

Abstract
Addictive drugs usurp neural plasticity mechanisms that normally serve reward-related learning and memory, primarily by evoking changes in glutamatergic synaptic strength in the mesocorticolimbic dopamine circuitry. Here, we show that repeated cocaine exposure in vivo does not alter synaptic strength in the mouse prefrontal cortex during an early period of withdrawal, but instead modifies a Hebbian quantitative synaptic learning rule by broadening the temporal window and lowers the induction threshold for spike-timing-dependent LTP (t-LTP). After repeated, but not single, daily cocaine injections, t-LTP in layer V pyramidal neurons is induced at +30 ms, a normally ineffective timing interval for t-LTP induction in saline-exposed mice. This cocaine-induced, extended-timing t-LTP lasts for ∼1 week after terminating cocaine and is accompanied by an increased susceptibility to potentiation by fewer pre–post spike pairs, indicating a reduced t-LTP induction threshold. Basal synaptic strength and the maximal attainable t-LTP magnitude remain unchanged after cocaine exposure. We further show that the cocaine facilitation of t-LTP induction is caused by sensitized D1-cAMP/protein kinase A dopamine signaling in pyramidal neurons, which then pathologically recruits voltage-gated l-type Ca2+ channels that synergize with GluN2A-containing NMDA receptors to drive t-LTP at extended timing. Our results illustrate a mechanism by which cocaine, acting on a key neuromodulation pathway, modifies the coincidence detection window during Hebbian plasticity to facilitate associative synaptic potentiation in prefrontal excitatory circuits. By modifying rules that govern activity-dependent synaptic plasticity, addictive drugs can derail the experience-driven neural circuit remodeling process important for executive control of reward and addiction.
SIGNIFICANCE STATEMENT It is believed that addictive drugs often render an addict's brain reward system hypersensitive, leaving the individual more susceptible to relapse. We found that repeated cocaine exposure alters a Hebbian associative synaptic learning rule that governs activity-dependent synaptic plasticity in the mouse prefrontal cortex, characterized by a broader temporal window and a lower threshold for spike-timing-dependent LTP (t-LTP), a cellular form of learning and memory. This rule change is caused by cocaine-exacerbated D1-cAMP/protein kinase A dopamine signaling in pyramidal neurons that in turn pathologically recruits l-type Ca2+ channels to facilitate coincidence detection during t-LTP induction. Our study provides novel insights on how cocaine, even with only brief exposure, may prime neural circuits for subsequent experience-dependent remodeling that may underlie certain addictive behavior.
Keywords: addiction, cocaine, dopamine, prefrontal cortex, reward, spike timing-dependent plasticity
Introduction
Drugs of abuse are believed to hijack neural plasticity mechanisms that normally serve reward-related learning and memory in the mesocorticolimbic dopamine (DA) circuits (Nestler, 2001; Hyman et al., 2006). The prefrontal cortex (PFC) receives DA innervations from the ventral tegmental area (VTA), reciprocally connects with both the VTA and nucleus accumbens (NAc), and is a key component of the mesocorticolimbic circuits regulating executive function, memory, motivation, and reward (Hyman et al., 2006; Koob and Volkow, 2010; Kolb et al., 2012). PFC is particularly implicated in executive control of drug seeking, craving, and relapse because it is activated both in human addicts during craving (Childress et al., 1999) and in animals when drug-associated cues are presented (Rebec and Sun, 2005). Restoring cocaine-impaired PFC activity prevents compulsive cocaine seeking in rats (Chen et al., 2013). A current view considers the PFC a “brake” in executive control of motivation and choice, alterations to which are proposed to cause overpowering motivational strength and decreased ability to control the desire to obtain drugs (Kalivas et al., 2005).
A predominant hypothesis posits that drug experience evokes lasting changes in synaptic strength in mesocorticolimbic circuits (Hyman et al., 2006; Lüscher and Malenka, 2011). Extensive studies have established that exposure to cocaine, even acutely, causes persistent alterations on the strength of glutamatergic synapses in the VTA and NAc (Lüscher and Malenka, 2011). Limited studies have examined drug-evoked synaptic plasticity in the PFC (Huang et al., 2007; Lu et al., 2009; Lu et al., 2010; Kasanetz et al., 2013) and results suggest a lack of change in the efficacy of glutamatergic synapses onto layer V (L5) pyramidal neurons, the primary output neurons in the PFC, after short-term noncontingent or contingent cocaine exposure in rats; however, GABAergic transmission onto these neurons is suppressed. In comparison, more prolonged cocaine self-administration causes delayed glutamatergic synaptic potentiation in L5 PFC neurons in “addict-like” rats (Kasanetz et al., 2013). Therefore, drug-induced circuit remodeling in the PFC may occur at some later stages of addiction.
We hypothesize that, rather than modifying synaptic efficacy directly, drugs may disrupt the rules that govern activity-dependent plasticity, especially at early stages after drug exposure. We evaluated this hypothesis in the PFC by analyzing spike timing-dependent plasticity (STDP), a Hebbian associative synaptic plasticity that may underlie certain forms of learning and cognitive processes (Magee and Johnston, 1997; Markram et al., 1997; Bi and Poo, 1998; Caporale and Dan, 2008; Feldman, 2012). STDP is unique in that it permits key quantitative plasticity rules to be investigated, most notably the spike-timing window, which predicts the associability between presynaptic and postsynaptic stimuli during STDP induction and induction threshold. Using cocaine-induced locomotor sensitization in mice, a behavioral paradigm often used to investigate early drug-induced neuroadaptations in addiction (Wolf, 1998; Nestler, 2001; Hyman et al., 2006), we found that repeated cocaine exposure extends the t-LTP timing window markedly and reduces t-LTP induction threshold through an abnormally recruited D1-cAMP/protein kinase A (PKA)–l-type Ca2+ channel (LTCC) pathway in PFC pyramidal neurons. A lower t-LTP induction threshold shortly after drug discontinuation may prime PFC synapses for potentiation by future cue-associated activity, leading to remodeling of PFC circuits related to addiction.
Materials and Methods
Experiments.
All experiments involving animals were approved by Harvard Medical School or SUNY Upstate Medical University institutional animal care and use committees.
Electrophysiology.
Male or female C57BL/6J mice (RRID:IMSR_JAX:000664) at postnatal day 21–30 were randomly assigned into cocaine or saline groups. Saline (0.9% NaCl) or cocaine (2 mg/ml in saline) was administered to mice via intraperitoneal injections once per day for 7 consecutive days, during which time the mice were kept in their home cages without changing the bedding. After the last injection, mice were allowed 1, 7, or 14 d of rest (withdrawal) before they were killed for electrophysiological assessments. In Figure 1 and as indicated, some mice received only one cocaine or saline injection and were used the following day for electrophysiology. In a few experiments shown in Figures 4 and 5, mice were rested for 2 d before electrophysiology, but the results were indistinguishable from those killed after 1 d of rest.
Figure 1.
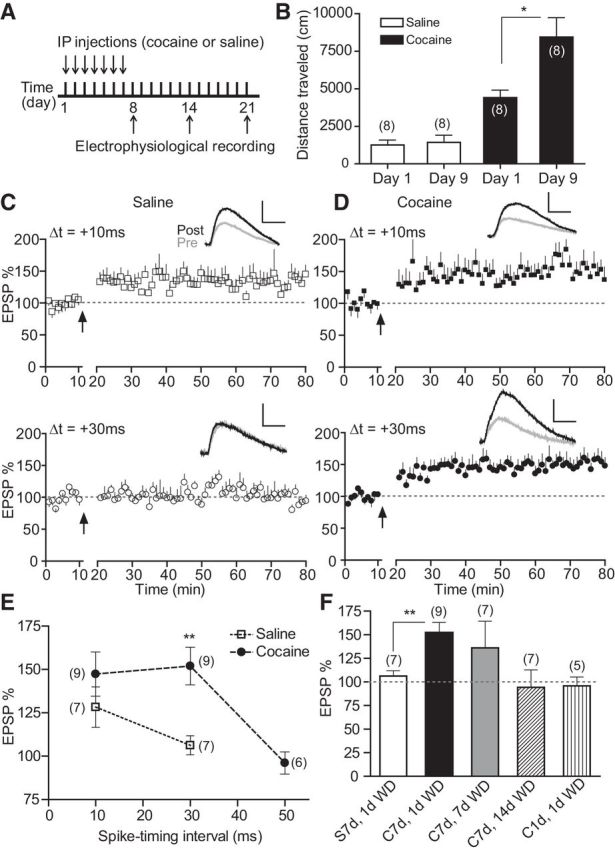
Repeat, but not single, cocaine exposure extends t-LTP timing window during an early period of withdrawal. A, Schematic of repeated cocaine or saline injection protocol. B, Cocaine-induced locomotor sensitization. Total distance traveled during a 30 min period after saline or cocaine challenge on day 1 and day 9 was measured. C, t-LTP induction by 60 EPSP–AP pairs at Δt = +10 ms, but not +30 ms, in slices from saline-treated mice. D, t-LTP induction by 60 EPSP–AP pairs at both +10 ms and +30 ms in slices from repeated cocaine-treated mice. E, Summary of t-LTP induction at different Δt's in repeated saline and cocaine conditions. F, Summary of t-LTP induction at +30 ms under different treatment and withdrawal conditions. S, Saline; C, cocaine; WD, withdrawal. Insets, Representative EPSPs 5 min before and 30 min after t-LTP induction. Scale bars, 2 mV, 20 ms. *p < 0.05, **p < 0.01, unpaired two-tailed t tests. Arrows indicate the start of t-LTP induction. Values in parentheses indicate number of cells recorded from at least three mice in each group.
