

Abstract
We discovered a hypomorphic reelin (Reln) mutant with abnormal cortical lamination and no cerebellar hypoplasia. This mutant, RelnCTRdel, carries a chemically induced splice-site mutation that truncates the C-terminal region (CTR) domain of RELN protein and displays remarkably distinct phenotypes from reeler. The mutant does not have an inverted cortex, but cortical neurons overmigrate and invade the marginal zone, which are characteristics similar to a phenotype seen in the cerebral cortex of Vldlrnull mice. The dentate gyrus shows a novel phenotype: the infrapyramidal blade is absent, while the suprapyramidal blade is present and laminated. Genetic epistasis analysis showed that RelnCTRdel/Apoer2null double homozygotes have phenotypes akin to those of reeler mutants, while RelnCTRdel/Vldlrnull mice do not. Given that the receptor double knock-out mice resemble reeler mutants, we infer that RelnCTRdel/Apoer2null double homozygotes have both receptor pathways disrupted. This suggests that CTR-truncation disrupts an interaction with VLDLR (very low-density lipoprotein receptor), while the APOER2 signaling pathway remains active, which accounts for the hypomorphic phenotype in RelnCTRdel mice. A RELN-binding assay confirms that CTR truncation significantly decreases RELN binding to VLDLR, but not to APOER2. Together, the in vitro and in vivo results demonstrate that the CTR domain confers receptor-binding specificity of RELN.
SIGNIFICANCE STATEMENT Reelin signaling is important for brain development and is associated with human type II lissencephaly. Reln mutations in mice and humans are usually associated with cerebellar hypoplasia. A new Reln mutant with a truncation of the C-terminal region (CTR) domain shows that Reln mutation can cause abnormal phenotypes in the cortex and hippocampus without cerebellar hypoplasia. Genetic analysis suggested that CTR truncation disrupts an interaction with the RELN receptor VLDLR (very low-density lipoprotein receptor); this was confirmed by a RELN-binding assay. This result provides a mechanistic explanation for the hypomorphic phenotype of the CTR-deletion mutant, and further suggests that Reln mutations may cause more subtle forms of human brain malformation than classic lissencephalies.
Keywords: APOER2, mutagenesis, Reelin, VLDLR
Introduction
Reeler has been identified as a spontaneous mutant with ataxia (Falconer, 1951), and its brain phenotypes have been extensively studied. In the cerebral cortex, reeler has a preplate splitting defect (Caviness, 1982; Derer and Nakanishi, 1983; Nakanishi, 1983; Sheppard and Pearlman, 1997) and has a complex defect of cortical lamination (Boyle et al., 2011), often described as “outside-in” instead of the normal “inside-out” pattern (Angevine and Sidman, 1961; Caviness and Sidman, 1973; Caviness and Rakic, 1978; Caviness, 1982). In the hippocampus, the pyramidal cell layer of the Ammon's horn appears dispersed and the dentate gyrus does not form a compact granule cell layer (Stanfield and Cowan, 1979). The mutant cerebellum is small without foliation (Hamburgh, 1960; Rice and Curran, 2001).
Positional cloning analyses revealed that reeler carries a deletion of Reln, which encodes an extracellular matrix protein secreted by Cajal-Retzius cells located near the pial surfaces (D'Arcangelo et al., 1995; Hirotsune et al., 1995; Ogawa et al., 1995). There are two main RELN receptors, Apoer2 and Vldlr, and the double knock-out mice have a phenotype indistinguishable from that of reeler mice (D'Arcangelo et al., 1999; Hiesberger et al., 1999; Trommsdorff et al., 1999). Upon binding to receptors, an adaptor protein DAB1 is recruited to the cytoplasmic tails of the receptors and activated by phosphorylation by Src family tyrosine kinases, initiating downstream signaling events (Howell et al., 1997, 1999b; Rice et al., 1998; Trommsdorff et al., 1998; Arnaud et al., 2003a; Bock and Herz, 2003).
RELN contains an N-terminal F-spondin-like domain, a central region with eight reelin repeats, and a short positively charged C-terminal region (CTR) domain (D'Arcangelo et al., 1995). RELN is cleaved by proteases at two sites within the central region, generating multiple fragments (D'Arcangelo et al., 1999; Lambert de Rouvroit et al., 1999a; Ignatova et al., 2004). The N-terminal fragment contains an epitope for the function-blocking antibody CR-50 and is also important for homodimer formation (Ogawa et al., 1995; Miyata et al., 1997; Nakajima et al., 1997; Utsunomiya-Tate et al., 2000; Kubo et al., 2002). The central fragment includes the minimal receptor-binding unit and can rescue the preplate-splitting defect (Hiesberger et al., 1999; Jossin et al., 2003, 2004). The importance of the CTR domain was revealed by characterization of Orleans reeler (Relnrl-Orl) mice, which have a truncation of a part of the eighth reelin repeat and the CTR domain; Relnrl-Orl protein cannot be secreted, and the mutant has the identical phenotype as the null mutant (D'Arcangelo et al., 1997; de Bergeyck et al., 1997). While discordant results exist (Jossin et al., 2004; Nakano et al., 2007), recent in vitro studies specifically targeting the CTR domain suggest that it might have additional functions; specifically, a short CTR truncation did not completely prevent RELN secretion, but reduced its binding to Apoer2-transfected cells or the neuronal cell surface (Nakano et al., 2007; Kohno et al., 2015).
We have previously reported a hypomorphic allele of Reln, RelnCTRdel, which we discovered in an N-ethyl-N-nitrosourea (ENU)-induced mutagenesis screen for cortical lamination defects (Ha et al., 2015). This screen used an Rgs4-lacZ reporter, which revealed that the superficial layers II–III and deep layer V were not distinguishable in the mutants. A single nucleotide change was found at exon 63 splice donor site, and analysis of the mutant transcript predicted deletion of the CTR domain and replacement with 5 aa without affecting the eighth reelin repeat. Surprisingly, the mutants did not show cerebellar malformation and ataxia, which are features of all previously reported null alleles of Reln. This suggested that the mutants might display variant phenotypes in other brain regions that would provide new insight into the mechanism of reelin function. In this paper, we demonstrate that RelnCTRdel has phenotypes distinct from those of reeler mice in the cortex and hippocampus. Importantly, we provide evidence that the CTR domain is required for binding of RELN to its receptor VLDLR (very low-density lipoprotein receptor), but not to APOER2.
Materials and Methods
Animals and genotyping.
Generation of RelnCTRdel mice has been previously described (Ha et al., 2015). The mice were maintained by outcrossing to C57BL/6N (Taconic). Genotyping was done using microsatellite markers (D5Mit193 and D5Mit386) to distinguish the mice carrying mutagenized A/J alleles (146 and 83 bp, respectively) from the wild-type C57BL/6N alleles (136 and 116 bp, respectively). Sequencing analysis was also performed, if necessary. In detail, PCR was performed using the following primers: 5′-GAAAGGTTCTCCTTACCATAAAAGG-3′ and 5′-GCATGCCAGGTAAAAGGAAC-3′. The sequencing reads were aligned with the following sequence near the site of mutation (bracket; t, wild type; c, mutant; uppercase, exon 63; lowercase, intron): GGAAGCTCGGATGAAAGGAGTTCTACTGCGCTGGTGGC AGCCACGCCACAATGGAACAGGTCATGATCAATGGGCTTTGGACCATGTGGAGGTCGTCCTg[t>c]gagtatctgctacttagcgtcct.
Reeler (Relnrl, MGI:1856398) and Vldlrnull (Vldlrtm1Her, MGI:1857267) mice were purchased from the Jackson Laboratory. Apoer2null (Lrp8tm1Her, MGI:1861964) mice were obtained from Dr. Joachim Herz at the University of Texas Southwestern Medical Center (Trommsdorff et al., 1999). Animals of both sexes were used for analyses. Animals were maintained in accordance with guidelines of National Institutes of Health and the Seattle Children's Hospital Institutional Animal Care and Use Committee.
Histology and immunohistochemistry.
Hematoxylin and eosin staining and Nissl staining were done on paraffin sections prepared as previously described (Ha et al., 2015). Nissl staining was performed using FD Thionin Solution from FD NeuroTechnologies following the manufacturer's instruction. For images of histological sections, a Leica DM4000B microscope was used. Immunohistochemistry was performed as previously described (Ha et al., 2015). If necessary, antigen enhancement was performed by brief boiling in 10 mm sodium citrate, pH 6.0. The primary antibodies used are as following: CUX1/CDP (Santa Cruz Biotechnology, sc-13024; 1:100), CTIP2 (Abcam, ab18465; 1:500), NeuN (EMD Millipore, MAB377; 1:1000), chondroitin sulfate proteoglycan (Sigma-Aldrich, C8035; 1:200), RELN clone G10 (EMD Millipore, MAB5364; 1:1000), and calbindin (EMD Millipore, AB1778; 1:1000). The secondary antibodies used are as following: Alexa Fluor 488-conjugated or 568-conjugated goat anti-mouse or rabbit IgG (Thermo Fisher Scientific, 1:600). Hoechst (Thermo Fisher Scientific) was used following the manufacturer's instruction.
Quantification of the cortical lamination defect.
