

Abstract
Three neuronal pentraxins are expressed in brain, the membrane-bound “neuronal pentraxin receptor” (NPR) and the secreted proteins NP1 and NARP (i.e., NP2). Neuronal pentraxins bind to AMPARs at excitatory synapses and play important, well-documented roles in the activity-dependent regulation of neural circuits via this binding activity. However, it is unknown whether neuronal pentraxins perform roles in synapses beyond modulating postsynaptic AMPAR-dependent plasticity, and whether they may even act in inhibitory synapses. Here, we show that NPR expressed in non-neuronal cells potently induces formation of both excitatory and inhibitory postsynaptic specializations in cocultured hippocampal neurons. Knockdown of NPR in hippocampal neurons, conversely, dramatically decreased assembly and function of both excitatory and inhibitory postsynaptic specializations. Overexpression of NPR rescued the NPR knockdown phenotype but did not in itself change synapse numbers or properties. However, the NPR knockdown decreased the levels of NARP, whereas NPR overexpression produced a dramatic increase in the levels of NP1 and NARP, suggesting that NPR recruits and stabilizes NP1 and NARP on the presynaptic plasma membrane. Mechanistically, NPR acted in excitatory synapse assembly by binding to the N-terminal domain of AMPARs; antagonists of AMPA and GABA receptors selectively inhibited NPR-induced heterologous excitatory and inhibitory synapse assembly, respectively, but did not affect neurexin-1β-induced synapse assembly as a control. Our data suggest that neuronal pentraxins act as signaling complexes that function as general trans-synaptic organizers of both excitatory and inhibitory synapses by a mechanism that depends, at least in part, on the activity of the neurotransmitter receptors at these synapses.
SIGNIFICANCE STATEMENT Neuronal pentraxins comprise three neuronal proteins, neuronal pentraxin receptor (NPR) which is a type-II transmembrane protein on the neuronal surface, and secreted neuronal pentraxin-1 and NARP. The general functions of neuronal pentraxins at synapses have not been explored, except for their basic AMPAR binding properties. Here, we examined the functional role of NPR at synapses because it is the only neuronal pentraxin that is anchored to the neuronal cell-surface membrane. We find that NPR is a potent inducer of both excitatory and inhibitory heterologous synapses, and that knockdown of NPR in cultured neurons decreases the density of both excitatory and inhibitory synapses. Our data suggest that NPR performs a general, previously unrecognized function as a universal organizer of synapses.
Keywords: AMPAR, cell adhesion molecule, neuronal pentraxin receptor, synaptogenesis
Introduction
Neuronal pentraxins (NPs) were described as presynaptic receptors for taipoxin snake neurotoxins (Schlimgen et al., 1995) and independently identified in a screen of neuronally expressed immediate-early genes (Tsui et al., 1996). The NP family comprises two secreted proteins, NP1 and NARP (for “neuronal activity-regulated pentraxin”; also named NP2), and one membrane-anchored protein, NPR (for “neuronal pentraxin receptor”). All NPs share a homologous N-terminal sequence of low complexity, a short central coiled-coil sequence, and a large C-terminal pentraxin domain. In addition, NPR contains an N-terminal transmembrane region that anchors NPR on the membrane. Similar to other pentraxins, NPs form homopentamers and heteropentamers via their pentraxin domains (Kirkpatrick et al., 2000). As a result, NPR can nucleate surface-exposed pentraxin pentamers containing NP1 and/or NARP, thus effectively recruiting these proteins to the membrane. NPs bind to, and cluster, AMPA-type glutamate receptors (AMPARs) (O'Brien et al., 1999, 2002; Xu et al., 2003). Via this activity, NPs recruit the AMPAR GluA4 to synapses on parvalbumin-positive (Pv) interneurons (Sia et al., 2007; Chang et al., 2010; Gu et al., 2013; Pelkey et al., 2015). However, the fundamental functions of NPs remain uncharacterized; even the mechanism of their AMPAR binding remains unclear because the AMPAR-associated proteome does not appear to contain NPs (Schwenk et al., 2012, 2014).
