

Abstract
Relapse to drug use can be initiated by drug-associated cues. The intensity of cue-induced relapse is correlated with the induction of transient synaptic potentiation (t-SP) at glutamatergic synapses on medium spiny neurons (MSNs) in the nucleus accumbens core (NAcore) and requires spillover of glutamate from prefrontal cortical afferents. We used a rodent self-administration/reinstatement model of relapse to show that cue-induced t-SP and reinstated cocaine seeking result from glutamate spillover, initiating a metabotropic glutamate receptor 5 (mGluR5)-dependent increase in nitric oxide (NO) production. Pharmacological stimulation of mGluR5 in NAcore recapitulated cue-induced reinstatement in the absence of drug-associated cues. Using NO-sensitive electrodes, mGluR5 activation by glutamate was shown to stimulate NO production that depended on activation of neuronal nitric oxide synthase (nNOS). nNOS is expressed in ∼1% of NAcore neurons. Using a transgene strategy to express and stimulate designer receptors that mimicked mGluR5 signaling through Gq in nNOS interneurons, we recapitulated cue-induced reinstatement in the absence of cues. Conversely, using a transgenic caspase strategy, the intensity of cue-induced reinstatement was correlated with the extent of selective elimination of nNOS interneurons. The induction of t-SP during cued reinstatement depends on activating matrix metalloproteinases (MMPs) and selective chemogenetic stimulation of nNOS interneurons recapitulated MMP activation and t-SP induction (increase in AMPA currents in MSNs). These data demonstrate critical involvement of a sparse population of nNOS-expressing interneurons in cue-induced cocaine seeking, revealing a bottleneck in brain processing of drug-associated cues where therapeutic interventions could be effective in treating drug addiction.
SIGNIFICANCE STATEMENT Relapse to cocaine use in a rat model is associated with transient increases in synaptic strength at prefrontal cortex synapses in the nucleus accumbens. We demonstrate the sequence of events that mediates synaptic potentiation and reinstated cocaine seeking induced by cocaine-conditioned cues. Activation of prefrontal inputs to the accumbens by cues initiates spillover of synaptic glutamate, which stimulates metabotropic glutamate receptor 5 (mGluR5) on a small population of interneurons (∼1%) expressing neuronal nitric oxide synthase. Stimulating these glutamate receptors increases nitric oxide (NO) production, which stimulates matrix metalloprotease-2 (MMP-2) and MMP-9 activity in the extracellular space. Manipulating the interaction between mGluR5, NO production, or MMP-2 and MMP-9 pharmacologically or genetically is sufficient to recapitulate transient synaptic potentiation and reinstate cocaine seeking.
Keywords: cocaine, glutamate, metabotropic glutamate receptor, MMP, nitric oxide, relapse
Introduction
Addiction is a chronic relapsing disorder characterized by loss of control over drug use. Drug-associated cues can initiate drug use, even after protracted abstinence, and the nucleus accumbens core (NAcore) serves as a portal whereby cue-induced activity in cortical and limbic projections initiates goal directed behaviors, including drug seeking (Cardinal and Everitt, 2004; Koob and Volkow, 2010). Accordingly, synaptic plasticity within these glutamatergic projections to the NAcore strongly contributes to drug relapse vulnerability (Conrad et al., 2008; Ma et al., 2014; Pascoli et al., 2014). A common rodent model of cocaine relapse is the self-administration reinstatement model, in which rats or mice are trained to press a lever to receive an intravenous infusion of cocaine (Shaham et al., 2003). During self-administration, drug infusions are paired to discrete light and tone cues and these cues become conditioned stimuli through Pavlovian learning. After self-administration, the rodent enters a period of extinction training in which lever pressing has no programmed consequences and drug-seeking behavior decreases rapidly. Relapse is then modeled by reintroducing the conditioned stimulus, robustly reinstating drug-seeking behavior without achieving drug access. Using this model, after extinction of cocaine or nicotine use, cortico-accumbens synapses exist in a persistently potentiated state and show impaired LTP or LTD under some experimental conditions (Conrad et al., 2008; Moussawi et al., 2009; Gipson et al., 2013b). However, presenting drug-associated cues elicits transient synaptic potentiation (t-SP) in NAcore medium spiny neurons (MSNs) that is required for cues to reinstate drug, but not sucrose seeking (Anderson et al., 2008; Shen et al., 2011; Gipson et al., 2013a; Gipson et al., 2013b). Importantly, the magnitude of t-SP is positively correlated with the extent of cocaine-seeking behavior (Gipson et al., 2013a). Recently, we found that, akin to electrically stimulated LTP in the hippocampus (Huntley, 2012), cue-induced t-SP and reinstated drug seeking require activating matrix metalloproteinase-2 (MMP-2) and MMP-9 (Smith et al., 2014). However, it remains unknown how presenting drug-conditioned cues activates MMPs to induce the t-SP necessary for reinstating drug seeking.
The induction of t-SP and drug seeking by conditioned cues requires activity in the prelimbic prefrontal cortex (PL-PFC; Gipson et al., 2013a; Stefanik et al., 2016), and reinstated drug-seeking is associated with the spillover of synaptic glutamate in the NAcore that is prevented by inhibiting neuronal activity in the PL-PFC (McFarland et al., 2003; LaLumiere and Kalivas, 2008). This poses the possibility that the spillover of synaptic glutamate in the NAcore activates MMPs and thereby induces t-SP and reinstated behavior. Consistent with this possibility, synaptic glutamate spillover is a mechanism for extrasynaptic communication and recruiting interneuronal microcircuits (Coddington et al., 2013) and can stimulate extrasynaptic metabotropic glutamate receptors (mGluRs) known to regulate drug seeking (Mitrano et al., 2010; Kupchik et al., 2012). Supporting a mechanistic linkage between glutamate spillover and t-SP as a critical mediator of relapse, many of the cue-induced synaptic events (e.g., glutamate spillover, t-SP, and/or MMP activation) are shared across classes of addictive drugs, including cocaine, methamphetamine, heroin, alcohol (Smith et al., 2011), and nicotine (Van Den Oever et al., 2010; Kupchik et al., 2012; Natarajan et al., 2013; Shen et al., 2014; Smith et al., 2014).
Here, we sought to further characterize the sequence of cellular events initiated in the NAcore by a drug-conditioned cue that leads to reinstated drug seeking. Specifically, we explored the mechanistic linkage between cue-induced synaptic glutamate spillover and activation of MMPs and t-SP in rats and mice trained to self-administer cocaine. We found that cue-induced glutamate spillover caused transient mGluR5-dependent production of nitric oxide (NO) in the sparse population of NAcore interneurons expressing neuronal nitric oxide synthase (nNOS). Moreover, the increase in NO activated MMP-2 via S-nitrosylation and thereby initiated t-SP and reinstated cocaine seeking.
Materials and Methods
Animal housing
Both rats and mice were single housed in a humidity- and temperature-controlled environment. Male Sprague-Dawley rats (250 g; Charles River Laboratories) and NOS1-Cre-transgenic mice (∼25 g; Jackson Laboratories #017526 B6.129-Nos1tm1(cre_Mgmj/J; RRID MGI:4947130) were individually housed with a 12:12 h dark/light cycle. All experiments occurred during the dark phase and rats/mice were allowed to acclimate to the vivarium environment for at least 4 d before surgery.
Rat surgery, drug self-administration, and reinstatement
Rats were anesthetized with a combination of ketamine HCl and xylazine and received ketorolac for analgesia. All rats received intrajugular catheters and, for microinjection experiments, intracranial cannula were targeted 2 mm above the NAcore for rats (mm relative to bregma +1.8 A/P, +2.1 M/L, −5.5 D/V; Paxinos and Watson, 2007). Rats were food restricted to 25 g of rat chow per day, given an NSAID (ketorolac 2 mg/kg, i.p.) and antibiotics (Cefazolin 200 mg/kg, i.v., and topical triple antibiotic ointment), and allowed to recover for 4–6 d. All methods used comply with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina. After 5 d of recovery from surgery, rats underwent one overnight (12 h) food training session. The next day, rats began daily 2 h self-administration (SA) sessions. During SA, cocaine was delivered using an FR1 schedule with a 20 s timeout after each infusion. Active lever presses that resulted in cocaine infusion simultaneously resulted in presentation of a compound light (above the active lever) and tone (2900 Hz) conditioning stimulus. An inactive lever was also provided to control for nonmotivated responding. After 10 SA sessions at ≥10 infusions/d, rats began extinction training, during which all programmed consequences were removed from lever pressing. Extinction training lasted at least 10 d, or until two consecutive days ≤25 active lever presses. Reinstatement was induced by presentation of light/tone cues after an active lever press. Microinjections of N-propyl l-arginine hydrochloride (NPLA), (RS)-2-chloro-5-hydroxyphenylglycine (CHPG; 10 μg), clozapine N-oxide (CNO) (1 μg), or vehicle were given 15 min before beginning reinstatement in most cases, or 15 min before gel infusion for zymography experiments. For behavioral experiments, a within-subject crossover design was used. In this paradigm, each rat received each condition according to a Latin square design. Rats were required to meet extinction criteria before each reinstatement test. Reinstatement sessions lasted 120 min in the behavioral experiment, and for zymography experiments reinstatement sessions were 15 min long, at which point rats were anesthetized with 100 mg of pentobarbital, then transcardially perfused with 4% paraformaldehyde for further measurements. For Western blotting experiments, rats underwent 15 min reinstatement sessions and then were killed via rapid decapitation.
Mouse surgery, drug self-administration, and reinstatement
Mice (25–30 g) were anesthetized with isofluorane (induction 3–5% v/v, maintenance 1–2% v/v) and implanted with an indwelling jugular catheter connected to a head mounted entry port as described previously (Griffin et al., 2007). AAV viral vector (AAV2-hSyn-DIO-hM4Dq-mCherry; titer 6 × 1012 or AAV2-Flex-taCasp3-TEVp; titer 1.8 × 1012) was infused in the nucleus accumbens (in mm, relative to bregma: +1.5 A/P, ±1.3 M/L, −4.4 D/V) based on (Paxinos and Franklin, 2012). For microinjection experiments in mice intracranial cannula were targeted 1 mm above the NAcore (+1.5 A/P, +1.3 M/L, −3.6 D/V). Mice were given NSAID (carprofen 5 mg/kg s.c.) and antibiotics (Cefazolin 200 mg/kg i.v. and topical triple antibiotic ointment) and were allowed to recover for 4–6 d. Catheters were maintained by flushing twice daily with heparinized saline throughout the procedure and catheter patency was checked at the end of the self-administration phase for all mice using Brevital (0.2 mg, i.v.). Mice in which apparent signs of hypnosis were not observed within 3 s were excluded from the study. Mice were trained for 10–15 d on active cocaine self-administration until they reached the criterion of at least 3 d of 15 cocaine infusions. During the first two sessions, mice were mildly food restrained to stimulate exploration of the operant box. During daily 2 h training sessions, presses on the active lever resulted in the infusion of cocaine (∼1 mg/kg/infusion in saline) paired with a compound cue stimulus (2 s tone + light, 20 s time out). After self-administration, mice were extinguished for at least 6 d until criterion was reached (2 d <15 presses on the active lever) and given the following tests in a counterbalanced within-subject design with at least 2 d of additional extinction in between. Before each reinstatement test, mice were given an injection of CNO or vehicle (3 mg/kg, i.p., dissolved in 0.5% DMSO) and placed into their home cages for 30 min. Cue induced reinstatement tests involved the reintroduction of the compound stimulus and extinction tests involved placing the mice back into the extinguished context.
Drugs
Drugs used include NPLA (Tocris Bioscience; nNOS Ki = 57 nm 3158-fold selectivity over iNOS, 149-fold specificity over eNOS); CHPG (Tocris Bioscience); and CNO, 3-((2-methyl-4-thiazolyl)ethynyl)pyridine (MTEP; Tocris Bioscience).