Analysis of immunofluorescence-labeled cells was performed using a cell-counter function of ImageJ as previously described (Mihalas et al., 2016). Single-plane images were acquired using a Zeiss LSM-710 confocal microscope. The cortex was divided into 10 bins, and the cell counts were reported as number of cells per area (mm2) for each bin. Six sections per mouse were analyzed. The mice analyzed were wild type (n = 3), RelnCTRdel (n = 3), compound heterozygote (n = 1), and reeler (n = 1). Data were reported as mean ± SEM.
Immunoprecipitation and Western blotting.
The samples were harvested flash frozen, and stored at −80°. The brain lysates were prepared as previously described (Ha et al., 2015). For immunoprecipitation, ∼3 mg of protein was used for immunoprecipitation using 1 μl of anti-DAB1 antibodies (EMD Millipore, AB5840) and PureProteome Protein A and G magnetic beads (EMD Millipore). Western blotting was performed as previously described using anti-RELN clone G10 (EMD Millipore, MAB5364; 1:4000), anti-RELN clone 12 and 14 (obtained from Dr. Jonathan Cooper; 1:600 each), anti-DAB1 (EMD Millipore, AB5840; 1:2000), anti-phosphorylated tyrosine clone 4G10 (EMD Millipore, 05-321; 1:1000), and anti-GAPDH (Cell Signaling Technologies, #5174; 1:2000) antibodies. For quantitative Western blotting, Odyssey Western kit (Li-Cor Biosciences) was used, including goat anti-mouse or rabbit secondary antibodies conjugated with IRDye, and data analysis was performed using Image Studio Lite Software (Li-Cor Biosciences). Statistical analysis of Western blotting results was performed using Prism7 (Graphpad).
Plasmids.
pCrlM, which was described previously (D'Arcangelo et al., 1997), was obtained from Dr. Jonathan Cooper at the Fred Hutchinson Cancer Research Center. Forty-two bases of the intronic sequence were inserted after 10,283th base of Reln cDNA to make a construct resembling RelnCTRdel transcript (Ha et al., 2015), which was named pCrlM42ins. Briefly, a DNA fragment flanked by PspXI sites, including the insertion, was synthesized. pCrlM and the synthesized fragment were digested, and the pCrlM fragment was replaced with the synthesized mutant fragment. Myc epitope at the BspEI site was removed from pCrlM and pCrlM42ins to generate pCrl and pCrl42ins, which encode wild-type RELN and CTR-truncated RELN, respectively. Apoer2 and Vldlr constructs were obtained from Dr. Joachim Herz. Murine Apoer2 Δexon5 construct, which encodes 869 aa protein with only five cysteine-rich repeats, was used (Beffert et al., 2005). Human Vldlr construct, which encodes 873 aa protein from 19 exons, including an intact exon 16, was used. These RELN receptor clones are equivalent to the ones used in the original RELN-binding assay (D'Arcangelo et al., 1999).
RELN-binding assay.
RELN-binding assay was performed as previously described (D'Arcangelo et al., 1999). Briefly, empty vector, pCrl (wild type), or pCrl42ins (RelnCTRdel) plasmids were transfected overnight into 293T cells using Fugene6 transfection reagent (Promega). The media were changed to Opti-MEM the next day, and the culture supernatants were collected after 2 d. The concentration of RELN in the media was quantified by Western blotting using anti-RELN G10 antibodies. The wild-type culture supernatant was diluted in Opti-MEM based on the quantification result. The culture supernatant from pCrl42ins-transfected cells was concentrated ∼9–10× using a 30K Macrosep advance centrifugal device (Pall Life Sciences). Murine Apoer2 Δexon5 or human Vldlr construct was transfected into 293T cells overnight. The cells were harvested in 1×PBS, and 1 × 106 cells were aliquoted from the same dish and used for each binding reaction. The cells were centrifuged to remove PBS, and then the cell pellets were resuspended in 100 μl of the RELN-containing culture supernatants. The mixture was incubated while being rotated at 4° for 1 h. Then, the cells were centrifuged to remove unbound RELN and washed with 1 × PBS once. The cell pellets containing bound RELN were resuspended and lysed in RIPA lysis buffer for 30 min on ice. The lysates were centrifuged at 14,000 rpm at 4° for 10 min, and the supernatants were used for Western blotting.
Results
Defective migration of cortical neurons without Reeler-like layer inversion
To investigate the lamination defect in more detail, layer marker analysis was performed on postnatal day (P) 21 brains using CUX1 and CTIP2, markers for layer II–IV and layer V/VI, respectively (Fig. 1A). Unlike reeler brains, RelnCTRdel brains did not have an inversion of the cortical layers, and a CUX1-positive cell layer was formed on top of the CTIP2-positive layer, suggesting that the later-born neurons were able to migrate past layers of the earlier-born neurons in a normal fashion. On the other hand, the mutants do have a mild lamination defect, in which neurons overmigrated beyond the upper boundary of the layers that they normally are destined for (Fig. 1A). Many CUX1-positive cells invaded the marginal zone, which is usually a cell-sparse layer (Fig. 1A). In addition, some CUX1-positive cells undermigrated and formed small ectopic clusters within deep layers of cortex (Fig. 1A). CTIP2-positive deep-layer neurons also overmigrated beyond their normal boundaries, and they were loosely distributed (Fig. 1A). This observation is consistent with the originally described RelnCTRdel phenotype visualized using the Rgs4-lacZ reporter, in which there is no gap between the two X-gal-stained stripes that mark layers II/III and V (Ha et al., 2015). Compound heterozygotes of RelnCTRdel and reeler (Relnrl) showed the identical lamination defect as that of a RelnCTRdel homozygote (Fig. 1A). The lamination defect was quantified to document the lack of inversion, overmigration, and less compact deep layers in RelnCTRdel mice (Fig. 1B).
Figure 1.
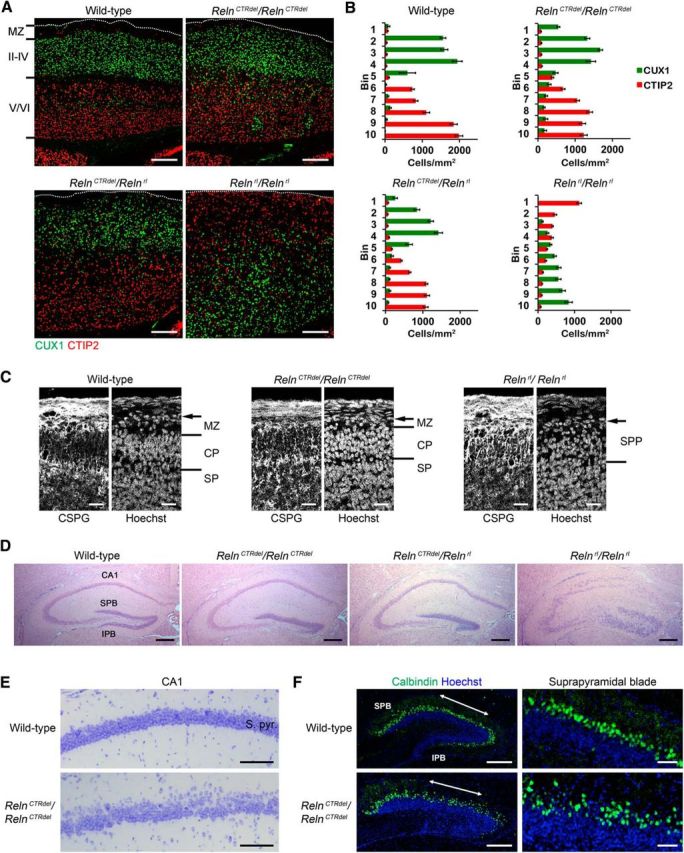
RelnCTRdel has hypomorphic phenotypes in the cortex and hippocampus. A, An inside-out lamination pattern in RelnCTRdel/RelnCTRdel mutants and RelnCTRdel/Relnrl mutants differs from the outside-in pattern in Relnrl/Relnrl mutants.CUX1-positive neurons (green) invaded the marginal zone (MZ) in RelnCTRdel/RelnCTRdel mutants and RelnCTRdel/Relnrl mutants, while CTIP2-positive cells invaded the MZ in Relnrl/Relnrl mutants. CTIP2-positive deep-layer neurons (red) also overmigrated beyond their destined layer in RelnCTRdel/RelnCTRdel and RelnCTRdel/Relnrl mutants. P21 brains were analyzed. White dotted lines indicate the pial surface. Scale bar, 250 μm. B, Quantification of the cell density (cells/mm2) from the results shown in A. The cortex is divided into 10 bins, and mean ± SEM for each bin is plotted. Bin 1 corresponds to the MZ. Increased cell density in bin 1 (green bar, CUX1) and bin 5 (red bar, CTIP2) demonstrates overmigration in RelnCTRdel/RelnCTRdel and RelnCTRdel/Relnrl mutants. C, Preplate splitting occurs in RelnCTRdel/RelnCTRdel mutants in contrast to Relnrl/Relnrl mutants. Embryonic day 14.5 brains were immunostained with anti-chondroitin sulfate proteoglycan antibodies. Arrow, Pial surface; CP, cortical plate; SP, subplate; SPP, superplate. Scale bar, 25 μm. D, IPB the of dentate gyrus does not develop in RelnCTRdel/RelnCTRdel and RelnCTRdel/Relnrl mutants. H and E staining of P21 brains. Note that the Ammon's horn pyramidal cell layer and dentate gyrus SPB are laminated better than in Relnrl/Relnrl mutants. Scale bar, 250 μm. E, The pyramidal cell layer (S. pyr., Stratum pyramidale) is less compact in RelnCTRdel/RelnCTRdel mutants. Nissl stain images of the CA1 region at P21 are shown. Scale bar, 100 μm. F, Calbindin staining (green) reveals distribution of the mature granule neurons at P7. Scale bar, 200 μm. Enlarged images of the SPBs (double-headed arrows) are shown on the right. Scale bar, 50 μm. Lamination defect of the superficial granule cell layer is apparent. Nuclei were stained using Hoechst (blue).