Figure 4.
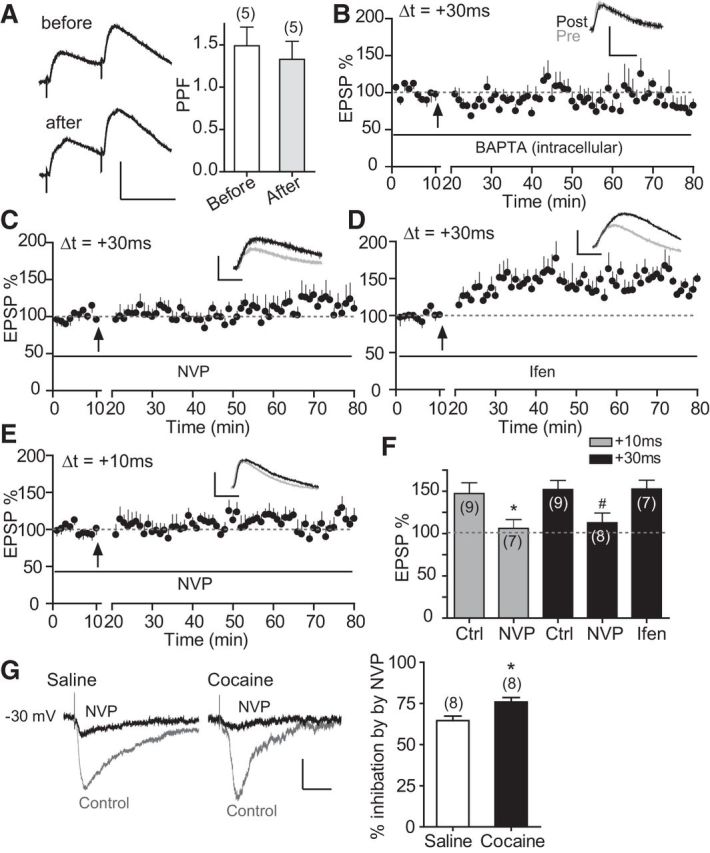
Cocaine-induced extended-timing t-LTP is expressed postsynaptically and depends on GluN2A-NMDARs. A, PPF before and 30 min after t-LTP induction (60 EPSP–AP pairs, 0.1 Hz, +30 ms). Left, Representative traces with a 50 ms interpulse interval. Scale bars, 2 mV, 50 ms. Right, Summary graph. B, Effect of intracellularly loaded BAPTA (15 mm) on t-LTP induction at +30 ms. C, D, Effects of bath-applied NVP-AAM077 (NVP, 0.4 μm, C) and ifenprodil (Ifen, 3 μm, D) on t-LTP induction at +30 ms. E, Effects of bath-applied NVP-AAM077 (0.4 μm) on t-LTP induction at +10 ms. Scale bars (B–E), 2 mV, 20 ms. Drugs were applied to the extracellular bath during EPSP–AP pairings. F, t-LTP summary. *p < 0.05 versus +10 ms-Ctrl, unpaired two-tailed t test; #p < 0.05 versus +30 ms Ctrl, one-way ANOVA with post hoc Dunnett's test. All experiments in A–F were conducted in slices prepared from repeated cocaine-treated mice. Ctrl t-LTP at +10 and +30 ms in F are replotted from Figure 1 for direct comparisons. G, Repeated cocaine exposure enhances NVP-AAM077-sensitive, GluN2A-mediated NMDAR current. Left, Representative EPSCNMDA recorded at −30 mV in the absence followed by the presence of NVP-AAM077 (0.4 μm) from mice exposed to either saline or cocaine. Right, Summary of NVP-AAM077 inhibition of EPSCNMDA. NVP-AAM077 was applied to the bath to block GluN2A-mediated component after 10–15 min recording of baseline total EPSCNMDA. *p < 0.05, Student's t test. Scale bars, 50 pA, 50 ms.
Figure 5.
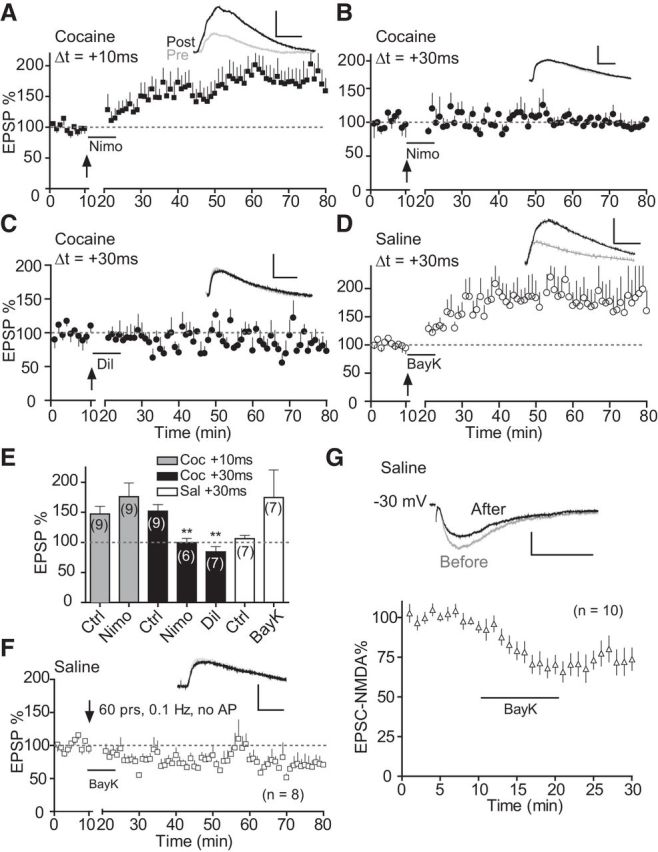
LTCCs regulate t-LTP induction selectively at +30 ms, but not +10 ms, in repeated cocaine-treated mice. A, B, Effects of nimodipine (Nimo, 10 μm) on t-LTP induction at +10 ms (A) and +30 ms (B) in cocaine (Coc) slices. C, Effect of diltiazem (Dil, 100 μm) on t-LTP induction at +30 ms in cocaine slices. D, Enabling effect of t-LTP at +30 ms in saline slices by Bay K8644 (BayK, 3 μm). E, t-LTP summary. Drugs were applied to the extracellular bath during EPSP–AP pairings. **p < 0.01 versus Coc +30 ms Ctrl, one-way ANOVA with post hoc Dunnett's test. Ctrl data in E are replotted from Figure 1 for direct comparisons. F, Lack of effect of Bay K8644 on basal EPSP in saline mice. Bay K8644 was applied during 60 EPSPs (0.1 Hz) without AP pairings. Scale bars (A–D, F), 2 mV, 20 ms. G, Effect of Bay K8644 on isolated EPSCNMDA. Bay K8644 was applied at the 10 min mark for 10 min after a stable baseline was obtained. Top, Representative EPSCNMDA before and after Bay K8644 application. Scale bars, 50 pA, 100 ms.
Acute medial PFC (mPFC) slices were prepared as described previously (Xu et al., 2009; Xu and Yao, 2010; Ruan et al., 2014) with minimal modifications. Briefly, brains were quickly removed into ice-cold artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 2.5 KCl, 2.5 CaCl2, 1.2 MgCl2, 25 NaHCO3, 1.2 NaH2PO4, and 11 d-glucose preoxygenated by bubbling with 95% O2 and 5% CO2. Cooled brains were mounted on a Leica S1200 vibratome and ∼300 μm slices containing the mPFC were cut. Slices were transferred to an incubator (Warner Instruments) and allowed to recover at 32°C for ∼30 min, followed by at least another 1 h of incubation at room temperature (21–23°C) before electrophysiology recording.
After incubation, slices were transferred to a recording chamber (Warner Instruments) mounted onto an Olympus BX51WI upright microscope. The chamber was perfused with continuously oxygenated ACSF maintained at 32°C with an automatic temperature controller (Warner Instruments). The GABAA receptor blocker picrotoxin (100 μm) was included in ACSF to block the GABAergic inhibitory transmission. Individual L5 pyramidal neurons were identified visually with DIC microscopy. Glass electrodes with a final resistance of ∼5–8 MΩ were prepared with a P-87 micropipette puller (Sutter Instrument). For current clamping, electrodes were filled with the following (in mm): 130 K-gluconate, 8 NaCl, 10 HEPES, 0.4 EGTA, 2 Mg-ATP, and 0.25 GTP-Tris, pH 7.25. For voltage clamping, electrodes were filled with the following (in mm): 142 Cs-gluconate, 8 NaCl, 10 HEPES, 0.4 EGTA, 2.5 QX-314, 2 Mg-ATP, and 0.25 GTP-Tris, pH 7.25 with CsOH. The BAPTA intracellular solution contained the following (in mm): 15 BAPTA, 115 K-gluconate, 8 NaCl, 10 HEPES, 2 Mg-ATP, and 0.25 GTP-Tris, pH 7.25 with KOH.
Whole-cell patch-clamp recordings were performed using an Axoclamp 2B or a Multiclamp 700B amplifier (Molecular Devices). Presynaptic stimuli (0.033 Hz, 200 μs) were delivered at layer II/III with a concentric electrode (FHC) to evoke EPSPs or currents (EPSCs). In t-LTP experiments, cells were held at their resting membrane potentials (∼−67 mV; Xu and Yao, 2010) under current-clamp conditions. Postsynaptic action potentials (APs) were evoked by current injection to the soma through the patch electrode. The STDP protocol consisted of repetitive pairing at 0.1 Hz of a single EPSP followed by a single AP at a specific time interval (Δt) between the onset of presynaptic stimulation and the postsynaptic AP. Input resistance was monitored throughout the experiment from the voltage response to a −200 pA hyperpolarizing current. LTP was measured as the percentage of the average EPSP between 50 and 60 min after induction to the average EPSP during a 10 min baseline recording. In AP firing experiments, AP threshold was determined by step current injection (500 ms, 10 pA increments) to the soma through the patch electrode.
