medium green), or 500 µg/ml (dark green) initially significantly inhibited M. tuberculosis growth in 2D cell cultures compared to that in untreated controls. However, bacterial regrowth occurred, with M. tuberculosis luminescence returning to control levels by day 28. (C) PZA is bactericidal in the 3D system. M. tuberculosis growth was inhibited slightly by a low dose of PZA, while 500 µg/ml (dark green) was bactericidal. Gray lines with × symbols indicate the background levels of luminescence. Black arrows indicate points of antibiotic addition. Data are the mean ± the standard error of the mean of an experiment performed in triplicate and are representative of three separate experiments. (D) Bacteria were plated on 7H11 agar on day 28. CFU counts confirm that the luminescence data reflect bacterial loads. Note that dilutions of control and 2D PZA plates start from a 1:10 dilution, while other plates start without dilution. Representative plates are shown. Download FIG S3, EPS file, 1.6 MB (1.6MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Second-line antibiotics kill M. tuberculosis more rapidly in 3D microspheres than in 2D cell cultures when added on day 1. (A) M. tuberculosis growth in 7H9 broth. d-Cycloserine at low (20 µg/ml) and high (200 µg/ml) concentrations (light and dark purple, respectively) inhibit M. tuberculosis growth. However, bacterial regrowth occurred with the lower dose. INH (blue, 0.25 µg/ml) had a moderate effect on M. tuberculosis growth, and regrowth occurred. Moxifloxacin (brown, 5 µg/ml) and linezolid (magenta, 24 µg/ml) were equally effective but with delayed killing in comparison with high-dose d-cycloserine. M. tuberculosis growth was unaffected in control samples (black; gray with the diluent DMSO). (B) M. tuberculosis growth in 2D cell cultures. d-Cycloserine at a higher dose (dark purple) killed M. tuberculosis with an efficacy equal to that of the other antibiotics tested (5 µg/ml moxifloxacin [brown], 24 µg/ml linezolid [magenta], and 0.25 µg/ml INH [blue]). d-Cycloserine at a lower concentration inhibited M. tuberculosis growth but with a delay in comparison with the other antibiotics. (C) d-Cycloserine kills M. tuberculosis more rapidly in 3D cell cultures. All of the antibiotics investigated were similarly effective against M. tuberculosis, with rapid killing and no bacterial regrowth. Gray lines with × symbols indicate background luminescence. Black arrows indicate points of antibiotic addition. Data are the mean ± the standard error of the mean of an experiment performed in triplicate and are representative of three separate experiments. Download FIG S4, EPS file, 1.5 MB (1.5MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Antibiotics do not cause cytotoxicity within microspheres. Cellular cytotoxicity was investigated at day 21 within the 3D system with the CellTiter-Glo 3D cell viability assay. No antibiotic significantly changed cellular survival either in the absence of infection (A) or after M. tuberculosis infection (B). Similarly, cytotoxicity, analyzed by LDH release, is not different in infected cells treated with antibiotics (C). Download FIG S5, EPS file, 1.2 MB (1.3MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
High-dose rifampin for 6 h/day has efficacy equal to that of a constant standard antibiotic concentration. (A) M. tuberculosis growth inhibition in the microfluidic system at different concentrations of rifampin (0.25 [salmon], 1 [bright red], and 4 [dark red] µg/ml). A constant 1 µg/ml rifampin (blue) or addition of rifampin at a concentration of 1 µg/ml without daily washes (checkered box) produced inhibition of M. tuberculosis growth equivalent to that of a 4 µg/ml temporary peak concentration of rifampin, but no overnight regrowth occurred. Controls with DMSO (black) and without DMSO (white squares) were included. (B) Colony counts on 7H11 agar confirm that the luminescence data reflect the total M. tuberculosis loads within microspheres. Download FIG S6, EPS file, 2 MB (2MB, eps) .
Copyright © 2017 Bielecka et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.