The lack of layer inversion suggested that preplate splitting might happen normally in RelnCTRdel mice. Chondroitin sulfate proteoglycan immunostaining (Sheppard et al., 1991) was done on embryonic day 14.5 brains to visualize preplate splitting (Fig. 1C). The RelnCTRdel mutation did not prevent preplate splitting, and cortical plate formation was apparent in the mutant brain. Of note, the marginal zone was already thinner than in wild-type mice at this age. Altogether, these results indicated that an intact CTR domain is required for proper neuronal migration in the cortex.
The infrapyramidal blade of dentate gyrus does not develop
A Reeler brain has a pronounced lamination defect in the hippocampus (Stanfield and Cowan, 1979) as well as in the cerebral cortex and cerebellum. Examination of a RelnCTRdel brain also revealed a milder but striking hippocampal malformation. The infrapyramidal blade (IPB) of the dentate gyrus, including both the granule cell layer and molecular layer, was either absent or severely truncated in the mutant and compound heterozygote (Fig. 1D). In contrast to the reeler hippocampus, lamination of the Ammon's horn does occur in the RelnCTRdel hippocampus, and the suprapyramidal blade (SPB) of the dentate gyrus develops a relatively compact granule cell layer. However, subtle lamination defects are present. The CA1 pyramidal cell layer was less compact in the RelnCTRdel hippocampus (Fig. 1E). The pyramidal cell layer of RelnCTRdel mice was 47.6% thicker than that of wild type. Mean ± SD measurements were 51.90 ± 4.10 μm in wild type and 76.63 ± 6.32 μm in the mutants from n = 3 animals each (p = 0.0074, two-tailed unpaired Student's t test). Calbindin staining, which highlights the mature granule cells during development (Goodman et al., 1993), revealed that the mutant has a mild lamination defect, with increased thickness and irregular boundaries of the SPB (Fig. 1F). This may account for our observation that the mutant SPB displays an uneven border between the superficial granule cell layer and the molecular layer (Fig. 1D).
Reduced RELN secretion, intracellular accumulation, and signaling defect
Truncation of the CTR domain and a part of the eighth reelin repeat in Orleans reeler mice prevents RELN secretion and results in intracellular accumulation of RELN protein (D'Arcangelo et al., 1997; de Bergeyck et al., 1997). However, it has been reported that certain forms of CTR-disrupted RELN could be secreted from the cells transfected with Reln constructs as well as from primary cultured neurons (Lambert de Rouvroit et al., 1999b; Nakano et al., 2007; Kohno et al., 2015). To assess RELN secretion in vivo, quantitative Western blot analysis was performed using serum samples (Fig. 2A,B). RelnCTRdel mice showed a 58.5% serum RELN level compared with the wild type. Given that heterozygote reeler (Reln+/Relnrl) mice with a similar level of serum RELN (47.5%) are normal, it is unlikely that the RelnCTRdel phenotype is due to the dosage effect rather than a specific disruption of protein function. This is also supported by the observation that the phenotype of RelnCTRdel/Relnrl mice is no more severe than that of RelnCTRdel/RelnCTRdel mice, although the former have even less secreted RELN (40%).
Figure 2.
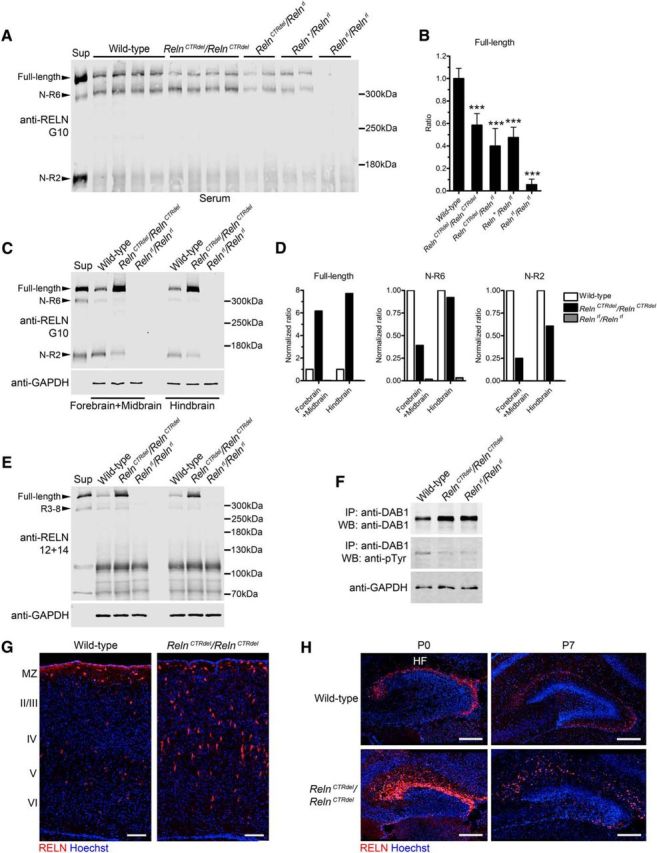
RELN protein is expressed and secreted in RelnCTRdel mice, but DAB1 degradation by a negative feedback is disrupted. A, Anti-RELN (clone G10) Western blot analysis of the serum samples from P7 mice. One microliter of serum was loaded to each well. The culture supernatant (Sup) from wild-type Reln-transfected 293T cells was used as a control. Arrowheads indicate full-length RELN and its cleavage products, N-R6 and N-R2. B, Quantification of data shown in A. Asterisks indicate statistical significance for each group compared with the wild type (1-way ANOVA and Dunnett's multiple-comparisons test; ***p < 0.001). P values from comparisons to the wild type are 0.0008, 0.0002, 0.0006, and 0.0001 for RelnCTRdel/RelnCTRdel, RelnCTRdel/Relnrl, Reln+/Relnrl, and Relnrl/Relnrl mutants, respectively. C, Western blot analysis of the brain homogenates using anti-RELN (G10) antibodies. The mutant has more full-length RELN, but less cleavage product. GAPDH was used as a control. D, Quantification of the representative data shown in C. E, Western blot analysis of the brain homogenates using anti-RELN clone 12 and 14 antibodies. Arrowheads indicate full-length RELN and its cleavage product, R3–8. F, DAB1 was immunoprecipitated from E18.5 brain lysates. Increased DAB1 protein level in RelnCTRdel/RelnCTRdel mutants is comparable to that in Relnrl/Relnrl mutants. Tyrosine phosphorylation of DAB1 was not increased. G, RELN expression in the cortex at P7. The mutant displays brighter staining in both marginal zone and middle layers of the cortex. Scale bar, 100 μm. H, Distribution of RELN-expressing cells (red) in the dentate gyrus at P0 and P7. HF, Hippocampal fissure. Scale bar, 200 μm.
In homogenates of brain tissue, full-length RELN protein is increased in the RelnCTRdel mutant (Fig. 2C–E). In contrast, intensities of cleavage product bands (N-R6, N-R2) were decreased in the mutant. This is probably due to the secretion defect, which would result in intracellular accumulation of full-length RELN, and reduced substrates for cleavage in the extracellular matrix.
To test whether expression of RelnCTRdel causes a signaling defect, the level of DAB1 protein was measured. DAB1 protein is degraded when it is phosphorylated by Src family kinases upon RELN-receptor binding (Arnaud et al., 2003b; Bock et al., 2004; Kerjan and Gleeson, 2007). In reeler and RELN receptor knock-out mice, the disruption of reelin signaling hinders this negative-feedback regulation of DAB1 protein level (Rice et al., 1998; Howell et al., 1999a, 2000; Trommsdorff et al., 1999; Kuo et al., 2005). Therefore, DAB1 accumulation can reflect a reelin signaling defect in vivo. We found that RelnCTRdel mice have elevated DAB1 protein expression to the same extent as reeler mice (Fig. 2F). These results suggested that secreted CTR-truncated RELN does not activate reelin signaling effectively enough to mediate subsequent DAB1 degradation by a negative-feedback mechanism. In addition, in RelnCTRdel mice, DAB1 tyrosine phosphorylation did not increase as in reeler mice (Fig. 2F).
