

Abstract
Moyamoya disease (MMD) is an idiopathic disease associated with recurrent stroke. However, the pathogenesis of MMD remains unknown. Therefore, we performed long noncoding RNA (lncRNA) and messenger RNA (mRNA) expression profiles in blood samples from MMD patients (N = 15) and healthy controls (N = 10). A total of 880 differentially expressed lncRNAs (3649 probes) and 2624 differentially expressed mRNAs (2880 probes) were obtained from the microarrays of MMD patients and healthy controls (P < 0.05; Fold Change >2.0). Gene ontology (GO) and pathway analyses showed that upregulated mRNAs were enriched for inflammatory response, Toll-like receptor signaling pathway, chemokine signaling pathway and mitogen-activated protein kinase (MAPK) signaling pathway among others, while the downregulated mRNAs were enriched for neurological system process, digestion, drug metabolism, retinol metabolism and others. Our results showed that the integrated analysis of lncRNA-mRNA co-expression networks were linked to inflammatory response, Toll-like signaling pathway, cytokine-cytokine receptor interaction and MAPK signaling pathway. These findings may elucidate the pathogenesis of MMD, and the differentially expressed genes could provide clues to find key components in the MMD pathway.
Moyamoya disease (MMD) is a congenital disease that is characterized by stenosis of terminal internal carotid arteries and a hazy network of basal collaterals1. Recurrent stroke is common among MMD patients, and the standard treatment for MMD is revascularization surgery2,3. Although genome-wide and locus-specific association studies identified RNF213 as an important susceptibility gene of MMD4, few studies focus on the dysregulated genes and the pathogenesis of MMD still remains unknown.
Long noncoding RNAs (LncRNAs) are RNA molecules longer than 200 nucleotides without protein-coding ability5. Many studies have revealed a wide range of functional activities of lncRNAs6,7, including chromatin remodeling, transcriptional control and post-transcriptional processing. The dysregulation of lncRNAs might contribute to inflammatory response8, and several studies have reported that lncRNAs are associated with various inflammatory conditions9,10,11,12. TUG1 could decrease inflammation in vivo13, and ANRIL could regulate inflammatory responses through the NF-κB pathway14. JMJD1A and MALAT1 can reduce the activity of mitogen-activated protein kinase (MAPK) signaling in glioma cells and gastric cancer15,16, respectively. Genetic and environmental factors may play important roles in MMD development17. In a previous study, lncRNA expression profiles produced completely different clusters, and the MAPK signaling pathway was found to play a core role in this pathway network18. Co-expression analysis is widely used to elucidate the relationship between lncRNAs and messenger RNAs (mRNAs)19,20. It can elucidate the key lncRNAs and help to find a new regulation mechanism.
Understanding dysregulated lncRNAs and mRNAs is important for the diagnosis and treatment of patients with MMD. Therefore, we performed a microarray to examine lncRNA and mRNA expression profiles in blood samples from MMD patients and healthy controls. A total of 880 differentially expressed lncRNAs (3649 probes) and 2624 mRNAs (2880 probes) were identified. We further performed four co-expression networks in inflammatory response, the Toll-like signaling pathway, cytokine-cytokine receptor interaction and the MAPK signaling pathway. The integrated analysis of the differentially expressed lncRNAs and mRNAs may provide clues to find genes with active roles in pathogenesis of MMD.
Results
The clinical characteristics of included MMD patients
We included 15 MMD patients and 10 healthy controls in our study. The clinical characteristics of the included patients are shown in Table 1. There were 6 patients with intraventricular hemorrhage (IVH), 1 with subarachnoid hemorrhage (SAH), 3 with transient ischemic attack (TIA) and 5 with infarction as their initial clinical findings. Based on that, we grouped the patients into the hemorrhagic group (HG), the ischemic group (IG) and the ischemic and hemorrhagic group (IHG). The MMD and Control groups had similar age and sex distributions (Table 1).
