

Abstract
Alzheimer's disease (AD) is characterized by the presence of parenchymal amyloid-β (Aβ) plaques, cerebral amyloid angiopathy (CAA) and neurofibrillary tangles. Currently there are no effective treatments for AD. Immunotherapeutic approaches under development are hampered by complications related to ineffectual clearance of CAA. Genome-wide association studies have demonstrated the importance of microglia in AD pathogenesis. Microglia are the primary innate immune cells of the brain. Depending on their activation state and environment, microglia can be beneficial or detrimental. In our prior work, we showed that stimulation of innate immunity with Toll-like receptor 9 agonist, class B CpG (cytosine–phosphate–guanine) oligodeoxynucleotides (ODNs), can reduce amyloid and tau pathologies without causing toxicity in Tg2576 and 3xTg-AD mouse models. However, these transgenic mice have relatively little CAA. In the current study, we evaluated the therapeutic profile of CpG ODN in a triple transgenic mouse model, Tg-SwDI, with abundant vascular amyloid, in association with low levels of parenchymal amyloid deposits. Peripheral administration of CpG ODN, both before and after the development of CAA, negated short-term memory deficits, as assessed by object-recognition tests, and was effective at improving spatial and working memory evaluated using a radial arm maze. These findings were associated with significant reductions of CAA pathology lacking adverse effects. Together, our extensive evidence suggests that this innovative immunomodulation may be a safe approach to ameliorate all hallmarks of AD pathology, supporting the potential clinical applicability of CpG ODN.
SIGNIFICANCE STATEMENT Recent genetic studies have underscored the emerging role of microglia in Alzheimer's disease (AD) pathogenesis. Microglia lose their amyloid-β-clearing capabilities with age and as AD progresses. Therefore, the ability to modulate microglia profiles offers a promising therapeutic avenue for reducing AD pathology. Current immunotherapeutic approaches have been limited by poor clearance of a core AD lesion, cerebral amyloid angiopathy (CAA). The present study used Tg-SwDI mice, which have extensive CAA. We found that stimulation of the innate immune system and microglia/macrophage activation via Toll-like receptor 9 using CpG (cytosine–phosphate–guanine) oligodeoxynucleotides (ODNs) leads to cognitive improvements and CAA reduction, without associated toxicity. Our data indicate that this novel concept of immunomodulation represents a safer method to reduce all aspects of AD pathology and provide essential information for potential clinical use of CpG ODN.
Keywords: Alzheimer's disease, amyloid β, immunomodulation, innate immunity, Toll-like receptor 9, transgenic mice
Introduction
Alzheimer's disease (AD), a progressive neurodegenerative disorder, is characterized by parenchymal amyloid-β (Aβ) plaque deposition, cerebral amyloid angiopathy (CAA), and neurofibrillary tangle formation (Schellenberg and Montine, 2012). Overwhelming experimental evidence points to a significant role of neuroinflammation in AD pathology and its association with cognitive impairment (Lampron et al., 2013a; Lee et al., 2013). Genome-wide association studies have shown the linkage of several innate immunity-related genes in late-onset Alzheimer's disease (Zhang et al., 2013; Karch et al., 2014). Recent genetic studies demonstrated the importance of microglia in AD by identifying several AD-associated genes that are expressed primarily in microglial cells. Microglia are critical regulators of innate immune responses in the brain. However, depending on the circumstances, their activation can be either beneficial or detrimental (ElAli and Rivest, 2016). Innate immune cells, including both CNS resident microglia and peripheral bone marrow-derived macrophages (BMDMs), can exhibit a dysfunctional/senescent profile characterized by impaired migration and phagocytosis in AD (Michaud and Rivest, 2015; Heneka et al., 2016). Therefore, modification of the macrophage/microglia activation state has strong therapeutic promise (Guillot-Sestier et al., 2015b; Heneka et al., 2016). Signaling via Toll-like receptors (TLRs) is known to influence the phenotypic status of microglia. TLRs are well characterized pattern-recognition receptors triggered by pathogen-associated molecular patterns expressed by bacteria, viruses, fungi, and protozoa. TLRs may mediate different pathways in microglia leading to either neuroprotective or neurotoxic phenotypes (Iliev et al., 2004; Cunningham et al., 2005; Hanke and Kielian, 2011).
Stimulation of innate immunity via TLR signaling pathways has been shown to be beneficial in modulating AD pathology (Richard et al., 2008; Doi et al., 2009; Michaud et al., 2013; Su et al., 2016). Nonetheless, manipulation of TLRs can also exert adverse effects in AD models (Heikenwalder et al., 2004; Lee et al., 2008; Campbell et al., 2009; Su et al., 2016). Discrepancies between studies may be the consequence of variations in types and concentrations of TLR ligands used, as well as differences in administration frequencies. We have focused on reconciling the age-related defects in immune-cell function and on tackling AD via Toll-like receptor 9 (TLR9). TLR9 recognizes the unmethylated CpG (cytosine–phosphate–guanine) motifs present at high frequency in bacterial and viral DNA and at low frequency in human DNA. Oligodeoxynucleotides (ODNs) containing these unmethylated CpG motifs trigger cells that express TLR9 (including monocytes, plasmacytoid dendritic cells, and B cells) to mount an innate immune response. TLR9 is localized in late endosomal and lysosomal compartments of the intracellular milieu (Krieg, 2002; Vollmer and Krieg, 2009). Several CpG ODNs have shown excellent safety profiles in humans and have been explored in numerous human clinical trials as antitumor, antimicroglial agents and as vaccine adjuvants (Vollmer and Krieg, 2009; Krieg, 2012).
Immunotherapy has emerged as an attractive approach for disease intervention in AD. However, major concerns are the occurrence of cerebral microhemorrhages associated with increased CAA and excessive neuroinflammation (Wilcock and Colton, 2009; Sakai et al., 2014; Wisniewski and Goñi, 2015). Our initial findings using the Tg2576 and 3xTg-AD mouse models document that stimulation of innate immunity via TLR9 ligand class B CpG ODN has the advantage of concurrently ameliorating Aβ and tau pathologies, in association with behavioral improvements (Scholtzova et al., 2009, 2014). However, these mouse models have relatively little vascular amyloid. The current study evaluated the therapeutic profile of CpG ODN in Tg-SwDI mice, which have extensive vascular amyloid in association with low levels of parenchymal amyloid deposits (Davis et al., 2004; Van Vickle et al., 2008). We tested the hypothesis that CpG ODN can harness innate immunity to prevent the age-dependent accumulation of CAA pathology in young mice and can rescue the CAA phenotype in aged mice. A growing number of studies have identified macrophages/microglia as highly dynamic cells with the coexistence of multiple activation states and a broad spectrum of overlapping polarization phenotypes with diverse functions (Brothers and Wilcock, 2013; Latta et al., 2015; Wes et al., 2016). Therefore, we conducted a short-term acute study in a separate cohort of old Tg-SwDI mice to further clarify the effect of CpG ODN on the recruitment and activation of beneficial macrophage/microglia phenotypes that may contribute to clearance of CAA pathology.
Materials and Methods
Animals and treatment
The treatment was performed in triple transgenic (Tg) mice expressing human Swedish K670N/M671L and vasculotropic Dutch/Iowa E693Q/D694N mutations under the control of the mouse Thy1.2 promoter (Tg-SwDI). These mice exhibit early and robust cerebral microvascular accumulation of Aβ in the form of fibrillar vascular Aβ deposits (starting at the age of 3–4 months), and mainly diffuse Aβ deposition in the brain parenchyma (Davis et al., 2004; Miao et al., 2005). Both Tg-SwDI mice and wild-type (Wt) control mice used in this study were bred internally and aged at the New York University School of Medicine on a pure C57BL/6 background. Breeding pairs of Tg-SwDI mice were obtained from the Jackson Laboratory (www.jax.org) where this transgenic (Tg) line was originally generated on a C57BL/6 background. The animals were maintained on a 12 h light/dark cycle, and animal care was in accordance with institutional guidelines in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care. All experimental procedures were approved by the Institutional Animal Care and Use Committee at the New York University School of Medicine. Mice were injected with either TLR9 agonist CpG ODN 1826 (2.5 mg/kg, i.p.) or vehicle (saline) at monthly intervals. The mice were divided into two study groups treated from 4 to 14 months of age, before significant pathology development, and from 8 to 18 months of age, at a point when pathology is well established. Control Wt mice, matched for age and sex to Tg-SwDI groups, were given CpG ODN or saline. Treatment and vehicle groups included 14 mice per group, and equal numbers of males and females were in each experimental group. A short-term acute study was conducted in a separate cohort of aged Tg-SwDI mice (18 months old; matched for age and sex to our 8–18-month study group at the end of the treatment period). Animals (n = 8) were acutely injected with CpG ODN or saline to further evaluate the effects of TLR9 signaling on the formation of dynamic range of macrophage/microglia activation states with the ability to promote phagocytosis. CpG ODN 1826 [5′-TCC ATG ACG TTC CTG ACG TT-3′ (CpG motifs italicized)], with a complete phosphorothioate backbone, was purchased from Integrated DNA Technologies. During the treatment, animals were monitored for signs of toxicity, such as differences in total body weight, altered behavior, and changes in physical appearance.
Behavioral testing
At the end of the treatment, mice were subjected to behavioral testing. Behavioral analyses consisted of both cognitive assessment and measurement of sensorimotor (locomotor) abilities. The latter tests were included to verify that the performance on cognitive testing was not influenced by sensorimotor abnormalities.
Locomotor activity test.
Locomotor activity was recorded in a circular open-field activity chamber measuring 70 cm in diameter. A Hamilton-Kinder Smart-Frame Photobeam System was used to make a computerized recording of animal activity, as we have previously described (Yang et al., 2011; Scholtzova et al., 2014). A video camera mounted above the chamber automatically recorded horizontal movements in the open field in each dimension. Before testing, mice were adapted to the room with lights on for a designated period. Animals were allowed to explore the environment for 15 min. Results were reported based on distance traveled (cm), mean resting time (sec), and velocity (mean and maximum; cm/sec) of the animal.
Novel object recognition test.
The novel object recognition test is based on the natural tendency of rodents to investigate a novel object instead of a familiar one (Asuni et al., 2007; Scholtzova et al., 2008). This test was used to measure deficits in short-term memory and did not require food or water deprivation. The object-recognition test was conducted in a square open-field box (48 cm square with 18-cm-high walls) constructed from black Plexiglas. The light intensity of the arena was set to 30 lux. The test consisted of a familiarization session (day 1, 15 min) in which mice explored the open-field arena containing two identical, symmetrically placed objects. During training sessions (day 2), two novel objects were placed at diagonal corners in the open field. The animal was allowed to explore for 15 min. The time spent exploring each object was recorded by a tracking system (San Diego Instruments) and, at the end of the training phase, the mouse was removed from the box for the duration of the retention delay (retention delay, 3 h; Chen et al., 2000; Dodart et al., 2002; Asuni et al., 2007; Scholtzova et al., 2008; Boutajangout et al., 2010). During the retention session, the animals were placed back into the same box, in which one of the previous familiar objects used during training was replaced by a second novel object. They were allowed to explore the objects for 6 min. The time spent exploring the novel and familiar objects was recorded for the duration of the test. The data are presented as a novel object preference index, a ratio of the amount of time spent exploring the novel object (retention session) over the total time spent exploring both objects. All objects used for our analysis have been previously tested for investigatory preference and have been used in earlier published studies (Scholtzova et al., 2008; Boutajangout et al., 2010, 2012). The two objects differed in shape, color, and reflectance. The shape and height of the objects prevented the mice from climbing onto them. The type of familiar or novel object as well as the relative position was counterbalanced and randomly selected for each experimental animal during both the training phase and the retention session.
Radial arm maze.
Spatial learning (working memory) was evaluated using an eight-arm radial maze as previously reported (Yang et al., 2011; Goñi et al., 2013; Scholtzova et al., 2014). The apparatus consisted of eight radial 30-cm-long arms originating from the central space with a water well at the end of each arm (0.1% saccharine solution). Plexiglas guillotine doors, operated by a remote pulley system, controlled access to the arms. Animals entered and exited all arms of the apparatus from a central area. After 4 days of adaptation, water-restricted mice (2 h daily access to water) were subjected to testing for 12 consecutive days (one test session per day). For each session, animals were permitted to enter all arms until the eight rewards had been consumed. Our criteria for termination of a session was whether the mouse had either successfully visited each of the distinct eight arms at least once or 900 sec session time had elapsed. The number of working-memory errors (entries to previously visited arms) and time to complete each session were recorded. The behavioral testing was performed by an individual blinded to the animal's treatment status.