In vivo microdialysis
Microdialysis probes were constructed as described previously (Pierce et al., 1996) except both the inlet and outlet tubing consisted of fused silica. The active region of the dialysis membrane was between 2 mm in length and ∼0.25 mm in diameter. After cocaine self-administration and extinction, the probes were inserted through the guide cannulae into the NAcore the night before the reinstatement trial. Subjects were housed overnight in the operant chamber after insertion of the dialysis probe. Dialysis buffer [artificial CSF (aCSF); 5 mm glucose, 140 mm NaCl, 1.4 mm CaCl2, 1.2 mm MgCl2, and 0.15% phosphate buffer saline, pH 7.4) was advanced through the microdialysis probes at a rate of 0.2 μl/min overnight and advanced to 2 μl/min in the morning. Two hours later, four 20 min baseline samples were collected (samples 1–4). Two additional 20 min samples were then collected with the addition of MTEP (100 μm) or LY379268 (100 μm) to the dialysis buffer (samples 5–6). After these six samples were collected, the cue (light/tone) presentation was restored to active lever presses (i.e., the animal underwent a cued reinstatement trial) or active lever pressing continued to have no programmed consequence (i.e., the animal underwent an extinction trial). Once the reinstatement or extinction trial commenced, another six 20 min dialysis samples were collected (samples 7–12). After a 2 h operant session, an additional three samples were collected (samples 13–15). In some cases, a liquid switch was turned back to aCSF from either LY379368 or MTEP. After collecting the last sample, the dialysis probe was removed and the rat underwent at least three daily extinction trials before another dialysis experiment was conducted with a dialysis probe being inserted in the contralateral NAcore from the first trial. The treatment order of dialysis within animal was randomized and each animal underwent a maximum of two dialysis experiments (one experiment in each brain hemisphere). A total of 28 rats were used for data analysis and, of a possible 56 experiments, data were used from 39. For the remainder of the experiments, the data were not included for one of the following four reasons: (1) one of the guide cannula was blocked, (2) the HPLC quantification failed, (3) histology was absent, or (4) the dialysis probe failed.
Dialysis sample quantification
Glutamate levels were analyzed using an HPLC system with electrochemical detection. The mobile phase consisted of MeOH (15% v/v), acetylnitrile (2.5% v/v), and 100 mm sodium dihydrogen phosphate monobasic, pH 6.0. Precolumn derivatization with o-phthalaldehyde was performed using an ESA Model 540 autosampler. Separation was done with a Shiseido column and glutamate was detected by a spectrophotometer (ESA). The glutamate content in each sample was analyzed by area under the curve and compared with an external standard curve for quantification. Of a total of 615 samples quantified from the 41 individual dialysis experiments (i.e., 15 samples/experiment), the value of 17 samples was interpolated from the value of adjacent samples due to the samples missing.
Locomotor activity
Locomotor activity was monitored over 5 min intervals in a photocell apparatus (Omnitech). Motor activity was quantified as distance traveled (determined by consecutive breaking of adjacent photobeams). Each rat was examined only once after a bilateral microinjection of aCSF, CHPG, NPLA, or cyclosomatostatin (c-SRIF) into the NAcore. For the CHPG study, rats were habituated to the activity chambers the day before testing because we hypothesized that CHPG might increase activity. Conversely, because we hypothesized that NPLA and c-SRIF would decrease motor activity, animal activity was quantified the first time they were exposed to the environment. Microinjections were made 5 min before placing the rats into the chamber and photocell beam breaks were quantified for a total of 60 min.
Measurement of MMP activity
Because MMPs are secreted in inactive proforms and catalytically activated within the ECM, activity assays are preferable to immunoblotting for protein content for assessing changes in MMP function (Kupai et al., 2010). We used an in vivo zymography assay to measure MMP activity directly. Dye-quenched gelatin is an MMP-2/MMP-9 substrate containing intramolecularly quenched FITC fluorophores that cannot fluoresce until they are proteolytically processed by MMP-2 or MMP-9 (Bozdagi et al., 2007). The amount of fluorescence produced forms a linear relationship with incubation time and MMP activity (see Figs. 2; 3; 4; 5; 6, and 7). Dye-quenched FITC-gelatin (Invitrogen) was reconstituted in PBS at 1 mg/ml, pH 7.2–7.4. Then, 3.0 μl of gel (1.5 μl/side) was microinjected 15 min before administering an overdose of pentobarbital (100 mg/kg, i.p.) and beginning transcardial perfusion of 4% paraformaldehyde (PFA). Brains were removed, placed in 4% PFA for 90 min for additional fixation, and a vibratome was used to obtain 50 μm sections through the nucleus accumbens. Sections were mounted and coverslipped. Fluorescence was excited with a 488 nm argon laser, emissions filtered to 515–535 nm, and images obtained through a 10× objective with a 0.3 numerical aperture (Leica confocal microscope). Only slices in which the injection site and anterior commissure could be visualized in the same frame were imaged. ImageJ software was used to quantify images. All quantified images contained the anterior commissure, which was masked to prevent being quantified, but provided a landmark for the NAcore. MMP activity is induced as part of the acute inflammatory response to tissue damage from the microinjector, so the microinjector tract was readily visible in all quantified sections due to equivalent high fluorescence in all treatment groups. This tract was also masked to eliminate quantifying any MMP activity caused by microinjection-induced acute damage. Fluorescence was quantified bilaterally as integrated density from four sections per rat and the integrated densities were averaged within each rat and normalized to yoked-saline control values.
Figure 2.
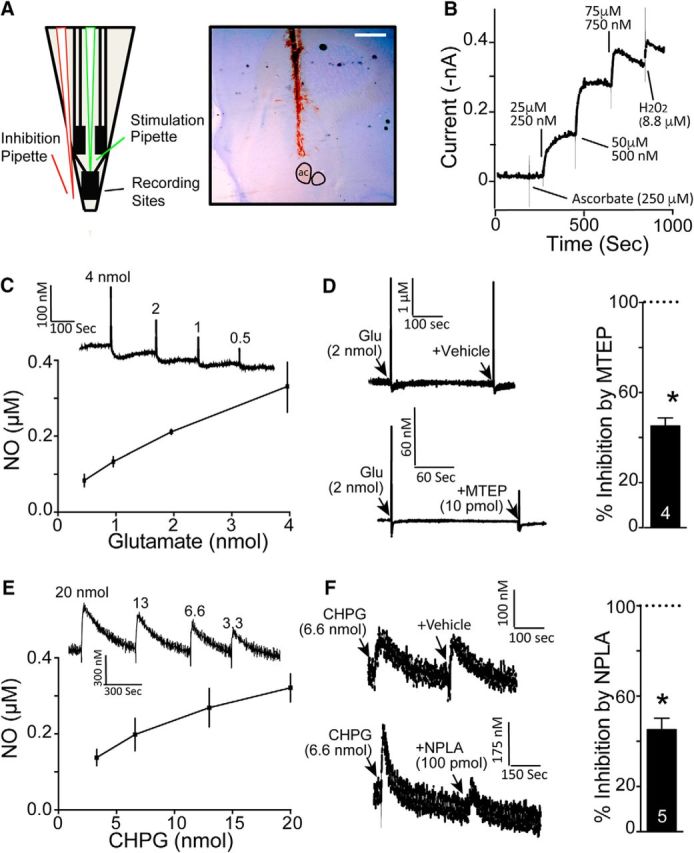
In vivo electrochemical evidence of glutamate acting via mGluR5 to activate nNOS. A, Schematic of the NO-sensitive electrode, the tip of which was implanted into the NAcore. The glass pipettes were waxed to the electrode assembly such that the tips were ∼100 μm from the surface of the electrode to pressure puff various compounds adjacent to the recording sites. The adjacent cresyl-violet-stained tissue shows an example of electrode placement into NAcore (note blood in electrode track). ac, Anterior commissure. Scale bar, 1 mm. B, In vitro calibration of NO electrodes performed after stabilization of baseline electrodes by measuring the amperometric response at +0.7 V versus Ag/AgCl of a nafion/o-PD microelectrode to AA (interferent), followed by increasing concentrations of DETA/NO (NO donor) and finally to H2O2 (positive control). C, Puffing glutamate adjacent to an NO electrode produced a dose-dependent increase in NO (n = 3). D, The effect of glutamate was antagonized by the mGluR5-negative allosteric modulator MTEP (10 pmol). Left shows example traces and right shows group data normalized to peak amplitude of the glutamate signal in each animal (*p < 0.05). E, Puffing the mGluR5 agonist CHPG produced a dose-dependent increase in NO (n = 3). F, Increase by CHPG (20 nmol) was inhibited by the nNOS antagonist NPLA (100 pmol, *p < 0.05).
Figure 3.
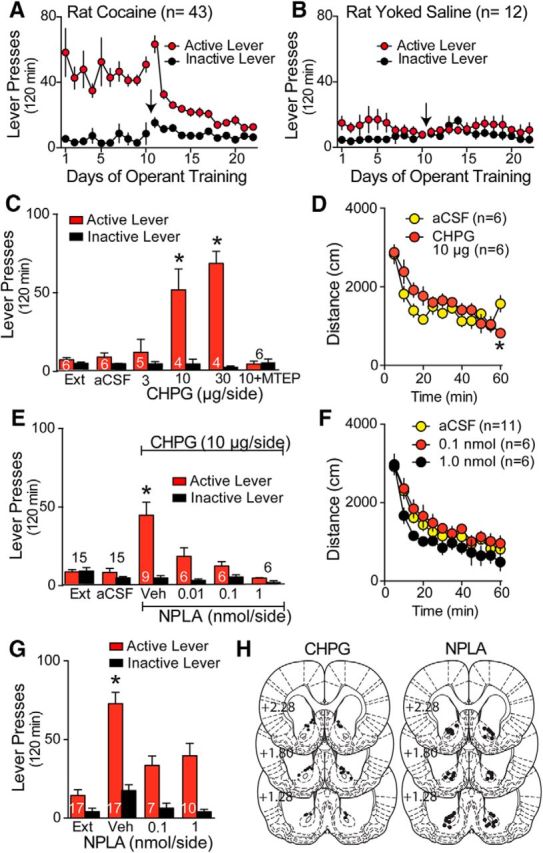
mGluR5 agonist in NAcore recapitulates cue-induced cocaine seeking by activating nNOS. A, Cocaine self-administration and extinction responding for rats used in Figures 3, 4, and 6. Arrow indicates the switch from cocaine or yoked saline infusions to extinction training. B, Yoked saline and extinction training for rats used to generate data in Figures 6, C–E, and 7, A and B. C, Microinjecting the mGluR5 agonist CHPG into the NAcore produced dose-dependent reinstatement of active lever pressing in extinguished rats in the absence of cocaine-conditioned cues. The increase in lever pressing by CHPG (10 μg/side) was abolished by coadministration of MTEP (1 μg/side). n is shown in bars. *p < 0.05, compared extinction (Ext). D, CHPG (10 μg/side) or aCSF was microinjected into the NAcore 5 min before placing the rat into a photocell box to which the animal was preadapted. The time course revealed a significant interaction between time and treatment, but no effect of treatment. E, Reinstated lever pressing by intra-NAcore microinjection of CHPG (10 μg/side) was abolished by coadministration of the nNOS antagonist NPLA into the NAcore. F, Intra-NAcore microinjection of 0.1 or 1 nmol of NPLA did not alter distance traveled compared with vehicle. G, Intra-NAcore microinjection of 0.1 or 1 nmol of NPLA reduced cue-induced active lever pressing compared with vehicle. +p < 0.05 compared with 0 NPLA. H, Histological localization of microinjection cannula tips in the NAcore for the behavioral data shown in Figure 3.
Figure 4.