Table 1. Clinical characteristics of included patients.
| Items | MMD | Control | |
|---|---|---|---|
| Number | 15 | 10 | |
| Sex | Male | 8 | 5 |
| Female | 7 | 5 | |
| Age (Mean ± SD) | Male | 32 ± 11.44 | 31.80 ± 10.30 |
| Female | 32.86 ± 10.57 | 32.52 ± 10.42 | |
| Initial Clinical | IVH | 6 | ND |
| SAH | 1 | ||
| TIA | 3 | ||
| Infarction | 5 | ||
| Subgroups | HG | 7 | ND |
| IG | 6 | ||
| HIG | 2 | ||
| mRS Score (Mean ± SD) | 1.8 ± 0.68 | ND | |
MMD, Moyamoya disease; IVH, intraventricular hemorrhage; SAH, subarachnoid hemorrhage; TIA, transient ischemic attack; HG, hemorrhagic group; IG, ischemic group; IHG, ischemic and hemorrhagic group; mRs, modified Rankin scale; ND, no data.
Identification of differentially expressed lncRNAs and mRNAs
The list of lncRNAs and their expression profiles were extracted in our previous study18. In brief, we identified 880 differentially expressed lncRNAs (3649 probes) and 2624 mRNAs (2880 probes) from the microarrays of MMD patients and healthy controls (P < 0.05; Fold Change >2.0) (Supplementary Table S1). Of those, 1746 upregulated mRNAs and 878 downregulated mRNAs were identified. A volcano plot was created and scatter analyses were conducted to identify differences among mRNAs (Fig. 1a,b). We further created a heat map of differentially expressed mRNAs (Fig. 2).
Figure 1. mRNA expression profile in MMD patients and healthy controls.

(a) Volcano plots of mRNAs expression levels between MMD and control group. (b) Scatter plots of mRNAs expression levels between the MMD and control groups. The red dots represented upregulated mRNAs, and the green dots represented downregulated mRNAs (P < 0.05; Fold Change >2.0).
Figure 2. Heat map of differentially expressed mRNAs of MMD patients and healthy controls.

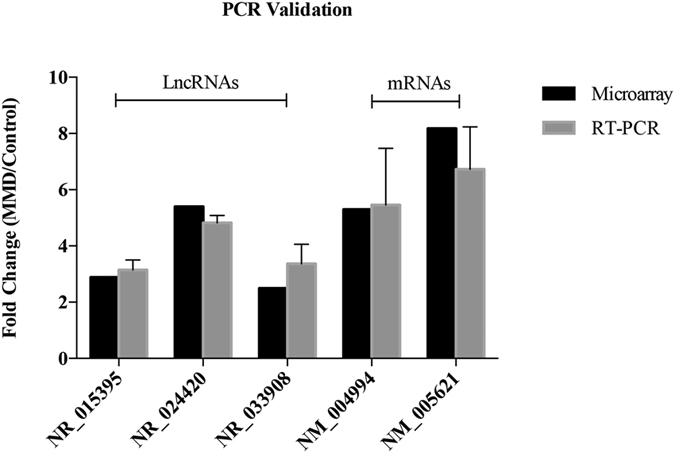
We also selected several differentially expressed genes randomly and further performed quantitative real-time polymerase chain reaction (qRT-PCR) to examine their expression levels. The qRT-PCR results suggest that the fold changes observed in the microarray analysis were robust (Fig. 3).
Figure 3. Validation of microarray results using quantitative real-time polymerase chain reaction (qRT-PCR).
Examination of the function of differentially expressed mRNAs
Gene ontology (GO) and KEGG pathway analyses were conducted to explore the function of the 2624 differentially expressed mRNAs using DAVID (The Database for Annotation, Visualization and Integrated Discovery). The results showed that upregulated genes were enriched for inflammatory response, response to wounding and defense response, etc. (Fig. 4a), while the downregulated genes were enriched for neurological system process, digestion and positive regulation of transcription from RNA polymerase II promoter, etc. (Fig. 4b). Moreover, the KEGG pathway analysis showed that the upregulated genes were enriched for Toll-like receptor signaling pathway, chemokine signaling pathway and MAPK signaling pathway, etc. (Fig. 4c), while the downregulated genes were enriched for drug metabolism, retinol metabolism and olfactory transduction, etc. (Fig. 4d).