Histological studies
The mice were anesthetized with sodium pentobarbital (150 mg/kg, i.p.) and perfused transaortically with 0.1 m PBS, pH 7.4. The right brain hemisphere was immersion-fixed in periodate-lysine-paraformaldehyde for 24 h, whereas the left hemisphere was snap-frozen on dry ice and stored at −80°C for biochemical analyses (measurements of Aβ levels and oligomers). After fixation, brains were placed in 2% DMSO/20% glycerol in PBS and stored at 4°C until sectioned. Serial coronal brain sections (40 μm) were cut from each experimental group on a freezing microtome and 10 series of sections were saved for histological and immunohistochemical analysis of staining with the following: (1) anti-Aβ antibodies 6E10/4G8, (2) thioflavin-S, (3) anti-CD45 antibody, (4) anti-CD11b antibody, (5) anti-CD206 antibody, (6) anti-Iba1 antibody, (7) anti-GFAP antibody, and (8) anti-CD3 antibody. For each antibody immunostain, we sampled every 10th section from the mouse brain. The remaining series were placed in ethylene glycol cryoprotectant (30% sucrose/30% ethylene glycol in 0.1 mol/l phosphate buffer) and stored at −20°C until used. Aβ deposits were stained either with a mixture of mouse monoclonal antibodies 6E10/4G8 (total amyloid) or thioflavin-S for fibrillar amyloid (both parenchymal and CAA).
Microglial activation is a complex process that can result in several phenotypes. Hence, in the present study we used four different markers to identify distinct macrophage/microglia activation states. CD11b [member of β-integrin family of adhesion molecules; also known as MAC-1 or complement receptor 3 (CR3)] and CD45 (protein-tyrosine phosphatase) are commonly used as markers for microglial activation at the earlier and later stages of plaque development, respectively (Morgan et al., 2005). CD11b labels macrophage/microglia in both M1 (classical) and M2 (alternative) activation states, while CD45 (highly expressed on peripherally derived macrophages) is often associated with the M1 classical activation state (Cao et al., 2012; Guerrero et al., 2012). In addition, histological staining for the alternative M2 macrophage activation marker mannose receptor 1 CD206 was performed. The mannose receptor is a phagocytic receptor of the C-type lectin family with known expression on activated macrophages/microglia (Gordon, 2003; Guerrero et al., 2012; He et al., 2012). Bone marrow-derived perivascular macrophages have been shown to be phagocytic and to express CD206 (Galea et al., 2005; Hawkes and McLaurin, 2009). Iba1 (ionized calcium-binding adaptor molecule) labels the entire microglia population (activated, resting), and is often used as a marker of generic microglia (Streit et al., 2009; Zotova et al., 2013). GFAP is a component of the glial intermediate filaments that form part of the cytoskeleton and is found predominantly in astrocytes.
Immunohistochemistry was performed on free-floating coronal brain sections as previously described (Scholtzova et al., 2009, 2014). Briefly, serial sections were incubated in mouse-on-mouse diluents containing different mouse monoclonal primary antibodies at 4°C overnight (anti-Aβ antibodies 6E10 and 4G8, 1:1000; Covance Research Products). CD206 (rabbit polyclonal, 1:200; 4°C overnight; Abcam)], Iba1 (rabbit polyclonal, 1:1000; 4°C overnight; Wako Chemicals), GFAP (rabbit polyclonal, 1:1000; 4°C overnight; Dako), and T-cell antibody CD3 (rabbit polyclonal, 1:100; 4°C overnight; Ventana Medical Systems) immunostaining was performed with a primary antibody diluent composed of 0.3% Triton X-100, 0.1% sodium azide, 0.01% bacitracin, 1% bovine serum albumin, and 10% normal goat serum in PBS, and secondary biotinylated goat anti-rabbit antibody (Vector Laboratories) reacted for 1 h at 1:1000 dilution. Moreover, T-cell immunohistochemistry was performed using the citrate buffer antigen retrieval protocol [10 mm sodium citrate, pH 6.0, 95°C water bath (10 min incubation)]. CD45 (rat anti-mouse, 1:1000; 4°C overnight; Bio-Rad Laboratories)] and CD11b [rat anti-mouse, 1:500; 4°C overnight; Bio-Rad Laboratories) immunohistochemistry was performed in a way similar to that for GFAP staining except that the secondary antibody was goat anti-rat (Vector Laboratories) diluted 1:1000. Antibody staining was revealed with 3,3-diaminobenzidine tetrahydrochloride (Sigma-Aldrich) with nickel ammonium sulfate color intensification. In addition, nuclear fast red (Vector Laboratories) was applied after completion of T-cell immunohistochemical staining.
To further evaluate the effects of TLR9 signaling on diverse range of macrophage/microglia activation states, we next assessed CD45, CD11b, CD206, and Iba1 immunohistochemistry on a separate set of brains from aged (18 months) Tg-SwDI mice acutely injected (12 h before perfusion) with CpG ODN or saline. Double immunofluorescence for macrophage/microglia markers (CD45 or CD206) and Aβ deposits (6E10/4G8) was done on selected sections to identify the location of acutely activated microglial cells in relationship to Aβ deposits. Subsequent series of double staining (CD206/CD45, CD206/CD11b, CD206/Iba1, and CD45/Iba1) were performed to examine distribution of distinct macrophage/microglia cell types. Secondary antibodies were Cy3-conjugated goat anti-mouse, Cy3-conjugated goat anti-rabbit Cy3, Alexa Fluor 488-conjugated goat anti-rat, and Alexa Fluor 488-conjugated goat anti-rabbit (1:500; room temperature 2 h; Jackson ImmunoResearch Laboratories). Fluorescent imaging was executed on a Zeiss LSM 700 inverted confocal microscope. ZEN 2011 software was used for data collection and analyses. Thioflavine-S staining for parenchymal and vascular fibrillar amyloid deposits was performed on mounted sections (Yang et al., 2011; Scholtzova et al., 2014). Microhemorrhages (small microscopic bleeds) were detected by Perls' Prussian blue staining for ferric iron in hemosiderin (a degradation product of hemoglobin) on another set of sections, as previously published (Yang et al., 2011; Scholtzova et al., 2014). Equally spaced defatted and hydrated sections were mounted and stained in a solution containing 10% potassium ferrocyanide and 20% hydrochloric acid for 45 min. The slides were then rinsed in distilled water, and the sections were dehydrated and coverslipped. Fifteen Perls-stained sections were examined per mouse and the average number of iron-positive profiles per section was calculated by an assessor blinded to treatment assignments.
Image analysis; quantification of amyloid and CD45 burden.
Quantitative image analyses of immunohistochemistry were performed according to our previously published protocols, using a Bioquant stereology semiautomated image analysis system (Bioquant Image Analysis) and an unbiased random-sampling scheme (Scholtzova et al., 2008, 2014; Liu et al., 2014). Fifteen evenly spaced coronal sections were analyzed per animal. All measurements were performed by an individual blinded to group assignment. Total cortical and hippocampal Aβ burden (defined as the percentage of test area occupied by Aβ) was quantified separately on sections stained with the monoclonal anti-Aβ antibodies 6E10 and 4G8. Intensification with nickel ammonium sulfate blackened the Aβ deposits, which facilitated threshold detection. Thresholds were established with accurately identified objects on a standard set of slides and these thresholds remained constant throughout the whole analysis. The cortical area analyzed was dorsomedial from the cingulate cortex and extended ventrolaterally to the rhinal fissure within the right hemisphere. Test areas (640 × 480 μm) were randomly selected by applying a grid (800 × 800 μm) over the traced contour. Hippocampal measurements (grid area, 600 × 600 μm) were performed similar to the way they were performed in the cortical analysis. Fibrillar Aβ burden was evaluated separately in sections stained with thioflavin-S, using our established methods (Yang et al., 2011; Scholtzova et al., 2014). The CD45 microglia burden (the percentage of area in the measurement field occupied by CD45-immunoreactive microglia) was quantified in a manner analogous to that used to measure the Aβ burden.
Rating of microgliosis.
Semiquantitative analysis of the extent of microgliosis in brain sections was based on the severity of CD11b, CD206, and Iba1 immunoreactivity. Immunostained sections were evaluated on scales (0–4) devised by a blinded individual (0, a few resting microglia; 1, rare ramified and/or phagocytic microglia; 2, a few ramified/phagocytic microglia; 3, a moderate ramified/phagocytic microglia; 4, numerous ramified/phagocytic microglia), as we have reported previously (Yang et al., 2011; Scholtzova et al., 2014). Approximately 13 cortical and seven hippocampal sections were analyzed per animal at 10× magnifications.
Rating of astrocytosis.
The assessment of GFAP-immunostained sections was based on a semiquantitative analysis of the extent of GFAP immunoreactivity (number of GFAP-immunoreactive cells and complexity of astrocytic branching), as published (Yang et al., 2011; Goñi et al., 2013). Reactive astrocytosis was rated on a scale from 0 to 4 (0 for 1–5 resting astrocytes; 1 for 6–15 resting astrocytes/rare processes; 2 for 16–25 resting astrocytes/few processes; 3 for 26–35 reactive astrocytes/moderate branching; 4 for ≥36 reactive astrocytes/extensive branching). Approximately thirteen cortical and seven hippocampal sections were analyzed per animal at 10× magnifications by an investigator blinded to treatment group assignment.
Rating of CD3-positive T cells.
The rating of T lymphocytes was based on a semiquantitative analysis of CD3 immunoreactivity (number of CD3-positive cells). Scores were assigned by a blinded individual using a scale from 0 to 4 (0 for no positive cells; 1 for 1–4 positive cells; 2 for 5–8 positive cells; 3 for 9–12 cells; 4 for ≥13 positive cells). Approximately thirteen cortical and seven hippocampal sections were analyzed per animal at 10× magnifications.
Biochemical assessment of Aβ pathology in the brain
Tissue homogenization.
Before tissue fractionations and extraction of Aβ from brain tissue, 10% (w/v) homogenates were prepared in tissue homogenization buffer (20 mm Tris base, pH 7.4, 250 mm sucrose, 1 mm EDTA, 1 mm EGTA) with 100 mm phenylmethylsulphonyl fluoride, protease inhibitors [protease inhibitors mixture Complete, Roche Diagnostic) and pepstatin A (Sigma-Aldrich) added immediately before homogenization, as we have previously published (Schmidt et al., 2005; Asuni et al., 2006; Liu et al., 2014). Subsequently, brain homogenates were aliquoted, frozen, and stored at −80°C until further use for extraction of soluble and insoluble fractions of Aβ and for biochemical analyses (ELISA).
Sandwich ELISA for Aβ levels.
Extraction of Aβ from left hemispheres was performed as previously described (Scholtzova et al., 2009, 2014; Yang et al., 2011; Goñi et al., 2013). In summary, for extraction of soluble Aβ, brain homogenates were thoroughly mixed with an equal volume of cold 0.4% diethylamine (DEA)/100 mm NaCl, and subsequently centrifuged at 135,000 × g for 1 h at 4°C. Then the supernatant was neutralized with 1:10 volume of 0.5 m Tris, pH 6.8. The samples were then aliquoted, flash-frozen on dry ice, and stored at −80°C until used for measurements of soluble Aβ fraction. Similarly for extraction of the total Aβ, homogenates (200 μl) were added to 440 μl of cold formic acid (FA) and sonicated for 1 min on ice. Subsequently, 400 μl of this solution was spun at 100,000 × g for 1 h at 4°C. Then, 210 μl of the resulting supernatant was diluted into 4 ml of FA neutralization solution (1 m Tris base, 0.5 m Na2HPO4, 0.05% NaN3), flash frozen on dry ice, and stored at −80°C until used for total Aβ measurements.
The total and soluble Aβ levels were measured using a combination of mouse monoclonal antibody 6E10 (specific to an epitope present on amino acid residues 1–16 of Aβ) and two different rabbit polyclonal antibodies specific for Aβ40 (R162) and Aβ42 (R165), in a double antibody sandwich ELISA, as we have previously reported (Yang et al., 2011; Scholtzova et al., 2014). Optical density (OD) was measured at 450 nm. The relationship between OD and Aβ peptide concentration was determined by a four-parameter logistic log function. The assay was performed by an investigator blinded to group assignment.
Sandwich ELISA for aggregated/oligomeric Aβ.
The levels of Aβ oligomers, within the soluble brain fraction, were determined using the Human Aggregated Aβ ELISA kit (Invitrogen), according to the manufacturer's instructions and as published previously (Goñi et al., 2013; Scholtzova et al., 2014). This ELISA method is specific for aggregated Aβ species. As this is being measured in a soluble brain fraction, the measured levels are equivalent to Aβ oligomer levels. In summary, the levels of aggregated/oligomeric Aβ in each sample were measured against a standard containing aggregated Aβ. Brain homogenates (10%) were centrifuged at 100,000 × g for 1 h at 4°C. Samples diluted in the provided standard diluent buffer (1:5 dilutions) were incubated for 2 h at room temperature, allowing the Aβ to bind the capture antibody (a monoclonal antibody specific for the N terminus of human Aβ precoated to each well), followed by extensive washing and incubation for 1 h at room temperature with biotin-conjugated detection antibody (same as the capture antibody), which binds to the immobilized aggregated Aβ. After removal of excess antibody, horseradish peroxidase-labeled streptavidin was added to incubate for 30 min, followed by washing, and a tetramethylbenzidine (TMB) substrate incubation, which resulted in a colometric solution. The intensity of the colored product was directly proportional to the concentration of oligomeric Aβ in the sample. The TMB reaction was stopped and the absorbance of each well was read at 450 nm. The standards provided a linear curve and the best-fit line determined by linear regression were used to calculate the concentration of Aβ oligomers in our samples.