Immunohistochemistry analysis also detected a higher level of RELN expression in RelnCTRdel mice, which is likely due to intracellular accumulation. The Cajal-Retzius cells in the marginal zone of the cerebral cortex were more brightly stained, and we found in the middle layers some bright RELN-positive cells (Fig. 2G), which are likely interneurons (Alcántara et al., 1998). Stronger RELN expression was also detected in the striatum and piriform cortex (data not shown). In the hippocampus, RELN-expressing cells were most prominent in the marginal zone, especially in the depths of the hippocampal fissure, at P0 and P7 (Fig. 2H), as previously known (Soriano et al., 1994; Schiffmann et al., 1997; Alcántara et al., 1998). The distribution of RELN-expressing cells in the mutant hippocampus appeared more dispersed than in the control hippocampus, and the intensity of RELN immunoreactivity appeared higher (Fig. 2H). It might appear as if there are more RELN-expressing cells in ectopic locations in both the cortex and hippocampus. However, as previous immunohistochemistry and in situ hybridization studies have reported that Reln mRNA is present in all these regions (Schiffmann et al., 1997; Alcántara et al., 1998; Pesold et al., 1998), it is likely that these signals represent cells that normally express low levels of RELN, but accumulate high intracellular RELN immunoreactivity in the mutant mice.
RelnCTRdel mice display differential genetic interactions with receptor knock-out mice
We have previously reported that, unlike reeler mutants, RelnCTRdel mutants have normal cerebellar foliation and are not ataxic (Ha et al., 2015). In this study, we report that the mutants displayed hypomorphic phenotypes in the cerebral cortex and hippocampus that differ from those found in reeler mice (Fig. 1). An explanation for this was suggested by the lamination pattern in the cerebral cortex. The RelnCTRdel mutant had marginal zone invasion without layer inversion, which resembles a phenotype caused by deficiency of the RELN receptor Vldlr (Hack et al., 2007). In contrast, mice null for the RELN receptor Apoer2 displayed a distinct phenotype; specifically, they have a partial layer inversion without marginal zone invasion (Trommsdorff et al., 1999; Benhayon et al., 2003; Hack et al., 2007). Both VLDLR and APOER2 are necessary for RELN function, and it is thought that these two receptors have some redundant functions, as well as some that are nonoverlapping. This is evident because Vldlr or Apoer2 knock-out mice have a less severe phenotype compared with reeler mice in all three brain regions, but double knock-out mice appear indistinguishable from reeler mice (Trommsdorff et al., 1999; Beffert et al., 2006). Importantly, single-receptor knock-out mice had relatively well foliated cerebella compared with reeler mice and were not ataxic (Trommsdorff et al., 1999; Beffert et al., 2006). These results led us to hypothesize that the RelnCTRdel mutation might differentially disrupt activation of VLDLR and APOER2.
To test this hypothesis in vivo, we performed a genetic epistasis test by generating double homozygotes that carried RelnCTRdel and either Vldlrnull or Apoer2null alleles. Importantly, we found that RelnCTRdel/Apoer2null double-homozygote mice displayed phenotypes similar to reeler mice in all three brain regions. First, RelnCTRdel/Apoer2null double-homozygote mice displayed an inversion of layer organization in the cerebral cortex (Fig. 3A), which appeared identical to the reeler phenotype (Fig. 1A). RelnCTRdel/Apoer2null mice also displayed marginal zone invasion by CTIP2-positive neurons (Fig. 3A,B), as also seen in reeler mice. Second, the hippocampal phenotype of RelnCTRdel/Apoer2null mice was identical to that of reeler mice; Ammon's horn pyramidal cell layer and the dentate gyrus granule cell layer were dispersed (Fig. 3C). Finally, RelnCTRdel/Apoer2null double homozygotes displayed a smaller and less foliated cerebellum, albeit not as severe as that found in reeler mice (Fig. 4A,B). This result was striking since neither RelnCTRdel nor Apoer2null mice have a cerebellar foliation defect. RelnCTRdel/Apoer2null double homozygotes and Apoer2null cerebellum shared some common traits. The area of the white matter at the level of the vermis appeared wider than that in the wild type, and calbindin-positive Purkinje cells were ectopically localized in the white matter (Fig. 4C). These phenotypes appeared to be slightly more severe in the double homozygote than Apoer2null mice, contributing to their reeler-like appearance.
Figure 3.
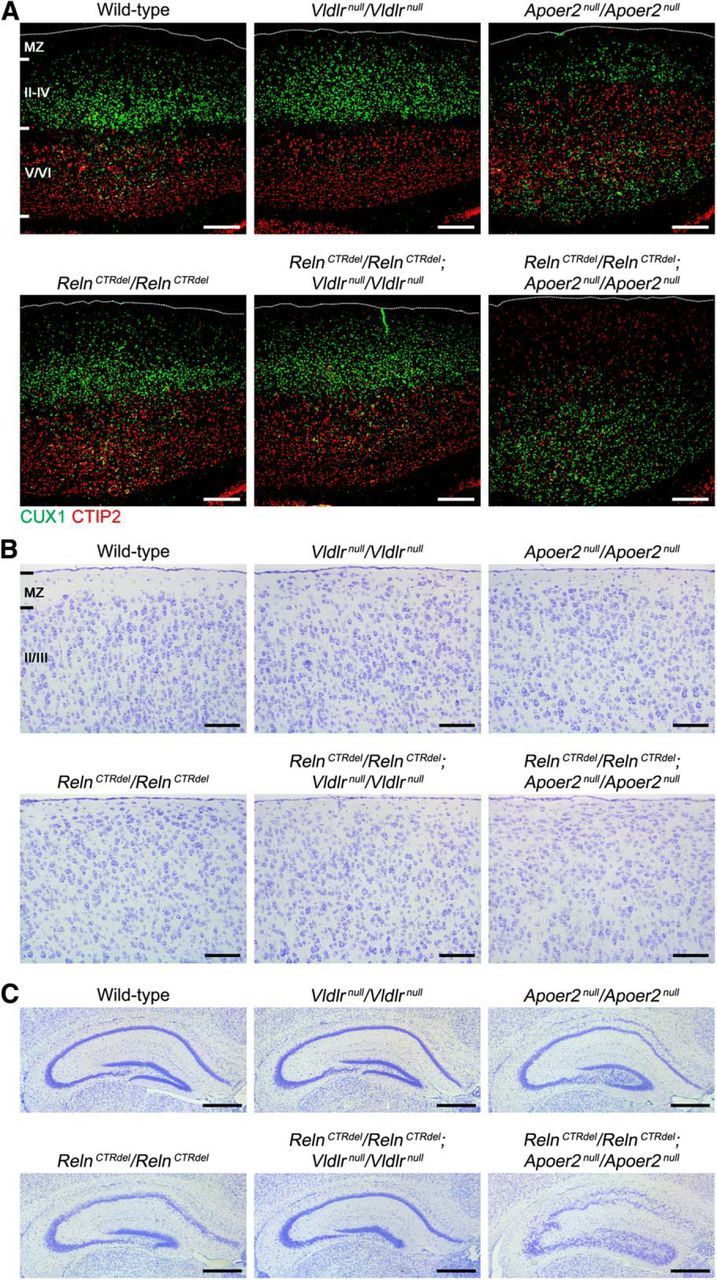
Differential genetic interaction with Apoer2null and Vldlrnull mice in the cerebral cortex and hippocampus. A, RelnCTRdel/Apoer2null double-homozygote mice, but not RelnCTRdel/Vldlrnull double-homozygote mice, display reeler-like cortical layer inversion. RelnCTRdel/Vldlrnull mice appear similar to RelnCTRdel and Vldlrnull mice without inversion. Sagittal sections of P21 brains were immunostained with anti-CUX1 and anti-CTIP2 antibodies. White dotted lines indicate the pial surface. Scale bar, 250 μm. B, RelnCTRdel/Apoer2null double-homozygote mice display the marginal zone invasion. RelnCTRdel/Vldlrnull, RelnCTRdel, and Vldlrnull mice appear similar to each other with the marginal zone invasion. Scale bar, 100 μm. C, RelnCTRdel/Apoer2null double-homozygote mice, but not RelnCTRdel/Vldlrnull double-homozygote mice, display reeler-like hippocampus. Nissl staining images of P21 brain coronal sections are shown. Scale bar, 500 μm.
Figure 4.