Figure 4. Gene ontology and KEGG Pathway analysis of 2624 differentially expressed mRNAs.

(a) The top 10 GO terms upregulated in MMD patients compared with healthy controls. (b) The top 10 GO terms downregulated in MMD patients compared with healthy controls. (c) The top 10 pathways upregulated in MMD patients compared with healthy controls. (d) The top 10 pathways downregulated in MMD patients compared with healthy controls.
LncRNA-mRNA co-expression networks
We performed lncRNA-mRNA co-expression network analysis including 3649 lncRNAs probes and 2880 mRNAs probes. Our results showed that the co-expression networks were linked to inflammatory response, Toll-like signaling pathway, cytokine-cytokine receptor interaction and MAPK signaling pathway (Fig. 5). Thirty-two lncRNAs interacted with 11 mRNAs in the GO term of inflammatory response (Fig. 5a), 26 lncRNAs interacted with 2 mRNAs in the Toll-like signaling pathway (Fig. 5b), 41 lncRNAs interacted with 6 mRNAs in the cytokine-cytokine receptor interaction (Fig. 5c), and 15 lncRNAs interacted with 6 mRNAs in the MAPK signaling pathway (Fig. 5d).
Figure 5. LncRNA-mRNA co-expression network.

(a) 32 lncRNAs interacted with 11 mRNAs in the GO term of inflammatory response. (b) 26 lncRNAs interacted with 2 mRNAs in the Toll-like signaling pathway. (c) 41 lncRNAs interacted with 6 mRNAs in the Cytokine-cytokine receptor interaction. (d) 15 lncRNAs interacted with 6 mRNAs in the MAPK signaling pathway.
Discussion
MMD is a chronic occlusive cerebrovascular disease of unknown etiology21, and it is usually diagnosed by radiological findings, such as computed tomography (CT) perfusion and magnetic resonance imaging (MRI)22. It has been shown that MMD has a high prevalence in Asian countries, such as China, Japan and South Korea23,24,25,26,27. However, there are fewer studies focused on lncRNA-mRNA co-expression in MMD, and the molecular mechanisms behind MMDs remain poorly understood. It has been reported that lncRNAs play important roles in a wide range of functional activities6,7. The dysregulation of lncRNAs might contribute towards MMD. Therefore, the integrated analysis of the differentially expressed lncRNAs and mRNAs could help to reveal the pathogenesis of MMD.
Many studies have associated single nucleotide polymorphisms (SNPs) of genes with MMD28,29,30,31,32,33,34. RNF213 and MMP3 were proposed to be susceptibility genes for MMD28,30. It was reported that RNF213 is associated with immune response and that it might act cooperatively with other molecules under inflammatory signals based on bioinformatics data. IFNG and TNFA synergistically activated transcription of RNF213 both in vitro and in vivo35. However, IFNG and RNF213 were not co-expressed based on the mRNA microarray. The presence of a heterozygous genotype in TIMP2 promoter could be a genetic factor for familial MMD. Moreover, it was reported that TGFB1 could be involved in vascular growth and transformation processes and may play an important role in the development of MMD34,36. SNPs may affect the expression of genes for MMD. Based on the microarray database, RNF213 was not differentially expressed between MMDs and controls. However, TGFB1 and TIMP2 were found to be differentially expressed. These findings need to be validated in further studies including more samples.
The co-expression results were based on the expression of lncRNAs and mRNAs for MMD. It was reported that HIF1A could directly bind to the promoter of HOTAIR, which has been identified in a variety of carcinomas37. CXCR2 is involved in migration and activation of leukocytes and plays a key role in several inflammatory diseases38,39. MALAT1 could downregulate the expression of CXCR2 via miR-22–3p40. These were consistent with the results that HOTAIR and MALAT1 were highly associated with HIF1A and CXCR2, respectively (correlation coefficient −0.76, 0.87; P < 0.01), based on our microarray. To our knowledge, this is the first study of lncRNA-mRNA co-expression network analysis for patients with MMD. Dai et al. have conducted a serum miRNA signature in MMD, and it identified 94 differential expressed miRNAs41. Non-coding RNAs and mRNAs compete for binding to miRNAs by sharing one or more miRNA response elements (MREs) to regulate gene expression42, and the differentially expressed lncRNAs and mRNAs in our study partly correlate with these miRNAs. Therefore, future studies could focus on finding a competing endogenous RNAs (ceRNA) network with a key role in the pathogenesis of MMDs
Next, we investigated whether mRNAs tend to be neighbors with lncRNAs in MMD. We have performed all lncRNA-mRNA (63431 lncRNAs, 39887 mRNAs) co-expression analyses and lncRNA/mRNA reciprocal expression pattern analyses for MMD patients. Surprisingly, the genomic position and orientation of lncRNAs and mRNAs do have relativity with their correlation coefficient.