Immune response analyses
Measurements of cytokine production to evaluate the subset of T helper cells (Th cells) induced and of autoantibody responses toward Aβ peptides were performed in plasma, using the New York University Langone Medical Center Immune Monitoring Core (http://www.med.nyu.edu/ocs/immune-monitoring-core). Animals were bled before immunization and every month thereafter, including a sacrifice bleed.
Cytokine assays.
Cytokine induction profiles in plasma from CpG ODN-treated and control animals were analyzed using a MilliplexMAP mouse cytokine/chemokine magnetic bead panel (EMD Millipore), after the intraperitoneal injection of either CpG ODN or saline at 4 h postinjection every month throughout the treatment period (Scholtzova et al., 2014). Additionally, cytokine/chemokine levels were assessed in plasma samples collected at the time of death, 4 weeks after the last injection. These assays allow quantification of an array of cytokines in a small-volume sample. A custom nine-plex detection kit, which measured IL1β, IL4, IL6, IL10, MCP1, TNFα, IFNγ, IP10, and IL12p70, was used and the assays were performed as per the manufacturer's protocol. Briefly, plasma samples (1:3 dilution–serum matrix) were incubated with a mixture of antibodies conjugated with fluorescent beads overnight. Following the biotinylated detection antibody and streptavidin phycoerytherin-conjugate incubations, levels of each cytokine were measured using the Luminex 200 analyzer (New York University Langone Medical Center Immune Monitoring Core). Median fluorescent intensity data were analyzed with ExPONENT software using a five-parameter logistic curve-fitting method for calculating cytokine/chemokine concentrations in samples. Concentrations were calculated from standard curves and are expressed in picograms per milliliter.
Aβ autoantibody response.
Plasma samples were evaluated for the presence of autoantibodies using ELISA as described previously (Scholtzova et al., 2009, 2014; Goñi et al., 2013). Plasma at dilutions of 1:200 was applied to 96-well microtiter plates coated with 5 μg of the Aβ40/42 peptides (Immulon 2HB, Thermo Fisher Scientific). The bound antibodies in plasma were detected by a goat anti-mouse IgG or a goat anti-mouse IgM linked to a horseradish peroxidase conjugate (IgG: GE Healthcare Bio-Sciences; IgM: KPL) at 1:3000 dilution. The development substrate was TMB (Bio-Rad Laboratories) and the readings were made at 450 nm.
Statistical analysis
The radial arm maze data (working-memory errors) were analyzed by two-way repeated-measures ANOVA followed by a Bonferroni's post hoc test. Wt animals' learning/improvement across 12 d (radial arm maze) was analyzed by one-way repeated-measures ANOVA, within-subjects contrast/trend tests, Mauchly's test of sphericity, and Student's paired two-tailed t test. A Student's unpaired two-tailed t test or one-tailed t test was used to analyze data collected from locomotor-activity test and novel object recognition test. Group differences in total amyloid burden, fibrillar/CAA amyloid burden, levels of extracted Aβ, levels of aggregated/oligomeric Aβ in brain homogenates, microgliosis (CD45, CD11b, CD206, Iba1), astrogliosis (GFAP), T-cell infiltration, brain microhemorrhages, autoantibody levels, and cytokine profiles were analyzed using a Student's unpaired two-tailed t test or one-tailed t test. Wt animals' learning statistical tests (radial arm maze) were performed using SPSS Advanced Statistics version 21 (IBM) and remaining statistical tests were performed using Prism version 6.07 (Graphpad).
Results
Behavioral studies
The mice underwent a battery of behavioral tests during the final month of treatment. Before cognitive assessment, all study groups were subjected to a sensorimotor (locomotor) activity test to verify that any CpG ODN treatment effects observed in the cognitive tasks could not be explained by differences in sensorimotor abilities. Locomotor tests eliminated the confounding factor of different motor functions among the Tg treatment groups by showing no statistical differences in any of the four parameters measured [distance traveled, velocity (average and maximum), and mean resting time; Fig. 1A–H]. Tg-SwDI mice performed comparably to Wt littermates. Furthermore, no differences were observed between both Wt treatment groups in their locomotor performance (data not shown).
Figure 1.

Locomotor activity test. A–H, No statistical differences between CpG ODN-treated and control Tg mice were discerned in any of the four locomotor parameters measured (A, E, average speed; B, F, maximum speed; C, G, resting time; D, H, distance traveled) in both age groups (A–D, 4–14 months; E–H, 8–18 months). The error bars show SEM. This applies to all subsequent figures. VEH, Vehicle.
In addition to sensorimotor evaluation, the mice underwent a cognitive assessment. Short-term memory deficits were assessed based on an animal's exploratory behavior using the novel object recognition test. Rodents generally exhibit a preference for the novel object, whereas animals with cognitive deficits will not exhibit any exploratory preference. We found that TLR9 stimulation was effective at preventing and improving short-term memory deficits in both age cohorts of Tg-SwDI mice. The recognition ability of the novel object was expressed as a preference index. The CpG ODN-treated Tg mice performed similarly to age-matched Wt littermates and demonstrated a significantly greater preference for interaction with the novel object than the vehicle (VEH)-treated Tg mice (4–14-month study group, two-tailed t test, Tg-CpG vs Tg-VEH, ***p = 0.0001; Fig. 2A; 8–18-month study group, Tg-CpG vs Tg-VEH, ***p = 0.0006; Fig. 2B). Both Wt groups spent more time interacting with the novel object, indicating their exploratory preference for the novel object. As shown in Figure 2C, there was no significant difference in the preference index for recognition of the novel object among Wt groups (two-tailed t test, Wt-CpG vs Wt-VEH, p = 0.39), confirming that chronic CpG ODN administration in Wt animals was not associated with toxicity leading to cognitive deficits. The total exploration time for novel versus familiar objects (retention session) was automatically recorded and is presented in Figure 2D–F. The CpG ODN-treated Tg mice spent significantly more time interacting with the novel object (4–14-month study group, one-tailed t test, novel vs familiar, *p = 0.04; Fig. 2D; 8–18-month study group, novel vs familiar, *p = 0.04; Fig. 2E), whereas our Tg controls spent substantially more time with the familiar object (4–14-month study group, two-tailed t test, novel vs familiar, ***p = 0.0008; Fig. 2D; 8–18-month study group, novel vs familiar, **p = 0.0017; Fig. 2E). As shown in Figure 2F, both Wt groups (CpG ODN Wt group, two-tailed t test, novel vs familiar, ****p < 0.0001; VEH Wt group, novel vs familiar, ****p < 0.0001) spent significantly more time exploring the novel object.
Figure 2.

Novel object recognition test. Peripheral administration of TLR9 agonist CpG ODN prevented and improved short-term memory deficits in both age cohorts of Tg-SwDI mice. The CpG ODN-treated Tg mice performed comparably to age-matched Wt littermates and spent more time exploring the novel object, indicating their novel-object preference, whereas our Tg control mice did not show any preference for the novel object. Data were reported as the preference index, i.e., the total time spent investigating the novel object was divided by total time the animal spent investigating both objects. The preference index was significantly different between CpG ODN-treated and vehicle (VEH)-treated Tg mice. A, Treatment from 4 to 14 months: Tg-CpG vs Tg-VEH, ***p = 0.0001. B, Treatment from 8 to 18 months: Tg-CpG vs Tg-VEH, ***p = 0.0006. C, Preference index in both Wt groups was >50% indicating their novel object preference, and no significant differences were observed in preference index among Wt groups (Wt-CpG vs Wt-VEH, p = 0.39). D–F, Total exploration time of each object within a defined zone of the arena during the retention session. The CpG ODN-treated mice spent significantly more time interacting with the novel object. D, Treatment from 4 to 14 months: novel vs familiar, *p = 0.04. E, Treatment from 8 to 18 months: novel vs familiar, *p = 0.04. VEH-treated Tg mice spent significantly more time near the familiar object. D, Treatment from 4 to 14 months: novel vs familiar, ***p = 0.0008. E, Treatment from 8 to 18 months: novel vs familiar, **p = 0.0017. F, Both Wt CpG ODN-treated and Wt VEH-treated groups spent significantly more time exploring the novel object, CpG ODN Wt group: novel vs familiar ****p < 0.0001; VEH Wt group, novel vs familiar, ****p < 0.0001.
Spatial/working memory was evaluated using the radial arm maze test. The overall performance (number of working-memory errors) of the mice differed significantly between Tg groups. The CpG ODN-treated Tg groups were better at navigating the maze than the VEH-treated Tg groups (Fig. 3A,B). All Tg animals made significantly more errors than Wt animals. However, the performance of CpG ODN-treated Tg mice was more comparable to that of our Wt littermates [4–14-month study group: two-way repeated-measures ANOVA, group (treatment) effect, p < 0.0001; sessions (days) effect, p = 0.0005; interaction (group vs days), p = 0.96. Bonferroni's post hoc test showed Tg-CpG vs Tg-VEH, p < 0.0001; Tg-CpG vs Wt, p = 0.006; Tg-VEH vs Wt, p < 0.0001; Fig. 3A; 8–18-month study group: two-way repeated-measures ANOVA, group (treatment) effect, p < 0.0001; sessions (days) effect, p = 0.37; interaction (group vs days), p = 0.26; Bonferroni's post hoc test showed Tg-CpG vs Tg-VEH, p = 0.0009; Tg-CpG vs Wt, p = 0.0004; Tg-VEH vs Wt, p < 0.0001; Fig. 3B]. No significant differences were detected between CpG ODN and VEH-treated Wt animals (data not shown). Moreover, our Wt animals showed evidence of learning across sessions (days) as measured by improvements in performance for both total time to complete each session (Fig. 3C) and total number of working-memory errors (Fig. 3D) committed within each session. One-way repeated-measures ANOVA on performance data of the Wt group (averaged across 12 days) indicated a significant main effect of sessions (days) for both time to complete the session (F(11,187) = 3.17, p = 0.001) and total number of errors (F(11,187) = 5.75, p < 0.0001). Within-subjects contrast/trend analyses confirmed both a significant linear trend of decreasing time to complete the session (F(1,17) = 15.34, p = 0.001) and a linearly decreasing number of errors (F(1,17) = 32.14, p < 0.0001) across sessions. For these analyses, Mauchly's sphericity test indicated no significant effects of sphericity on either the session time (p = 0.16) or the number of errors (p = 0.12). Furthermore, a comparison of the total session time and total errors between the first (day 1) versus last (day 12) sessions confirmed significant reductions in both of these variables (paired two-tailed t test: session time, p < 0.0001; number of errors, p = 0.002). Together, present data provide evidence that Wt mice exhibit characteristic monotonic learning/improvement curves using our radial arm maze procedure, suggesting good execution of the task protocol.
Figure 3.

Radial arm maze test. Tg-SwDI mice treated with CpG ODN navigated the radial arm maze with significantly fewer working-memory errors than control Tg mice. Spatial/working-memory improvements were observed in both study groups. A, Treatment from 4 to 14 months: two-way repeated-measures ANOVA, group (treatment) effect, p < 0.0001; sessions (days) effect, p = 0.0005; interaction (group vs days), p = 0.96. Bonferroni's post hoc test revealed Tg-CpG vs Tg-vehicle (Tg-VEH), p < 0.0001; Tg-CpG vs Wt, p = 0.006; Tg-VEH vs Wt, p < 0.0001. B, Treatment from 8 to 18 months: two-way repeated-measures ANOVA, group (treatment) effect, p < 0.0001; sessions (days) effect, p = 0.37; interaction (group vs days), p = 0.26. Bonferroni's post hoc test revealed Tg-CpG vs Tg-VEH, p = 0.0009; Tg-CpG vs Wt, p = 0.0004; Tg-VEH vs Wt, p < 0.0001. The Wt groups did not differ significantly in their performance on the radial arm maze; hence, CpG ODN-treated and VEH-treated Wt mice are presented as one group. C, D, Furthermore, our Wt mice showed evidence of learning across sessions (days) as measured by both the total time to complete (C) and total number of working memory errors (D) committed within each session. One-way repeated-measures ANOVA indicated a significant main effect of sessions (days) for both time to complete the session (F(11,187) = 3.17, p = 0.001) and total number of errors (F(11,187) = 5.75, p < 0.0001). Within-subjects contrast/trend analyses confirmed both a significant linear trend of decreasing time to complete the session (F(1,17) = 15.34, p = 0.001) and a linearly decreasing number of errors (F(1,17) = 32.14, p < 0.0001) across sessions. Mauchly's sphericity test indicated no significant effects of sphericity on either the session time (p = 0.16) or the number of errors (p = 0.12). Moreover, a comparison of the total session time and total errors between the first (day 1) versus last (day 12) sessions confirmed significant reductions in both of these variables (two-tailed t test: session time, p < 0.0001; number of errors, p = 0.002). Overall, these data indicate that our Wt mice exhibit characteristic monotonic learning-improvement curves using our radial arm maze protocol.