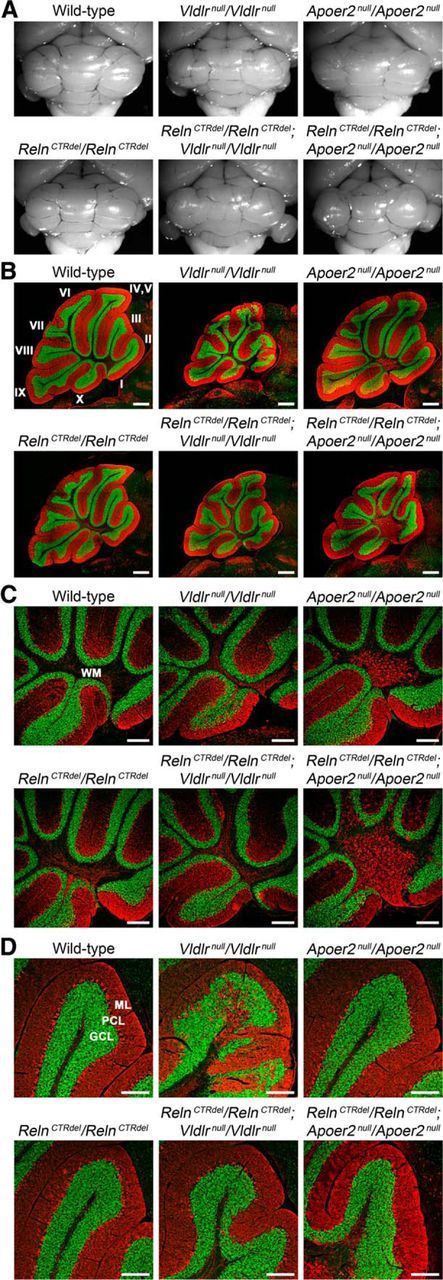
Differential genetic interaction with Apoer2null and Vldlrnull mice in the cerebellum. A, Gross morphology of the cerebellum. B–D, Immunohistochemistry images of P21 brain sagittal sections at the level of the vermis are shown. Anti-calbindin (red, Purkinje cells) and anti-NeuN (green, granule cells) antibodies were used. B, RelnCTRdel/Apoer2null double-homozygote mice display a smaller and less foliated cerebellum than either mutant alone. RelnCTRdel/Vldlrnull double-homozygote display a foliation pattern similar to that of Vldlrnull mice. Ten cerebellar lobules (I–X) are labeled. Scale bar, 500 μm. C, High-magnification images of the cerebellar white matter (WM) region taken from the same section shown in B. RelnCTRdel/Apoer2null double homozygote mice, as well as Apoer2null mice, have ectopically localized calbindin-expressing Purkinje cells (red) at the level of the vermis. Scale bar, 250 μm. D, High-magnification images of the cerebellar granule cell layer (GCL) taken from the same section shown in B. Disorganized layers of the granule cells and Purkinje cells (PCL) seen in Vldlrnull mice are less severe in RelnCTRdel/Vldlrnull mice. ML, molecular layer. Scale bar, 200 μm.
In contrast to RelnCTRdel/Apoer2null double homozygotes, the phenotype of RelnCTRdel/Vldlrnull double-homozygote mice was clearly unlike that of reeler mice. Instead, their phenotypes were similar to either RelnCTRdel or Vldlrnull mice. The cerebral cortex was not inverted (Fig. 3A), but marginal zone invasion was observed (Fig. 3B). This appearance is identical to that of RelnCTRdel and Vldlrnull mice. The hippocampus appeared similar to that of RelnCTRdel mice lacking the IPB (Fig. 3C) although without dispersion of CA1 pyramidal cell layer. The cerebellar foliation pattern, at least at the level of vermis, was similar to that of Vldlrnull mice. That is, lobule IV was larger and lobules VI and VII were simpler than comparable features of normal mice (Fig. 4B). RelnCTRdel/Vldlrnull double-homozygote mice displayed a phenotype that is actually less severe than that of Vldlrnull mice with respect to cerebellar lamination; the cerebellar granule neuron layer and Purkinje cell layer appear less disorganized than in Vldlrnull mice (Fig. 4D). While unexpected, this phenotype is still unlike that of reeler mice.
CTR domain is required for an interaction with VLDLR
RelnCTRdel/Apoer2null double-homozygote mice appeared similar to reeler mice, while RelnCTRdel/Vldlrnull did not (Fig. 5A). These results from epistasis analysis suggested the possibility that the CTR truncation differentially affected binding of RELN to its receptors. A model for this hypothesis is illustrated in Figure 5B. The epistasis analysis implies that activation of both APOER2 and VLDLR pathways is disrupted in RelnCTRdel/Apoer2null double-homozygote mice, which suggests the possibility that CTR truncation disrupted binding to the VLDLR receptor. APOER2 signaling appears to be active in RelnCTRdel/Vldlrnull double mutants, as they do not have a reeler-like phenotype, so CTR truncation appears not to preclude binding to the APOER2 receptor.
Figure 5.
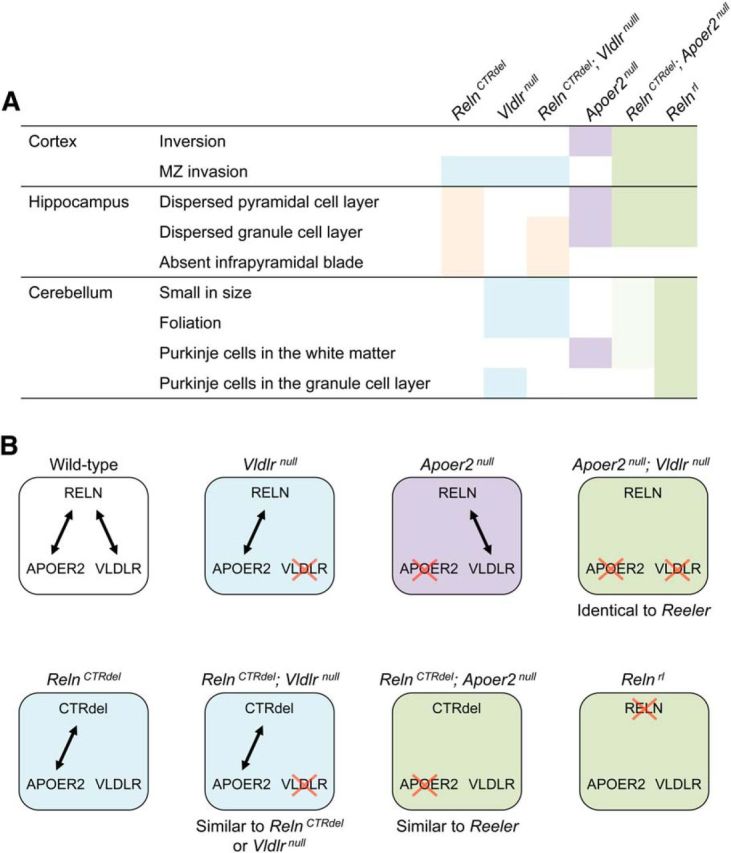
Summary and interpretation of the epistasis analysis. A, A table illustrating phenotypic comparison. Blue, Resemblance to Vldlrnull phenotype; purple, resemblance to Apoer2null phenotype; pink, resemblance to RelnCTRdel phenotype; green, resemblance to reeler phenotype. Lighter shade of green indicates less severe cerebellar phenotype of RelnCTRdel/Apoer2null mice. B, Hypothetical model of RELN and receptor interactions. Interactions between RELN and its receptors (double-headed arrows) are illustrated in boxes. RELN, Wild-type full-length RELN protein; CTRdel, mutant RELN protein; red crosses, gene deletion. The mice are color-coded based on similarities in their hypothetical RELN-receptor interaction patterns. The color scheme is consistent with the table in A. Blue, Mice with disrupted interaction with VLDLR; purple, disrupted interaction with APOER2; green, disrupted interaction with both receptors.
It has been shown that modifying the CTR domain can reduce binding to Apoer2-transfected cell membrane or primary cultured cortical neurons (Nakano et al., 2007; Kohno et al., 2015). However, an interaction between CTR-truncated RELN and VLDLR has not been previously tested. To test our hypothesis, we performed a RELN-binding assay as previously described (D'Arcangelo et al., 1999). The culture supernatant of 293T cells transfected with wild-type Reln (pCrl) or RelnCTRdel (pCrl42ins) constructs were analyzed by quantitative Western blotting. CTR-truncated RELN was secreted, but not as efficiently as wild type RELN, consistent with what we observed in serum and brain lysate. To normalize for this, the binding assay was performed at two input concentrations of RELN; the culture supernatant from wild-type Reln-transfected cells was diluted to match the lower RELN concentration in the culture supernatant from RelnCTRdel-transfected cells, and the culture supernatant from RelnCTRdel-transfected cells was concentrated to match the higher RELN concentration in the wild-type Reln-transfected cell culture supernatant (Fig. 6A). These culture supernatants were incubated with mock-transfected, Apoer2-transfected, or Vldlr-transfected 293T cells, and the RELN proteins bound on the cell surface were detected by Western blotting. Strikingly, CTR-truncated RELN binds to VLDLR significantly less efficiently than wild-type RELN at both low and high concentrations, while there was little effect on binding to APOER2 (Fig. 6B). Although quantitative analysis revealed that the wild-type RELN actually binds to VLDLR less than to APOER2 under our assay condition, the difference was markedly greater for mutant RELN (Fig. 6C). This is clear when the ratio of VLDLR-bound RELN to APOER2-bound RELN was compared; CTR-truncated RELN displayed significantly smaller ratios at both low and high concentrations (Fig. 6D). These results demonstrate that the CTR domain is required for an interaction with VLDLR, but not APOER2, and confirmed our model (Fig. 5B) based on the results from epistasis analysis.
Figure 6.