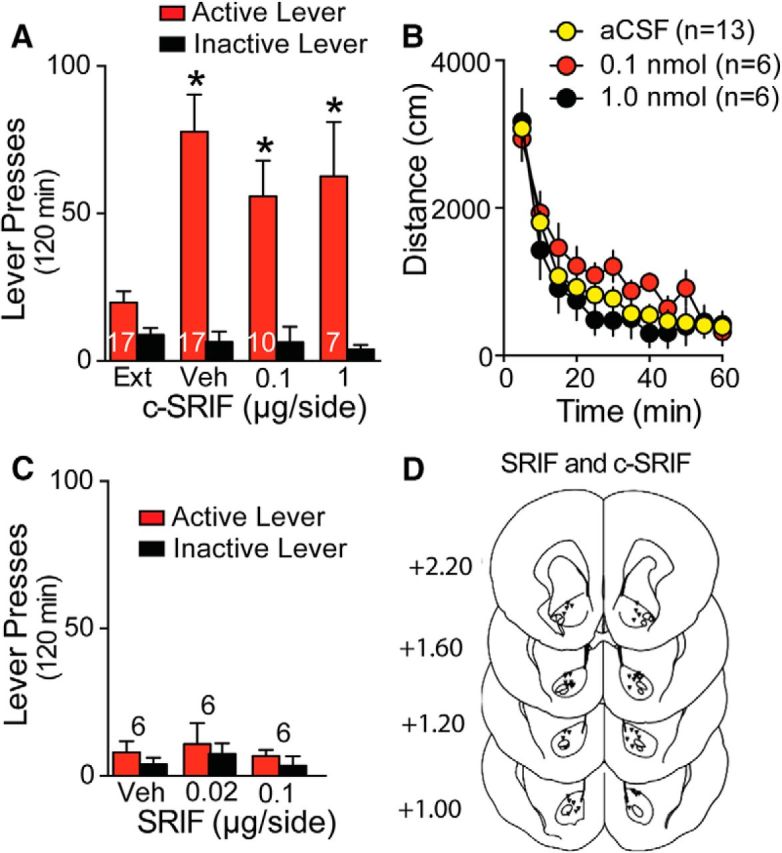
Lack of effect by SRIF transmission on reinstated cocaine seeking. A, Intra-NAcore microinjection of 0.1 or 1 μg of c-SRIF did not alter cue-induced active lever pressing compared with vehicle. B, Intra-NAcore microinjection of 0.1 or 1 μg of c-SRIF did not alter motor activity. C, Intra-NAcore microinjection of SRIF did not reinstate lever pressing. D, Histological localization of microinjection cannula tips in the NAcore for the behavioral data shown in Figure 4.
Figure 5.

Gq-DREADD stimulation of nNOS activity. A, Representative micrographs showing the sparse distribution of nNOS-expressing interneurons in NAcore and labeling of nNOS interneurons with Gq-DREADD in NOS1-Cre mice (left). Also shown is double labeling of Gq-DREADD and Cre immunoreactivity (right). Arrowhead indicates double-labeled nNOS+Gq-DREADD; asterisk, single-labeled Gq-DREADD. Scale bar, 30 μm. B, Gq-DREADD-transfected nNOS neurons near a track made by the in vivo NO electrode. ac, Anterior commissure. Scale bar, 30 μm. C, CNO puffed onto Gq-DREADD-transfected nNOS neurons in NAcore in drug-naive NOS1-Cre mice produced a dose-dependent elevation of NO (n = 5). D, Voltage changes induced by −100 pA hyperpolarizing (blue) or +60 pA depolarizing (red) current pulses in Gq-DREADD-transfected (mCherry+) neuron compared with a neuron that was not transfected (mCherry−). The differences in RMP, amplitude of hyperpolarization, and firing pattern are typical of nNOS interneurons (mCherry+) compared with MSNs. E, CNO (10 μm) depolarized nNOS+ neurons (n = 8), but not nNOS− neurons (n = 5). F, Analyzing the change in RMP between 5 min before and 5 min after bath application of drug revealed CNO (10 μm) and DHPG (100 μm) depolarized nNOS+ neurons and this was prevented by blocking potassium channels with cesium (Ce) or mGluR5 with MTEP (10 μm), respectively. Number of cells/mice is shown in bars. *p < 0.05 compared with cell baseline RMP before drug application.
Figure 6.

Chronic cocaine exposure increases nNOS activity, S-nitrosylation of MMP-2, and MMP activity. A, Representative examples of gelantinolytic fluorescence corresponding to MMP-2 and MMP-9 activity. Each of two groups, extinguished from cocaine self-administration or cocaine-extinguished plus 15 min of cued reinstatement were microinjected bilaterally into the NAcore with FITC-quenched gelatin in combination with either NPLA (1 nmol) or vehicle into contralateral hemispheres. Tissue was obtained 15 min after microinjection and 100 μm sections were analyzed for fluorescence. Scale bar, 1 mm. Dashed lines outline microinjection track and anterior commissure (ac), both of which were masked out for quantification. B, NPLA reduced both constitutive MMP-2 activity in extinguished (Ext) and transient MMP-9 activity (Smith et al., 2014). C, nNOS serine-847 was dephosphorylated after extinction and cued reinstatement of cocaine seeking. D, E, MMP-2 S-nitrosylation was increased after 15 min of cued reinstatement (D), but MMP-9 was not (E). *p < 0.05 compared with vehicle (B) or yoked saline (C–E). n is shown inside each bar.
Figure 7.

Chemogenetic stimulation of nNOS-expressing interneurons induces MMP activity and t-SP. A, Representative micrographs showing FITC-gelatin fluorescence in the vicinity of the cannula track in NAcore. Drug naive NOS1-Cre mice were transfected with Gq-DREADD and microinjected with vehicle or CNO (3 nmol) into opposite hemispheres in combination with FITC-gelatin and tissue was obtained 15 min later. In a second experiment, CNO was microinjected into both hemispheres in combination with vehicle or NPLA (1 nmol) and FITC-gelatin. Dashed lines indicate anterior commissure (ac) and cannula track that were masked out for quantification. Scale bar, 1 mm. B, Mice were microinjected into contralateral hemispheres with CNO or vehicle (left) or CNO + NPLA (right) and data were evaluated within each panel using a paired Student's t test comparing CNO with vehicle or CNO + NPLA between hemispheres. Data are shown normalized to the CNO treatment. CNO increased MMP-2 and MMP-9 activity in the NAcore of drug-naive mice compared with vehicle (left) and this effect was abolished by treatment with NPLA (right). C, CNO stimulation of Gq-DREADD in nNOS-expressing interneurons potentiated the AMPA/NMDA ratio at glutamatergic synapses on NAcore MSNs. n shown in bar represents the number of cells/mice.
Protein measurements
Western blotting.
Rats were rapidly decapitated after extinction of cocaine self-administration or yoked saline or 15 or 45 min after cued or cocaine-primed reinstatement. The NAcore was dissected and homogenized in RIPA lysis buffer containing 1.0% SDS and protease/phosphatase inhibitors. Homogenate was centrifuged at 4°C for 5 min at 10,000 × g. Supernatant was collected and protein concentration was determined via a bicinchoninic acid assay (Thermo Scientific). Then, 30 μg of protein was added to each lane of 10% Bis-Tris gels (Bio-Rad) and transferred to nitrocellulose membranes via the Invitrogen iBlot transfer system. Primary antibodies were used for nNOS (1:1000; Millipore #AB5380; RRID AB_91824), phospho-Ser847 nNOS (1:1000; Abcam #16650; RRID AB_443418) and HRP-conjugated goat anti-rabbit secondary antibody was used at 1:10,000. GAPDH was used as a loading control. A Kodak Image Station was used to visualize and quantify protein expression.
Immunoprecipitation.
Protein-A dynabeads were washed in cold PBS, and MMP-2 (Millipore #19015, RRID AB_91089) or MMP-9 antibody (Millipore #19016, RRID AB_91090) were added at 1:1000 dilution and allowed to incubate for 1 h at room temperature. Beads were washed 3 times for 5 min each in cold PBS containing 0.2% Triton X-100 (PBST). Protein A was cross-linked to antibodies with 5 mm bis(sulfosuccinimidyl)suberate (BS3) for 30 min and the cross-linking reaction was quenched via 1 m Tris-HCl for 15 min. NAcore samples were prepared as whole-cell lysates in RIPA buffer with protease and phosphatase inhibitors. Then, 45 μg of protein was added for detection of MMP-2 or 100 μg of protein for MMP-9 in 200 μl of RIPA buffer containing protease and phosphatase inhibitors. Beads were washed 3 min for 5 min each in cold PBST. Elution occurred via 50 mm glycine, pH 2.8, and samples were heated to 50°C for 10 min. Western blotting was performed as above and membranes were probed with an antibody against S-nitrosocysteine (Sigma-Aldrich #N5411, RRID AB_260785). Membranes were then stripped via RestorePLUS Western stripping buffer (Life Technologies #46428) for 10 min at 37°C and reprobed using the same antibody that was used for the immunprecipitation. SNO–cysteine content was normalized to total protein for quantification.
Immunohistochemistry.
Mice were deeply anesthetized with pentobarbital and perfused with PBS followed by 4% paraformaldehyde. Brains were postfixed for 24 h, after which 100 μm brain sections were cut using a vibratome. Free-floating sections were rinsed in PBS/Triton X-100 (0.1%) and incubated in normal goat serum and primary antibodies (Anti-Cre Recombinase; Millipore #MAB3120 Clone 2D8, RRID 2085748, 1:1000; Anti-nNOS; Millipore #AB5380, RRID AB_91824, 1:1000) overnight at 4°C. Afterward, sections were rinsed in PBS-Triton X-100 and incubated in Alexa Fluor-conjugated secondary antibodies (Life Technologies, 1:1000). Images were taken on a confocal microscope (Leica) using a 63× objective and deconvolved using AutoQuant X2 (MediaCybernetics, RRID SCR_002465). Cell counts were done using ImageJ software (RRID SCR_003070).
Slice preparation for electrophysiology
Rats and mice were anesthetized with ketamine HCl (100 and 150 mg/kg Ketaset, respectively; Fort Dodge Animal Health) and decapitated. The brain was removed from the skull and 220-μm-thick coronal sections were obtained using a vibratome (VT1200S Leica vibratome). Slices were immediately placed into a vial with aCSF containing the following (in mm): 126 NaCl, 1.4 NaH2PO4, 25 NaHCO3, 11 glucose, 1.2 MgCl2, 2.4 CaCl2, 2.5 KCl, 2.0 NaPyruvate, 0.4 ascorbic acid (AA), bubbled with 95% O2 and 5% CO2) and a mixture of 5 mm kynurenic acid and 50 μm d-(-)-2-amino-5-phosphonopentanoic acid. Slices were incubated at room temperature until recording.
In vitro whole-cell recording
All recordings were collected at 32°C (controlled by TC-344B; Warner Instruments) in the dorsomedial NAcore with a Multiclamp 700 B amplifier (Molecular Devices). Data were acquired at 10 kHz and filtered at 2 kHz using AxoGraph X software (AxoGraph Scientific, RRID SCR_014284). Inhibitory synaptic transmission was blocked with picrotoxin (100 μm). Multiclamp 700B (Molecular Devices) was used to record EPSCs in whole-cell patch-clamp configuration. Glass microelectrodes (1.5–2.5 MΩ) were filled with cesium-based internal solution containing the following (in mm): 128 cesium methanesulfonate, 10 HEPES potassium, 1 EGTA, 1 MgCl2, 10 NaCl, 2.0 MgATP, and 0.3 NaGTP, 1 QX-314, pH 7.2–7.4, 275 mOsm. Data were acquired at 10 kHz and filtered at 2 kHz using AxoGraph X software (AxoGraph Scientific). To evoke EPSCs, a bipolar stimulating electrode (FHC) was placed ∼300 μm dorsomedial to the recorded cell to maximize chances of stimulating prelimbic afferents. The stimulation intensity was set to evoke an EPSC of 200–500 pA, which was usually 30–70% of maximal EPSC. Recordings were collected every 20 s. Rs measured with a 2 mV hyperpolarizing step (10 ms) given with each stimulus and holding current was always monitored online. Recordings with unstable Rs or when Rs exceeded 20 MΩ were aborted.