We further performed GO and KEGG pathway analyses, which could help us understand the pathogenesis of MMD and could provide new potential therapeutic targets. Moreover, the lncRNA-mRNA co-expression analysis showed that 32 lncRNAs interacted with 11 mRNAs in the GO of inflammatory response and 15 lncRNAs interacted with 6 mRNAs in the MAPK signaling pathway (Fig. 5a,d). Association of differentially expressed lncRNAs and mRNAs with pathways relevant to MMD pathogenesis may partly explain the etiology of MMD. Inflammatory response leads to the hyperplasia of intimal vascular smooth muscle cells (VSMCs), which causes lumen stenosis of MMDs43,44. The MAPK signaling pathway played important roles in vascular pathological processes45,46,47 and vascular inflammation48. It was reported that many cytokines induce the proliferation of VSMCs via MAPK signaling pathway49,50,51. MAPK signaling pathway inhibitors have been successfully used to treat atherosclerosis in vivo52. Our results can elucidate key lncRNAs and provide leads to further understand the pathogenesis of MMD.
There are limitations to our study. There were only 15 MMD patients and 10 healthy controls included in our analysis, and we could still identify valuable genes and pathways from our database. Further studies would enlarge the sample size. Moreover, the results were obtained from the bioinformatic analysis and microarray analysis. Therefore, further studies are needed to confirm these differentially expressed genes and pathway mechanisms with experiments. This manuscript is our preliminary work, and further work remains to be done.
In conclusion, we identified 880 differentially expressed lncRNAs (3649 probes) and 2624 differentially expressed mRNAs (2880 probes) from the microarray of MMD patients and healthy controls. The upregulated differentially expressed mRNAs were enriched for inflammatory response, the Toll-like receptor signaling pathway, chemokine signaling pathway and the MAPK signaling pathway. The integrated analysis also indicated interregulation in MMD patients. These findings may reveal the pathogenesis of MMD, and future studies should focus on the inflammatory response and MAPK signaling pathway for MMD patients.
Methods
Patients and Samples
We enrolled 15 MMD patients who had been diagnosed with MMD according to the characteristic angiographic findings with strict inclusion criteria in Beijing Tiantan Hospital and 10 healthy controls in our study. Informed consent was obtained at enrolment and the basic characteristics of all MMD patients and healthy controls are summarized in Table 1. Our study was approved by the Ethics Committee in Beijing Tiantan hospital. The study was carried out in accordance with the Declaration of Helsinki, and all methods were performed in accordance with the relevant guidelines and regulations.
We obtained the peripheral blood anticoagulated with ethylene diamine tetraacetic acid (EDTA) from all patients and healthy controls. Further, we extracted the RNA by using TRIzol reagent (Invitrogen, Grand Island, NY, USA) according to the manufacturer’s instructions and quality evaluations were performed using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
LncRNA and mRNA Microarrays
The Agilent Human 4 × 180 K lncRNA and mRNA Microarrays (Agilent, Santa Clara, CA, USA) were performed using a Gene Expression Hybridization Kit (Agilent, Santa Clara, CA, US) according to the manufacturer’s instructions. Slides were washed in staining dishes with a Gene Expression Wash Buffer Kit (Agilent, Santa Clara, CA, USA) and scanned by an Agilent Microarray Scanner (Agilent, Santa Clara, CA, USA) with default settings according to the manufacturer’s instructions. Raw data were normalized by Quantile algorithm using Gene Spring Software 12.6 (Agilent Technologies). The differentially expressed lncRNAs and mRNAs were identified by using R software (version 3.2.3) with the samr package53.