Amyloid pathology
Quantification of amyloid burden
Comprehensive immunohistochemistry and quantitative image analyses were performed to further assess whether the CpG ODN memory-enhancing effect correlated with its effect on Aβ pathology in Tg-SwDI mice. The mice were killed at 15 and 19 months of age after behavioral testing and their brains were processed for histology. Total amyloid burden in the cortical, hippocampal, and thalamic regions was quantified by stereological techniques using an unbiased, hierarchical sampling scheme and a semiautomated image analysis system on serial coronal sections immunostained with anti-Aβ 6E10/4G8 antibodies. Significant reductions in the total amyloid burden were noted in all three brain regions of CpG ODN-treated animals in both study groups (Fig. 4A–L). Quantitative assessment of the total cortical amyloid burden revealed a significant 22% reduction (two-tailed t test, *p = 0.025; Fig. 4M) in the 4–14-month study group. Reductions of 32% and 36% in the total amyloid burden were observed in the hippocampus and thalamus, respectively (hippocampus, two-tailed t test, ***p = 0.0007; Fig. 4N; thalamus, two-tailed t test, *p = 0.013; Fig. 4O). In the 8–18-month study group, peripheral administration of TLR9 agonist CpG ODN led to a 20% reduction (two-tailed t test, *p = 0.012; Fig. 4P) in the total cortical amyloid burden, a 37% reduction (two-tailed t test, ***p = 0.00032; Fig. 4Q) in total hippocampal amyloid burden, and a 33% reduction (two-tailed t test, *p = 0.045; Fig. 4R) in thalamic amyloid burden compared with age-matched VEH Tg animals. Thioflavine-S staining revealed fibrillar amyloid depositions, which in this model corresponds almost exclusively to vascular amyloid burden. The CAA burden (Aβ burden in the vasculature) was quantified by using the same principle of an unbiased sampling scheme as described previously (Scholtzova et al., 2009). Reductions in the fibrillar cortical and hippocampal vascular amyloid burden were observed in CpG ODN-treated animals compared with controls in both age groups (Fig. 5A–L). There was a 29% reduction (two-tailed t test, **p = 0.0062; Fig. 5M) in cortical CAA burden and 27% reduction (two-tailed t test, *p = 0.034; Fig. 5N) in hippocampal CAA burden in Tg-SwDI mice treated with CpG ODN in the 4–14-month study group. Similar effects were seen in the 8–18-month study group. Quantitative analysis within the cortex showed a significant 21% reduction (two-tailed t test, *p = 0.041; Fig. 5P) in treated mice. A 30% reduction (two-tailed t test, *p = 0.017; Fig. 5Q) was noted in the hippocampus. No statistical differences between treatment groups were observed in the thalamus in both age cohorts (4–14-month study group; Fig. 5O; 8–18-month study group; Fig. 5R). The integrity of cerebral vessels was examined using the Perls' stain method that detects hemosiderin, indicating cerebral bleeding. Significant reductions in the extent of CAA-associated (hippocampal and cortical) microhemorrhages were demonstrated in CpG ODN-treated animals in both study groups (Fig. 6A–H) (4–14-month study group, two-tailed t test, *p = 0.02; Fig. 6I; 8–18-month study group, two-tailed t test, *p = 0.04; Fig. 6J). Furthermore, we did not detect increased microhemorrhages in association with CpG ODN treatment in brains of Wt animals (data not shown), providing further evidence of the safety of CpG ODN administration.
Figure 4.

Total amyloid burden. A–L, 6E10/4G8 immunohistochemistry showed significant reduction in total amyloid burden in cortical, hippocampal, and thalamic brain regions in CpG ODN-treated Tg-SwDI mice compared with Tg control mice treated with vehicle (VEH) in both study groups. M–O, Quantitative stereological assessment of total amyloid burden revealed a 22% reduction (*p = 0.025) in the cortex (M), a 32% reduction (***p = 0.0007) in the hippocampus (N), and a 36% reduction (*p = 0.013) in the thalamus (O) in the 4–14-month study group. P–R, Similarly, there was a 20% reduction (*p = 0.012) in total cortical amyloid burden (P), a 37% reduction (***p = 0.00032) in total hippocampal amyloid burden (Q), and a 33% reduction (*p = 0.045) in total thalamic amyloid burden (R) in the 8–18-month study group. Scale bars: (in B, H) A, B, G, H, 100 μm; (in D, J) C, D, I, J, 100 μm; (in F, L) E, F, K, L, 100 μm.
Figure 5.
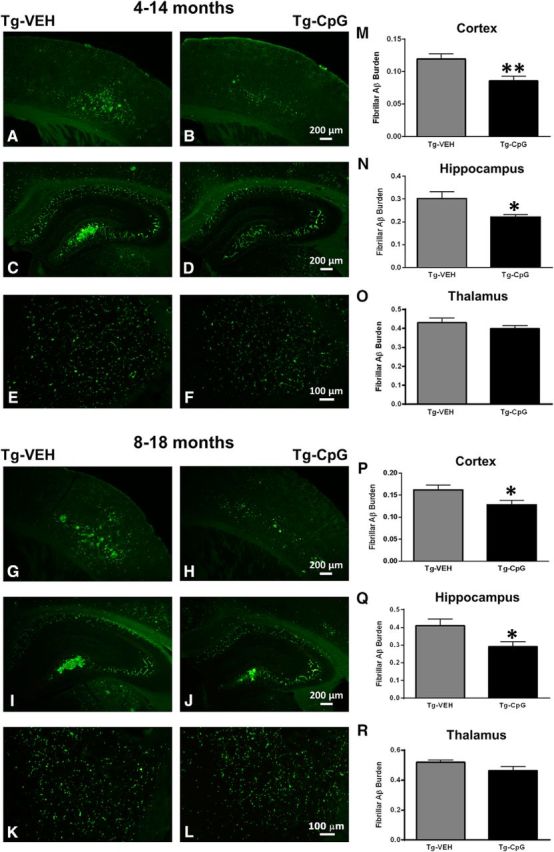
Fibrillar-vascular amyloid burden. A–L, Thioflavine-S staining revealed significant differences in fibrillar cortical and hippocampal vascular amyloid burden (CAA burden) between CpG ODN-treated and vehicle (VEH)-treated Tg-SwDI mice in both study groups. M, N, There was a 29% reduction (**p = 0.0062) in the cortical CAA burden (M) and a 27% reduction (*p = 0.034) in the hippocampal CAA burden (N) in the 4–14-month study group. P, Q, Likewise, cortical and hippocampal CAA burdens were reduced by 21% (*p = 0.041; P) and 30% (*p = 0.017; Q), respectively, in CpG ODN-treated mice in the 8–18-month study group. O, R, No statistical differences between our treated and control Tg-SwDI animals were detected in the thalamus in both age cohorts. Scale bars: (in B, H) A, B, G, H, 200 μm; (in D, J) C, D, I, J, 200 μm; (in F, L) E, F, K, L, 100 μm.
Figure 6.

Brain microhemorrhages. A–H, Representative cortical and hippocampal brain sections stained with Perls' stain for ferric iron in hemosiderin, indicating cerebral bleeding. I, J, Quantification revealed a significant reduction in the extent of CAA-associated hippocampal and cortical microhemorrhages in the CpG ODN-treated animals in both the 4–14-month study group (*p = 0.02; I) and the 8–18-month study group (*p = 0.04; J). Scale bars: (in B, F) A, B, E, F, 25 μm; (in D, H) C, D, G, H, 100 μm.
Assessment of Aβ levels, Aβ oligomers in the brain
To further examine the extent of amyloid pathology in Tg-SwDI mice after CpG ODN administration, we performed multiple biochemical analyses. Stimulation of TLR9 signaling significantly decreased total (FA extract) and soluble (DEA extract) brain Aβ levels in Tg-SwDI mice in both study groups. ELISA measurements revealed a statistically significant decrease in the levels of total (FA extracted) Aβ40 by 43% (two-tailed t test, **p = 0.004) and Aβ42 by 64% (two-tailed t test, **p = 0.0016) after the CpG ODN treatment in the 4–14-month study group (Fig. 7A). The levels of soluble (DEA extracted) Aβ40 and Aβ42 fractions were significantly reduced by 44% (two-tailed t test, *p = 0.03) and 47% (two-tailed t test, *p = 0.013), respectively, in CpG ODN-treated mice in the 4–14-month study group (Fig. 7B). Treatment in the 8–18-month study group significantly lowered total Aβ42 by 32% (one-tailed t test, *p = 0.04), while there was a strong trend for reduction in the levels of total Aβ40 (one-tailed t test, p = 0.09; Fig. 7C). Soluble Aβ40 and Aβ42 brain levels were reduced by 20% (one-tailed t test, *p = 0.041) and 23% (two-tailed t test, *p = 0.015), respectively, in CpG ODN-treated animals compared with controls in the 8–18-month study group (Fig. 7D).
Figure 7.

Brain Aβ levels and Aβ oligomers. A, CpG ODN treatment significantly lowered total (FA extracted) Aβ40 levels by 43% (**p = 0.004) and Aβ42 levels by 64% (**p = 0.0016) in the 4–14-month study group. B, There was also a reduction in the levels of soluble (DEA extracted) Aβ40 (44%, *p = 0.03) and Aβ42 (47%, *p = 0.013). C, Treatment in the 8–18-month study group significantly decreased total Aβ42 by 32% (*p = 0.04), and there was a strong trend for reduced total Aβ40 levels (p = 0.09). D, Soluble Aβ40 and Aβ42 brain levels were reduced by 20% (*p = 0.041) and 23% (*p = 0.015), respectively. E, F, Furthermore, TLR9 stimulation led to a significant decrease in soluble oligomeric Aβ ligands in both treatment groups (E, 4–14-month study group, *p = 0.01; F, 8–18-month study group,*p = 0.03).
Soluble oligomeric Aβ ligands (also known as Aβ-derived diffusible ligands) may account for AD neuropathology and memory loss, thus presenting a significant therapeutic target (Lesné et al., 2013). Pathogenic Aβ oligomers in 10% brain homogenates were assessed by ELISA specific to aggregated/oligomeric Aβ. CpG ODN treatment led to a significant decrease in Aβ oligomers in Tg-SwDI mice in both age cohorts (4–14- month study group, two-tailed t test, *p = 0.01; Fig. 7E; 8–18-month study group, two-tailed t test, *p = 0.03; Fig. 7F). Overall, biochemical analysis of Aβ levels in brain homogenates provided additional validity to shifts in amyloid burden observed in our CpG ODN-treated Tg-SwDI mice.
Associated histopathology: assessment of microgliosis and astrogliosis
Potential signs of cerebral inflammatory toxicity were assessed by quantification of reactive gliosis as described in our earlier studies (Scholtzova et al., 2009, 2014; Yang et al., 2011). To determine CpG ODN treatment effect on the development of a diverse range of macrophage/microglia activation states, we immunohistochemically evaluated sections from CpG ODN-treated and VEH-treated Tg-SwDI mice using four different markers. Quantitative stereological analysis of macrophage/microglia marker CD45, which is typically expressed on peripherally derived macrophages and in association with more mature plaques (Morgan et al., 2005), indicated 41% cortical reduction (two-tailed t test, *p = 0.03; Fig. 8M) and 30% hippocampal reduction (two-tailed t test, *p = 0.034; Fig. 8N) in CD45 burden in CpG ODN-treated Tg-SwDI mice compared with VEH-treated mice in the 4–14-month study group at the end of the study. Analysis in the thalamus did not reveal group differences (Fig. 8O). No significant differences in CD45 immunoreactivity were detected between CpG ODN mice and control animals in the 8–18-month study group (Fig. 8P–R). The assessment of another well established mononuclear phagocyte marker, CD206 (alternative M2 activation marker), was based on semiquantitative analyses (Gordon, 2003; He et al., 2012). No apparent differences were observed between groups in the extent of CD206 immunoreactivity in both age cohorts at the end of the treatment period (Fig. 9A–L). The staining intensity of the CD206 marker was very low in the thalamus (data not shown). Similarly, no group differences were found in CD11b-reactive microglia in all brain regions from both study groups (Fig. 10A–R). Furthermore, Iba1 has been reported to label all microglia populations, whether resting or activated (Streit et al., 2009; Zotova et al., 2013). Our immunohistochemical observations demonstrated no apparent differences in Iba1-immunoreactive cells between the Tg CpG ODN-treated and VEH-treated animals in both age groups (data not shown). Subsequent analysis for astroglial marker GFAP was performed. No differences in astrogliosis were detected between treatment groups in both study cohorts (Fig. 11A–R). To further evaluate the safety profile of CpG ODN, assessment of microgliosis in Wt mice was performed. Minimal microglial immunoreactivity was observed in our Wt animals. No increase in microgliosis was found in age-matched Wt littermates treated with CpG ODN (Fig. 12A–L). CD3 immunohistochemistry indicated the presence of T lymphocytes in all Tg groups at the end of the treatment period (Fig. 13A–D). However, the number of CD3-positive cells did not differ significantly between CpG ODN-treated Tg mice and our Tg controls in both study groups. Parenchymal (hippocampal, cortical, and thalamic) and perivascular lymphocytes were included in the semiquantitative analysis (Fig. 13E,F). Moreover, there was no evidence of increased T-cell infiltration in any brain regions of CpG ODN-treated Wt mice (data not shown). Hence, stimulation of innate immunity with TLR9 agonist CpG ODN did not enhance neuroinflammation.
Figure 8.