In voltage-clamp experiments, miniature EPSCs (mEPSCs) were recorded at −60 mV with TTX (1 μm) and APV (50 μm) present in the bath to block voltage-gated sodium channels and NMDARs, respectively. Two methods were used to measure NMDA/AMPA ratios. First, evoked EPSCs were recorded at −60 mV and then at +40 mV. After averaging the EPSCs at each potential, the NMDA/AMPA ratio was measured as the amplitude of the NMDAR component 80 ms after stimulation at +40 mV divided by the peak AMPAR component at −60 mV. Second, EPSCs were recorded at −30 mV in the absence (to derive total EPSC) and then the presence (to derive EPSCNMDA) of CNQX, followed by subtracting the EPSCNMDA from the total EPSC to obtain EPSCAMPA. The NMDA/AMPA ratio was then calculated using the peaks of EPSCNMDA and EPSCAMPA. In experiments assessing the potential effect of Bay K8644 on EPSCNMDA, glycine was included in the bath to boost the NMDA currents. To estimate relative contributions of GluN2A and GluN2B subunits to total EPSCNMDA, EPSCs were first recorded at −30 mV in the presence of CNQX and, after obtaining a stable EPSCNMDA baseline, NVP-AAM077 was added to record the remaining EPSCs (mainly mediated by the GluN2B-dependent EPSCNMDA). Input resistance in voltage-clamp experiments was monitored throughout the experiment to a −5 mV hyperpolorization.
Signals were filtered at 1 kHz, digitized at 10 kHz, and analyzed with pClamp version 10.2 (Molecular Devices) or Mini Analysis version 6 (Synaptosoft) software.
Locomotor assessment.
Age-matched adult male mice were assessed in an open-field activity chamber (Med Associates) and total distances traveled were measured in 5 min intervals. Mice were first placed in the chamber and habituated for 30 min. Each of the mice was then taken out of the chamber and received an intraperitoneal dose of saline or cocaine injection and their locomotor activities were measured for up to 60 min.
Drugs.
The following drugs were used: APV (Sigma-Aldrich), BAPTA (Sigma-Aldrich), Bay K8644 (Sigma-Aldrich), CNQX (Sigma-Aldrich), cocaine (Tocris Bioscience), diltiazem (Sigma-Aldrich), d-serine (Tocris Bioscience), ifenprodil (Sigma-Aldrich), nimodipine (Sigma-Aldrich), NVP-AAM077 (Novartis), picrotoxin (Sigma-Aldrich), protein kinase inhibitor (PKI) (6–22; PKA inhibitor 6–22 amide; Calbiochem), QX-314 (Sigma-Aldrich), RP-cAMPS (Sigma-Aldrich), SCH23390 (Tocris Bioscience), SKF81297 (Sigma-Aldrich), and tetrodotoxin (Sigma-Aldrich). Picrotoxin was dissolved directly in the ACSF used for perfusion. Nimodipine and diltiazem were dissolved in DMSO with a 1:1000 ratio as stock solutions, which were added directly to ASCF to a desired final concentration during experiments. All other drugs were dissolved in ddH2O. Drugs were delivered to the recording chamber with a gravity-driven system (Harvard Apparatus).
Data are expressed as mean ± SEM. Statistical comparisons were made with two-tailed t tests or ANOVA with appropriate post hoc tests.
Results
Repeated cocaine exposure extends t-LTP timing window within a 7 d withdrawal period
We examined t-LTP induction over a 50 ms pre–post timing window in L5 pyramidal cells in mPFC slices prepared from cocaine- or saline-treated mice. These neurons receive inputs from corticocortical, thalamocortical, and hippocampal pathways and project to other brain areas, exerting top-down control of behavior (Miller and Cohen, 2001). Mice were injected daily with cocaine (20 mg/kg, i.p.) or saline for 7 consecutive days and killed ∼24 h later for whole-cell patch-clamp analysis (Fig. 1A). As expected, this treatment resulted in a significant enhancement of locomotor activity in response to the same cocaine challenge (Fig. 1B), a characteristic of behavioral sensitization.
We recorded AMPA receptor (AMPAR)- and NMDA receptor (NMDAR)-mediated EPSPs, with GABAergic transmission blocked by picrotoxin, from visually identified neurons at their resting membrane potential (∼−67 mV) (Xu and Yao, 2010) while stimulating layer II/III. To induce t-LTP, we used an STDP protocol that involves pairing a presynaptically elicited EPSP and a postsynaptic AP at 0.1 Hz for 10 min (60 pairs) with a variable Δt (Xu and Yao, 2010; Ruan et al., 2014). This pre–post pairing protocol produced sustained increases in EPSP amplitude at Δt = +10 ms, but not +30 ms (Fig. 1C), in slices from saline-treated mice, consistent with the narrow t-LTP timing window (∼+10 ms) at these PFC synapses (Xu and Yao, 2010; Ruan et al., 2014). In comparison, the same protocol induced robust potentiation at both +10 ms and +30 ms in slices from cocaine-treated mice (Fig. 1D). At a more extended timing interval (+50 ms), this protocol was ineffective at inducing t-LTP, even in cocaine-injected mice (Fig. 1E; cf. Fig. 6E). These results indicate that the t-LTP timing window, an index of associability of pre–post coincident stimuli, is broadened significantly after repeated in vivo cocaine exposure.
Figure 6.
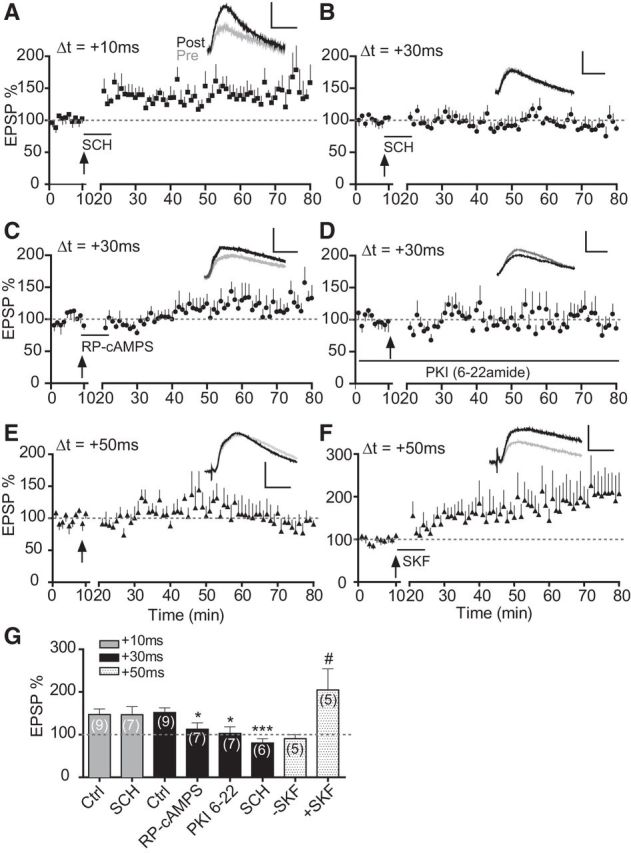
Excessive D1-cAMP/PKA signaling maintains the extended-timing t-LTP in cocaine mice. A, Effect, or lack thereof, of SCH23390 (SCH, 10 μm) on t-LTP induction at +10 ms in cocaine slices. B, Blocking effect of SCH23390 (SCH, 10 μm) on t-LTP at +30 ms in cocaine slices. C, Effect of bath-applied RP-cAMPS (50 μm) on t-LTP induction at +30 ms in cocaine slices. D, Lack of t-LTP induction at +30 ms in cocaine slices when PKI 6–22 (20 μm) was loaded in the patch pipette. E, Lack of t-LTP induction at +50 ms in slices prepared from cocaine mice. F, SKF81297 (SKF, 2 μm) enabled t-LTP induction at +50 ms in cocaine slices. All drugs (except PKI 6–22) were applied to the extracellular bath during EPSP–AP pairings. Scale bars, 2 mV, 20 ms. *p < 0.05, ***p < 0.001 versus Ctrl (+30 ms), unpaired two-tailed t tests; #p < 0.05 vs SKF, unpaired t test with Welch's correction. Ctrl data in G are replotted from Figure 1 for direct comparisons.
We further characterized the emergence and persistence of this cocaine-induced t-LTP timing window broadening. Mice injected with a single dose of cocaine and tested ∼24 h later did not exhibit t-LTP at +30 ms (Fig. 1F), suggesting that repeated treatments were necessary for the t-LTP timing window extension. Seven days after terminating repeated cocaine injections, t-LTP at +30 ms could be induced in less than half (3/7) of the cells examined and the mean difference did not reach significance (p = 0.09). On day 14, t-LTP was not inducible in any slices examined. These data indicate that the cocaine-induced t-LTP timing window extension lasted for ∼7 d. Together, our data demonstrate that repeated, but not single, cocaine exposure modifies a quantitative synaptic learning rule in PFC synapses during an early period of withdrawal.