Quantitative Real-time Polymerase Chain Reaction (qRT-PCR)
We selected several differentially expressed genes to detect their expression in MMD. Total RNAs were isolated from all samples using PAXgene Blood RNA Kit (Qiagen, Germany) and then reverse transcribed using an iScript cDNA synthesis Kit (Bio-Rad, USA) according to the manufacturer’s instructions. qRT-PCR was performed using SYBR Select Master Mix (Applied Biosystems, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control, and all the primer sequences are shown below:
NR_015395-F: 5′-CTAATTTGCCACCACCCTGT-3′
NR_015395-R: 5′-AAGACCCAGATGCCGTTTTA-3′
NR_024420-F: 5′-CTGCAACGAATCCCAAAAGT-3′
NR_024420-R: 5′-ACCACTTTCCAGAGGCTGAA-3′
NR_033908-F: 5′-CGAGCTGTAAAAGCCAAAGG-3′
NR_033908-R: 5′-CCTGGGCGATAAGAGTGAAA-3′
NM_004994-F: 5′-TGTACCGCTATGGTTACACTCG-3′
NM_004994-R: 5′-GGCAGGGACAGTTGCTTCT-3′
NM_005621-F: 5′-ATTGAGGGGTTAACATTAGGCTG-3′
NM_005621-R: 5′-GATATTCTTGATGGTGTTTGCAAGC-3′
GAPDH-F: 5′-AGGGCTGCTTTTAACTCTGGT-3′
GAPDH-R: 5′-CCCCACTTGATTTTGGAGGGA-3′
Statistical Analysis
All statistical data were analyzed by using SPSS (version 22; SPSS Inc., Chicago, IL, USA) and R software (version 3.2.3). Genes with a two-sided P value of <0.05 and Fold Change >2.0 were regarded as statistically significant genes. The P value was false discovery rate (FDR) corrected. LncRNAs-mRNAs co-expression networks were constructed by Cytoscape software54 (version 3.4.0; The Cytoscape Consortium, San Diego, CA, USA).
Additional Information
How to cite this article: Wang, W. et al. Integrated Analysis of LncRNA-mRNA Co-Expression Profiles in Patients with Moyamoya Disease. Sci. Rep. 7, 42421; doi: 10.1038/srep42421 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Material
Acknowledgments
This work was supported by grants from National High Technology Research and Development Program (No. 2012AA02A508), National Natural Science Foundation of China (No. 81371292), the “13th Five-Year Plan” National Science and Technology supporting plan (No. 2015BAI09B04).
Footnotes
The authors declare no competing financial interests.
Author Contributions W.W. and F.L.G. wrote the main manuscript text. Z.Z., H.Y.W., L.Z., D.Z. and Y.Z. prepared Figures 1–5. Q.L. and J.F.W. collected clinical samples. J.Z.Z. designed the protocol of the study. All authors reviewed the manuscript.