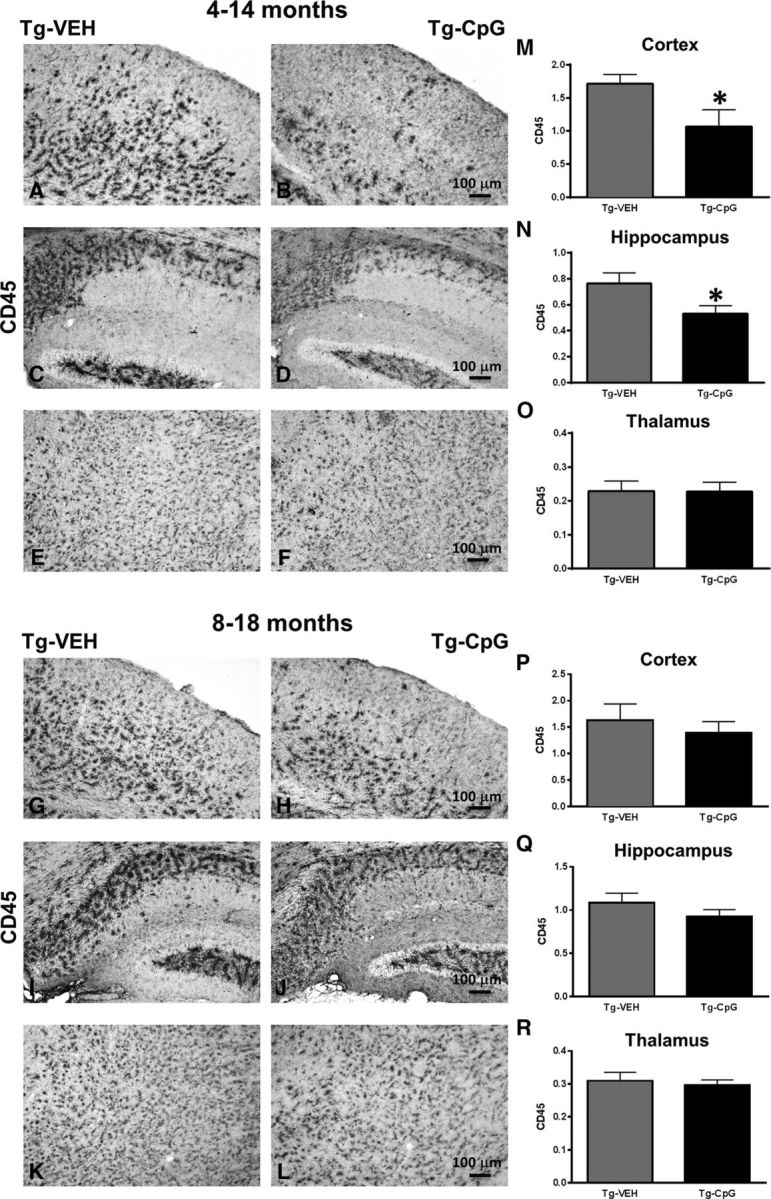
Assessment of CD45 immunoreactivity. A–F, Quantitative stereological analysis of macrophage/microglia marker CD45 revealed an overall reduction in Tg-SwDI mice treated with CpG ODN compared with control Tg mice in the 4–14-month study group. M–O, There was a 41% reduction (*p = 0.03) in cortical CD45 burden (M) and a 30% reduction (*p = 0.034) in hippocampal CD45 burden (N), whereas no changes were noted in the thalamus (O) at the end of the treatment. G–L, P–R, No differences in CD45 immunoreactivity were detected between our treated and control animals in the 8–18-month study group. Scale bars: (in B, H) A, B, G, H, 100 μm; (in D, J) C, D, I, J, 100 μm; (in F, L) E, F, K, L, 100 μm.
Figure 9.

Assessment of CD206 immunoreactivity. A–L, Representative cortical and hippocampal immunostained images with perivascular macrophage activation marker (BMDM marker) CD206 and subsequent semiquantitative analysis showed no differences between CpG ODN-treated Tg mice compared with control Tg mice in both age cohorts at the end of the treatment. The degree of CD206 immunoreactivity was graded on a scale of 0–4. CD206 staining intensity in thalamus was minimal. Scale bars: (in B, F) A, B, E, F, 100 μm; (in D, H) C, D, G, H, 100 μm.
Figure 10.
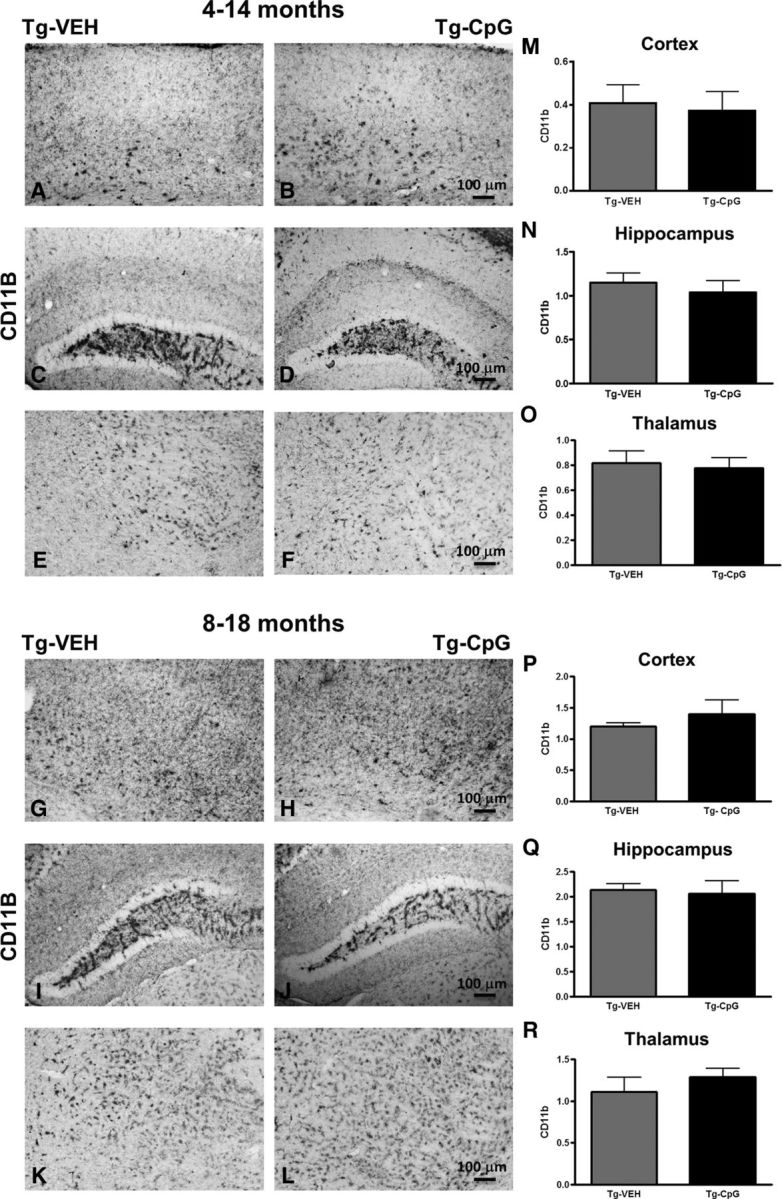
Assessment of CD11b immunoreactivity. A–R, Histological observation and semiquantitative rating of CD11b-reactive microglia did not reveal group differences in the degree of CD11b immunoreactivity in all three brain regions analyzed in both age groups at the end of the treatment. The degree of CD11b immunoreactivity was analyzed on a scale of 0–4. Scale bars: (in B, H) A, B, G, H, 100 μm; (in D, J) C, D, I, J, 100 μm; (in F, L) E, F, K, L, 100 μm.
Figure 11.
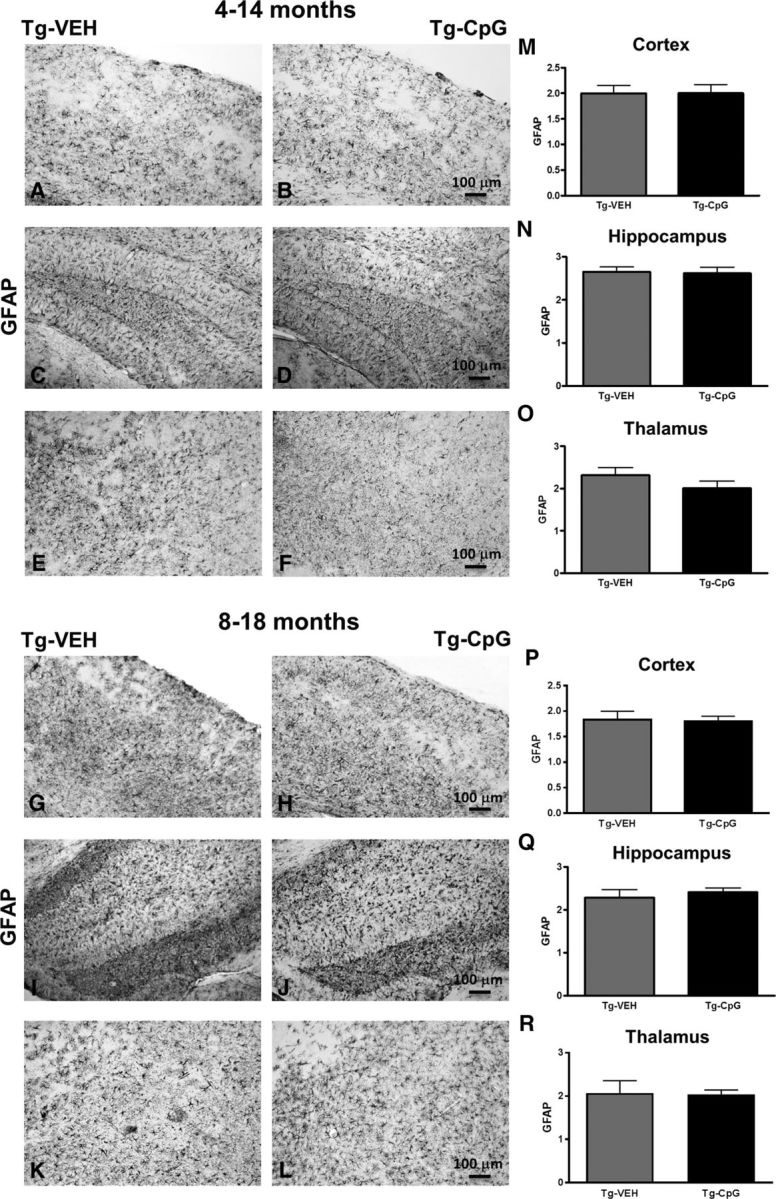
Assessment of GFAP immunoreactivity. A–R, Subsequent semiquantitative analysis for astroglial marker GFAP showed no differences between our treatment groups in both study cohorts at the end of the treatment period. GFAP immunoreactivity was rated on a scale of 0–4. Scale bars: (in B, H) A, B, G, H, 100 μm; (in D, J) C, D, I, J, 100 μm; (in F, L) E, F, K, L, 100 μm.
Figure 12.

Assessment of CD206, CD45, CD11b, and Iba1 immunoreactivity in Wt animals. A–L, CD206 (A, B), CD45 (C, D), CD11b (E, F), and Iba1 (G, H) macrophage/microglial immunoreactivity observed in our Wt animals was minimal and no apparent differences were detected between our treated and control animals at the end of the study (I–L). Hence, microgliosis was not affected by CpG ODN treatment in age-matched Wt littermates, providing further evidence of the safety of our approach. Scale bars: (in B, D, F, H) A–H, 100 μm.
Figure 13.

Assessment of CD3 immunoreactivity. A–D, Representative hippocampal brain sections stained with CD3 marker for positive T cells. The degree of CD3 immunoreactivity was semiquantitatively graded on a scale of 0–4. E, F, Quantification of CD3-positive T lymphocytes in all three brain regions did not reveal differences between CpG ODN-treated and vehicle (VEH)-treated Tg animals in both the 4–14-month study group (E) and the 8–18-month study group (F). Scale bars: (in B, D) A–D, 100 μm.
Our short-term acute study was conducted in a separate cohort of aged (18 months) Tg-SwDI mice. An acute administration of CpG ODN was performed to further assess the effects of TLR9 signaling on the stimulation of transient macrophage/microglia activation states and subsequent formation of the dynamic range of their phenotypes with the ability to promote phagocytosis. Semiquantitative analysis revealed a significant increase in hippocampal CD45 (two-tailed t test, ***p = 0.0001; Fig. 14A–C) and CD11b (two-tailed t test, *p = 0.04; Fig. 14D–F) immunoreactivity in the brains of Tg-SwDI mice 12 h after CpG ODN intraperitoneal administration.
Figure 14.