Repeated cocaine exposure lowers t-LTP induction threshold without altering maximal LTP magnitude
The STDP temporal window is thought to reflect the LTP/LTD threshold directly; the smaller the window, the higher the threshold (Zhang et al., 2009). Therefore, we hypothesized that cocaine exposure reduces t-LTP threshold and that fewer spiking pairs might be needed to induce t-LTP in cocaine mice than in saline mice. Indeed, we found that 50 EPSP–AP pairs (0.1 Hz; 10 ms) induced t-LTP in slices from cocaine but not saline mice (Fig. 2A,D). Decreasing the spike pair number to 40 failed to induce t-LTP even in cocaine mice (Fig. 2C) while increasing the spike pairs to 60 or 70 successfully induced t-LTP in both saline and cocaine mice (Fig. 2B,C). Importantly, the maximal magnitude of potentiation was similar between saline and cocaine slices (p > 0.1; Fig. 2C). Therefore, repeat cocaine exposure made PFC neurons more sensitive to paired pre–post stimuli in t-LTP induction.
Figure 2.
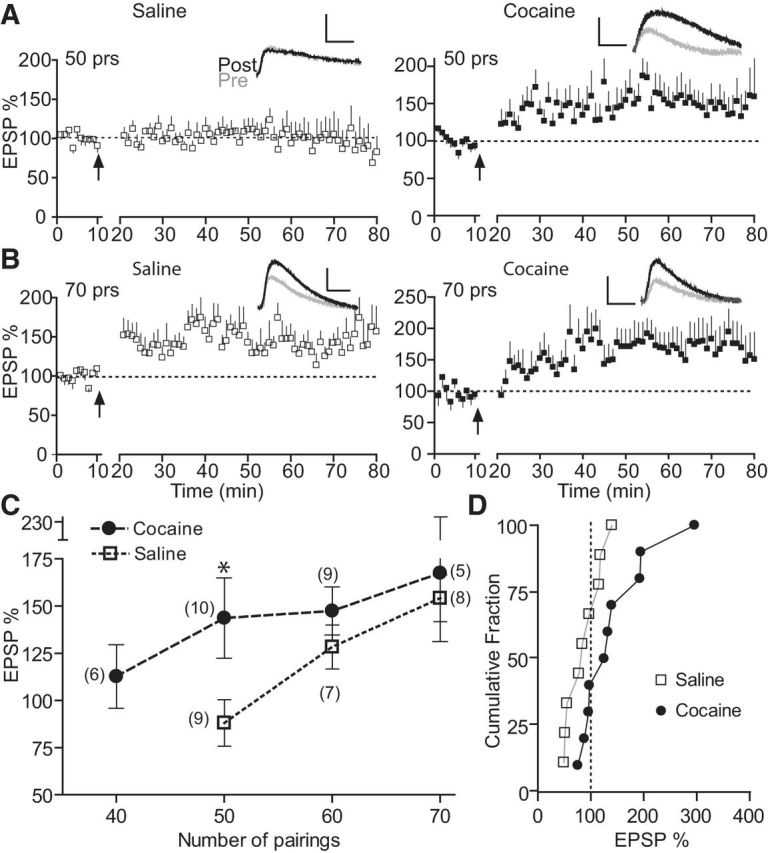
Repeated cocaine exposure lowers t-LTP induction threshold. A, t-LTP induction by 50 EPSP–AP pairs in cocaine- but not saline-treated mice. B, t-LTP induction by 70 EPSP–AP pairs in both cocaine- and saline-treated mice. C, Summary of t-LTP induction by various numbers of EPSP–AP pairs. D, Cumulative distribution of t-LTP magnitude by 50 EPSP–AP pairs from A and C in individual cells in cocaine and saline mice. EPSP–AP pairs were delivered at Δt = +10 ms, 0.1 Hz. Scale bars, 2 mV, 20 ms. *p < 0.05 unpaired two-tailed t test with Welch's correction.
Repeated cocaine exposure does not alter synaptic strength or postsynaptic AP waveform
We next investigated whether repeated cocaine exposure alters PFC synaptic efficacy, a hallmark in multiple other regions of reward circuits (Lüscher and Malenka, 2011). We found no difference in either the amplitude or the frequency of AMPAR-mediated mEPSCs between cocaine and saline mice (Fig. 3A,B), suggesting unaltered presynaptic transmitter release and postsynaptic AMPAR responsiveness. Similarly, the ratio of AMPAR- to NMDAR-mediated EPSCs (AMPA/NMDA ratio), measured by either a kinetically (Fig. 3C) or pharmacologically (Fig. 3D) based method, was indistinguishable between cocaine and saline slices. These experiments suggest that repeated cocaine exposure does not modify the synaptic strength of these neurons during early cocaine withdrawal.
Figure 3.
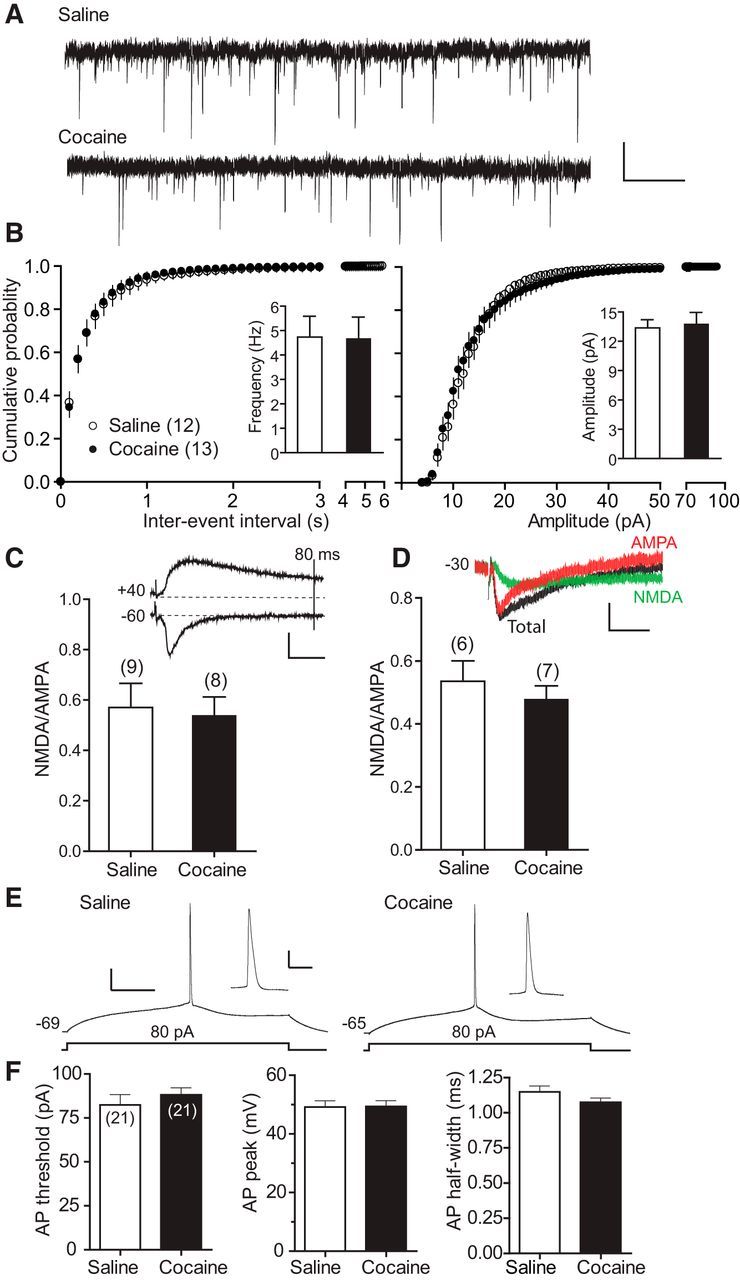
Repeated cocaine exposure does not alter either AMPAR-mediated synaptic strength or intrinsic AP waveform. A, Representative mEPSCs recorded at −60 mV from repeated cocaine- and saline-treated mice. Scale bars, 20 pA, 1 s. B, Cumulative probability summaries and mean (insets) mEPSC frequencies and amplitudes. C, NMDA/AMPA ratio estimated based on the differential kinetics of EPSCNMDA and EPSCAMPA currents. Inset, Representative EPSCs recorded at −60 mV (to record EPSCAMPA) and +40 mV (to record total EPSC). Vertical line indicates 80 ms poststimulation when EPSCAMPA has returned to the baseline, whereas EPSCNMDA has not. Scale bars, 100 pA, 20 ms. Bottom, Summary of mean NMDA/AMPA ratios. NMDA/AMPA ratio is defined as the amplitude of EPSCNMDA 80 ms after stimulation at +40 mV divided by the peak EPSCAMPA at −60 mV. D, NMDA/AMPA ratio measured based on pharmacologically isolated EPSCAMPA and EPSCNMDA currents. Inset, Representative total EPSC, EPSCNMDA (in the presence of CNQX), and EPSCAMPA (EPSCNMDA subtracted from total EPSC) recorded at −30 mV from saline or cocaine slices. Scale bars, 25 pA, 25 ms. Bottom, Summary of NMDA/AMPA ratios. E, Representative AP spikes in response to 80 pA depolarizing current recorded from repeated cocaine- and saline-treated mice. Scale bars, 20 mV, 100 ms. Insets, AP displayed at an amplified time scale. Scale bars, 20 mV, 10 ms. F, Summary of AP threshold, AP peak, and AP half-width.
Back-propagating postsynaptic APs can interact with coinciding EPSPs nonlinearly and affect dendritic integration and thus the STDP timing window (Caporale and Dan, 2008; Sjostrom et al., 2008; Feldman, 2012). We therefore investigated whether repeated cocaine exposure altered AP in mouse mPFC L5 neurons. APs elicited by depolarizing current injections into the recorded neurons showed statistically similar initiation threshold, amplitude, and duration in saline and cocaine slices (Fig. 3E,F). Therefore, repeated cocaine did not affect AP waveform characteristics in these neurons substantially, at least in the soma.