References
- Suzuki J. & Takaku A. Cerebrovascular “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch Neurol 20, 288–299 (1969). [DOI] [PubMed] [Google Scholar]
- Narisawa A., Fujimura M. & Tominaga T. Efficacy of the revascularization surgery for adult-onset moyamoya disease with the progression of cerebrovascular lesions. Clin Neurol Neurosurg 111, 123–126, doi: 10.1016/j.clineuro.2008.09.022 (2009). [DOI] [PubMed] [Google Scholar]
- Fukui M. Guidelines for the diagnosis and treatment of spontaneous occlusion of the circle of Willis (‘moyamoya’ disease). Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease) of the Ministry of Health and Welfare, Japan. Clin Neurol Neurosurg 99 Suppl 2, S238–240 (1997). [PubMed] [Google Scholar]
- Fujimura M. et al. Genetics and Biomarkers of Moyamoya Disease: Significance of RNF213 as a Susceptibility Gene. J Stroke 16, 65–72, doi: 10.5853/jos.2014.16.2.65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk M. S., Higgs D. R. & Gingeras T. R. Molecular biology: RNA discrimination. Nature 482, 310–311, doi: 10.1038/482310a (2012). [DOI] [PubMed] [Google Scholar]
- Mercer T. R., Dinger M. E. & Mattick J. S. Long non-coding RNAs: insights into functions. Nat Rev Genet 10, 155–159, doi: 10.1038/nrg2521 (2009). [DOI] [PubMed] [Google Scholar]
- Fang Y. & Fullwood M. J. Roles, Functions, and Mechanisms of Long Non-coding RNAs in Cancer. Genomics Proteomics Bioinformatics 14, 42–54, doi: 10.1016/j.gpb.2015.09.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heward J. A. & Lindsay M. A. Long non-coding RNAs in the regulation of the immune response. Trends Immunol 35, 408–419, doi: 10.1016/j.it.2014.07.005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. et al. The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL. Proc Natl Acad Sci USA 111, 1002–1007, doi: 10.1073/pnas.1313768111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhlmuller B. et al. Detection of oncofetal h19 RNA in rheumatoid arthritis synovial tissue. Am J Pathol 163, 901–911, doi: 10.1016/S0002-9440(10)63450-5 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. et al. Long noncoding RNA related to cartilage injury promotes chondrocyte extracellular matrix degradation in osteoarthritis. Arthritis Rheumatol 66, 969–978, doi: 10.1002/art.38309 (2014). [DOI] [PubMed] [Google Scholar]
- Pearson M. J. et al. Long Intergenic Noncoding RNAs Mediate the Human Chondrocyte Inflammatory Response and Are Differentially Expressed in Osteoarthritis Cartilage. Arthritis Rheumatol 68, 845–856, doi: 10.1002/art.39520 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S. et al. Overexpression of the long noncoding RNA TUG1 protects against cold-induced injury of mouse livers by inhibiting apoptosis and inflammation. FEBS J 283, 1261–1274, doi: 10.1111/febs.13660 (2016). [DOI] [PubMed] [Google Scholar]
- Zhou X. et al. Long non-coding RNA ANRIL regulates inflammatory responses as a novel component of NF-kappaB pathway. RNA Biol 13, 98–108, doi: 10.1080/15476286.2015.1122164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y. et al. Tumor-suppressive function of long noncoding RNA MALAT1 in glioma cells by downregulation of MMP2 and inactivation of ERK/MAPK signaling. Cell Death Dis 7, e2123, doi: 10.1038/cddis.2015.407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. et al. Elevated JMJD1A is a novel predictor for prognosis and a potential therapeutic target for gastric cancer. Int J Clin Exp Pathol 8, 11092–11099 (2015). [PMC free article] [PubMed] [Google Scholar]
- Bang O. Y., Fujimura M. & Kim S. K. The Pathophysiology of Moyamoya Disease: An Update. J Stroke 18, 12–20, doi: 10.5853/jos.2015.01760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F. et al. Long Noncoding RNAs and Their Regulatory Network: Potential Therapeutic Targets for Adult Moyamoya Disease. World Neurosurgery 93, 111–119, doi: 10.1016/j.wneu.2016.05.081 (2016). [DOI] [PubMed] [Google Scholar]