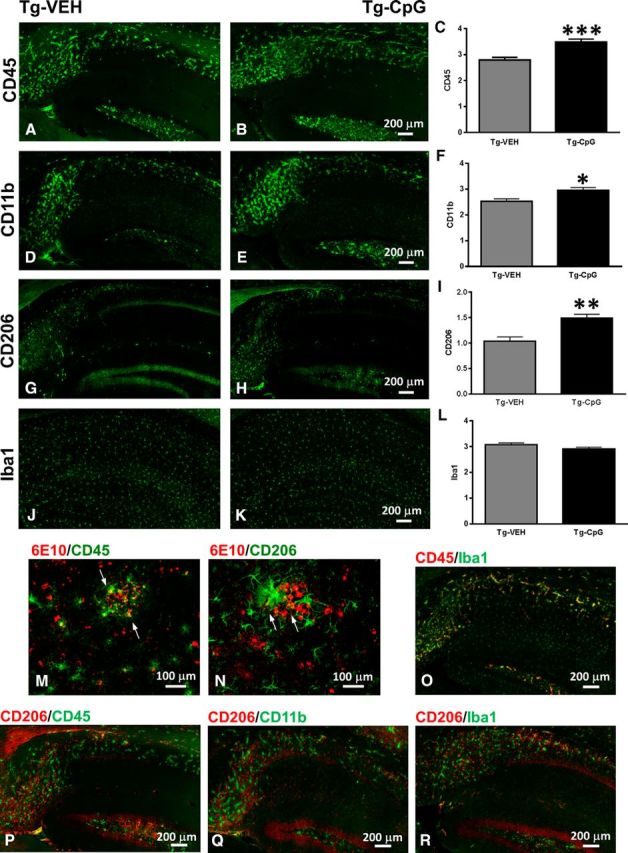
Acute study assessment of macrophage/microglia activation states. An acute administration of CpG ODN elicited an induction of beneficial macrophage/microglia activation in a separate cohort of aged Tg-SwDI mice. A–I, Hippocampal semiquantitative analysis of the extent of macrophage/microglia density and activation revealed a significant increase in CD45-positive (***p = 0.0001; A–C), CD11b-positive (*p = 0.04; D–F), and CD206-positive (**p = 0.001;G–I) cells 12 h after single intraperitoneal CpG ODN injection. Cortical comparisons showed similar effects (data not shown). J–L, Quantitative assessment of Iba1 marker did not reveal group differences in the degree of hippocampal (J–L) and cortical (data not shown) Iba1 immunoreactivity. Double immunofluorescence labeling indicated dense gathering of activated CD45-immunoreactive and CD206-immunoreactive cells, likely to be infiltrating peripherally derived macrophages, around amyloid deposits in CpG ODN-injected mice. M, N, Confocal microscopy confirmed partial colocalization between 6E10/4G8-positive Aβ deposits (red) and CD45 and CD206 favorable macrophage/microglia markers (green). Arrows indicate colocalization. Further series of double staining were performed to determine distribution of distinct activated macrophage/microglia cell types in Tg-SwDI mice acutely injected with CpG ODN. O, Clusters of colocalized CD45-activated (red) and Iba1-activated (green) cells were detected around Aβ deposits. P–R, Histological evaluation of CD206 (red) and CD45 (green; P), CD206 (red) and CD11b (green; Q), and CD206(red) and Iba1(green; R) confocal images did not reveal colocalization. M–R, Representative hippocampal confocal images from 18-month-old Tg-SwDI mice. Scale bars: (in B, E, H, K) A, B, D, E, G, H, J, K, 200 μm; M, N, 100 μm; O–R, 200 μm.
Subsequent staining with CD206 M2 macrophage marker was conducted to further characterize distinct macrophage/microglia phenotypes. Moreover, perivascular macrophages expressing CD206 continuously turn over in the CNS and have been shown to be phagocytic (Galea et al., 2005; Hawkes and McLaurin, 2009). Immunohistochemical examination showed an increase in CD206 hippocampal microglial activation in CpG ODN-injected animals (two-tailed t test, **p = 0.001; Fig. 14G–I) when compared with controls that received saline only. Cortical CD45, CD206, and CD11b immunohistochemical analyses also revealed significant increase (CD45, two-tailed t test, p = 0.0001; CD206, two-tailed t test, p = 0.0001; CD11b, two-tailed t test, p = 0.002) in macrophage/microglia density in mice injected with CpG ODN 12 h before perfusion (data not shown). On the other hand, we found that acute administration of CpG ODN did not alter Iba1 immunoreactivity in our separate cohort of old Tg-SwDI animals. No differences were detected in the number of Iba1-positive cells in the hippocampus (Fig. 14J–L) and cortex (data not shown) between groups. Subsequently, we costained the brain sections with anti-Aβ 6E10/4G8. Double immunofluorescence for 6E10/4G8-positive Aβ deposits and both macrophage/microglial markers (CD45 and CD206) demonstrated dense clustering of acutely activated microglia around Aβ deposits in mice injected with CpG ODN compared with saline controls. Partial colocalization of CD45-positive or CD206-positive microglia and 6E10/4G8-positive Aβ deposits was often observed in CpG ODN group (Fig. 14M,N). CD45-positive and CD206-positive cells, commonly representing subsets of infiltrating peripheral BMDMs, were examined as possible contributors to the elimination of Aβ by phagocytosis.
Furthermore, we performed series of double staining to examine the distribution and relationship between CD45-reactive, CD11b-reactive, Iba1-reactive, and CD206-reactive macrophages/microglia in old Tg-SwDI mice acutely injected with CpG ODN. Interestingly, direct colocalization between activated Iba1 and CD45 cells was found in areas adjacent to amyloid deposits (Fig. 14O). However, no colocalization was noted between CD206-immunoreactive cells and CD45 (Fig. 14P), CD11b (Fig. 14Q), Iba1 (Fig. 14R) markers, further confirming the CpG ODN treatment effect on recruitment and activation of distinct macrophage/microglia cell types. Our observations highlight the importance of assessing macrophage/microglia activation states responsible for the formation of unique phenotypes that may have contributed to clearance of CAA pathology.
Characterization of immune responses
Cytokine responses (plasma)
We next examined whether peripheral administration of CpG ODN in Tg-SwDI mice was effective in inducing beneficial immunostimulatory response in the absence of any inflammatory toxicity. Cytokine profiles in plasma collected 4 h after an intraperitoneal injection of CpG ODN or saline were analyzed at monthly intervals using the Th1/Th2 Luminex mouse cytokine/chemokine kit. As shown in Figure 15A–P, significant differences in cytokine/chemokine levels in plasma collected before and after CpG ODN administration were detected in both study groups. Higher peak plasma levels of IP10 (interferon gamma-induced protein 1, also known as CXCL10), and MCP1 (monocyte chemotactic protein-1, also known as CCL2) chemokines were observed after CpG ODN administration in the 4–14-month study group [IP10, two-tailed t test (applies to all subsequent t tests for cytokine responses), ****p < 0.0001; Fig. 15A; MCP1, ***p = 0.0002; Fig. 15B]. Similar effects were seen in the 8–18-month study group (IP10, **p = 0.0016; Fig. 15I; MCP1, *p < 0.02; Fig. 15J). There was a strong induction of plasma IL6 levels (4–14 months, ***p = 0.0001; Fig. 15C; 8–18 months, *p = 0.04; Fig. 15K) 4 h post-CpG ODN injection compared with saline animals. In addition, our subjects showed an evident increase in IL12p70 plasma levels in response to stimulation with CpG ODN in both age groups (4–14 months, *p = 0.04; Fig. 15D; 8–18 months, *p = 0.02; Fig. 15L). Slightly more modest, but significant elevation was observed in circulating levels of TNFα (4–14 months, ***p = 0.0002; Fig. 15E; 8–18 months, **p = 0.007; Fig. 15M) and IFNγ (4–14 months, **p = 0.0085; Fig. 15F; 8–18 months, *p = 0.04; Fig. 15N). The involvement of the IL1β signaling cascade as one of the key disease mechanism for AD has been well documented (Mrak and Griffin, 2001; Kitazawa et al., 2011). Of interest to us, IL1β, a potent inflammatory cytokine, was detectable at very low levels, and no significant changes were observed between the groups (4–14 months, p = 0.13; Fig. 15G; 8–18 months, p = 0.46; Fig. 15O). The levels of IL10 (a potent anti-inflammatory cytokine) were also significantly induced in both CpG ODN groups (4–14 months, ***p = 0.0005; Fig. 15H; 8–18 months, **p = 0.002; Fig. 15P). IL4 cytokine levels were near the detection limit and exhibited no differences between the treatment groups (data not shown).
Figure 15.
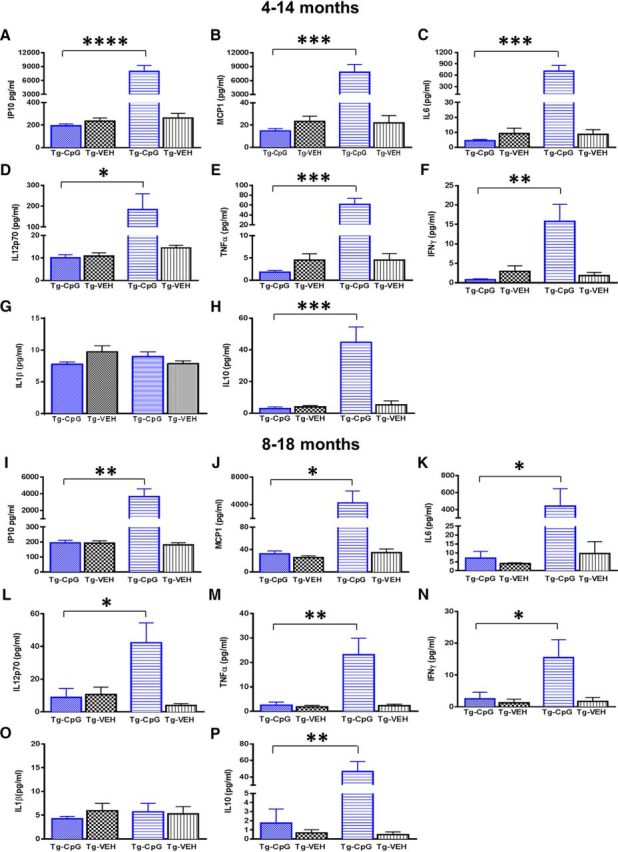
Plasma cytokine/chemokine analysis. Immunostimulatory responses induced by CpG ODN were evaluated using the Th1/Th2 Luminex mouse cytokine/chemokine detection kit. Results are shown as picograms per milliliter of IP10, MCP1, IL6, IL12p70, TNFα, IFNγ, IL1β, and IL10. A–P, Significant differences in cytokine/chemokine profiles were detected in plasma collected before and 4 h after CpG ODN administration. See results section for p values. An apparent elevation in most measured plasma cytokines/chemokines was observed in both study groups. However, strong inflammatory cytokine IL1β was detected at very low levels with no significant changes noted between all treatment groups (G, O). Data are presented as cytokine/chemokine responses in plasma samples collected after the representative injection of CpG ODN or saline. As expected, lower cytokine levels were observed in plasma collected 4 weeks after the last intraperitoneal CpG ODN or saline injection and no differences were found between the groups in both age cohorts. Hence, stimulation of innate immunity with CpG ODN was effective in inducing immunostimulatory response in the absence of prolonged inflammatory environment in both study groups.
Separate plasma cytokine analyses were performed at the time of death, 4 weeks after the last intraperitoneal CpG ODN or saline injection in both study cohorts. As expected, the cytokine/chemokine levels subsided over time and no significant differences were found between the Tg-SwDI CpG ODN-treated and control groups (data not shown). Overall, CpG ODN elevated the levels of various Th1/Th2 cytokines/chemokines, but the absolute levels were lower than what was observed in previous reports with TLR4 agonist LPS (Jaeger et al., 2009; Michaud et al., 2013). Present findings demonstrate that stimulation of innate immunity with TLR9 agonist CpG ODN induces a suitable degree of innate immunity stimulation, without producing a sustained neuroinflammatory environment, therefore leading to beneficial modulation of immune profiles resulting in restriction of AD-related pathology.
Aβ autoantibody levels
Furthermore, stimulation of TLR9 signaling did not lead to increased generation of anti-Aβ antibodies in CpG ODN-treated Tg-SwDI mice. No differences were detected in the levels of IgG or IgM Aβ autoantibodies in plasma obtained at the end of the study, compared with the beginning of the study, in both study groups (data not shown). This indicates that the effects of CpG ODN on reducing vascular amyloid burden cannot be attributed to stimulation of a humoral response against Aβ.
Discussion
Multiple immunotherapeutic approaches for AD have been developed and tested in clinical trials. However, major impediments include the limited evidence of significant clinical benefits, the occurrence of excessive neuroinflammation, and CAA-related complications (Wisniewski and Drummond, 2016). CAA, as a common neuropathological finding in both AD patients and cognitively normal elderly individuals, has been shown to be more resistant to clearance than parenchymal amyloid (Wilcock and Colton, 2009; Yamada, 2015). Current evidence points to a key role of CAA in the pathophysiology leading to development of amyloid-related imaging abnormalities. These MRI abnormalities include parenchymal vasogenic edema and sulcal effusion and abnormalities due to microhemorrhages (Sperling et al., 2012; Piazza and Winblad, 2016). Our initial findings from AD mouse models provide the first in vivo evidence that stimulation of innate immunity with TLR9 agonist class B CpG ODN appears to reduce behavioral deficits and restrict all pathological hallmarks of AD, including amyloid plaques, tau pathology, and CAA, without any adverse reactions (Scholtzova et al., 2009, 2014). However, the levels of vascular amyloid in these models of neurodegeneration were low. Given the independent correlation of the degree of CAA pathology with the severity of cognitive impairment, as well as the importance of CAA-linked complications associated with immunotherapy, the present study was designed to determine whether stimulation of TLR9 signaling with CpG ODN could have beneficial effects in the Tg-SwDI mice with extensive CAA (Davis et al., 2004; Yamada, 2015; Vidoni et al., 2016). We assessed the association of our treatment approach with potential adverse effects (such as inflammation and microhemorrhages), which have been reported with many immunotherapeutic paradigms (Wisniewski and Goñi, 2015).