Postsynaptic NVP-AAM077-sensitive, GluN2A-NMDARs mediate both normal and extended-timing t-LTP
We next investigated the induction and expression mechanisms of the cocaine-induced extended timing t-LTP. There was no significant difference in paired-pulse facilitation (interpulse interval = 50 ms) before and after t-LTP induction (Δt = +30 ms; Fig. 4A), suggesting no change in presynaptic release probability associated with t-LTP expression. In comparison, loading pyramidal cells with the Ca2+ chelator BAPTA blocked the induction of +30 ms t-LTP in cocaine slices (Fig. 4B), suggesting that this extended-timing t-LTP required an increase of postsynaptic Ca2+ levels. Further experiments found that bath application of the NMDAR antagonist APV (50 μm) blocked the +30 ms t-LTP in cocaine slices (data not shown). Together, the cocaine-induced extended-timing t-LTP is postsynaptic in nature and involved Ca2+ influx, likely through NMDARs, similar to the postsynaptic NMDAR-dependent mechanism known to mediate conventional LTP and classical STDP.
Cortical NMDARs are heterotetramers composed of the obligatory GluN1 subunit, along with various GluN2 subunits that differ in channel gating, ion permeability, and intracellular signaling (Lau and Zukin, 2007). Given the differential roles of GluN2A and GluN2B in STDP induction at different timings (Gerkin et al., 2007; Ruan et al., 2014), we investigated which GluN2 subunits might mediate the cocaine-induced extended-timing t-LTP. We found that including the GluN2B-specific antagonist ifenprodil in the bath did not affect t-LTP at +30 ms in cocaine slices (Fig. 4D,F), suggesting that GluN2B is not involved. In contrast, bath inclusion of the GluN2A-preferred competitive antagonist NVP-AAM077 abolished t-LTP at +30 ms (Fig. 4C,F). NVP-AAM077 also blocked t-LTP at +10 ms (Fig. 4E,F), suggesting that GluN2A-containing NMDARs mediate both the normal and extended timing t-LTP nonselectively in cocaine slices. Despite concerns about the specificity of NVP-AAM077 (Neyton and Paoletti, 2006; Frizelle et al., 2006), other studies, including ours (Ruan et al., 2014), have used this compound convincingly to suppress synaptic GluN2A EPSCNMDA currents in cultured neurons (Gerkin et al., 2007) and PFC slices (Zhao et al., 2005). Importantly, even if NVP-AAM077 inhibits some synaptic GluN2B-NMDARs at the concentration used (0.4 μm), which we cannot rule out, our conclusion that GluN2A-NMDARs mediates primarily the +30 ms-t-LTP should stand true regardless because blocking GluN2B selectively with ifenprodil at a sufficient concentration (3 μm) did not affect +30 ms-t-LTP (Fig. 4D,F).
Voltage-clamp analysis further showed that isolated NMDAR-mediated EPSCs were significantly more sensitive to NVP-AAM077 in cocaine slices compared with the saline control (Fig. 4G), suggesting that GluN2A-NMDAR currents were increased after repeated cocaine exposure. Together, these results indicate that the extended-timing t-LTP after cocaine exposure is mediated by GluN2A-NMDARs, likely a result of altered NMDAR subunit composition or assembly at PFC synapses.
LTCCs mediate extended-timing t-LTP selectively in cocaine mice
In search of mechanisms that can account for the extended-timing t-LTP selectively in cocaine-treated mice, we turned our attention to LTCCs, a class of high-voltage-activated Ca2+ channels (Catterall, 2000; Lipscombe et al., 2004) that regulate Ca2+ influx, membrane excitability, and synaptic plasticity, including STDP (Bi and Poo, 1998; Seamans and Yang, 2004; Sjostrom et al., 2008). Of particular relevance, LTCCs have emerged as an important player in psychostimulant addiction (Nasif et al., 2005; Rajadhyaksha and Kosofsky, 2005; Giordano et al., 2010). Nimodipine, an LTCC blocker, did not reduce t-LTP induction at +10 ms (Fig. 5A,E) when applied to the bath during pre–post pairings in cocaine slices, suggesting that t-LTP at this normal timing did not require the opening of LTCCs. In contrast, blockade of LTCCs by nimodipine during pairings completely abolished t-LTP at +30 ms in cocaine slices (Fig. 5B,E). Diltiazem, a structurally different LTCC blocker, also blocked t-LTP induction at +30 ms in cocaine slices (Fig. 5C,E). Importantly, applying the LTCC agonist Bay K8644 to the bath during pre–post pairings in saline slices enabled t-LTP induction at +30 ms (Fig. 5D,E), a normally ineffective timing interval. Application of Bay K8644 without EPSP–AP pairings did not increase, and in fact decreased, EPSPs (Fig. 5F) in saline slices. Bay K8644 also suppressed isolated EPSCNMDA (Fig. 5G), consistent with earlier observations that Ca2+ entry via voltage-dependent Ca2+ channels generally suppresses NMDARs (Legendre et al., 1993; Tong et al., 1995). These results suggest that Bay K8644 alone did not cause the +30 ms t-LTP in saline slices. Together, these data indicate that the opening of LTCCs is essential for the cocaine-induced extended-timing t-LTP.
Sensitized D1-cAMP/PKA pathway maintains the extended-timing t-LTP in cocaine mice
We have shown previously that selective activation of the D1-cAMP/PKA signaling in PFC pyramidal cells broadens the t-LTP timing window (Xu and Yao, 2010; Ruan et al., 2014). It has also been reported that the D1-cAMP/PKA/signaling becomes enhanced in the rat PFC after repeated in vivo cocaine administration (Dong et al., 2005; Ford et al., 2009), raising the possibility that the extended t-LTP timing window in cocaine mice might be due to an overactivation of the D1-cAMP/PKA pathway in pyramidal neurons. Indeed, bath application of the D1 antagonist SCH23390 during pre–post pairings completely blocked t-LTP induction at +30 ms in cocaine slices (Fig. 6B,G). Interestingly, SCH23390 did not affect t-LTP induction at +10 ms in cocaine slices (Fig. 6A,G), suggesting that normal-timing t-LTP does not require D1 activation. Application of the cAMP inhibitor RP-cAMPS during pre–post pairings also prevented t-LTP at +30 ms in cocaine slices (Fig. 6C,G). Finally, loading patched cells with PKI 6–22 (a membrane-impermeable PKI) blocked the +30 ms t-LTP (Fig. 6D,G). These results indicate that cocaine-induced t-LTP timing window extension at PFC synapses is caused by a constitutively hypersensitive D1-cAMP/PKA signaling in pyramidal neurons.
We then investigated whether, with the already elevated D1-cAMP/PKA signaling, stimulation of D1 receptors could further modulate t-LTP induction in cocaine slices. Applying the D1 receptor agonist SKF81297 during pre–post pairings enabled t-LTP induction at +50 ms, an ineffective timing in cocaine slices (Fig. 6E–G). This result indicates that, although PFC D1 receptor signaling is heightened after cocaine exposure, it is not saturated; these receptors still respond to agonist stimulation and can drive the t-LTP induction at an even wider window.
LTCCs synergize with GluN2A-NMDARs to mediate SKF81297-enabled extended-timing t-LTP in normal mice
LTCCs are known substrates of PKA (Catterall, 2000; Rajadhyaksha and Kosofsky, 2005). We thus investigated whether LTCCs and D1-cAMP/PKA work in the same pathway to control the timing window of t-LTP. Consistent with our earlier work, selective activation of D1 receptors in saline slices by bath application of SKF81297 during pre–post pairings enabled t-LTP at +30 ms (Fig. 7A), which is otherwise nonexistent (Xu and Yao, 2010). Coapplication of nimodipine with SKF81297 abolished this SKF81297-enabled +30 ms t-LTP (Fig. 7B,D). This suggests that LTCCs are indeed downstream of D1 signaling in driving the +30 ms t-LTP. Interestingly, coapplication of NVP-AAM077 also blocked the SKF81297-enabled +30 ms t-LTP (Fig. 7C,D), which is consistent with the idea that GluN2A is also a downstream target of the D1 signaling pathway. Therefore, both GluN2A and LTCCs downstream of D1-cAMP/PKA pathway are required for SKF81297-enabled +30 ms t-LTP in saline slices.
Figure 7.