Constitutive knock-out (KO) mice of NPs presented discrete and interesting phenotypes that have been analyzed in detail at the level of neural circuits. The NARP KO impaired excitatory synaptic plasticity in Pv interneurons in the hippocampus (Chang et al., 2010) and decreased the excitatory drive on Pv interneurons in the visual cortex, thereby altering ocular dominance plasticity (Gu et al., 2013). Furthermore, double KO of NARP and NPR decreased the levels of GluA4 at excitatory synapses in hippocampal Pv interneurons, delayed circuit maturation during development, and caused dysregulation of circuit functions (Pelkey et al., 2015). NP1/NARP double KO mice, conversely, exhibited altered segregation of eye-specific retinal ganglion cell projections to the dorsal lateral geniculate nucleus (Bjartmar et al., 2006). The NPR KO, on the other hand, impaired mGluR1/5-dependent LTD in the hippocampus, leading to the hypothesis that NPR enables LTD by clustering AMPARs and facilitating endocytosis of AMPAR clusters after NPR cleavage by TNF-converting enzyme (TACE) protease (Cho et al., 2008). Studies with zebrafish NARP mutants, finally, suggested that NPs enhance synapse function (Elbaz et al., 2015).
These elegant studies documented the importance of NPs for brain function. However, since few cellular, more basic studies on NPs were performed, these results were difficult to interpret with a single unifying mechanistic hypothesis. Here, we have examined the fundamental functions of NPR as the only NP that is membrane-anchored. Our data show that NPR potently induces both excitatory and inhibitory synapses in neurons that are cocultured with HEK293-expressing NPR, and that both types of synapses are impaired after knockdown of NPR in cultured neurons. NPR directly binds to the N-terminal domain (NTD) of AMPARs, and chronic incubation of NPR-expressing HEK293 cells cocultured with neurons in the presence of AMPAR or GABA receptor (GABAR) antagonists inhibits induction of excitatory or inhibitory synapses, respectively. Thus, NPR acts in cultured neurons as a general organizer of excitatory and inhibitory synapses, using a novel mechanism that involves a trans-synaptic interaction of presynaptic NPR with postsynaptic neurotransmitter receptors.
Materials and Methods
Constructs of expression vectors.
The constructs encoding cDNAs of hNPR (HsCD00083063), hNP1 (HsCD00296108), and hNARP (HsCD00040444) were obtained from PlasmID (http://plasmid.med.harvard.edu/PLASMID/). The constructs encoding cDNAs of GABRG2 (clone ID: 5288256; MHS6278-202808792 glycerol stock) and mNPR (clone ID: 6809324; MMM1013-202732095 glycerol stock) were obtained from Dharmacon. The cDNA of hNPR-Flag was constructed from the HsCD00083063 by PCR-based method and cloned into the HindIII and XbaI of pCMV5 to yield pCMV5-NPR-Flag. The multicloning site of pEBMulti-Neo (Wako) was swapped with new multicloning site containing KpnI-XhoI-BamHI-SalI-BsrGI-NotI to yield pEB1. The cDNA of IRES-GFP was transferred from pL316 plasmid to pEB1 using BamHI and BsrGI to yield pEB1;IRES-GFP. For secretory protein production and for the elimination of transmembrane domain, 105 bp cDNA of mNPR from the clone 6809324 was swapped with the 66 bp of mNP1 to yield NPR-ECD-Flag. The NPR-ECD-Flag was cloned into between XhoI and BamHI of pEB1;IRES-GFP to yield pEB1-NPR-ECD-Flag;IRES-GFP. The His tag (6X histidines) was added after Flag tag by PCR into the pEB-NPR-ECD-Flag;IRES-GFP to yield pEB-NPR-ECD-Flag-His;IRES-GFP. The cDNA of hNP1-Flag-His and hNARP2-Flag-His was constructed from HsCD00296108 and HsCD00040444 by PCR-based method and cloned into the SalI and BamHI of pEBMulti-Neo to yield pEB-NP1-Flag-His and pEB-NARP-Flag-His, respectively. The cDNA of hNARP from HsCD00040444 was transferred to pLX304 by Gateway cloning method to yield pLX304-NARP-V5. The cDNA of hNPR was amplified from pCMV5-NPR-Flag by PCR and cloned into the EcoRI of pFSW67; IRES-GFP, which contains the human synapsin promoter to drive expression, to yield pFSW67-NPR-Flag;IRES-GFP. The NTD of GluA1 were amplified from pCIneo-GluA1-NTD-Fc by PCR and cloned into the NheI-HindIII of pAP5-tag5 (GenHunter) and into the XmaI-SalI of pDisplay (Invitrogen) to yield pAP5-GluA1-NTD-AMH and pDis-GluA1-NTD, respectively. The expression vectors for NPR-LC-Flag, NPR-LC,CC-Flag, and NPR-PTX-Flag were constructed from pCMV5-NPR-Flag by PCR-based domain deletion and cloned into the SacI-XbaI of pCMV5 to yield pCMV5-NPR-LC-Flag, pCMV5-NPR-LC,CC-Flag, and pCMV5-NPR-PTX-Flag, respectively.