The efficacy of TLR9 agonist was verified in animals treated before and after the onset of CAA pathology. Specifically, peripheral administration of CpG ODN negated short-term memory deficits as assessed by the novel object recognition test and was effective at improving spatial and working memory evaluated using a radial arm maze in both age cohorts of Tg-SwDI mice. Further analysis of Wt mice provided evidence of learning (improvements in performance) across 12 sessions (days) using our radial arm maze procedure, hence validating proper execution of the test. Moreover, cognitive improvements were not confounded by any differences in locomotor functions among the Tg treatment groups. Detailed neuropathological evaluation accompanied by quantitative image analyses demonstrated significant reductions in total amyloid burden in CpG ODN-treated Tg animals compared with Tg controls. Even though fibrillar deposits are less amenable to clearance, quantification of thioflavine-S-stained sections confirmed a significant reduction in fibrillar vascular amyloid burden without associated microhemorrhages in CpG ODN groups. Furthermore, we did not detect any microhemorrhages in Wt animals after CpG ODN administration, thus providing additional evidence of the safety of our approach. These favorable histological findings were corroborated by measurements of Aβ levels in the brain homogenates, which revealed a significant decrease in the levels of total and soluble Aβ40/42 fractions and Aβ oligomer levels in CpG ODN-treated Tg-SwDI mice.
With aging and particularly in the presence of AD pathology, macrophages/microglia become senescent, displaying decreased migration, phagocytosis, and inability to restrict AD pathology (Streit et al., 2014). Therefore, modulating macrophage/microglia profiles represents a potential therapeutic avenue for AD. Prior reports have demonstrated that microglia become significantly more efficient in Aβ uptake and degradation when stimulated with TLR9 agonists in vitro (Iribarren et al., 2005; Tahara et al., 2006; Doi et al., 2009). We and others have shown that microglial activation associated with TLR9 is beneficial in AD mouse models (Scholtzova et al., 2009, 2014; Su et al., 2016). It has been reported that TLR9 ligands drive macrophages/microglia into a phagocytic, long-lived state with a decreased capacity for antigen presentation resembling M2-like macrophages, which are associated with the production of regulatory or anti-inflammatory cytokines (Celhar et al., 2016). These lines of evidence led us to an evaluation of macrophage/microglia responses, as well as potential signs of treatment-associated toxicity. Reduction in CD45 microgliosis paralleled Aβ clearance in the 4–14-month study group at the end of the treatment period. No group differences were detected in the 8–18-month study group. No increases in the extent of CD11b, CD206, Iba1 gliosis, and GFAP astrogliosis were observed in CpG ODN-treated animals in both age cohorts at the end of the treatment. A CpG ODN immunostimulatory effect was not associated with activation of T-cell response in the brains of Tg-SwDI mice in both study groups. Hence, there was no evidence of encephalitis in the brains of CpG ODN-treated animals. Additionally, no alteration in macrophage/microglia responses or T-cell infiltration was detected immunohistochemically in our Wt animals treated with CpG ODN, further supporting the viability of our approach. A short-term acute study was performed to assess the effect of CpG ODN on the dynamic range of macrophage/microglia phenotypes that may have contributed to CpG ODN therapeutic outcomes. Transient increases in favorable CD45, CD206, and CD11b immunoreactivity were observed 12 h after CpG ODN administration in a separate cohort of old Tg-SwDI mice. Interestingly, we noted that costaining for CD206-immunoreactive cells and CD45, CD11b markers, did not reveal colocalization, confirming the effect of TLR9 signaling on the recruitment of distinct macrophage/microglia populations with unique activation states. On the other hand, accumulation and distribution of Iba1 immunoreactivity (marker of both resting and activated microglia) was not affected by the acute injection of CpG ODN. No colocalization between CD206 and Iba1 markers was found. However, we noted abundant activated CD45/Iba1-coimmunoreactive macrophages/microglia at sites of amyloid deposits. In a recent postmortem study of 299 aged subjects, the increased presence of Iba1-immunoreactive microglia correlated positively with better cognition, as measured by the Mini-Mental State Examination (Minett et al., 2016). Similarly, APP/PS1 mice treatment with IL33, which skews microglia toward an alternative activation state with enhanced phagocytic capacity, resulted in AD pathology amelioration in association with Iba1 microglia in the vicinity of amyloid deposits (Fu et al., 2016). The role of macrophages/microglia in Aβ clearance was also elucidated by documenting the upregulation and increased density of CD45-positive and CD206-positive cells (typically expressed on peripheral BMDMs) around amyloid plaques (Guillemin and Brew, 2004; Feng et al., 2011; Durafourt et al., 2012; Lai and McLaurin, 2012). Confocal double-fluorescence microscopy analyses confirmed partial colocalization of CD45 and CD206 markers and Aβ deposits. CD206-expressing perivascular macrophages continuously undergo turnover in the CNS and may contribute to removal of CAA pathology as they are located near vascular amyloid and are capable of more efficient phagocytosis (Galea et al., 2005; Hawkes and McLaurin, 2009). CpG ODN does not cross an intact blood–brain barrier (BBB); however, we cannot exclude the possibility that activated resident microglia was involved at later stages of AD, when some BBB breakdown may occur. We postulate that TLR9 stimulation primarily potentiates recruitment of peripherally derived macrophages. BMDMs, as a more accessible therapeutic target compared with resident microglia, have been found to enter the brain in AD mouse models and are known to be more effective in eliminating amyloid deposits (Jucker and Heppner, 2008; Lai and McLaurin, 2012; Lampron et al., 2013b). Hence, we suggest that BMDMs activated with CpG ODN can play a critical role in restricting AD pathology. The dependence of this amyloid-clearance capacity on appropriate stimulation is consistent with observations that unstimulated peripheral myeloid cells are unable to reduce Aβ-related pathology in APP/PS1 AD mice (Prokop et al., 2015).
Since excessive and dysregulated release of proinflammatory markers can contribute to neuronal degeneration, subsequent cytokine/chemokine analyses were performed. Measurements in plasma collected 4 h following injections demonstrated elevations in both Th1/Th2 cytokines and chemokines, but the absolute levels were lower than plasma levels observed in previous studies after exposure to LPS challenge (Jaeger et al., 2009; Michaud et al., 2013). The levels of IL1β, a potent inflammatory cytokine, which has been proposed to play a role in AD pathogenesis, were low and no differences were observed between groups. Significant increases were observed in the anti-inflammatory cytokine IL10 in our CpG ODN groups; however, elevations were modest and comparable to circulating levels of proinflammatory cytokines (i.e., IFNγ and TNFα). Recent evidence suggests that abnormally elevated anti-inflammatory cytokines, such as IL10, could prevent a proper Aβ clearance by inhibiting microglial phagocytosis (Guillot-Sestier et al., 2015a; Michaud and Rivest, 2015). At the same time, to benefit from macrophage/microglia ability to clear Aβ pathology, phagocytosis must be stimulated without prolonged and excessive activation of inflammatory processes (Michaud and Rivest, 2015; Rego et al., 2016). Importantly, our results indicate that peripheral administration of CpG ODN is not associated with induction of sustained inflammatory responses, which was confirmed by reduced cytokine/chemokine levels analyzed 4 weeks after the last CpG ODN injection. Moreover, we confirmed the effectiveness of CpG ODN to elicit efficient immunostimulatory responses in aged Tg-SwDI mice, where the immune system might be compromised.
Overall, peripheral administration of TLR9 agonist class B CpG ODN successfully triggered a targeted immune response polarizing macrophages/microglia toward beneficial states of activation with improved phagocytic function, resulting in restriction of AD pathology in the absence of apparent toxicity. TLR9 ligands are being developed for use in humans for a variety of clinical indications and have shown favorable safety profiles in humans (Vollmer and Krieg, 2009; Krieg, 2012). Hence, the present findings together with our earlier experimental evidence validate this novel concept of immunomodulation as a safe method to successfully prevent and ameliorate AD-related pathologies and support the potential future testing of CpG ODN in AD patient clinical trials.
Notes
Supplemental material for this article is available at https://nyumc.box.com/v/ScholtzovaSupplementaryFig1: Selected object pair used for the novel object recognition test. This material has not been peer reviewed.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants AG08051 (T.W.), AG20245 (T.W.) and NS073502 (T.W.), as well as Alzheimer's Association Grants NIRG-10-173242 (H.S.) and IIRG-12-239474 (H.S.).The New York University Langone Medical Center Immune Monitoring Core is supported in part by Grant UL1 TR00038 from the National Center for Advancing Translational Sciences, NIH, and Grant P30CA016087 from the National Cancer Institute, NIH. We also thank the following students for their assistance: Helen Keizhen Lyo, Charlotte Herber, and Monica Lee. We also thank Dr. Adam C. Mar of the New York University Langone Medical Center Rodent Behavior Core for his advice and contributions to the analysis of the behavioral data in this manuscript.
The authors declare no competing financial interests.