- Xu J. et al. Differentially expressed lncRNAs and mRNAs identified by microarray analysis in GBS patients vs healthy controls. Sci Rep 6, 21819, doi: 10.1038/srep21819 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J. et al. Microarray Analysis of lncRNA and mRNA Expression Profiles in Patients with Neuromyelitis Optica. Mol Neurobiol, doi: 10.1007/s12035-016-9754-0 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. S. Moyamoya Disease: Epidemiology, Clinical Features, and Diagnosis. J Stroke 18, 2–11, doi: 10.5853/jos.2015.01627 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda S. & Houkin K. Moyamoya disease: current concepts and future perspectives. Lancet Neurol 7, 1056–1066, doi: 10.1016/S1474-4422(08)70240-0 (2008). [DOI] [PubMed] [Google Scholar]
- Kuriyama S. et al. Prevalence and clinicoepidemiological features of moyamoya disease in Japan: findings from a nationwide epidemiological survey. Stroke 39, 42–47, doi: 10.1161/STROKEAHA.107.490714 (2008). [DOI] [PubMed] [Google Scholar]
- Han D. H. et al. A co-operative study: clinical characteristics of 334 Korean patients with moyamoya disease treated at neurosurgical institutes (1976–1994). The Korean Society for Cerebrovascular Disease. Acta Neurochir (Wien) 142, 1263–1273; discussion 1273–1264 (2000). [DOI] [PubMed]
- Hung C. C., Tu Y. K., Su C. F., Lin L. S. & Shih C. J. Epidemiological study of moyamoya disease in Taiwan. Clin Neurol Neurosurg 99 Suppl 2, S23–25 (1997). [DOI] [PubMed] [Google Scholar]
- Miao W. et al. Epidemiological and clinical features of Moyamoya disease in Nanjing, China. Clin Neurol Neurosurg 112, 199–203, doi: 10.1016/j.clineuro.2009.11.009 (2010). [DOI] [PubMed] [Google Scholar]
- Duan L. et al. Moyamoya disease in China: its clinical features and outcomes. Stroke 43, 56–60, doi: 10.1161/STROKEAHA.111.621300 (2012). [DOI] [PubMed] [Google Scholar]
- Kamada F. et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet 56, 34–40, doi: 10.1038/jhg.2010.132 (2011). [DOI] [PubMed] [Google Scholar]
- Kang H. S. et al. Single nucleotide polymorphisms of tissue inhibitor of metalloproteinase genes in familial moyamoya disease. Neurosurgery 58, 1074–1080; discussion 1074–1080, doi: 10.1227/01.NEU.0000215854.66011.4F (2006). [DOI] [PubMed] [Google Scholar]
- Li H. et al. Association of a functional polymorphism in the MMP-3 gene with Moyamoya Disease in the Chinese Han population. Cerebrovasc Dis 30, 618–625, doi: 10.1159/000319893 (2010). [DOI] [PubMed] [Google Scholar]
- Liu C. et al. Analysis of TGFB1 in European and Japanese Moyamoya disease patients. Eur J Med Genet 55, 531–534, doi: 10.1016/j.ejmg.2012.05.002 (2012). [DOI] [PubMed] [Google Scholar]
- Liu W. et al. A rare Asian founder polymorphism of Raptor may explain the high prevalence of Moyamoya disease among East Asians and its low prevalence among Caucasians. Environ Health Prev Med 15, 94–104, doi: 10.1007/s12199-009-0116-7 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y. S. et al. Age-specific eNOS polymorphisms in moyamoya disease. Childs Nerv Syst 27, 1919–1926, doi: 10.1007/s00381-011-1504-z (2011). [DOI] [PubMed] [Google Scholar]
- Roder C. et al. Polymorphisms in TGFB1 and PDGFRB are associated with Moyamoya disease in European patients. Acta Neurochir (Wien) 152, 2153–2160, doi: 10.1007/s00701-010-0711-9 (2010). [DOI] [PubMed] [Google Scholar]
- Ohkubo K. et al. Moyamoya disease susceptibility gene RNF213 links inflammatory and angiogenic signals in endothelial cells. Sci Rep 5, 13191, doi: 10.1038/srep13191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojo M. et al. Role of transforming growth factor-beta1 in the pathogenesis of moyamoya disease. J Neurosurg 89, 623–629, doi: 10.3171/jns.1998.89.4.0623 (1998). [DOI] [PubMed] [Google Scholar]
- Zhou C. et al. Long noncoding RNA HOTAIR, a hypoxia-inducible factor-1alpha activated driver of malignancy, enhances hypoxic cancer cell proliferation, migration, and invasion in non-small cell lung cancer. Tumour Biol 36, 9179–9188, doi: 10.1007/s13277-015-3453-8 (2015). [DOI] [PubMed] [Google Scholar]