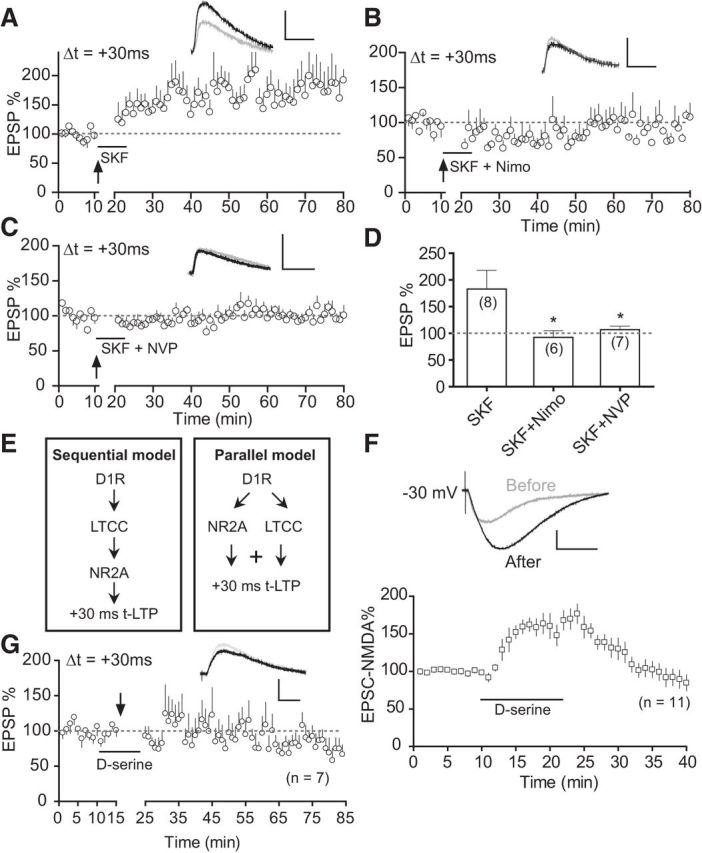
SKF81297-enabled +30 ms-t-LTP in saline mice is mediated by both LTCCs and GluN2A-NMDARs in a parallel, synergistic manner. A, Enabling effect of SKF812907 (SKF, 2 μm) on t-LTP at +30 ms in saline slices. B, C, Blockade of SKF81297-enabled t-LTP at +30 ms in saline slices by coapplied nimodipine (3 μm; B) or NVP-AAM077 (0.4 μm; C). D, t-LTP summary. Drugs were applied to the extracellular bath during EPSP–AP pairings. *p < 0.05 versus SKF, unpaired t test with Welch's correction. E, Two possible models in which LTCCs and GluN2A-NMDARs may act together to drive the +30 ms-t-LTP. F, d-serine (100 μm) potentiates EPSCNMDA in saline slices. d-serine was applied at the 10 min mark for 10 min after a stable baseline was obtained. Top, Representative EPSCNMDA before and after d-serine application. Scale bars, 50 pA, 100 ms. G, Failure of d-serine to enable t-LTP at +30 ms in saline slices when applied during EPSP–AP pairings. Scale bars (A–D, G), 2 mV, 20 ms.
GluN2A-NMDARs and LTCCs may act either sequentially or in parallel (Fig. 7E) to drive the extended timing t-LTP. To distinguish these possibilities, we used d-serine, a potent coagonist of NMDARs at the glycine binding site of the GluN1 subunit (Kuryatov et al., 1994) to enhance EPSCNMDA selectively in the absence of D1-cAMP/PKA or LTCC activation. As expected, application of d-serine potently and reversibly enhanced EPSCNMDA (Fig. 7F). If the “sequential” model were true, we would expect that solely activating NMDARs downstream of LTCCs without SKF81297 application would enable t-LTP at +30 ms in saline slices. This was not the case: d-serine applied during EPSP–AP pairings at +30 ms did not enable t-LTP (Fig. 7G). These results therefore favored a parallel model of NMDAR–LTCC synergy (Fig. 7E): these two distinct Ca2+ sources act in parallel to drive the abnormal +30 ms t-LTP.
Cocaine-recruited signaling mechanism broadens t-LTP timing window in the PFC
Our results support a model for cocaine-induced t-LTP timing window extension in PFC excitatory circuits (Fig. 8). Activation of the D1-cAMP/PKA-LTCC pathway is unnecessary for the normal-timing t-LTP (e.g., +10 ms), which is mediated by GluN2A-NMDARs alone (Fig. 8A). In comparison, repeated cocaine exposure exacerbates D1 signaling in pyramidal neurons, which then recruits downstream LTCCs to enable t-LTP induction at markedly extended, normally ineffective timing intervals and at a lower threshold (Fig. 8B). Importantly, GluN2A-NMDARs are also obligatory for the +30 ms t-LTP in both cocaine slices and saline slices treated with SKF81297, suggesting that these receptors are also engaged by the D1-cAMP/PKA activation and synergize with LTCCs to extend the t-LTP timing window. Recruitment of these signaling pathways by cocaine sensitizes PFC synapses for potentiation by less correlated pre–post spiking activity. Our results thus identify a pathological signaling axis modulating coincidence detection, a novel form of drug-induced associative synaptic plasticity.
Figure 8.

Working model for cocaine-recruited signaling axis that broadens t-LTP timing window in the PFC. A, Opening of the classical coincidence detector GluN2A-NMDARs alone without the need for activating D1-cAMP/PKA-LTCC pathway is sufficient to drive t-LTP within the normal timing window. B, Repeated cocaine exposure sensitizes D1 DA receptors, elevates cAMP/PKA signaling, and recruits downstream voltage-gated LTCCs, which then synergizes with GluN2A-NMDARs to drive t-LTP induction at +30 ms, a normally ineffective timing interval. Key pharmacological agents used in this study are indicated in parentheses.
Discussion
We show that repeated cocaine exposure in vivo broadens the timing window markedly and reduces the induction threshold of t-LTP in the mouse mPFC, rendering synapses more susceptible for potentiation. Our study highlights a novel concept in addiction biology: drugs can hijack the reward circuits by disrupting Hebbian quantitative synaptic learning rules without altering synapse strength. Our study suggests an early cocaine withdrawal period when PFC circuits may be particularly vulnerable for experience-dependent remodeling.
Several forms of cocaine-evoked synaptic plasticity have been recognized. Exposure to cocaine alters the strength of glutamatergic synapses in the VTA (Ungless et al., 2001) and NAc (Thomas et al., 2001; Pascoli et al., 2011), as well as strength of GABAergic synapses in the VTA (Liu et al., 2005) and PFC (Lu et al., 2010). Cocaine also alters AMPAR composition at NAc and VTA excitatory synapses (Bellone and Lüscher, 2006; Conrad et al., 2008), characterized by the insertion of GluA2-lacking, Ca2+-permeable AMPARs shown to drive a NMDAR-independent anti-Hebbian LTP (Mameli et al., 2011). In vivo cocaine experience also generates NMDAR-containing and AMPAR-lacking silent synapses de novo in the NAc (Huang et al., 2009), a neural-circuit-remodeling mechanism believed to contribute to cocaine craving and relapse (Dong and Nestler, 2014). Finally, after cocaine withdrawal, glutamatergic synapses onto DA neurons become sustainably sensitive to potentiation by weak presynaptic stimuli and display a nonassociative form of plasticity caused by an elevated BDNF signaling (Pu et al., 2006). Our study uncovers a distinct form of cocaine-induced plasticity that disrupts a key Hebbian associative plasticity process. Because the STDP timing window reflects how closely presynaptic and postsynaptic spikes converge at the synapse, our data suggest that cocaine compromises the temporal stringency of the STDP that leads to a gain in sensitivity and a decrease in threshold for t-LTP induction. Modifications of rules that govern associative synaptic plasticity may represent an important mechanism that can dictate how environmental cues and drugs interact with and remodel reward circuits.
DA remains central in addiction neurobiology; all addictive drugs increase the synaptic level of DA pathologically, which is believed to elicit hedonic responses, enhance behavioral saliency, and promote reward-related learning and memories in DA circuits (Nestler, 2001; Schultz, 2002; Hyman et al., 2006). We have shown previously that DA, by acting on the D1-cAMP/PKA signaling, facilitates t-LTP induction by extending its timing window by ∼six-fold in PFC excitatory synapses (Xu and Yao, 2010; Ruan et al., 2014), providing a potential mechanism by which DA can drive reward learning and consolidate associative long-term memories. Those studies, along with others (Lin et al., 2003; Seol et al., 2007; Pawlak and Kerr, 2008; Zhang et al., 2009; Yang and Dani, 2014), have helped to derive the modified tricomponent STDP learning rule (Pawlak et al., 2010; Feldman, 2012), in which neuromodulators such as DA are proposed as potent coincidence modulators that influence markedly STDP polarity, timing window, and ultimately coincidence detection during STDP induction. Our present findings demonstrate that cocaine exacerbates this DA neuromodulatory process to alter t-LTP rules in the PFC. Importantly, though sensitized, the D1 receptors are not saturated—they still respond to agonist stimulation and broaden the t-LTP timing window to an even greater level, raising the possibility that PFC synapses can be even more susceptible for potentiation when synaptic DA level is higher, such as during reward behavior or motivational arousal. Our study suggests that cocaine, in essence, can hijack coincidence detection and processing during STDP, altering associative plasticity mechanisms that may underlie reward.
Our study identifies LTCCs as a potential dendritic/synaptic effector that can broaden the t-LTP timing window in both normal and cocaine conditions. LTCCs are activated by modest to high depolarizing voltages (Lipscombe et al., 2004), which are localized in dendrites of PFC neurons (Seamans and Yang, 2004) and involved in the regulation of STDP (Bi and Poo, 1998; Froemke et al., 2005; Zhou et al., 2005). LTCCs conduct dendritic Ca2+ currents, regulate dendritic integrations (Johnston et al., 1996; Sjostrom et al., 2008; Spruston, 2008), and are coupled to various downstream Ca2+-signaling mechanisms (Greengard et al., 1999), all of which can influence t-LTP timing window (Caporale and Dan, 2008). Importantly, LTCCs are not required for t-LTP at +10 ms, but for +30 ms when D1 signaling is exogenously stimulated in normal condition or intrinsically heightened in cocaine conditions, suggesting that LTCCs at an “activated” state regulate t-LTP timing window selectively. Modulations of LTCCs after D1-cAMP/PKA activation can enhance the conductance, slow the deactivation, and shift the voltage dependence of these channels negatively to alter dendritic Ca2+ currents, excitability, and integration (Catterall, 2000; Seamans and Yang, 2004). DA/D1-recruited LTCCs may allow PFC neurons to be more responsive to NMDAR activation (Seamans and Yang, 2004) during STDP induction to favor t-LTP at extended timings. Through this and/or other mechanisms, LTCCs serve as a modulator, rather than a mediator, and synergize in parallel with the more obligatory coincidence detector GluN2A-NMDARs to drive extended-timing t-LTP.