CTR Truncation of RELN decreased binding to VLDLR but not APOER2. A, Western blot analysis to compare RELN protein amount in the culture supernatant used for RELN-binding assay (input). Arrowheads indicate full-length RELN and its cleavage products, N-R6 and N-R2. B, Bound RELN protein on the surface of the receptor-transfected 293T cells. M, Mock; A, Apoer2; V, Vldlr-transfected cells; Sup, culture supernatant from wild-type Reln-transfected cells as a control. C, Quantification of the RELN-binding assay in B (n = 4). Analysis of full-length RELN bands is shown. Mean ratios and SDs of each group compared with the wild-type RELN bound to APOER2 are plotted. Asterisks indicate statistical significance (2-way ANOVA and Tukey's multiple-comparisons test; *p < 0.05; **p < 0.01; ***p < 0.001). P values for comparisons of RELN bound to Apoer2 versus Vldlr-transfected cells are 0.2546, 0.20 × 106, 0.0119, and 0.07 × 1011 for diluted wild type, wild type, RelnCTRdel, and concentrated RelnCTRdel, respectively. D, Ratios of VLDLR-bound RELN and APOER2-bound RELN were calculated from the same dataset shown in C. Means and SDs are plotted. Asterisks indicate statistical significance (1-way ANOVA and Tukey's multiple-comparisons test; **p < 0.01; ***p < 0.001). P values are 0.0053 and 0.0005, for comparisons between concentration-matched samples (diluted wild type vs RelnCTRdel, and wild type vs concentrated RelnCTRdel), respectively.
Discussion
The RELN CTR domain is required to arrest cortical neuronal migration
It has been suggested that RELN might be both an attractive or permissive signal and a stop signal for cortical neurons (Ogawa et al., 1995; Frotscher, 1997; Schiffmann et al., 1997; Sheppard and Pearlman, 1997; Dulabon et al., 2000; Herrick and Cooper, 2002; Olson et al., 2006; Hack et al., 2007; Zhao and Frotscher, 2010). In reeler mice, the absence of the attractive or permissive signal explains why later-born neurons cannot go past earlier-born neurons. Reeler also shows marginal zone invasion phenotype, suggesting that RELN acts as a stop signal. Our results provide evidence supporting the notion that RELN does play a role as a stop signal during the final stage of cortical neuronal migration, and this is dissociable from its function as an attractive or permissive signal (Fig. 1A,B). Of note, analysis of the cortical lamination defect phenotypes in knock-out mice suggested that VLDLR mediates a stop signal, while APOER2 is responsible for an attractive or permissive signal (Frotscher, 1997; Herrick and Cooper, 2002; Hack et al., 2007; Zhao and Frotscher, 2010). In addition, expression patterns of the RELN receptors were consistent with this interpretation; Apoer2 is expressed throughout the developing brain, while Vldlr is selectively expressed in neurons near the marginal zone where the stop signal is needed (Trommsdorff et al., 1999; Perez-Garcia et al., 2004; Hack et al., 2007). Our observations demonstrating that RelnCTRdel fails to interact with VLDLR support this model.
The RelnCTRdel phenotype is due to differential receptor interaction
Biochemical and cell biological analyses have established that reelin signaling is mediated via the VLDLR and APOER2 receptors (D'Arcangelo et al., 1999; Hiesberger et al., 1999; Trommsdorff et al., 1999). Genetic studies support this; while Vldlrnull and Apoer2null phenotypes are milder than those of reeler mice and distinct from each other, the phenotype of the double-receptor mutants appears indistinguishable from that of reeler mice (Trommsdorff et al., 1999; Beffert et al., 2006). Briefly, the Vldlrnull phenotype features cortical marginal zone invasion without layer inversion, relatively normal hippocampal lamination, and abnormally foliated cerebellum with disorganized granule and Purkinje cell layers. The Apoer2null phenotype shows partial cortical layer inversion, a split CA1 pyramidal layer, a less compact dentate gyrus granule cell layer, and normally foliated cerebellum with ectopic Purkinje cells in the white matter. The evidence that the receptors mediate different aspects of reelin signaling suggested the possibility that the hypomorphic phenotype of RelnCTRdel is a consequence of a differential interaction of the mutant protein with its receptors. Our genetic analysis showed that a RelnCTRdel/Apoer2null double mutant has a phenotype in the cerebral cortex, hippocampus, and cerebellum that is closer to that of reeler mice than either mutant alone (Figs. 3, 4). These results suggest that the CTR domain is required for signaling through VLDLR. In contrast, the RelnCTRdel/Vldlrnull double mutant does not show a reeler-like phenotype, implying APOER2 signaling in the RelnCTRdel mutant is intact. This hypothesis was confirmed by RELN-binding assay (Fig. 6). CTR-truncated RELN showed significantly reduced binding to VLDLR, but binding to APOER2 was unaffected. This is the first report demonstrating that CTR-domain modification can affect receptor-binding specificity.
These results provide a mechanistic explanation for the hypomorphic phenotype of RelnCTRdel, which is that CTR truncation has a differential effect on the interaction of RELN with its receptors. However, some puzzling aspects remain. One of them is the cerebellar phenotype. Vldlr mutations in mice (Trommsdorff et al., 1999) and humans (Moheb et al., 2008; Ozcelik et al., 2008; Türkmen et al., 2008) affect cerebellar development drastically, but the RelnCTRdel mutation does not (Fig. 4). One possible explanation is that CTR-truncated RELN retains some residual VLDLR-binding affinity (Fig. 6B,C), and this might be able to activate reelin signaling sufficiently for cerebellar development. Residual binding affinity can also explain why the RelnCTRdel/Apoer2null double-mutant cerebellar phenotype is milder than that of reeler mice. Also surprising is our observation that the dentate gyrus malformation in RelnCTRdel mice is more severe than that in Vldlrnull mice (Fig. 3C). Certain forms of CTR modification can decrease APOER2-binding affinity (Nakano et al., 2007; Kohno et al., 2015). While our results show that APOER2 binding is grossly normal, a subtle change in the interaction of CTR-truncated RELN and APOER2 might contribute to the hippocampal abnormality. However, these hypotheses will require additional study.
Comparison with ΔC-KI mice
RelnCTRdel displays an interesting novel phenotype in the dentate gyrus; the IPB does not develop (Fig. 1D). This phenotype makes RelnCTRdel extraordinary, since another CTR-domain mutant (ΔC-KI) recently reported does not have this phenotype (Kohno et al., 2015). ΔC-KI displays a more reeler-like hippocampal phenotype with a split pyramidal cell layer near the CA1 region and a loosely packed granule cell layer in the dentate gyrus. This phenotype is somewhat similar to that of Apoer2null mice. Since this form of CTR modification (ΔC-FLAG) has been shown to disrupt interaction with APOER2 receptor (Nakano et al., 2007; Kohno et al., 2015), ΔC-KI may interfere with signaling through APOER2 at least in the hippocampus. In addition, the cortical marginal zone invasion phenotype appears at earlier age than in ΔC-KI mice (Fig. 1C), and deep-layer neurons, not only superficial neurons, overmigrate in RelnCTRdel mice (Fig. 1A,B). It is unclear yet why these very similar mutants show different phenotypes.
We focused our cerebellar analysis (Fig. 4) at the level of the cerebellar vermis to facilitate comparison with published studies of RELN receptor mutants. It has recently been reported that ΔC-KI mice display focal ectopic localization of Purkinje cells in more lateral regions of lobules VI and VII (Nakamura et al., 2016). Our observation of ectopic Purkinje cells in the Vldlrnull mice near lobules VI and VII even at the level of the vermis (Fig. 4B) is comparable to this result, and this phenotype could be considered another resemblance of ΔC-KI mice to Vldlrnull mice.
The difference between these two alleles is in the sequence that replaces the CTR domain. ΔC-KI mice have exogenous FLAG, which was added to increase secretion efficiency, and a stop codon that replaces exon 65. This would result in a longer C-terminal tail after L3428 (the last amino acid translated from exon 63), VRDYKDDDDK* with the second amino acid switched from S to R, or just FLAG (DYKDDDDK*) in an alternative transcript with microexon 64 spliced out. In contrast, RelnCTRdel is predicted to have only 5 aa (RVSAT*) added after L3428 (Ha et al., 2015). The phenotypic difference indicates that RelnCTRdel and ΔC-KI mice differentially disrupt reelin signaling, and suggests that even a small change in the sequence and length of the CTR can significantly affect reelin function. Another difference is the presence or absence of the stop codon at the junction of exon 63. This stop codon is used when a minor form of Reln transcript, which is expected to produce an endogenous RELN protein with CTR truncation right after L3428, is generated by alternative polyadenylation (Lambert de Rouvroit et al., 1999b). RelnCTRdel mutation disrupts this stop codon and is expected to force all major and minor transcripts to adopt the RelnCTRdel form. In contrast, ΔC-KI modification does not alter this stop codon and leaves alternative transcripts intact. Therefore, ΔC-KI mice would express both endogenous and exogenous CTR-truncated RELN protein.
Potential implications of RELN CTR truncation
Alternatively spliced variants of Reln transcripts have been previously studied, and one of them is predicted to encode a CTR-truncated RELN protein due to the inclusion of a stop codon at the junction of exon 63 (Lambert de Rouvroit et al., 1999b). While the protein expression of a CTR-truncated protein in vivo was reported (Kohno et al., 2009), its function is unknown. Our study suggests two potential implications of CTR truncation. First, the endogenous CTR-truncated RELN might play a role in vivo, selectively activating APOER2 receptor when and where specificity is required. Studying timing and localization of expression of this alternative spliced form of Reln during development may reveal an additional complexity of how RELN regulates brain development. Second, there may be pathological consequences for mutations that affect the RELN CTR. While RELN mutation has been associated with type II lissencephaly with cerebellar malformation (Hong et al., 2000), the phenotype of the RelnCTRdel mouse suggests RELN should not be excluded as a candidate in human brain malformations without cerebellar malformation.