Electrophysiology: voltage clamp
Inhibitory synaptic transmission was blocked with picrotoxin (50 μm) to isolate EPSCs in whole-cell patch-clamp configuration. Glass microelectrodes (1–2 MΩ) were filled with cesium-based internal solution containing the following (in mm): 124 cesium methanesulfonate, 10 HEPES potassium, 1 EGTA, 1 MgCl2, 10 NaCl, 2.0 MgATP, and 0.3 NaGTP, 1 QX-314, pH 7.2–7.3, 275 mOsm. To evoke EPSCs, a bipolar stimulating electrode (FHC) was placed ∼300 μm dorsomedial to the recorded cell to maximize chances of stimulating prelimbic afferents. The stimulation intensity was set to evoke an EPSC of 200–500 pA, which was usually 30–70% of maximal EPSC. Recordings were collected every 20 s. Rs measured with a 2 mV hyperpolarizing step (10 ms) given with each stimulus and holding current was always monitored online. Recordings with unstable Rs or when Rs exceeded 10 MΩ were aborted.
AMPA/NMDA ratio measurement.
Recordings started no earlier than 10 min after the cell membrane was ruptured to allow diffusion of the internal solution into the cell. AMPA currents were first measured at −80 mV to ensure stability of response. The membrane potential was then gradually increased to +40 mV. Recording of currents resumed 5 min after reaching +40 mV to allow stabilization of cell parameters. Currents composed of both AMPA and NMDA were then obtained. D-AP5 (50 μm) was bath-applied to block NMDA currents and recording of AMPA currents at +40 mV was started after 2 min. NMDA currents were obtained by subtracting the AMPA currents from the total current at +40 mV.
Electrophysiology: current clamp
Glass microelectrodes (1–2 MΩ) were filled with potassium-gluconate-based internal solution containing the following (in mm): 130 potassium d-gluconate, 0.2 EGTA, 10 HEPE-K, 20 KCl, 1 MgCl, 2.0 MgATP, and 0.3 NaGTP, pH 7.2–7.3, 275 mOsm. To block potassium channels, the same cesium-based internal solution as for the voltage-clamp experiments was used. To perform current–voltage curves and to test the excitability of MSNs, a series of hyperpolarizing and depolarizing current steps was applied for 5 min after breaking in the cell. The resting membrane potential was measured every 20 s.
Measurement of NO
Enzyme-based microelectrode array.
R2 ceramic-based microelectrode arrays (MEAs) were prepared for in vivo recordings as described previously (Burmeister et al., 2002; Ferreira et al., 2005; Quintero et al., 2011; Onifer et al., 2012). Briefly, recording sites were coated with Nafion (Sigma-Aldrich) and baked at 170°C for 10 min. After a 24 h room temperature incubation, platinum sites were electroplated (through the enzyme layer) with an O-phenylenediamine dihydrochloride size exclusion layer (Acros Organics) to block potential interferents such as dopamine and AA. When a potential of +0.7 V versus a silver/silver chloride reference electrode is applied to the MEA, NO is oxidized, resulting in the transfer of electrons to the platinum recording surface. The resulting change in current was amplified and digitized by a Fast Analytical Sensing Technology (FAST16 MKIII recording system; Quanteon). The FAST16 MKIII recording system saved amperometric data, time, and experimenter ejection marks. All traces from the MEAs were analyzed using FAST Analysis software (Jason Burmeister Consulting).
Electrode calibration.
MEAs were calibrated to determine their sensitivity to NO and selectivity against AA using constant potential amperometry with a FAST16 MKIII system as described previously (Burmeister et al., 2002; Ferreira et al., 2005; Quintero et al., 2011; Onifer et al., 2012). Briefly, the MEA was submerged into 40 ml of a continuously stirred solution of 0.05 m PBS titrated to pH 7.4 and allowed to reach a stable baseline for ∼30 min before calibration. Phosphate buffer temperature was maintained at 37°C using a circulating water bath (Gaymar Industries). Aliquots of freshly made 20 mm AA and 10 mm DETA-NO (a compound that degrades resulting in the release of NO) were used to obtain final concentrations of 250 μm AA and 0.25, 0.5, and 0.75 μm NO for calibrations. Selectivity ratios for NO over AA were calculated in addition to the limit of detection and linearity (R2) for all NO MEAs. The MEAs were also tested to compare the recording capability among the platinum recording sites using H2O2 (8.8 μm, final concentration) as a test substance. Electrodes that had R2 values <0.8 and did not respond to H2O2 were not used for recordings.
Electrode placement and recording procedures.
Before implantation, MEAs were fitted with single-barrel glass capillaries with an inner tip diameter of ∼10 μm. Pipettes were embedded in modeling clay and covered with molten wax to stabilize the assembly. The tip of the stimulating pipette was positioned 50–100 μm from the surface of the MEA directly in front of the platinum recording sites (Burmeister et al., 2002; Michael and Borland, 2007). After attachment, the pipette was filled with 10 mm glutamate, 134 mm CHPG or 875 μm CNO solution (0.3 mg/ml, 0.5% DMSO in 0.9% sterile saline). For MTEP and NPLA inhibition experiments, a second pipette was positioned ∼300 μm lateral to the central pipette, also 50–100 μm from the surface of the MEA (Fig. 2A). Pipettes poised to deliver inhibitors were filled with 100 μm MTEP, 1 mm NPLA, or vehicle. In all cases, pipettes were connected via tubing to a Picospritzer III (Parker Instruments) for delivery of solutions.
Rats were anesthetized with 30% urethane (5 mg/kg, i.p.) and placed in a stereotaxic apparatus (David Kopf Instruments) on a heating pad (37°C). After removal of the scalp tissue, rats underwent a craniotomy to remove the skull above the NAcore on both the right and left side, leaving bregma intact. Rats were then implanted with a NO-selective electrode into either the left or right NAcore (in millimeters relative to bregma: AP, +1.8 mm; ML, ±1.5 mm; DV, −7 mm vs bregma). A small hole was drilled slightly anterior to lambda and used for placement of the silver/silver chloride reference electrode. Mouse procedures were performed in a similar fashion with the following exceptions. Mice were anesthetized with 12.5% urethane (5 mg/kg, i.p.) and different stereotaxic coordinates used for electrode placement in the NAcore were used (in millimeters relative to bregma: +1.5 A/P; ±1.2 M/L; −4.2 D/V) based on Paxinos and Franklin (2012).
In dose–response experiments, after establishment of a stable baseline, increasing amounts of glutamate, CHPG, and CNO (60–200 nl) were given to determine whether NO responses evoked by glutamate, CHPG, or CNO were dose dependent. After each application of glutamate CHPG, or CNO, the recording was allowed to reach baseline for 3–5 min. In inhibition experiments, after establishment of a stable baseline, 100–200 nl of glutamate or CHPG was given to elicit an “uninhibited” NO response. The system was allowed to reach baseline again for 3–5 min and then the same volume of MTEP or NPLA was administered immediately, followed by a second pulse (again using the same volume) of glutamate or CHPG for the “inhibited” response. Maximum amplitude of the responses was used to calculate the percentage inhibition of glutamate- or CHPG-evoked NO by MTEP and NPLA, respectively. Three to four paired observations (uninhibited vs inhibited peaks) were averaged for each recording across three to five rats/mice.
Statistics
All statistics were performed using GraphPad Prism (RRID SCR_002798) version 7 software. Two-hour reinstatement sessions were analyzed using a one-way ANOVA followed by a Sidak's test for post hoc multiple comparisons. Microdialysis data were evaluated using a two-way ANOVA with a Sidak's test for post hoc comparisons. Zymography data were analyzed using paired t tests, with opposite hemispheres forming pairs. Western blots were analyzed using a one-way ANOVA with a Sidak's post hoc. Electrophysiology and electrochemistry were analyzed using Student's t tests or two-way ANOVAs.
Results
Cue-induced glutamate spillover and mGluRs
Reinstated drug seeking is associated with an increase in extracellular glutamate that depends on synaptic activity in PL-PFC afferents to the NAcore (McFarland et al., 2003; LaLumiere and Kalivas, 2008). This reinstatement-induced increase in synaptic glutamate spillover allows glutamate to access and stimulate extrasynaptic mGluRs. Interestingly, the pharmacological regulation of nonsynaptic group 1 and group 2 mGluRs produces opposing effects on reinstated drug seeking. Therefore, either blockade of mGluR5 or stimulation of mGluR2/3 in accumbens prevents reinstated cocaine seeking (Peters and Kalivas, 2006; Sinclair et al., 2012; Schmidt et al., 2013). In the first series of experiments, we used microdialysis in the NAcore to determine whether reducing cue-induced cocaine seeking by either stimulating mGluR2/3 or blocking mGluR5 also reduced cue-induced glutamate spillover. We hypothesized that mGluR2/3 stimulation would block cue-induced reinstatement by inhibiting glutamate spillover through activating mGluR2/3 presynaptic autoreceptors and thereby reducing synaptic glutamate release probability (Moussawi and Kalivas, 2010). Conversely, we hypothesized that blocking mGluR5 would not affect glutamate spillover because cue-induced synaptic glutamate spillover stimulates extrasynaptic mGluR5 to reinstate cocaine seeking.
Rats were trained to self-administer cocaine, followed by extinction training (Fig. 1A). Relapse was modeled in a trial in which cocaine-associated cues were restored to the active lever without drug delivery to reinstate cocaine seeking simultaneously with recording extracellular glutamate via a unilateral microdialysis probe inserted into the NAcore (Fig. 1B). After collecting six baseline samples (20 min each), a portion of rats underwent cued reinstatement and an additional nine 20 min samples were collected to quantify extracellular glutamate. Restoring conditioned cues to active lever pressing increased extracellular glutamate significantly in the NAcore over the first 60 min after initiating the reinstatement trial compared with rats undergoing dialysis during an extinction trial without cue presentation (Fig. 1C). Adding the mGluR2/3 agonist LY379268 (100 μm) to the dialysis buffer 40 min before beginning the reinstatement trial prevented the cue-induced increase in extracellular glutamate. Conversely, adding the mGluR5 negative allosteric modulator MTEP (100 μm) to the dialysis buffer potentiated the cue-induced rise in extracellular glutamate slightly over the first 20 min after cue presentation (two-way ANOVA with repeated measures over time, treatment F(3,35) = 5.50, p = 0.003, time F(14,490) = 8.36, p < 0.001, interaction F(42,490) = 4.01, p < 0.001). Neither LY379268 nor MTEP in the dialysis buffer altered basal levels of extracellular glutamate significantly in the 20 min before beginning the reinstatement trial (extinction = 0.23 ± 0.06 pmol/μl, n = 13; cue = 0.29 ± 0.06, n = 17; LY379268 = 0.24 ± 0.04, n = 6; MTEP = 0.33 ± 0.12, n = 5).
Figure 1.