Neuronal LTCCs containing mainly the Cav1.2 and Cav1.3 subunits have emerged as a key player in psychostimulant addiction (Rajadhyaksha and Kosofsky, 2005). Repeated exposure to cocaine upregulates LTCC protein levels and enhances the channel activity in rodent mPFC (Nasif et al., 2005; Ford et al., 2009). LTCCs are required for both the development and expression of behavioral sensitization, perhaps in a subunit-dependent manner (Giordano et al., 2010). It has been suggested that, in the NAc, psychostimulant-activated D1 receptors couple with LTCCs to elevate the intracellular Ca2+ level, which, via CREB, ERK, and other signaling mechanisms, induces complex activity-dependent gene expression and molecular adaptations (Rajadhyaksha and Kosofsky, 2005). Our data indicate that, despite potentially widespread adaptations triggered by LTCCs after cocaine exposure, acute manipulations of D1 signaling or LTCCs in saline slices can mimic the t-LTP window extension in cocaine slices. Therefore, LTCCs may facilitate t-LTP induction at extended timings via cAMP/PKA-dependent regulations of biophysical properties or functional recruitment of the channel.
Neuronal excitability plays an important role in synaptic integration and STDP (Sjostrom et al., 2008). In particular, back-propagating APs attenuate along dendrites in both hippocampal CA1 and L5 neurons and require boosting by inactivating A-type K+ (Watanabe et al., 2002) and recruiting dendritic Na+ channels (Stuart and Hausser, 2001), respectively, to elicit t-LTP. DA/D1 activation exerts complex modulations on PFC Na+ currents (Seamans and Yang, 2004), but generally suppresses voltage-gated K+ currents, especially a slowly inactivating component (Yang and Seamans, 1996; Dong and White, 2003), increasing neuronal excitability (Seamans and Yang, 2004). Interestingly, DA/D1 affects neither the AP waveform (Yang and Seamans, 1996; Gulledge and Stuart, 2003; Xu and Yao, 2010) nor dendritic back-propagation (Gulledge and Stuart, 2003) in these neurons. Repeated cocaine administration persistently decreases ID via a heightened D1-cAMP/PKA signaling and inward-rectifying K+ currents (Dong et al., 2005), enhancing excitability without altering AP waveform (Nasif et al., 2005a; this study). Because our t-LTP protocol depends on precisely timed EPSP/single AP pairings, AP waveform but not AP number or somatic excitability plays a more direct role in dendritic integration and t-LTP. Therefore, cocaine-induced adaptations in K+ or other somatic ion currents are unlikely to contribute significantly to the extended-timing t-LTP in cocaine slices. However, we cannot rule out potential cocaine effects on dendritic Na+ and K+ conductance, which can affect AP back-propagation and t-LTP induction.
It is hypothesized that addictive drugs sensitize reward systems (Robinson and Berridge, 2001) and render neural circuits more plastic (Bellone and Lüscher, 2012; Dong and Nestler, 2014). Our study provides a potential mechanism for this hypothesis: drugs can lower the threshold for associative synaptic plasticity such that cues associated with the drug can trigger experience-dependent reward circuit remodeling more effectively than do natural stimuli. Consistently, self-administered cocaine, but not natural rewards, produces persistent LTP in the VTA (Chen et al., 2008). Reexposure to cues previously associated with heroin, but not natural reinforcers, triggers AMPAR endocytosis in the PFC (Van den Oever et al., 2008). A recent study (Kasanetz et al., 2013) showed that, after cocaine self-administration, the AMPA/NMDA ratio in mPFC L5 neurons is increased in “addict-like” rats. Interestingly, this synaptic strength increase does not develop at early stages (17 d) of cocaine self-administration, consistent with the lack of synaptic efficacy change during the early withdrawal observed here, but becomes established after prolonged (∼2 months) cocaine self-administration. The delayed potentiation could be a result of impaired LTD concurrently observed in these animals, which may prevent synapses to undergo necessary weakening. Alternatively, a decrease in LTP threshold, together with an increase in neuronal excitability mentioned above, may provide a “permissive function” mechanism (Kourrich et al., 2015) for experience-dependent synaptic potentiation to develop. Once developed, persistently potentiated synapses and impaired LTD could render PFC circuits resistant to modulation by an ever-changing environment, reducing cognitive flexibility that can culminate in the loss of executive control over drug seeking, a clinical hallmark of addiction known as hypofrontality (Goldstein and Volkow, 2011). The Hebbian plasticity rule change reported here may represent a form of drug usurpation of reward circuits that predisposes PFC executive circuits to hypofrontality.
Footnotes
This work was supported by the National Institutes of Health (Grant DA032283 to W.-D.Y. and Grant OD011103 to the New England Primate Research Center, Harvard University). We thank Dr. Y.P. Auberson at Novartis Pharma AG, Basel, for the generous gift of NVP-AAM077.
The authors declare no competing financial interests.
References
- Bellone C, Lüscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- Bellone C, Lüscher C. Drug-evoked plasticity: do addictive drugs reopen a critical period of postnatal synaptic development? Front Mol Neurosci. 2012;5:75. doi: 10.3389/fnmol.2012.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporale N, Dan Y. Spike timing-dependent plasticity: a Hebbian learning rule. Annu Rev Neurosci. 2008;31:25–46. doi: 10.1146/annurev.neuro.31.060407.125639. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BT, Yau HJ, Hatch C, Kusumoto-Yoshida I, Cho SL, Hopf FW, Bonci A. Rescuing cocaine-induced prefrontal cortex hypoactivity prevents compulsive cocaine seeking. Nature. 2013;496:359–362. doi: 10.1038/nature12024. [DOI] [PubMed] [Google Scholar]
- Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O'Brien CP. Limbic activation during cue-induced cocaine craving. Am J Psychiatry. 1999;156:11–18. doi: 10.1176/ajp.156.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, Marinelli M, Wolf ME. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Nestler EJ. The neural rejuvenation hypothesis of cocaine addiction. Trends Pharmacol Sci. 2014;35:374–383. doi: 10.1016/j.tips.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, White FJ. Dopamine D1-class receptors selectively modulate a slowly inactivating potassium current in rat medial prefrontal cortex pyramidal neurons. J Neurosci. 2003;23:2686–2695. doi: 10.1523/JNEUROSCI.23-07-02686.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Nasif FJ, Tsui JJ, Ju WY, Cooper DC, Hu XT, Malenka RC, White FJ. Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci. 2005;25:936–940. doi: 10.1523/JNEUROSCI.4715-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. The spike-timing dependence of plasticity. Neuron. 2012;75:556–571. doi: 10.1016/j.neuron.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford KA, Wolf ME, Hu XT. Plasticity of L-type Ca2+ channels after cocaine withdrawal. Synapse. 2009;63:690–697. doi: 10.1002/syn.20651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frizelle PA, Chen PE, Wyllie DJ. Equilibrium constants for (R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydroquinoxalin-5-yl)-methyl]-phosphonic acid (NVP-AAM077) acting at recombinant NR1/NR2A and NR1/NR2B N-methyl-D-aspartate receptors: Implications for studies of synaptic transmission. Mol Pharmacol. 2006;70:1022–1032. doi: 10.1124/mol.106.024042. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Poo MM, Dan Y. Spike-timing-dependent synaptic plasticity depends on dendritic location. Nature. 2005;434:221–225. doi: 10.1038/nature03366. [DOI] [PubMed] [Google Scholar]
- Gerkin RC, Lau PM, Nauen DW, Wang YT, Bi GQ. Modular competition driven by NMDA receptor subtypes in spike-timing-dependent plasticity. J Neurophysiol. 2007;97:2851–2862. doi: 10.1152/jn.00860.2006. [DOI] [PubMed] [Google Scholar]
- Giordano TP, Tropea TF, Satpute SS, Sinnegger-Brauns MJ, Striessnig J, Kosofsky BE, Rajadhyaksha AM. Molecular switch from L-type Ca v 1.3 to Ca v 1.2 Ca2+ channel signaling underlies long-term psychostimulant-induced behavioral and molecular plasticity. J Neurosci. 2010;30:17051–17062. doi: 10.1523/JNEUROSCI.2255-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. 2011;12:652–669. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greengard P, Allen PB, Nairn AC. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–447. doi: 10.1016/S0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Action potential initiation and propagation in layer 5 pyramidal neurons of the rat prefrontal cortex: absence of dopamine modulation. J Neurosci. 2003;23:11363–11372. doi: 10.1523/JNEUROSCI.23-36-11363.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Yang PC, Lin HJ, Hsu KS. Repeated cocaine administration impairs group II metabotropic glutamate receptor-mediated long-term depression in rat medial prefrontal cortex. J Neurosci. 2007;27:2958–2968. doi: 10.1523/JNEUROSCI.4247-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Lin Y, Mu P, Lee BR, Brown TE, Wayman G, Marie H, Liu W, Yan Z, Sorg BA, Schlüter OM, Zukin RS, Dong Y. In vivo cocaine experience generates silent synapses. Neuron. 2009;63:40–47. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Johnston D, Magee JC, Colbert CM, Cristie BR. Active properties of neuronal dendrites. Annu Rev Neurosci. 1996;19:165–186. doi: 10.1146/annurev.ne.19.030196.001121. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, Seamans J. Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron. 2005;45:647–650. doi: 10.1016/j.neuron.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Kasanetz F, Lafourcade M, Deroche-Gamonet V, Revest JM, Berson N, Balado E, Fiancette JF, Renault P, Piazza PV, Manzoni OJ. Prefrontal synaptic markers of cocaine addiction-like behavior in rats. Mol Psychiatry. 2013;18:729–737. doi: 10.1038/mp.2012.59. [DOI] [PubMed] [Google Scholar]