In summary, our study characterized an ENU-induced Reln mutant, which carries a splice-site mutation that could be similar to naturally occurring mutations in humans. Using both an in vivo epistasis analysis and an in vitro RELN-binding assay, we uncovered a molecular mechanism by which this mutation causes a hypomorphic phenotype. Results from both approaches agree with our model; CTR truncation selectively disrupts an interaction between RELN and VLDLR. Our discovery assigned a novel function to the CTR domain of RELN, a critical domain for an interaction with VLDLR and a regulator of receptor-binding specificity. Future study will be required for a mechanistic explanation for dentate gyrus malformation and discordances in epistasis analysis.
Footnotes
This work was supported by National Institutes of Health Grants R01MH081187 and R01HD36404. We thank Dr. Joachim Herz for providing Apoer2null mice and RELN receptor constructs; Dr. Matthew B. Johnson for help with cortical layer marker selection; Dr. Christopher A. Walsh for consultation about epistasis analysis; Dr. Jonathan A. Cooper and Sergi Simó for providing a Reln construct, antibodies, and consultation about reelin signaling; Dr. Kathleen J. Millen for consultation about cerebellar analysis; and Anna M. Lindsay, Dr. Mingyue Lun, Haiyan Qiu, and George C. Talbott for technical assistance.
The authors declare no competing financial interests.
References
- Alcántara S, Ruiz M, D'Arcangelo G, Ezan F, de Lecea L, Curran T, Sotelo C, Soriano E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. J Neurosci. 1998;18:7779–7799. doi: 10.1523/JNEUROSCI.18-19-07779.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angevine JB, Jr, Sidman RL. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- Arnaud L, Ballif BA, Förster E, Cooper JA. Fyn tyrosine kinase is a critical regulator of disabled-1 during brain development. Curr Biol. 2003a;13:9–17. doi: 10.1016/S0960-9822(02)01397-0. [DOI] [PubMed] [Google Scholar]
- Arnaud L, Ballif BA, Cooper JA. Regulation of protein tyrosine kinase signaling by substrate degradation during brain development. Mol Cell Biol. 2003b;23:9293–9302. doi: 10.1128/MCB.23.24.9293-9302.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beffert U, Weeber EJ, Durudas A, Qiu S, Masiulis I, Sweatt JD, Li WP, Adelmann G, Frotscher M, Hammer RE, Herz J. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron. 2005;47:567–579. doi: 10.1016/j.neuron.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Beffert U, Durudas A, Weeber EJ, Stolt PC, Giehl KM, Sweatt JD, Hammer RE, Herz J. Functional dissection of Reelin signaling by site-directed disruption of Disabled-1 adaptor binding to apolipoprotein E receptor 2: distinct roles in development and synaptic plasticity. J Neurosci. 2006;26:2041–2052. doi: 10.1523/JNEUROSCI.4566-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benhayon D, Magdaleno S, Curran T. Binding of purified Reelin to ApoER2 and VLDLR mediates tyrosine phosphorylation of Disabled-1. Brain Res Mol Brain Res. 2003;112:33–45. doi: 10.1016/S0169-328X(03)00032-9. [DOI] [PubMed] [Google Scholar]
- Bock HH, Herz J. Reelin activates SRC family tyrosine kinases in neurons. Curr Biol. 2003;13:18–26. doi: 10.1016/S0960-9822(02)01403-3. [DOI] [PubMed] [Google Scholar]
- Bock HH, Jossin Y, May P, Bergner O, Herz J. Apolipoprotein E receptors are required for reelin-induced proteasomal degradation of the neuronal adaptor protein Disabled-1. J Biol Chem. 2004;279:33471–33479. doi: 10.1074/jbc.M401770200. [DOI] [PubMed] [Google Scholar]
- Boyle MP, Bernard A, Thompson CL, Ng L, Boe A, Mortrud M, Hawrylycz MJ, Jones AR, Hevner RF, Lein ES. Cell-type-specific consequences of Reelin deficiency in the mouse neocortex, hippocampus, and amygdala. J Comp Neurol. 2011;519:2061–2089. doi: 10.1002/cne.22655. [DOI] [PubMed] [Google Scholar]
- Caviness VS., Jr Neocortical histogenesis in normal and reeler mice: a developmental study based upon [3H]thymidine autoradiography. Brain Res. 1982;256:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Sidman RL. Time of origin or corresponding cell classes in the cerebral cortex of normal and reeler mutant mice: an autoradiographic analysis. J Comp Neurol. 1973;148:141–151. doi: 10.1002/cne.901480202. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Rakic P. Mechanisms of cortical development: a view from mutations in mice. Annu Rev Neurosci. 1978;1:297–326. doi: 10.1146/annurev.ne.01.030178.001501. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Curran T. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J Neurosci. 1997;17:23–31. doi: 10.1523/JNEUROSCI.17-01-00023.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–479. doi: 10.1016/S0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- de Bergeyck V, Nakajima K, Lambert de Rouvroit C, Naerhuyzen B, Goffinet AM, Miyata T, Ogawa M, Mikoshiba K. A truncated Reelin protein is produced but not secreted in the ‘Orleans’ reeler mutation (Relnrl-Orl) Brain Res Mol Brain Res. 1997;50:85–90. doi: 10.1016/S0169-328X(97)00166-6. [DOI] [PubMed] [Google Scholar]
- Derer P, Nakanishi S. Extracellular matrix distribution during neocortical wall ontogenesis in “normal” and “Reeler” mice. J Hirnforsch. 1983;24:209–224. [PubMed] [Google Scholar]
- Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, Anton ES. Reelin binds alpha3beta1 integrin and inhibits neuronal migration. Neuron. 2000;27:33–44. doi: 10.1016/S0896-6273(00)00007-6. [DOI] [PubMed] [Google Scholar]
- Falconer DS. Two new mutants, ‘trembler’ and ‘reeler’, with neurological actions in the house mouse (Mus musculus L.) J Genet. 1951;50:192–201. doi: 10.1007/BF02996215. [DOI] [PubMed] [Google Scholar]
- Frotscher M. Dual role of Cajal-Retzius cells and reelin in cortical development. Cell Tissue Res. 1997;290:315–322. doi: 10.1007/s004410050936. [DOI] [PubMed] [Google Scholar]
- Goodman JH, Wasterlain CG, Massarweh WF, Dean E, Sollas AL, Sloviter RS. Calbindin-D28k immunoreactivity and selective vulnerability to ischemia in the dentate gyrus of the developing rat. Brain Res. 1993;606:309–314. doi: 10.1016/0006-8993(93)90999-4. [DOI] [PubMed] [Google Scholar]
- Ha S, Stottmann RW, Furley AJ, Beier DR. A forward genetic screen in mice identifies mutants with abnormal cortical patterning. Cereb Cortex. 2015;25:167–179. doi: 10.1093/cercor/bht209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack I, Hellwig S, Junghans D, Brunne B, Bock HH, Zhao S, Frotscher M. Divergent roles of ApoER2 and Vldlr in the migration of cortical neurons. Development. 2007;134:3883–3891. doi: 10.1242/dev.005447. [DOI] [PubMed] [Google Scholar]
- Hamburgh M. Observations on the neuropathology of “reeler”, a neurological mutation in mice. Experimentia. 1960;16:460–461. doi: 10.1007/BF02171154. [DOI] [Google Scholar]
- Herrick TM, Cooper JA. A hypomorphic allele of dab1 reveals regional differences in reelin-Dab1 signaling during brain development. Development. 2002;129:787–796. doi: 10.1242/dev.129.3.787. [DOI] [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/S0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- Hirotsune S, Takahara T, Sasaki N, Hirose K, Yoshiki A, Ohashi T, Kusakabe M, Murakami Y, Muramatsu M, Watanabe S. The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat Genet. 1995;10:77–83. doi: 10.1038/ng0595-77. [DOI] [PubMed] [Google Scholar]
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND, Walsh CA. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93–96. doi: 10.1038/79246. [DOI] [PubMed] [Google Scholar]
- Howell BW, Gertler FB, Cooper JA. Mouse disabled (mDab1): a Src binding protein implicated in neuronal development. EMBO J. 1997;16:121–132. doi: 10.1093/emboj/16.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Cooper JA. Reelin-induced tyrosine phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 1999a;13:643–648. doi: 10.1101/gad.13.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Lanier LM, Frank R, Gertler FB, Cooper JA. The disabled 1 phosphotyrosine-binding domain binds to the internalization signals of transmembrane glycoproteins and to phospholipids. Mol Cell Biol. 1999b;19:5179–5188. doi: 10.1128/MCB.19.7.5179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BW, Herrick TM, Hildebrand JD, Zhang Y, Cooper JA. Dab1 tyrosine phosphorylation sites relay positional signals during mouse brain development. Curr Biol. 2000;10:877–885. doi: 10.1016/S0960-9822(00)00608-4. [DOI] [PubMed] [Google Scholar]