Cue-induced increases in extracellular glutamate are antagonized by stimulating mGluR2/3, but not by blocking mGluR5. A, Self-administration and extinction lever pressing for all rats used in the microdialysis studies. Arrow indicates switch from 10 d of self-administration to 14 d of extinction training. B, Histological localization of the active dialysis membrane in the NAcore, which often traversed striatal areas dorsal or ventral to NAcore. C, Cue presentation increased extracellular glutamate significantly in the NAcore. The increase in glutamate was abolished by introducing the mGluR2/3 agonist LY379268 (100 μm), but not the mGluR5 negative allosteric modulator MTEP (100 μm) 40 min before initiating cued reinstatement. Data are shown as the percentage change from the average baseline value (0–80 min). n in each group is shown in bars in D. D, Reverse microdialysis of either LY379268 or MTEP prevented cue-induced reinstatement. Conditioned cues were restored to active lever presses in all groups except the extinction group. n is shown in bars and is the same for C and D. All data shown as mean ± SEM. *p < 0.05, compared with extinction using a Sidak test for multiple comparisons; +p < 0.05 comparing cue with MTEP.
Rats infused with only dialysis buffer that had cues restored to active lever pressing (the cue group) showed an increase in active lever pressing compared with rats not presented with cues that underwent an extinction trial (Fig. 1D). Inactive lever pressing was not altered in response to restoring cocaine-paired cues. The reduction in cue-induced extracellular glutamate produced by reverse dialysis of LY379268 was accompanied by blockade of cue-induced active lever pressing. Even though MTEP did not reduce cue-induced extracellular glutamate (Fig. 1C), it produced a similar reduction in active lever pressing as LY379268 (Fig. 1D, two-way ANOVA with repeated measures over lever, treatment F(3,35) = 28.09, p < 0.001, lever F(1,35) = 61.25, p < 0.001, interaction F(3,35) = 25.02, p < 0.001). The fact that LY379268 inhibited both the rise in glutamate and active lever pressing is consistent with the hypothesized outcome that stimulating mGluR2/3 presynaptic autoreceptors reduces synaptic glutamate release probability and spillover. The dissociation between behavior and extracellular glutamate by MTEP is consistent with the hypothesis that the inhibition of cued reinstatement produced by antagonizing mGluR5 is not due to inhibiting glutamate release, but rather that cue-induced glutamate stimulates extrasynaptic mGluR5 to reinstate cocaine seeking.
Glutamate spillover stimulates mGluR5 to produce NO and an nNOS-dependent reinstatement of cocaine seeking
Given the data presented above, we focused on the activation of mGluR5 by glutamate spillover as an essential component of the mechanism for cued reinstatement of cocaine seeking. mGluR5 is a Gq-coupled receptor that promotes the release of calcium from intracellular stores, and is expressed by most neurons in the NAcore (Lüscher and Huber, 2010). However, we reasoned that activation of mGluR5 by glutamate spillover might be particularly important on nNOS interneurons. This reasoning is supported by several observations, including that the activation of nNOS is calcium dependent (Hayashi et al., 1999) and that the product of nNOS activity, NO, regulates synaptic plasticity (Hardingham et al., 2013) and could therefore contribute to the t-SP mediating cued reinstatement. In addition, repeated noncontingent cocaine administration increases NO efflux in the dorsal striatum (Lee et al., 2010) and induces the S-nitrosylation of the AMPA glutamate receptor trafficking protein stargazin in the accumbens shell (Selvakumar et al., 2014). Finally, cocaine treatment downregulates mGluR5 on the most abundant neuron type in NAcore, MSNs, and desensitizes D1-expressing MSNs to mGluR5-endocannabinoid LTD (Fourgeaud et al., 2004; Knackstedt et al., 2010). To examine the hypothesis that mGluR5 stimulation of nNOS is mediating reinstated cocaine seeking, we first determined whether stimulating mGluR5 in the NAcore could promote NO production. We made in vivo measurements of the extracellular concentration of NO by implanting an NO-sensitive electrochemical electrode (Fig. 2A) into the NAcore of drug-naive urethane-anesthetized rats that had been calibrated in vitro using an NO donor (Fig. 2B; Barbosa et al., 2008). Glutamate was puffed near the electrode to mimic synaptic glutamate spillover and elicited a transient dose-dependent elevation in NO that was reduced by prepuffing MTEP, but not the MTEP vehicle (paired Student's t test comparing glutamate with glutamate + MTEP; t(3) = 8.08, p = 0.004; Fig. 2C,D). We then showed that stimulating mGluR5 by puffing the mGluR5 agonist CHPG into the NAcore induced a dose-dependent increase in NO that was reduced by antagonizing nNOS with the selective nNOS inhibitor NPLA, but not by the NPLA vehicle (paired Student's t test comparing CHPG with CHPG + NPLA; t(4) = 4.42, p = 0.012; Fig. 2E,F).
After demonstrating that mGluR5 stimulation promoted in vivo production of NO, we determined whether CHPG microinjection into the NAcore could substitute for a cocaine cue and reinstate active lever pressing in rats extinguished from cocaine self-administration (Fig. 3A). Bilateral microinjection CHPG into the NAcore 10 min before placing a rat into the extinguished environment without discrete conditioned cues elicited a dose-dependent increase in active lever pressing that was abolished by co-microinjecting MTEP (Fig. 3B; two-way ANOVA with repeated measures over lever, treatment F(5,24) = 8.89, p < 0.001, lever F(1,24) = 71.66, p < 0.001, interaction F(5,24) = 14.62, p < 0.001). The microinjection of CHPG produced a selective effect on active lever pressing because there was no difference in inactive lever pressing between any of the treatment groups (Fig. 3B) and a behaviorally effective dose of CHPG (10 μg) did not increase locomotor activity in a familiar open-field environment compared with microinjection of aCSF (Fig. 3C; two-way ANOVA with repeated measures over time, time F(11,110) = 18.27, p < 0.001; treatment F(1,10) = 1.97, p = 0.191; interaction F(11.110) = 2.66, p = 0.005). Linking CHPG-induced reinstatement to nNOS, the increase in lever pressing produced by CHPG was abolished in a dose-dependent manner by coadministration of NPLA into the NAcore (Fig. 3D; two-way ANOVA with repeated measures over lever, treatment F(5,54) = 8.60, p < 0.001, lever F(1,54) = 21.57, p < 0.001, interaction F(5,54) = 8.53, p < 0.001). The effect of NPLA was selective for the increase in active lever pressing by CHPG because NPLA did not alter inactive lever pressing or locomotor activity significantly (Fig. 3D,E; two-way ANOVA, treatment F(2,20) = 3.90, p = 0.037, time F(11,220) = 69.28, p < 0.001, interaction F(22,220) = 0.71, p = 0.825). Similarly, microinjection of NPLA into the NAcore produced a dose-dependent blockade of cue-induced reinstatement (Fig. 3F; two-way ANOVA treatment F(3,47) = 11.93, p < 0.001, lever F(1,47) = 103.5, p < 0.001, interaction F(3,47) = 11.93, p < 0.001), supporting the possibility that mGluR5 activation of nNOS mediates cued reinstatement of cocaine seeking. Figure 3G shows the histological localization of microinjections within the NAcore for the CHPG and NPLA experiments.
Finally, the majority of nNOS interneurons express SRIF that could also be released in response to CHPG microinjection or synaptic spillover of glutamate (Figueredo-Cardenas et al., 1996). To test the possible involvement of SRIF, the SRIF antagonist c-SRIF was microinjected into the NAcore at doses shown previously to alter behavior (Ikeda et al., 2009). Compared with vehicle, neither dose of c-SRIF altered active or inactive lever pressing significantly (Fig. 4A; two-way ANOVA, treatment F(3,47) = 8.52, p < 0.001, lever F(1,47) = 93.70, p < 0.001, interaction F(3,47) = 10.40, p < 0.001) or locomotor activity (Fig. 4B; two-way ANOVA, treatment F(2,22) = 1.38, p = 0.276, time F(11,242) = 71.95, p < 0.001, interaction F(22,242) = 1.02, p = 0.438). In addition, unlike activating mGluR5 with CHPG (Fig. 3B), previously reported behaviorally effective doses of SRIF microinjected into the NAcore in extinguished rats did not reinstate cocaine seeking (Fig. 4C; two-way ANOVA, treatment F(2,15) = 2.76, p = 0.147, lever F(1,15) = 20.88, p < 0.001, interaction F(2,15) = 1.53, p = 0.248) and showed a trend toward decreasing locomotor activity (vehicle = 14,063 ± 1493, n = 6, photocell counts over 60 min; SRIF-100 ng = 11,224 ± 1102, n = 7; Raynor et al., 1993; Santis et al., 2009). Figure 4D shows the histological localization of microinjections within the NAcore for the SRIF and c-SRIF experiments.
mGluR5 and Gq stimulation in nNOS interneurons
The data so far support the hypothesis that glutamate spillover stimulates mGluR5 to promote nNOS activity. nNOS-expressing neurons constitute ∼1% of all neurons in the NAcore (Figueredo-Cardenas et al., 1996). To study selectively the role of nNOS-expressing interneurons without simultaneously activating other NAcore neurons, we used NOS1-Cre transgenic mice, which permitted viral expression of a Gq-coupled designer receptor exclusively activated by designer drug (Gq-DREADD; AAV2-hSyn-DIO-hM3Dq-mCherry) in NAcore nNOS interneurons (Sutton et al., 2014). This strategy resulted in Gq-DREADD expression in nNOS-expressing cells, as shown by double labeling for the Gq-DREADD reporter mCherry and immunoreactive nNOS or Cre expression (Fig. 5A). Interestingly, ∼20–25% of those neurons infected by DIO-Gq-DREADD did not show significant immunoreactivity for nNOS, indicating a low level of “leak” infection via this virus (Fig. 5A; see further discussion of leaky expression below). Stimulating Gq-DREADD increases intracellular calcium in a manner similar to mGluR5 (Agulhon et al., 2013), which can activate nNOS by promoting binding of activated calmodulin (Hayashi et al., 1999). We first evaluated transfected Gq-DREADD-mediated nNOS activation directly by using an NO electrode implanted in vivo into the transfection site in the NAcore to measure NO efflux in cocaine-naive urethane-anesthetized mice (Fig. 5B). The Gq-DREADD ligand CNO produced a dose-dependent increase in extracellular NO in the NAcore (Fig. 5C).
To examine directly the electrophysiological properties of nNOS-expressing interneurons and further evaluate the type of NAcore cell transfected, we made whole-cell patch recordings from nNOS neurons labeled with mCherry via Cre-dependent Gq-DREADD transduction. Examining resting membrane potential (RMP) of DREADD infected neurons (identified by mCherry fluorescence) provides further evidence for the relative specificity of the viral construct used, but also indicated ∼20–25% leak of mCherry expression in non-nNOS interneurons. The resting membrane potential (RMP) and input resistance distinguishes nNOS interneurons from MSNs that constitute ∼90% of all accumbens neurons (Kawaguchi, 1993; Fino et al., 2009). Of 19 Gq-DREADD-infected neurons (mCherry+) recorded, 4 had RMPs <−70 mV, indicating that 21% of infected cells are likely MSNs. In contrast, of 9 nonlabeled neurons (mCherry−) recorded, only 1 had an RMP >−70 mV (mCherry+ RMP = −58.11 ± 3.5 mV; mCherry− RMP = −76.63 ± 3.97 mV; t(26) = 3.373, p = 0.002), consistent with ∼10% of neurons being non-MSN in a random sample. Furthermore, also indicative of nNOS interneurons, mCherry+ cells had significantly higher input resistance (mCherry+ = 443.3 ± 75.6ΩM; mCherry− = 168.5 ± 39.1 ΩM; t(26) = 2.19, p = 0.023) and lower capacitance (mCherry+ = 46.2 ± 6.1 F; mCherry− = 75.8 ± 7.5 F; t(26) = 2.74, p = 0.011). These characteristics are reflected in the example voltage traces showing the differential responses of the cells types to hyperpolarizing and depolarizing pulses (Fig. 5D). Only mCherry+ fluorescent neurons showed membrane depolarization in response to CNO activation of Gq-DREADD, whereas nonfluorescent neurons were unresponsive (Fig. 5E, two-way repeated-measures ANOVA treatment F(1,258) = 71.54, p < 0.001). CNO-mediated depolarization was masked by blocking potassium channels cesium (Fig. 5F, paired t(7) = 0.02, p = 0.984 comparing pre-CNO vs post-CNO), consistent with previously reported Gq-dependent inhibition of leak potassium currents (Wilke et al., 2014). Supporting functional mGluR5 expression by nNOS interneurons, stimulating group I mGluR with bath-applied dihydroxyphenlglycol produced membrane depolarization (paired t(7) = 2.61, p = 0.035) that was antagonized by MTEP (paired t(3) = 1.45, p = 0.335; Fig. 5F).