- Kolb B, Mychasiuk R, Muhammad A, Li Y, Frost DO, Gibb R. Experience and the developing prefrontal cortex. Proc Natl Acad Sci U S A. 2012;109:17186–17193. doi: 10.1073/pnas.1121251109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourrich S, Calu DJ, Bonci A. Intrinsic plasticity: an emerging player in addiction. Nat Rev Neurosci. 2015;16:173–184. doi: 10.1038/nrn3877. [DOI] [PubMed] [Google Scholar]
- Kuryatov A, Laube B, Betz H, Kuhse J. Mutational analysis of the glycine-binding site of the NMDA receptor: structural similarity with bacterial amino acid-binding proteins. Neuron. 1994;12:1291–1300. doi: 10.1016/0896-6273(94)90445-6. [DOI] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Legendre P, Rosenmund C, Westbrook GL. Inactivation of NMDA channels in cultured hippocampal neurons by intracellular calcium. J Neurosci. 1993;13:674–684. doi: 10.1523/JNEUROSCI.13-02-00674.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YW, Min MY, Chiu TH, Yang HW. Enhancement of associative long-term potentiation by activation of beta-adrenergic receptors at CA1 synapses in rat hippocampal slices. J Neurosci. 2003;23:4173–4181. doi: 10.1523/JNEUROSCI.23-10-04173.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. J Neurophysiol. 2004;92:2633–2641. doi: 10.1152/jn.00486.2004. [DOI] [PubMed] [Google Scholar]
- Liu QS, Pu L, Poo MM. Repeated cocaine exposure in vivo facilitates LTP induction in midbrain dopamine neurons. Nature. 2005;437:1027–1031. doi: 10.1038/nature04050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Lim B, Poo MM. Cocaine exposure in utero alters synaptic plasticity in the medial prefrontal cortex of postnatal rats. J Neurosci. 2009;29:12664–12674. doi: 10.1523/JNEUROSCI.1984-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Cheng PL, Lim BK, Khoshnevisrad N, Poo MM. Elevated BDNF after cocaine withdrawal facilitates LTP in medial prefrontal cortex by suppressing GABA inhibition. Neuron. 2010;67:821–833. doi: 10.1016/j.neuron.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Mameli M, Bellone C, Brown MT, Lüscher C. Cocaine inverts rules for synaptic plasticity of glutamate transmission in the ventral tegmental area. Nat Neurosci. 2011;14:414–416. doi: 10.1038/nn.2763. [DOI] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Miller EK, Cohen JD. An integrative theory of prefrontal cortex function. Annu Rev Neurosci. 2001;24:167–202. doi: 10.1146/annurev.neuro.24.1.167. [DOI] [PubMed] [Google Scholar]
- Nasif FJ, Hu XT, White FJ. Repeated cocaine administration increases voltage-sensitive calcium currents in response to membrane depolarization in medial prefrontal cortex pyramidal neurons. J Neurosci. 2005;25:3674–3679. doi: 10.1523/JNEUROSCI.0010-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasif FJ, Sidiropoulou K, Hu XT, White FJ. Repeated cocaine administration increases membrane excitability of pyramidal neurons in the rat medial prefrontal cortex. J Pharmacol Exp Ther. 2005a;312:1305–1313. doi: 10.1124/jpet.104.075184. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Neyton J, Paoletti P. Relating NMDA receptor function to receptor subunit composition: limitations of the pharmacological approach. J Neurosci. 2006;26:1331–1333. doi: 10.1523/JNEUROSCI.5242-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Turiault M, Lüscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature. 2011;481:71–75. doi: 10.1038/nature10709. [DOI] [PubMed] [Google Scholar]
- Pawlak V, Kerr JN. Dopamine receptor activation is required for corticostriatal spike-timing-dependent plasticity. J Neurosci. 2008;28:2435–2446. doi: 10.1523/JNEUROSCI.4402-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak V, Wickens JR, Kirkwood A, Kerr JN. Timing is not everything: neuromodulation opens the STDP gate. Front Synaptic Neurosci. 2010;2:146. doi: 10.3389/fnsyn.2010.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L, Liu QS, Poo MM. BDNF-dependent synaptic sensitization in midbrain dopamine neurons after cocaine withdrawal. Nat Neurosci. 2006;9:605–607. doi: 10.1038/nn1687. [DOI] [PubMed] [Google Scholar]
- Rajadhyaksha AM, Kosofsky BE. Psychostimulants, L-type calcium channels, kinases, and phosphatases. Neuroscientist. 2005;11:494–502. doi: 10.1177/1073858405278236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV, Sun W. Neuronal substrates of relapse to cocaine-seeking behavior: role of prefrontal cortex. J Exp Anal Behav. 2005;84:653–666. doi: 10.1901/jeab.2005.105-04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Incentive-sensitization and addiction. Addiction. 2001;96:103–114. doi: 10.1046/j.1360-0443.2001.9611038.x. [DOI] [PubMed] [Google Scholar]
- Ruan H, Saur T, Yao WD. Dopamine-enabled anti-Hebbian timing-dependent plasticity in prefrontal circuitry. Front Neural Circuits. 2014;8:38. doi: 10.3389/fncir.2014.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Getting formal with dopamine and reward. Neuron. 2002;36:241–263. doi: 10.1016/S0896-6273(02)00967-4. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 2004;74:1–58. doi: 10.1016/j.pneurobio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Seol GH, Ziburkus J, Huang S, Song L, Kim IT, Takamiya K, Huganir RL, Lee HK, Kirkwood A. Neuromodulators control the polarity of spike-timing-dependent synaptic plasticity. Neuron. 2007;55:919–929. doi: 10.1016/j.neuron.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Rancz EA, Roth A, Häusser M. Dendritic excitability and synaptic plasticity. Physiol Rev. 2008;88:769–840. doi: 10.1152/physrev.00016.2007. [DOI] [PubMed] [Google Scholar]
- Spruston N. Pyramidal neurons: dendritic structure and synaptic integration. Nat Rev Neurosci. 2008;9:206–221. doi: 10.1038/nrn2286. [DOI] [PubMed] [Google Scholar]
- Stuart GJ, Häusser M. Dendritic coincidence detection of EPSPs and action potentials. Nat Neurosci. 2001;4:63–71. doi: 10.1038/82910. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Beurrier C, Bonci A, Malenka RC. Long-term depression in the nucleus accumbens: a neural correlate of behavioral sensitization to cocaine. Nat Neurosci. 2001;4:1217–1223. doi: 10.1038/nn757. [DOI] [PubMed] [Google Scholar]
- Tong G, Shepherd D, Jahr CE. Synaptic desensitization of NMDA receptors by calcineurin. Science. 1995;267:1510–1512. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- Van den Oever MC, Goriounova NA, Li KW, Van der Schors RC, Binnekade R, Schoffelmeer AN, Mansvelder HD, Smit AB, Spijker S, De Vries TJ. Prefrontal cortex AMPA receptor plasticity is crucial for cue-induced relapse to heroin-seeking. Nat Neurosci. 2008;11:1053–1058. doi: 10.1038/nn.2165. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Hoffman DA, Migliore M, Johnston D. Dendritic K+ channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998;54:679–720. doi: 10.1016/S0301-0082(97)00090-7. [DOI] [PubMed] [Google Scholar]
- Xu TX, Yao WD. D1 and D2 dopamine receptors in separate circuits cooperate to drive associative long-term potentiation in the prefrontal cortex. Proc Natl Acad Sci U S A. 2010;107:16366–16371. doi: 10.1073/pnas.1004108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu TX, Sotnikova TD, Liang C, Zhang J, Jung JU, Spealman RD, Gainetdinov RR, Yao WD. Hyperdopaminergic tone erodes prefrontal long-term potential via a D2 receptor-operated protein phosphatase gate. J Neurosci. 2009;29:14086–14099. doi: 10.1523/JNEUROSCI.0974-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Seamans JK. Dopamine D1 receptor actions in layers V–VI rat prefrontal cortex neurons in vitro: modulation of dendritic-somatic signal integration. J Neurosci. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K, Dani JA. Dopamine D1 and D5 receptors modulate spike timing-dependent plasticity at medial perforant path to dentate granule cell synapses. J Neurosci. 2014;34:15888–15897. doi: 10.1523/JNEUROSCI.2400-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JC, Lau PM, Bi GQ. Gain in sensitivity and loss in temporal contrast of STDP by dopaminergic modulation at hippocampal synapses. Proc Natl Acad Sci U S A. 2009;106:13028–13033. doi: 10.1073/pnas.0900546106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MG, Toyoda H, Lee YS, Wu LJ, Ko SW, Zhang XH, Jia Y, Shum F, Xu H, Li BM, Kaang BK, Zhuo M. Roles of NMDA NR2B subtype receptor in prefrontal long-term potentiation and contextual fear memory. Neuron. 2005;47:859–872. doi: 10.1016/j.neuron.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Zhou YD, Acker CD, Netoff TI, Sen K, White JA. Increasing Ca2+ transients by broadening postsynaptic action potentials enhances timing-dependent synaptic depression. Proc Natl Acad Sci U S A. 2005;102:19121–19125. doi: 10.1073/pnas.0509856103. [DOI] [PMC free article] [PubMed] [Google Scholar]