- Ignatova N, Sindic CJ, Goffinet AM. Characterization of the various forms of the Reelin protein in the cerebrospinal fluid of normal subjects and in neurological diseases. Neurobiol Dis. 2004;15:326–330. doi: 10.1016/j.nbd.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Jossin Y, Bar I, Ignatova N, Tissir F, De Rouvroit CL, Goffinet AM. The reelin signaling pathway: some recent developments. Cereb Cortex. 2003;13:627–633. doi: 10.1093/cercor/13.6.627. [DOI] [PubMed] [Google Scholar]
- Jossin Y, Ignatova N, Hiesberger T, Herz J, Lambert de Rouvroit C, Goffinet AM. The central fragment of Reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. J Neurosci. 2004;24:514–521. doi: 10.1523/JNEUROSCI.3408-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerjan G, Gleeson JG. A missed exit: Reelin sets in motion Dab1 polyubiquitination to put the break on neuronal migration. Genes Dev. 2007;21:2850–2854. doi: 10.1101/gad.1622907. [DOI] [PubMed] [Google Scholar]
- Kohno T, Nakano Y, Kitoh N, Yagi H, Kato K, Baba A, Hattori M. C-terminal region-dependent change of antibody-binding to the eighth Reelin repeat reflects the signaling activity of Reelin. J Neurosci Res. 2009;87:3043–3053. doi: 10.1002/jnr.22143. [DOI] [PubMed] [Google Scholar]
- Kohno T, Honda T, Kubo K, Nakano Y, Tsuchiya A, Murakami T, Banno H, Nakajima K, Hattori M. Importance of reelin C-terminal region in the development and maintenance of the postnatal cerebral cortex and its regulation by specific proteolysis. J Neurosci. 2015;35:4776–4787. doi: 10.1523/JNEUROSCI.4119-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo K, Mikoshiba K, Nakajima K. Secreted Reelin molecules form homodimers. Neurosci Res. 2002;43:381–388. doi: 10.1016/S0168-0102(02)00068-8. [DOI] [PubMed] [Google Scholar]
- Kuo G, Arnaud L, Kronstad-O'Brien P, Cooper JA. Absence of Fyn and Src causes a Reeler-like phenotype. J Neurosci. 2005;25:8578–8586. doi: 10.1523/JNEUROSCI.1656-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert de Rouvroit C, de Bergeyck V, Cortvrindt C, Bar I, Eeckhout Y, Goffinet AM. Reelin, the extracellular matrix protein deficient in reeler mutant mice, is processed by a metalloproteinase. Exp Neurol. 1999a;156:214–217. doi: 10.1006/exnr.1998.7007. [DOI] [PubMed] [Google Scholar]
- Lambert de Rouvroit C, Bernier B, Royaux I, de Bergeyck V, Goffinet AM. Evolutionarily conserved, alternative splicing of Reelin during brain development. Exp Neurol. 1999b;156:229–238. doi: 10.1006/exnr.1999.7019. [DOI] [PubMed] [Google Scholar]
- Mihalas AB, Elsen GE, Bedogni F, Daza RA, Ramos-Laguna KA, Arnold SJ, Hevner RF. Intermediate progenitor cohorts differentially generate cortical layers and require Tbr2 for timely acquisition of neuronal subtype identity. Cell Rep. 2016;16:92–105. doi: 10.1016/j.celrep.2016.05.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Nakajima K, Mikoshiba K, Ogawa M. Regulation of Purkinje cell alignment by Reelin as revealed with CR-50 antibody. J Neurosci. 1997;17:3599–3609. doi: 10.1523/JNEUROSCI.17-10-03599.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moheb LA, Tzschach A, Garshasbi M, Kahrizi K, Darvish H, Heshmati Y, Kordi A, Najmabadi H, Ropers HH, Kuss AW. Identification of a nonsense mutation in the very low-density lipoprotein receptor gene (VLDLR) in an Iranian family with dysequilibrium syndrome. Eur J Hum Genet. 2008;16:270–273. doi: 10.1038/sj.ejhg.5201967. [DOI] [PubMed] [Google Scholar]
- Nakajima K, Mikoshiba K, Miyata T, Kudo C, Ogawa M. Disruption of hippocampal development in vivo by CR-50 mAb against reelin. Proc Natl Acad Sci U S A. 1997;94:8196–8201. doi: 10.1073/pnas.94.15.8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Beppu M, Sakai K, Yagyu H, Matsumaru S, Kohno T, Hattori M. The C-terminal region of Reelin is necessary for proper positioning of a subset of Purkinje cells in the postnatal cerebellum. Neuroscience. 2016;336:20–29. doi: 10.1016/j.neuroscience.2016.08.039. [DOI] [PubMed] [Google Scholar]
- Nakanishi S. Extracellular matrix during laminar pattern formation of neocortex in normal and reeler mutant mice. Dev Biol. 1983;95:305–316. doi: 10.1016/0012-1606(83)90031-3. [DOI] [PubMed] [Google Scholar]
- Nakano Y, Kohno T, Hibi T, Kohno S, Baba A, Mikoshiba K, Nakajima K, Hattori M. The extremely conserved C-terminal region of Reelin is not necessary for secretion but is required for efficient activation of downstream signaling. J Biol Chem. 2007;282:20544–20552. doi: 10.1074/jbc.M702300200. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Miyata T, Nakajima K, Yagyu K, Seike M, Ikenaka K, Yamamoto H, Mikoshiba K. The reeler gene-associated antigen on Cajal-Retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron. 1995;14:899–912. doi: 10.1016/0896-6273(95)90329-1. [DOI] [PubMed] [Google Scholar]
- Olson EC, Kim S, Walsh CA. Impaired neuronal positioning and dendritogenesis in the neocortex after cell-autonomous Dab1 suppression. J Neurosci. 2006;26:1767–1775. doi: 10.1523/JNEUROSCI.3000-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcelik T, Akarsu N, Uz E, Caglayan S, Gulsuner S, Onat OE, Tan M, Tan U. Mutations in the very low-density lipoprotein receptor VLDLR cause cerebellar hypoplasia and quadrupedal locomotion in humans. Proc Natl Acad Sci U S A. 2008;105:4232–4236. doi: 10.1073/pnas.0710010105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garcia CG, Tissir F, Goffinet AM, Meyer G. Reelin receptors in developing laminated brain structures of mouse and human. Eur J Neurosci. 2004;20:2827–2832. doi: 10.1111/j.1460-9568.2004.03733.x. [DOI] [PubMed] [Google Scholar]
- Pesold C, Impagnatiello F, Pisu MG, Uzunov DP, Costa E, Guidotti A, Caruncho HJ. Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc Natl Acad Sci U S A. 1998;95:3221–3226. doi: 10.1073/pnas.95.6.3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DS, Curran T. Role of the reelin signaling pathway in central nervous system development. Annu Rev Neurosci. 2001;24:1005–1039. doi: 10.1146/annurev.neuro.24.1.1005. [DOI] [PubMed] [Google Scholar]
- Rice DS, Sheldon M, D'Arcangelo G, Nakajima K, Goldowitz D, Curran T. Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development. 1998;125:3719–3729. doi: 10.1242/dev.125.18.3719. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Bernier B, Goffinet AM. Reelin mRNA expression during mouse brain development. Eur J Neurosci. 1997;9:1055–1071. doi: 10.1111/j.1460-9568.1997.tb01456.x. [DOI] [PubMed] [Google Scholar]
- Sheppard AM, Pearlman AL. Abnormal reorganization of preplate neurons and their associated extracellular matrix: an early manifestation of altered neocortical development in the reeler mutant mouse. J Comp Neurol. 1997;378:173–179. doi: 10.1002/(SICI)1096-9861(19970210)378:2%3C173::AID-CNE2%3E3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Sheppard AM, Hamilton SK, Pearlman AL. Changes in the distribution of extracellular matrix components accompany early morphogenetic events of mammalian cortical development. J Neurosci. 1991;11:3928–3942. doi: 10.1523/JNEUROSCI.11-12-03928.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano E, Del Río JA, Martínez A, Supèr H. Organization of the embryonic and early postnatal murine hippocampus. I. Immunocytochemical characterization of neuronal populations in the subplate and marginal zone. J Comp Neurol. 1994;342:571–595. doi: 10.1002/cne.903420406. [DOI] [PubMed] [Google Scholar]
- Stanfield BB, Cowan WM. The morphology of the hippocampus and dentate gyrus in normal and reeler mice. J Comp Neurol. 1979;185:393–422. doi: 10.1002/cne.901850302. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Borg JP, Margolis B, Herz J. Interaction of cytosolic adaptor proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J Biol Chem. 1998;273:33556–33560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/S0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- Türkmen S, Hoffmann K, Demirhan O, Aruoba D, Humphrey N, Mundlos S. Cerebellar hypoplasia, with quadrupedal locomotion, caused by mutations in the very low-density lipoprotein receptor gene. Eur J Hum Genet. 2008;16:1070–1074. doi: 10.1038/ejhg.2008.73. [DOI] [PubMed] [Google Scholar]
- Utsunomiya-Tate N, Kubo K, Tate S, Kainosho M, Katayama E, Nakajima K, Mikoshiba K. Reelin molecules assemble together to form a large protein complex, which is inhibited by the function-blocking CR-50 antibody. Proc Natl Acad Sci U S A. 2000;97:9729–9734. doi: 10.1073/pnas.160272497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Frotscher M. Go or stop? Divergent roles of Reelin in radial neuronal migration. Neuroscientist. 2010;16:421–434. doi: 10.1177/1073858410367521. [DOI] [PubMed] [Google Scholar]