Increased NO production is required for t-SP
Because blockade of nNOS with NPLA microinjection into the NAcore inhibited cue-induced reinstatement of cocaine seeking (Fig. 3F), we next tested the hypothesis that NO production is required for the induction of t-SP. Cue-induced induction of t-SP and reinstated cocaine seeking requires MMP-2 and MMP-9 activity in the NAcore (Smith et al., 2014). We quantified cue-induced MMP-2 and MMP-9 activity using in vivo zymography. A FITC-quenched gelatin peptide that is proteolytically cleaved by MMP-2 and MMP-9 was microinjected into NAcore to generate a fluorescent signal that is linearly proportional to MMP-2 and MMP-9 activity (Nagy et al., 2007). We quantified MMP-2 and MMP-9 activity in the NAcore 15 min after bilateral microinjection of FITC-gelatin in combination with the nNOS inhibitor NPLA or vehicle in contralateral hemispheres. Rats were trained to self-administer cocaine and were extinguished and MMP-2 and MMP-9 activity quantified at 24 h after the last extinction session or after 15 min of cued reinstatement. NPLA reduced both constitutive MMP-2 and MMP-9 activity after extinction and the transient increase in MMP-2 and MMP-9 activity elicited by 15 min of cued reinstatement (Fig. 6A,B; extinction paired t test t(3) = 3.62, p = 0.036, reinstated paired t test t(3) = 5.09, p = 0.015). This indicated that nNOS activity was required for cue-induced stimulation of both MMP-2 and MMP-9 activity because MMP-2 is upregulated after extinction and MMP-9 after reinstatement (Smith et al., 2014).
We used Western blotting to evaluate the phosphorylation of nNOS and found evidence of a role for nNOS activity in both constitutive and transient MMP-2 and MMP-9 activity. Dephosphorylation of serine 847 increases NO production by facilitating calmodulin binding to nNOS (Rameau et al., 2004; Rao et al., 2008) and, after extinction or 15 min of cued reinstatement, Ser-847 phosphorylation was reduced in NAcore compared with yoked-saline controls (Fig. 6C, one-way ANOVA F(2,20) = 16.50, p < 0.001). The altered phosphorylation state of nNOS after cocaine self-administration and extinction is consistent with the constitutive increase in NO production previously observed in the striatum of rats pretreated with daily noncontingent cocaine injections (Lee et al., 2010). One mechanism for physiologically activating MMP-2 and MMP-9 is by S-nitrosylation of a cysteine residue within the protein prodomain (Gu et al., 2002). To measure S-nitrosylation, MMP-2 and MMP-9 were immunoprecipitated and probed for S-nitroso-cysteine via Western blotting. MMP-2 S-nitrosylation was increased after cued reinstatement (Fig. 6D; one-way ANOVA F(2,18) = 4.69, p = 0.023). Although no change was detected in the S-nitrosylation of MMP-9 (Fig. 6E), it is possible that S-nitrosylation and activation of MMP-2 induced the proteolytic activation of MMP-9 (Toth et al., 2003).
We next used in vivo zymography in cocaine-naive NOS1-Cre mice to show that nitrergic signaling induced by CNO in NAcore activated MMP-2 and MMP-9. NOS1-Cre mice were microinjected with Gq-DREADD in NAcore, followed by a bilateral microinjection of either CNO or vehicle in contralateral hemispheres 3 weeks later. CNO microinjection activated MMP-2 and MMP-9 robustly, as indicated by increased fluorescence around the injection site (Fig. 7A). Moreover, the increase was abolished by inhibiting nNOS with NPLA (Fig. 7B, left, CNO paired t(4) = 3.02, p = 0.039; right, paired t(4) = 2.97, p = 0.041). Cue-induced MMP-2 and MMP-9 activity drives t-SP in NAcore MSNs (Smith et al., 2014) and we used whole-cell patch-clamp electrophysiology to show that the nitrergic signaling and activation of MMP-2 and MMP-9 induced by CNO also elicited t-SP. As a measure of t-SP induction, the AMPA/NMDA ratio was quantified in MSNs 20 min after systemic injection of CNO (3 mg/kg, i.p.). CNO treatment elevated the AMPA/NMDA ratio significantly in Gq-DREADD-transfected, drug-naive NOS1-Cre mice compared with vehicle administration (Fig. 7C; unpaired t(19) = 2.21, p = 0.040), indicating that Gq activation of nNOS produced t-SP.
Gq stimulation of nNOS activity recapitulates cue-induced reinstatement of cocaine seeking
Finally, we used Gq-DREADD-transfected NOS1-Cre mice to show that stimulating nNOS interneurons was sufficient to reinstate cocaine seeking. After extinguishing mice from cocaine self-administration (Fig. 8A), systemic administration with CNO (3.0 mg/kg, i.p.) reinstated lever pressing in the absence of a conditioned cue (Fig. 8B; two-way repeated-measures ANOVA, treatment F(2,16) = 18.80, p < 0.001, lever F(1,8) = 9.61, p = 0.015, interaction F(2,16) = 7.35, p = 0.005). Moreover, CNO potentiated cue-induced reinstatement markedly (Fig. 8C; two-way repeated-measures ANOVA, treatment F(2,16) = 19.65, p < 0.001, lever F(1,8) = 19.25, p = 0.002, interaction F(2,16) = 9.96, p = 0.002). Although CNO treatment produced a trend toward increasing inactive lever pressing, the effect was not statistically significant and mice continued to distinguish significantly between the active and inactive lever.
Figure 8.

Selective activation or destruction of nNOS interneurons in NAcore produces opposite effects on reinstated cocaine seeking. A, NOS1-Cre transgenic mice were trained to self-administer cocaine for 10 d before undergoing extinction training for 10 d. B, Activation of nNOS-expressing interneurons with CNO (3 mg/kg, i.p.) reinstated cocaine seeking in extinguished NOS1-Cre mice in the absence of cocaine cues. C, CNO also potentiated cue-induced reinstatement markedly compared with vehicle. D, Immunostaining showing example animal with large nNOS depletion after genetic activation of the caspase 3 apoptosis cascade (taCasp3-TEVp). *nNOS immune-positive cell. ac, Anterior commissure. Scale bar, 100 μm. E, nNOS immunostaining for animal sustaining only partial nNOS depletion. F, Animal sustaining large depletion of nNOS-immunoreactive neurons (green) with blue counterstaining for DNA (TO-PRO-3 iodide) showing that the loss of nNOS-immunoreactive neurons did not alter overall cell staining. G, Number of cue-induced active lever presses was positively correlated with the number of surviving nNOS cells at the site of lesion (average over four sections per mouse). Letters by data point correspond to mice shown as examples in D and E. *p < 0.05 compared with extinction; +p < 0.05 comparing active with inactive lever presses.
We also conducted the converse experiment with NOS1-Cre mice and used a genetic strategy to ablate NAcore nNOS neurons selectively (Yang et al., 2013). Using the genetically engineered caspase-3 viral vector taCasp3-TEVp, the expression of which commits the cell to apoptosis, we successfully killed nNOS-expressing neurons in the site of virus injection (Fig. 8D–G). This specific construct limits toxicity to Cre-expressing cells by triggering cell-autonomous apoptosis. Indeed, immunoreactive nNOS neurons were reduced in number without altering the amount of signal obtained from surrounding cells using a nonspecific fluorescent signal for DNA, TO-PRO-3 iodide (Fig. 8F). The extent of nNOS neuron ablation was quantified by counting nNOS-immunoreactive cells in the NAcore from three to four sections containing evidence of a visible injection tract in each hemisphere to obtain an animal average (Fig. 8D,E). The loss of nNOS neurons did not affect cocaine self-administration and extinction, although the loss of nNOS neurons was variable across mice, ranging from no apparent loss to complete depletion. However, this extent of depletion was positively correlated with the amount of reinstated active lever pressing (Fig. 8G; r2 = 0.699, p = 0.019, n = 7). Therefore, mice with near complete loss of nNOS expression showed no reinstatement, whereas mice with the smallest lesions showed normal levels of cue-induced reinstatement.
Discussion
Cue-induced reinstatement of cocaine seeking is a widely used animal model of cocaine relapse that requires spillover of synaptically released glutamate and the activation of t-SP in NAcore. Here, we uncovered a cellular sequence of events whereby spillover of synaptic glutamate elicited by presenting cocaine-conditioned cues induces t-SP and reinstates cocaine seeking (Fig. 9). We show that extrasynaptic glutamate activated mGluR5 to stimulate NO production by the sparse population of NAcore interneurons that express nNOS. The increase in NO then caused S-nitrosylation and activation of MMP-2, which is required for inducing MMP activity and t-SP and ultimately for reinstating cocaine seeking (Gipson et al., 2013a; Smith et al., 2014).
Figure 9.

Model of how a cocaine cue induces synaptic glutamate (Glu) spillover from PL-PFC synapses in NAcore and reinstates cocaine seeking. (1) Cocaine-induced downregulation of glial glutamate transporters (GLT-1) allows cue-induced release of synaptic glutamate from PL-PFC afferents to (2) spillover into the extracellular space. The increase in extrasynaptic glutamate (3) stimulates mGluR5 in nNOS interneurons and activates nNOS to increase the production of NO. Diffusion of NO into the extracellular space (2) activates MMP-2 and MMP-9 (4), which is necessary to induce t-SP in NAcore MSNs and reinstate behavior.
Glutamate spillover
A shared characteristic of withdrawal from many classes of addictive drug is downregulation of glial glutamate uptake via GLT-1 in the NAcore (Sari et al., 2011; Fischer-Smith et al., 2012; Gipson et al., 2013b) or decreased coverage of synapses by synaptic astrocytic processes (Scofield et al., 2016). Glial glutamate uptake via GLT-1 is the prepotent physiological mechanism for clearing glutamate from the extracellular space (Danbolt, 2001). Therefore, constitutive downregulation of GLT-1 or reduced proximity of GLT-1 to glutamatergic synapses after using addictive drugs creates a scenario whereby cue-induced synaptic release of glutamate from PL-PFC afferents to the NAcore spills excessively outside of the synaptic cleft and gains access to extrasynaptic regions of the neuropil. In the extrasynaptic space, glutamate has access primarily to mGluR and NMDA glutamate receptors that are localized outside of the synaptic cleft. Stimulating group II (mGluR2/3) mGluRs is well characterized to produce a Gi-signaling mediated decrease in release probability at glutamatergic synapses (Moussawi and Kalivas, 2010). In contrast, stimulating either Gq-coupled group I mGluR1/5 or NMDA receptors increases intracellular calcium and thereby initiates a variety of cellular actions, including activating nNOS (Hayashi et al., 1999; Hardingham et al., 2013).