- Bizzarri C. et al. ELR + CXC chemokines and their receptors (CXC chemokine receptor 1 and CXC chemokine receptor 2) as new therapeutic targets. Pharmacol Ther 112, 139–149, doi: 10.1016/j.pharmthera.2006.04.002 (2006). [DOI] [PubMed] [Google Scholar]
- Muller-Edenborn B. et al. Volatile anaesthetics reduce neutrophil inflammatory response by interfering with CXC receptor-2 signalling. Br J Anaesth 114, 143–149, doi: 10.1093/bja/aeu189 (2015). [DOI] [PubMed] [Google Scholar]
- Tang Y. et al. The lncRNA MALAT1 protects the endothelium against ox-LDL-induced dysfunction via upregulating the expression of the miR-22-3p target genes CXCR2 and AKT. FEBS Lett 589, 3189–3196, doi: 10.1016/j.febslet.2015.08.046 (2015). [DOI] [PubMed] [Google Scholar]
- Dai D. et al. Serum miRNA signature in Moyamoya disease. PLoS One 9, e102382, doi: 10.1371/journal.pone.0102382 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmena L., Poliseno L., Tay Y., Kats L. & Pandolfi P. P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146, 353–358, doi: 10.1016/j.cell.2011.07.014 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takekawa Y., Umezawa T., Ueno Y., Sawada T. & Kobayashi M. Pathological and immunohistochemical findings of an autopsy case of adult moyamoya disease. Neuropathology 24, 236–242 (2004). [DOI] [PubMed] [Google Scholar]
- Weinberg D. G. et al. Moyamoya disease: a review of histopathology, biochemistry, and genetics. Neurosurg Focus 30, E20, doi: 10.3171/2011.3.FOCUS1151 (2011). [DOI] [PubMed] [Google Scholar]
- Park K. M., Chen A. & Bonventre J. V. Prevention of kidney ischemia/reperfusion-induced functional injury and JNK, p38, and MAPK kinase activation by remote ischemic pretreatment. J Biol Chem 276, 11870–11876, doi: 10.1074/jbc.M007518200 (2001). [DOI] [PubMed] [Google Scholar]
- Nordmeyer J. et al. Upregulation of myocardial estrogen receptors in human aortic stenosis. Circulation 110, 3270–3275, doi: 10.1161/01.CIR.0000147610.41984.E8 (2004). [DOI] [PubMed] [Google Scholar]
- Hu Y. et al. Icariin Attenuates High-cholesterol Diet Induced Atherosclerosis in Rats by Inhibition of Inflammatory Response and p38 MAPK Signaling Pathway. Inflammation 39, 228–236, doi: 10.1007/s10753-015-0242-x (2016). [DOI] [PubMed] [Google Scholar]
- Paudel K. R., Karki R. & Kim D. W. Cepharanthine inhibits in vitro VSMC proliferation and migration and vascular inflammatory responses mediated by RAW264.7. Toxicol In Vitro 34, 16–25, doi: 10.1016/j.tiv.2016.03.010 (2016). [DOI] [PubMed] [Google Scholar]
- Cui Y. et al. Platelet-derived growth factor-BB induces matrix metalloproteinase-2 expression and rat vascular smooth muscle cell migration via ROCK and ERK/p38 MAPK pathways. Mol Cell Biochem 393, 255–263, doi: 10.1007/s11010-014-2068-5 (2014). [DOI] [PubMed] [Google Scholar]
- Suwanabol P. A. et al. Transforming growth factor-beta increases vascular smooth muscle cell proliferation through the Smad3 and extracellular signal-regulated kinase mitogen-activated protein kinases pathways. J Vasc Surg 56, 446–454, doi: 10.1016/j.jvs.2011.12.038 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Garrido A. et al. Transforming growth factor beta inhibits platelet derived growth factor-induced vascular smooth muscle cell proliferation via Akt-independent, Smad-mediated cyclin D1 downregulation. PLoS One 8, e79657, doi: 10.1371/journal.pone.0079657 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M. et al. Activation of the p38 MAP kinase pathway is required for foam cell formation from macrophages exposed to oxidized LDL. APMIS 110, 458–468 (2002). [DOI] [PubMed] [Google Scholar]
- Huang X., Hao C., Bao H., Wang M. & Dai H. Aberrant expression of long noncoding RNAs in cumulus cells isolated from PCOS patients. J Assist Reprod Genet 33, 111–121, doi: 10.1007/s10815-015-0630-z (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504, doi: 10.1101/gr.1239303 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.