References
- Asuni AA, Boutajangout A, Scholtzova H, Knudsen E, Li YS, Quartermain D, Frangione B, Wisniewski T, Sigurdsson EM. Vaccination of Alzheimer's model mice with Abeta derivative in alum adjuvant reduces Abeta burden without microhemorrhages. Eur J Neurosci. 2006;24:2530–2542. doi: 10.1111/j.1460-9568.2006.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010;30:16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutajangout A, Li YS, Quartermain D, Sigurdsson EM. Cognitive and sensorimotor tasks for assessing functional impairments in mouse models of Alzheimer's disease and related disorders. Methods Mol Biol. 2012;849:529–540. doi: 10.1007/978-1-61779-551-0_35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brothers HM, Wilcock DM. Are inflammatory profiles the key to personalized Alzheimer's treatment? Neurodegener Dis Manag. 2013;3:343–351. doi: 10.2217/nmt.13.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JD, Cho Y, Foster ML, Kanzler H, Kachura MA, Lum JA, Ratcliffe MJ, Sathe A, Leishman AJ, Bahl A, McHale M, Coffman RL, Hessel EM. CpG-containing immunostimulatory DNA sequences elicit TNF-alpha-dependent toxicity in rodents but not in humans. J Clin Invest. 2009;119:2564–2576. doi: 10.1172/JCI38294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao T, Thomas TC, Ziebell JM, Pauly JR, Lifshitz J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience. 2012;225:65–75. doi: 10.1016/j.neuroscience.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celhar T, Pereira-Lopes S, Thornhill SI, Lee HY, Dhillon MK, Poidinger M, Connolly JE, Lim LH, Biswas SK, Fairhurst AM. TLR7 and TLR9 ligands regulate antigen presentation by macrophages. Int Immunol. 2016;28:223–232. doi: 10.1093/intimm/dxv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Doi Y, Mizuno T, Maki Y, Jin S, Mizoguchi H, Ikeyama M, Doi M, Michikawa M, Takeuchi H, Suzumura A. Microglia activated with the toll-like receptor 9 ligand CpG attenuate oligomeric amyloid {beta} neurotoxicity in in vitro and in vivo models of Alzheimer's disease. Am J Pathol. 2009;175:2121–2132. doi: 10.2353/ajpath.2009.090418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durafourt BA, Moore CS, Zammit DA, Johnson TA, Zaguia F, Guiot MC, Bar-Or A, Antel JP. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012;60:717–727. doi: 10.1002/glia.22298. [DOI] [PubMed] [Google Scholar]
- ElAli A, Rivest S. Microglia in Alzheimer's disease: a multifaceted relationship. Brain Behav Immun. 2016;55:138–150. doi: 10.1016/j.bbi.2015.07.021. [DOI] [PubMed] [Google Scholar]
- Feng Y, Li L, Sun XH. Monocytes and Alzheimer's disease. Neurosci Bull. 2011;27:115–122. doi: 10.1007/s12264-011-1205-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu AK, Hung KW, Yuen MY, Zhou X, Mak DS, Chan IC, Cheung TH, Zhang B, Fu WY, Liew FY, Ip NY. IL-33 ameliorates Alzheimer's disease-like pathology and cognitive decline. Proc Natl Acad Sci U S A. 2016;113:E2705–E2713. doi: 10.1073/pnas.1604032113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea I, Palin K, Newman TA, Van Rooijen N, Perry VH, Boche D. Mannose receptor expression specifically reveals perivascular macrophages in normal, injured, and diseased mouse brain. Glia. 2005;49:375–384. doi: 10.1002/glia.20124. [DOI] [PubMed] [Google Scholar]
- Goñi F, Herline K, Peyser D, Wong K, Ji Y, Sun Y, Mehta P, Wisniewski T. Immunomodulation targeting of both Abeta and tau pathological conformers ameliorates Alzheimer's disease pathology in TgSwDI and 3xTg mouse models. J Neuroinflammation. 2013;10:150. doi: 10.1186/1742-2094-10-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Guerrero AR, Uchida K, Nakajima H, Watanabe S, Nakamura M, Johnson WE, Baba H. Blockade of interleukin-6 signaling inhibits the classic pathway and promotes an alternative pathway of macrophage activation after spinal cord injury in mice. J Neuroinflammation. 2012;9:40. doi: 10.1186/1742-2094-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75:388–397. doi: 10.1189/jlb.0303114. [DOI] [PubMed] [Google Scholar]
- Guillot-Sestier MV, Doty KR, Gate D, Rodriguez J, Jr, Leung BP, Rezai-Zadeh K, Town T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron. 2015a;85:534–548. doi: 10.1016/j.neuron.2014.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillot-Sestier MV, Doty KR, Town T. Innate immunity fights Alzheimer's disease. Trends Neurosci. 2015b;38:674–681. doi: 10.1016/j.tins.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke ML, Kielian T. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin Sci (Lond) 2011;121:367–387. doi: 10.1042/CS20110164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2009;106:1261–1266. doi: 10.1073/pnas.0805453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Xu J, Warren CM, Duan D, Li X, Wu L, Iruela-Arispe ML. Endothelial cells provide an instructive niche for the differentiation and functional polarization of M2-like macrophages. Blood. 2012;120:3152–3162. doi: 10.1182/blood-2012-04-422758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikenwalder M, Polymenidou M, Junt T, Sigurdson C, Wagner H, Akira S, Zinkernagel R, Aguzzi A. Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat Med. 2004;10:187–192. doi: 10.1038/nm987. [DOI] [PubMed] [Google Scholar]
- Heneka M, Andreasson KI, Bachstetter AD, Colonna M, Ginhoux F, Holmes C, Lamb B, Landreth G, Lee DC, Low D, Lynch MA, Monsonego A, O'Banion MK, Pekny M, Puschmann T, Russek-Blum N, Sandusky LA, Selenica ML, Takata K, Teeling J, Town T, Van Eldik LJ, Schulz JB. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J Neurochem. 2016;138:653–693. doi: 10.1111/jnc.13667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliev AI, Stringaris AK, Nau R, Neumann H. Neuronal injury mediated via stimulation of microglial toll-like receptor-9 (TLR9) FASEB J. 2004;18:412–414. doi: 10.1096/fj.03-0670fje. [DOI] [PubMed] [Google Scholar]
- Iribarren P, Chen K, Hu J, Gong W, Cho EH, Lockett S, Uranchimeg B, Wang JM. CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid beta 1–42 peptide by up-regulating the expression of the G-protein-coupled receptor mFPR2. FASEB J. 2005;19:2032–2034. doi: 10.1096/fj.05-4578fje. [DOI] [PubMed] [Google Scholar]
- Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, Shah GN, Price TO, Fleegal-Demotta MA, Butterfield DA, Banks WA. Lipopolysaccharide alters the blood-brain barrier transport of amyloid beta protein: a mechanism for inflammation in the progression of Alzheimer's disease. Brain, Behav Immun. 2009;23:507–517. doi: 10.1016/j.bbi.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker M, Heppner FL. Cerebral and peripheral amyloid phagocytes—an old liaison with a new twist. Neuron. 2008;59:8–10. doi: 10.1016/j.neuron.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Cruchaga C, Goate AM. Alzheimer's disease genetics: from the bench to the clinic. Neuron. 2014;83:11–26. doi: 10.1016/j.neuron.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer's disease model. J Immunol. 2011;187:6539–6549. doi: 10.4049/jimmunol.1100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg AM. From A to Z on CpG. Trends Immunol. 2002;23:64–65. doi: 10.1016/s1471-4906(01)02150-0. [DOI] [PubMed] [Google Scholar]
- Krieg AM. CpG still rocks! Update on an accidental drug. Nucleic Acid Ther. 2012;22:77–89. doi: 10.1089/nat.2012.0340. [DOI] [PubMed] [Google Scholar]
- Lai AY, McLaurin J. Clearance of amyloid-beta peptides by microglia and macrophages: the issue of what, when and where. Future Neurol. 2012;7:165–176. doi: 10.2217/fnl.12.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampron A, Elali A, Rivest S. Innate immunity in the CNS: redefining the relationship between the CNS and its environment. Neuron. 2013a;78:214–232. doi: 10.1016/j.neuron.2013.04.005. [DOI] [PubMed] [Google Scholar]
- Lampron A, Pimentel-Coelho PM, Rivest S. Migration of bone marrow-derived cells into the central nervous system in models of neurodegeneration. J Comp Neurol. 2013b;521:3863–3876. doi: 10.1002/cne.23363. [DOI] [PubMed] [Google Scholar]
- Latta CH, Brothers HM, Wilcock DM. Neuroinflammation in Alzheimer's disease; a source of heterogeneity and target for personalized therapy. Neuroscience. 2015;302:103–111. doi: 10.1016/j.neuroscience.2014.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DC, Rizer J, Hunt JB, Selenica ML, Gordon MN, Morgan D. Review: experimental manipulations of microglia in mouse models of Alzheimer's pathology: activation reduces amyloid but hastens tau pathology. Neuropathol Appl Neurobiol. 2013;39:69–85. doi: 10.1111/nan.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, Hong JT. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37. doi: 10.1186/1742-2094-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, Ashe KH. Brain amyloid-beta oligomers in ageing and Alzheimer's disease. Brain. 2013;136:1383–1398. doi: 10.1093/brain/awt062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Breitbart A, Sun Y, Mehta PD, Boutajangout A, Scholtzova H, Wisniewski T. Blocking the apolipoprotein E/amyloid beta interaction in triple transgenic mice ameliorates Alzheimer's disease related amyloid beta and tau pathology. J Neurochem. 2014;128:577–591. doi: 10.1111/jnc.12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Xu F, Davis J, Otte-Höller I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. Am J Pathol. 2005;167:505–515. doi: 10.1016/S0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud JP, Rivest S. Anti-inflammatory signaling in microglia exacerbates Alzheimer's disease-related pathology. Neuron. 2015;85:450–452. doi: 10.1016/j.neuron.2015.01.021. [DOI] [PubMed] [Google Scholar]
- Michaud JP, Hallé M, Lampron A, Thériault P, Préfontaine P, Filali M, Tribout-Jover P, Lanteigne AM, Jodoin R, Cluff C, Brichard V, Palmantier R, Pilorget A, Larocque D, Rivest S. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer's disease-related pathology. Proc Natl Acad Sci U S A. 2013;110:1941–1946. doi: 10.1073/pnas.1215165110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minett T, Classey J, Matthews FE, Fahrenhold M, Taga M, Brayne C, Ince PG, Nicoll JA, Boche D, Boche D. Microglial immunophenotype in dementia with Alzheimer's pathology. J Neuroinflammation. 2016;13:135. doi: 10.1186/s12974-016-0601-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol. 2005;64:743–753. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. Interleukin-1, neuroinflammation, and Alzheimer's disease. Neurobiol Aging. 2001;22:903–908. doi: 10.1016/s0197-4580(01)00287-1. [DOI] [PubMed] [Google Scholar]
- Piazza F, Winblad B. Amyloid-related imaging abnormalities (ARIA) in immunotherapy trials for Alzheimer's disease: need for prognostic biomarkers? J Alzheimers Dis. 2016;52:417–420. doi: 10.3233/JAD-160122. [DOI] [PubMed] [Google Scholar]
- Prokop S, Miller KR, Drost N, Handrick S, Mathur V, Luo J, Wegner A, Wyss-Coray T, Heppner FL. Impact of peripheral myeloid cells on amyloid-beta pathology in Alzheimer's disease-like mice. J Exp Med. 2015;212:1811–1818. doi: 10.1084/jem.20150479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego Â, Viana SD, Ribeiro CA, Rodrigues-Santos P, Pereira FC. Monophosphoryl lipid-A: a promising tool for Alzheimer's disease toll. J Alzheimers Dis. 2016;52:1189–1202. doi: 10.3233/JAD-151183. [DOI] [PubMed] [Google Scholar]
- Richard KL, Filali M, Préfontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid beta 1-42 and delay the cognitive decline in a mouse model of Alzheimer's disease. J Neurosci. 2008;28:5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai K, Boche D, Carare R, Johnston D, Holmes C, Love S, Nicoll JA. Abeta immunotherapy for Alzheimer's disease: effects on apoE and cerebral vasculopathy. Acta Neuropathol. 2014;128:777–789. doi: 10.1007/s00401-014-1340-9. [DOI] [PubMed] [Google Scholar]
- Schellenberg GD, Montine TJ. The genetics and neuropathology of Alzheimer's disease. Acta Neuropathol. 2012;124:305–323. doi: 10.1007/s00401-012-0996-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt SD, Nixon RA, Mathews PM. ELISA method for measurement of amyloid-beta levels. Methods Mol Biol. 2005;299:279–297. doi: 10.1385/1-59259-874-9:279. [DOI] [PubMed] [Google Scholar]
- Scholtzova H, Wadghiri YZ, Douadi M, Sigurdsson EM, Li YS, Quartermain D, Banerjee P, Wisniewski T. Memantine leads to behavioral improvement and amyloid reduction in Alzheimer's-disease-model transgenic mice shown as by micromagnetic resonance imaging. J Neurosci Res. 2008;86:2784–2791. doi: 10.1002/jnr.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, Kerr DJ, Meeker HC, Mehta PD, Spinner DS, Wisniewski T. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer's disease-related pathology. J Neurosci. 2009;29:1846–1854. doi: 10.1523/JNEUROSCI.5715-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtzova H, Chianchiano P, Pan J, Sun Y, Goñi F, Mehta PD, Wisniewski T. Amyloid β and Tau Alzheimer's disease related pathology is reduced by Toll-like receptor 9 stimulation. Acta Neuropathol Commun. 2014;2:101. doi: 10.1186/s40478-014-0101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Lieberburg I, Arrighi HM, Morris KA, Lu Y, Liu E, Gregg KM, Brashear HR, Kinney GG, Black R, Grundman M. Amyloid-related imaging abnormalities in patients with Alzheimer's disease treated with bapineuzumab: a retrospective analysis. Lancet Neurol. 2012;11:241–249. doi: 10.1016/S1474-4422(12)70015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:475–485. doi: 10.1007/s00401-009-0556-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Xue QS, Tischer J, Bechmann I. Microglial pathology. Acta Neuropathol Commun. 2014;2:142. doi: 10.1186/s40478-014-0142-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F, Bai F, Zhou H, Zhang Z. Reprint of: Microglial toll-like receptors and Alzheimer's disease. Brain Behav Immun. 2016;55:166–178. doi: 10.1016/j.bbi.2016.05.016. [DOI] [PubMed] [Google Scholar]
- Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vickle GD, Esh CL, Daugs ID, Kokjohn TA, Kalback WM, Patton RL, Luehrs DC, Walker DG, Lue LF, Beach TG, Davis J, Van Nostrand WE, Castaño EM, Roher AE. Tg-SwDI transgenic mice exhibit novel alterations in AbetaPP processing, Abeta degradation, and resilient amyloid angiopathy. Am J Pathol. 2008;173:483–493. doi: 10.2353/ajpath.2008.071191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidoni ED, Yeh HW, Morris JK, Newell KL, Alqahtani A, Burns NC, Burns JM, Billinger SA. Cerebral beta-amyloid angiopathy is associated with earlier dementia onset in Alzheimer's disease. Neurodegener Dis. 2016;16:218–224. doi: 10.1159/000441919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer J, Krieg AM. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv Drug Deliv Rev. 2009;61:195–204. doi: 10.1016/j.addr.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Wes PD, Sayed FA, Bard F, Gan L. Targeting microglia for the treatment of Alzheimer's Disease. Glia. 2016;64:1710–1732. doi: 10.1002/glia.22988. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Colton CA. Immunotherapy, vascular pathology, and microhemorrhages in transgenic mice. CNS Neurol Disord Drug Targets. 2009;8:50–64. doi: 10.2174/187152709787601858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Drummond E. Developing therapeutic vaccines against Alzheimer's disease. Expert Rev Vaccines. 2016;15:401–415. doi: 10.1586/14760584.2016.1121815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Goñi F. Immunotherapeutic approaches for Alzheimer's disease. Neuron. 2015;85:1162–1176. doi: 10.1016/j.neuron.2014.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M. Cerebral amyloid angiopathy: emerging concepts. J Stroke. 2015;17:17–30. doi: 10.5853/jos.2015.17.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ji Y, Mehta P, Bates KA, Sun Y, Wisniewski T. Blocking the apolipoprotein E/amyloid-beta interaction reduces fibrillar vascular amyloid deposition and cerebral microhemorrhages in TgSwDI mice. J Alzheimers Dis. 2011;24:269–285. doi: 10.3233/JAD-2011-101401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153:707–720. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zotova E, Bharambe V, Cheaveau M, Morgan W, Holmes C, Harris S, Neal JW, Love S, Nicoll JA, Boche D. Inflammatory components in human Alzheimer's disease and after active amyloid-beta42 immunization. Brain. 2013;136:2677–2696. doi: 10.1093/brain/awt210. [DOI] [PubMed] [Google Scholar]