Activation of nNOS and MMPs
Here, we show that glutamate spillover stimulates nNOS interneurons bearing mGluR5. We also found that enduring dephosphorylation of nNOS at serine 847 was produced after extinction from cocaine self-administration and this posttranslational modification leaves the enzyme hyperresponsive to mGluR5-induced increases in intracellular calcium (Rao et al., 2008). Accordingly, stimulation of mGluR5 or Gq-DREADD expressed selectively in nNOS interneurons increases NO synthesis robustly. Finally, stimulation of this signaling sequence in nNOS interneurons induced MMP-2 and MMP-9 activity, at least in part via S-nitrosylating MMP-2, which in turn initiated t-SP in MSNs and reinstated cocaine seeking. In addition to activating MMP-2, withdrawal from noncontingent cocaine administration increases S-nitrosylation of stargazin constitutively in the shell subcompartment of the nucleus accumbens (Selvakumar et al., 2014). Although this study showed no change in NAcore, given its role to chaperone AMPA receptors into the postsynapse, it is tempting to speculate that S-nitrosylation of stargazin or another protein by cue-induced NO synthesis could contribute to the increase in AMPA signaling associated with t-SP.
DREADD activation of nNOS interneurons
Our data show that, even in the absence of conditioned cues, the intrinsic microcircuit shown in Figure 9 is sufficient for inducing cocaine seeking. Therefore, activating mGluR5- or Gq-DREADD-induced activity in nNOS-expressing interneurons reinstated active lever pressing in mice even in the absence of restoring conditioned cues to lever pressing. We propose that the behavioral effect of Gq-DREADD activation results from increased NO production because we measured the capacity of CNO to increase extracellular NO (Fig. 5C) alongside increased sensitivity of nNOS to intracellular calcium that results from enduring cocaine-induced dephosphorylation of serine-847. (Fig. 6C; Rao et al., 2008). Furthermore, stimulation of MMP2,9 activity is a prerequisite for cue-induced reinstatement (Smith et al., 2014) and antagonizing nNOS with NPLA prevented MMP-2 and MMP-9 activation by CNO (Fig. 6A,B), as well as cue-induced reinstatement (Fig. 3F). Finally, SRIF is a transmitter frequently colocalized in nNOS interneurons (Figueredo-Cardenas et al., 1996) and we found that neither activation nor inhibition of SRIF receptors in NAcore altered reinstated or extinguished lever pressing (Fig. 4A,C). Nonetheless, it remains possible that GABA release from nNOS cells in response to the Gq-mediated increase in cell firing could influence cue-induced reinstatement. In addition, the spillover of synaptic glutamate would be expected to stimulate other glutamate receptors on neurons and glia in addition to the 1% of interneurons expressing nNOS. Therefore, in addition to an untested role for GABA release from nNOS interneurons, the mGluR5-nNOS signaling described herein may not necessarily be a complete description of how the NAcore contributes to translating a cocaine-conditioned cue into reinstated drug seeking. Perhaps most revealing that a mechanism(s) parallel to activating nNOS interneurons may contribute to cued reinstatement is that, whereas NPLA completely abolished reinstatement and MMP-2 and MMP-9 activity elicited by stimulating mGluR5, it did not appear to reduce cue-induced reinstatement to extinction levels of lever pressing (Fig. 3F).
Alternative circuitry
Although we did not uncover this hypothesized parallel pathway for cued reinstatement, two possibilities are consistent with the literature. When mGluR5 was knocked down selectively in D1-expressing MSNs, cue-induced reinstatement of cocaine seeking was reduced, but not abolished, posing mGluR5 on this population of accumbens neurons as a parallel reinstatement pathway to nNOS interneurons (Novak et al., 2010). However, if this were the case, then NPLA should not have completely abolished reinstated cocaine seeking produced by CHPG microinjections (Fig. 3D). Alternatively, because reinstated cocaine seeking was not associated with measureable increases in S-nitrosylation of MMP-9, activation of MMP-9 may be an insertion point for a parallel signaling mechanism. For example, whereas MMP-9 can be stimulated by activated (S-nitrosylated) MMP-2 (Toth et al., 2003), MMP-9 is also activated indirectly by proteases such as tissue or urokinase plasminogen activator (tPA or uPA), which act independently of S-nitrosylation (Huntley, 2012). Interestingly, cocaine and other psychostimulants increase tPA or uPA synthesis in corticostriatal neurons (Hashimoto et al., 1998) and manipulating tPA or uPA pharmacologically or genetically alters the behavioral effects of acute and chronic cocaine use (Bahi and Dreyer, 2008).
Summary
The intrinsic nitrergic circuit revealed in our study identifies mGluR5 stimulation on nNOS interneurons as a key mechanism for activating NO production and stimulating t-SP and reinstating cocaine seeking. Although cue-induced glutamate spillover into the extracellular space from PL-PFC synapses in the NAcore is one likely mechanism for stimulating mGluR5-bearing nNOS neurons, it is possible that direct glutamatergic input to nNOS cells could also contribute. Particular intriguing along these lines, nNOS neurons receive glutamatergic synaptic input from the ventral hippocampus (French et al., 2005) and ventral hippocampal inputs to the NAcore are known to contribute to reinstated cocaine seeking (Rogers and See, 2007). Nonetheless, activation of the mGluR5-nNOS-MMP pathway by glutamate spillover explains how preventing cue-induced spillover in the NAcore pharmacologically by restoring the glial glutamate transporters with N-acetylcysteine or ceftriaxone inhibits cue-induced reinstatement to many addictive drugs (Kalivas and Volkow, 2011; Moussawi et al., 2011; Sari et al., 2011). Importantly, using N-acetylcysteine to restore glutamate transport has also been a successful strategy in pilot clinical trials for reducing cocaine and marijuana craving and use (Kalivas and Volkow, 2011; Gray et al., 2012). Our discovery here that a sparsely distributed population of nNOS-expressing interneurons is a bottleneck in how glutamate spillover induced by drug-conditioned cues stimulates relapse points to a potential new target for developing cell-specific therapeutic interventions for drug addiction.
Footnotes
This work was funded in part by the National Institutes of Health (Grants DA003906, DA012513, DA015369, and DA007135).
References
- Agulhon C, Boyt KM, Xie AX, Friocourt F, Roth BL, McCarthy KD. Modulation of the autonomic nervous system and behaviour by acute glial cell Gq protein-coupled receptor activation in vivo. J Physiol. 2013;591:5599–5609. doi: 10.1113/jphysiol.2013.261289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SM, Famous KR, Sadri-Vakili G, Kumaresan V, Schmidt HD, Bass CE, Terwilliger EF, Cha JH, Pierce RC. CaMKII: a biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat Neurosci. 2008;11:344–353. doi: 10.1038/nn2054. [DOI] [PubMed] [Google Scholar]
- Bahi A, Dreyer JL. Overexpression of plasminogen activators in the nucleus accumbens enhances cocaine-, amphetamine- and morphine-induced reward and behavioral sensitization. Genes Brain Behav. 2008;7:244–256. doi: 10.1111/j.1601-183X.2007.00346.x. [DOI] [PubMed] [Google Scholar]
- Barbosa RM, Lourenço CF, Santos RM, Pomerleau F, Huettl P, Gerhardt GA, Laranjinha J. In vivo real-time measurement of nitric oxide in anesthetized rat brain. Methods Enzymol. 2008;441:351–367. doi: 10.1016/S0076-6879(08)01220-2. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Nagy V, Kwei KT, Huntley GW. In vivo roles for matrix metalloproteinase-9 in mature hippocampal synaptic physiology and plasticity. J Neurophysiol. 2007;98:334–344. doi: 10.1152/jn.00202.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister JJ, Pomerleau F, Palmer M, Day BK, Huettl P, Gerhardt GA. Improved ceramic-based multisite microelectrode for rapid measurements of L-glutamate in the CNS. J Neurosci Methods. 2002;119:163–171. doi: 10.1016/S0165-0270(02)00172-3. [DOI] [PubMed] [Google Scholar]
- Cardinal RN, Everitt BJ. Neural and psychological mechanisms underlying appetitive learning: links to drug addiction. Curr Opin Neurobiol. 2004;14:156–162. doi: 10.1016/j.conb.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Coddington LT, Rudolph S, Vande Lune P, Overstreet-Wadiche L, Wadiche JI. Spillover-mediated feedforward inhibition functionally segregates interneuron activity. Neuron. 2013;78:1050–1062. doi: 10.1016/j.neuron.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/S0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Ferreira NR, Ledo A, Frade JG, Gerhardt GA, Laranjinha J, Barbosa RM. Electrochemical measurement of endogenously produced nitric oxide in brain slices using Nafion/o-phenylenediamine modified carbon fiber microelectrodes. Analytica Chimica Acta. 2005;535:1–7. doi: 10.1016/j.aca.2004.12.017. [DOI] [Google Scholar]
- Figueredo-Cardenas G, Morello M, Sancesario G, Bernardi G, Reiner A. Colocalization of somatostatin, neuropeptide Y, neuronal nitric oxide synthase and NADPH-diaphorase in striatal interneurons in rats. Brain Res. 1996;735:317–324. doi: 10.1016/0006-8993(96)00801-3. [DOI] [PubMed] [Google Scholar]
- Fino E, Paille V, Deniau JM, Venance L. Asymmetric spike-timing dependent plasticity of striatal nitric oxide-synthase interneurons. Neuroscience. 2009;160:744–754. doi: 10.1016/j.neuroscience.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Fischer-Smith KD, Houston AC, Rebec GV. Differential effects of cocaine access and withdrawal on glutamate type 1 transporter expression in rat nucleus accumbens core and shell. Neuroscience. 2012;210:333–339. doi: 10.1016/j.neuroscience.2012.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourgeaud L, Mato S, Bouchet D, Hémar A, Worley PF, Manzoni OJ. A single in vivo exposure to cocaine abolishes endocannabinoid-mediated long-term depression in the nucleus accumbens. J Neurosci. 2004;24:6939–6945. doi: 10.1523/JNEUROSCI.0671-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French SJ, Ritson GP, Hidaka S, Totterdell S. Nucleus accumbens nitric oxide immunoreactive interneurons receive nitric oxide and ventral subicular afferents in rats. Neuroscience. 2005;135:121–131. doi: 10.1016/j.neuroscience.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Gipson CD, Kupchik YM, Shen H, Reissner KJ, Thomas CA, Kalivas PW. Relapse induced by cues predicting cocaine depends on rapid, transient synaptic potentiation. Neuron. 2013a;77:867–872. doi: 10.1016/j.neuron.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gipson CD, Reissner KJ, Kupchik YM, Smith AC, Stankeviciute N, Hensley-Simon ME, Kalivas PW. Reinstatement of nicotine seeking is mediated by glutamatergic plasticity. Proc Natl Acad Sci U S A. 2013b;110:9124–9129. doi: 10.1073/pnas.1220591110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray KM, Carpenter MJ, Baker NL, DeSantis SM, Kryway E, Hartwell KJ, McRae-Clark AL, Brady KT. A double-blind randomized controlled trial of N-acetylcysteine in cannabis-dependent adolescents. Am J Psychiatry. 2012;169:805–812. doi: 10.1176/appi.ajp.2012.12010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WC, 3rd, Randall PK, Middaugh LD. Intravenous cocaine self-administration: individual differences in male and female C57BL/6J mice. Pharmacol Biochem Behav. 2007;87:267–279. doi: 10.1016/j.pbb.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- Hardingham N, Dachtler J, Fox K. The role of nitric oxide in pre-synaptic plasticity and homeostasis. Front Cell Neurosci. 2013;7:190. doi: 10.3389/fncel.2013.00190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Kajii Y, Nishikawa T. Psychotomimetic-induction of tissue plasminogen activator mRNA in corticostriatal neurons in rat brain. Eur J Neurosci. 1998;10:3387–3399. doi: 10.1046/j.1460-9568.1998.00343.x. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Nishio M, Naito Y, Yokokura H, Nimura Y, Hidaka H, Watanabe Y. Regulation of neuronal nitric-oxide synthase by calmodulin kinases. J Biol Chem. 1999;274:20597–20602. doi: 10.1074/jbc.274.29.20597. [DOI] [PubMed] [Google Scholar]
- Huntley GW. Synaptic circuit remodelling by matrix metalloproteinases in health and disease. Nat Rev Neurosci. 2012;13:743–757. doi: 10.1038/nrn3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Kotani A, Koshikawa N, Cools AR. Somatostatin receptors in the nucleus accumbens modulate dopamine-dependent but not acetylcholine-dependent turning behaviour of rats. Neuroscience. 2009;159:974–981. doi: 10.1016/j.neuroscience.2009.01.053. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow ND. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol Psychiatry. 2011;16:974–986. doi: 10.1038/mp.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J Neurosci. 1993;13:4908–4923. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knackstedt LA, Moussawi K, Lalumiere R, Schwendt M, Klugmann M, Kalivas PW. Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J Neurosci. 2010;30:7984–7992. doi: 10.1523/JNEUROSCI.1244-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupai K, Szucs G, Cseh S, Hajdu I, Csonka C, Csont T, Ferdinandy P. Matrix metalloproteinase activity assays: importance of zymography. J Pharmacol Toxicol Methods. 2010;61:205–209. doi: 10.1016/j.vascn.2010.02.011. [DOI] [PubMed] [Google Scholar]
- Kupchik YM, Moussawi K, Tang XC, Wang X, Kalivas BC, Kolokithas R, Ogburn KB, Kalivas PW. The effect of N-acetylcysteine in the nucleus accumbens on neurotransmission and relapse to cocaine. Biol Psychiatry. 2012;71:978–986. doi: 10.1016/j.biopsych.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaLumiere RT, Kalivas PW. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J Neurosci. 2008;28:3170–3177. doi: 10.1523/JNEUROSCI.5129-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DK, Koh WC, Shim YB, Shim I, Choe ES. Repeated cocaine administration increases nitric oxide efflux in the rat dorsal striatum. Psychopharmacology (Berl) 2010;208:245–256. doi: 10.1007/s00213-009-1724-z. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma YY, Lee BR, Wang X, Guo C, Liu L, Cui R, Lan Y, Balcita-Pedicino JJ, Wolf ME, Sesack SR, Shaham Y, Schlüter OM, Huang YH, Dong Y. Bidirectional modulation of incubation of cocaine craving by silent synapse-based remodeling of prefrontal cortex to accumbens projections. Neuron. 2014;83:1453–1467. doi: 10.1016/j.neuron.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland K, Lapish CC, Kalivas PW. Prefrontal glutamate release into the core of the nucleus accumbens mediates cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2003;23:3531–3537. doi: 10.1523/JNEUROSCI.23-08-03531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael AC, Borland LM. Electrochemical methods for neuroscience. Boca Raton, FL: CRC/Taylor and Francis; 2007. [PubMed] [Google Scholar]
- Mitrano DA, Pare JF, Smith Y. Ultrastructural relationships between cortical, thalamic, and amygdala glutamatergic inputs and group I metabotropic glutamate receptors in the rat accumbens. J Comp Neurol. 2010;518:1315–1329. doi: 10.1002/cne.22277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Kalivas PW. Group II metabotropic glutamate receptors (mGlu(2/3)) in drug addiction. Eur J Pharmacol. 2010;639:115–122. doi: 10.1016/j.ejphar.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, Kalivas PW. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–189. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussawi K, Zhou W, Shen H, Reichel CM, See RE, Carr DB, Kalivas PW. Reversing cocaine-induced synaptic potentiation provides enduring protection from relapse. Proc Natl Acad Sci U S A. 2011;108:385–390. doi: 10.1073/pnas.1011265108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy V, Bozdagi O, Huntley GW. The extracellular matrix protease matrix metalloproteinase-9 is activated by inhibitory avoidance learning and required for long-term memory. Learn Mem. 2007;14:655–664. doi: 10.1101/lm.678307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan R, Harding JW, Wright JW. A role for matrix metalloproteinases in nicotine-induced conditioned place preference and relapse in adolescent female rats. J Exp Neurosci. 2013;7:1–14. doi: 10.4137/JEN.S11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak M, Halbout B, O'Connor EC, Rodriguez Parkitna J, Su T, Chai M, Crombag HS, Bilbao A, Spanagel R, Stephens DN, Schütz G, Engblom D. Incentive learning underlying cocaine-seeking requires mGluR5 receptors located on dopamine D1 receptor-expressing neurons. J Neurosci. 2010;30:11973–11982. doi: 10.1523/JNEUROSCI.2550-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onifer SM, Quintero JE, Gerhardt GA. Cutaneous and electrically evoked glutamate signaling in the adult rat somatosensory system. J Neurosci Methods. 2012;208:146–154. doi: 10.1016/j.jneumeth.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Espallergues J, Valjent E, O'Connor EC, Lüscher C. Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature. 2014;509:459–464. doi: 10.1038/nature13257. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinate. Ed 4. Cambridge, MA: Elsevier; 2012. [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 6. Burlington, MA: Elsevier; 2007. [DOI] [PubMed] [Google Scholar]
- Peters J, Kalivas PW. The group II metabotropic glutamate receptor agonist, LY379268, inhibits both cocaine- and food-seeking behavior in rats. Psychopharmacology (Berl) 2006;186:143–149. doi: 10.1007/s00213-006-0372-9. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. J Neurosci. 1996;16:1550–1560. doi: 10.1523/JNEUROSCI.16-04-01550.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero JE, Pomerleau F, Huettl P, Johnson KW, Offord J, Gerhardt GA. Methodology for rapid measures of glutamate release in rat brain slices using ceramic-based microelectrode arrays: basic characterization and drug pharmacology. Brain Res. 2011;1401:1–9. doi: 10.1016/j.brainres.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rameau GA, Chiu LY, Ziff EB. Bidirectional regulation of neuronal nitric-oxide synthase phosphorylation at serine 847 by the N-methyl-D-aspartate receptor. J Biol Chem. 2004;279:14307–14314. doi: 10.1074/jbc.M311103200. [DOI] [PubMed] [Google Scholar]
- Rao YM, Chaudhury A, Goyal RK. Active and inactive pools of nNOS in the nerve terminals in mouse gut: implications for nitrergic neurotransmission. Am J Physiol Gastrointest Liver Physiol. 2008;294:G627–634. doi: 10.1152/ajpgi.00519.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynor K, Lucki I, Reisine T. Somatostatin receptors in the nucleus accumbens selectively mediate the stimulatory effect of somatostatin on locomotor activity in rats. J Pharmacol Exp Ther. 1993;265:67–73. [PubMed] [Google Scholar]
- Rogers JL, See RE. Selective inactivation of the ventral hippocampus attenuates cue-induced and cocaine-primed reinstatement of drug-seeking in rats. Neurobiol Learn Mem. 2007;87:688–692. doi: 10.1016/j.nlm.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santis S, Kastellakis A, Kotzamani D, Pitarokoili K, Kokona D, Thermos K. Somatostatin increases rat locomotor activity by activating sst(2) and sst (4) receptors in the striatum and via glutamatergic involvement. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:181–189. doi: 10.1007/s00210-008-0346-z. [DOI] [PubMed] [Google Scholar]
- Sari Y, Sakai M, Weedman JM, Rebec GV, Bell RL. Ceftriaxone, a beta-lactam antibiotic, reduces ethanol consumption in alcohol-preferring rats. Alcohol Alcoholism. 2011;46:239–246. doi: 10.1093/alcalc/agr023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Schassburger RL, Guercio LA, Pierce RC. Stimulation of mGluR5 in the accumbens shell promotes cocaine seeking by activating PKC gamma. J Neurosci. 2013;33:14160–14169. doi: 10.1523/JNEUROSCI.2284-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield MD, Li H, Siemsen BM, Healey KL, Tran PK, Woronoff N, Boger HA, Kalivas PW, Reissner KJ. Cocaine self-administration and extinction leads to reduced glial fibrillary acidic protein expression and morphometric features of astrocytes in the nucleus accumbens core. Biol Psychiat. 2016;80:207–215. doi: 10.1016/j.biopsych.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvakumar B, Campbell PW, Milovanovic M, Park DJ, West AR, Snyder SH, Wolf ME. AMPA receptor upregulation in the nucleus accumbens shell of cocaine-sensitized rats depends upon S-nitrosylation of stargazin. Neuropharmacology. 2014;77:28–38. doi: 10.1016/j.neuropharm.2013.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl) 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- Shen HW, Scofield MD, Boger H, Hensley M, Kalivas PW. Synaptic glutamate spillover due to impaired glutamate uptake mediates heroin relapse. J Neurosci. 2014;34:5649–5657. doi: 10.1523/JNEUROSCI.4564-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Moussawi K, Zhou W, Toda S, Kalivas PW. Heroin relapse requires long-term potentiation-like plasticity mediated by NMDA2b-containing receptors. Proc Natl Acad Sci U S A. 2011;108:19407–19412. doi: 10.1073/pnas.1112052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair CM, Cleva RM, Hood LE, Olive MF, Gass JT. mGluR5 receptors in the basolateral amygdala and nucleus accumbens regulate cue-induced reinstatement of ethanol-seeking behavior. Pharmacol Biochem Behav. 2012;101:329–335. doi: 10.1016/j.pbb.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AC, Kupchik YM, Scofield MD, Gipson CD, Wiggins A, Thomas CA, Kalivas PW. Synaptic plasticity mediating cocaine relapse requires matrix metalloproteinases. Nat Neurosci. 2014;17:1655–1657. doi: 10.1038/nn.3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AW, Nealey KA, Wright JW, Walker BM. Plasticity associated with escalated operant ethanol self-administration during acute withdrawal in ethanol-dependent rats requires intact matrix metalloproteinase systems. Neurobiol Learn Mem. 2011;96:199–206. doi: 10.1016/j.nlm.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanik MT, Kupchik YM, Kalivas PW. Optogenetic inhibition of cortical afferents in the nucleus accumbens simultaneously prevents cue-induced transient synaptic potentiation and cocaine-seeking behavior. Brain Struct Funct. 2016;221:1681–1689. doi: 10.1007/s00429-015-0997-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton AK, Pei H, Burnett KH, Myers MG, Jr, Rhodes CJ, Olson DP. Control of food intake and energy expenditure by Nos1 neurons of the paraventricular hypothalamus. J Neurosci. 2014;34:15306–15318. doi: 10.1523/JNEUROSCI.0226-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth M, Chvyrkova I, Bernardo MM, Hernandez-Barrantes S, Fridman R. Pro-MMP-9 activation by the MT1-MMP/MMP-2 axis and MMP-3: role of TIMP-2 and plasma membranes. Biochem Biophys Res Commun. 2003;308:386–395. doi: 10.1016/S0006-291X(03)01405-0. [DOI] [PubMed] [Google Scholar]
- Van den Oever MC, Lubbers BR, Goriounova NA, Li KW, Van der Schors RC, Loos M, Riga D, Wiskerke J, Binnekade R, Stegeman M, Schoffelmeer AN, Mansvelder HD, Smit AB, De Vries TJ, Spijker S. Extracellular matrix plasticity and GABAergic inhibition of prefrontal cortex pyramidal cells facilitates relapse to heroin seeking. Neuropsychopharmacology. 2010;35:2120–2133. doi: 10.1038/npp.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke BU, Lindner M, Greifenberg L, Albus A, Kronimus Y, Bünemann M, Leitner MG, Oliver D. Diacylglycerol mediates regulation of TASK potassium channels by Gq-coupled receptors. Nat Commun. 2014;5:5540. doi: 10.1038/ncomms6540. [DOI] [PubMed] [Google Scholar]
- Yang CF, Chiang MC, Gray DC, Prabhakaran M, Alvarado M, Juntti SA, Unger EK, Wells JA, Shah NM. Sexually dimorphic neurons in the ventromedial hypothalamus govern mating in both sexes and aggression in males. Cell. 2013;153:896–909. doi: 10.1016/j.cell.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]