Preparation of soluble recombinant proteins.
Soluble recombinant NPR-ECD-Flag, NPR-ECD-Flag-His, NP1-Flag-His, NARP-Flag-His, GluA1-NTD-AMH, and GluA1-NTD-Fc were prepared as described previously (Uemura and Mishina, 2008). Briefly, the cell medium was collected 5 d after the respective expression vectors were transfected into the FreeStyle 293 cells (Invitrogen). And NPR-ECD-Flag was purified by anti-Flag antibody (M2)-conjugated agarose (Sigma), eluted with Flag peptides and dialyzed with HBSS. His-tagged proteins were purified by Talon metal affinity resin (Clontech) with manufacturer's instructions and dialyzed against HBSS (Invitrogen). Fc-tagged proteins were purified by rProteinA Sepharose Fast Flow (GE Healthcare), eluted with 10 mm glycine, pH 2.5, and dialyzed with HBSS. Protein concentrations were estimated by SDS-PAGE and Coomassie staining together with BSA as the quantitative standard.
Heterologous synapse formation assay.
Assays were performed essentially as described previously (Uemura and Mishina, 2008). Expression vectors were transfected into HEK293 cells. Cultured hippocampal neurons (DIV16) from neonatal mice at P0 were cocultured with the transfected HEK293 cells. After 24 h, cells were fixed with 4% PFA and immunostained with mouse or rabbit anti-Flag (Sigma; 1:1000 both) together with mouse anti-PSD95 (Sysy; 1:500), mouse anti-Bassoon (Stressgen; 1:1000), mouse anti-panShank (NeuroMab; 1:500), rabbit anti-Homer (Sysy; 1:500), rabbit anti-Synapsin (EO28; 1:1000, Hosaka and Südhof, 1999), rabbit anti-GluA1 (Millipore; 1:500), mouse anti-GluA2 (Millipore; 1:500), mouse anti-Gephyrin (Sysy; 1:1000), or mouse anti-Collybistin (R&D Systems; 10 μg/ml) antibodies, respectively. Species-specific AlexaFluor-488-, -546-, and -633-conjugated antibodies (Invitrogen) were used as secondary antibodies. The same experiments were performed in the presence of soluble 100 μg/ml of GluA1-NTD-Fc. Immediately before seeding the HEK293 cells onto neurons, the culture medium of neurons was replaced with fresh culture medium containing the recombinant proteins. The same experiments were also performed in the presence of 20 μm of NBQX, 500 nm TTX, 50 μm of picrotoxin (PTX) or vehicles as controls of those. Immediately before seeding the HEK293 cells onto neurons, the culture medium of neurons was replaced with fresh culture medium containing the indicated drugs.
Cell surface binding assay.
Expression vectors for NPR-Flag, NARP-V5, GluA1-GFP, GluA2-GFP, GluD2-GFP, stargazin, Dis-GluA1-NTD, and domain mutants of NPR-Flag were transfected into HEK293 cells. Transfected cells expressing NPR-Flag were incubated with 100 μg/ml GluA1-NTD-Fc, the transfected cells expressing GluA1-GFP, GluA2-GFP, or GluD2-GFP with stargazin were incubated with 50 μg/ml NPR-ECD-Flag in the absence or the presence of 20 μm NBQX, and the transfected cells expressing Dis-GluA1-NTD were incubated with 50 μg/ml of NP1-Flag-His or NARP-Flag-His in HBSS containing 2 mm CaCl2 and 1 mm MgCl2 for 2 h at room temperature. After washing, cells were fixed with 4% PFA and immunostained with mouse or rabbit anti-Flag (sigma; 1:1000 both), mouse anti-V5 (Invitrogen; 1:1000), followed by incubation with 488-conjugated anti-human Fcγ and species-specific AlexaFluor-488-, -546-, or -633-conjugated secondary antibodies (Invitrogen).
Cell aggregation assay.
FreeStyle HEK cells were cotransfected with two indicated different expression vectors, pEGFP with pCMV5-NPR-PTX-Flag and pDsRed with pDis-GluA1-NTD, following the manufacturer's instructions. After 2 d, the cells were mixed together and incubated at 37°C with shaking on Orbi-Shaker (Benchmark) with 125 rpm for 2 h. Cells were carefully collected, cells were dropped onto microscope slides (Denville Scientific), and fluorescence images were taken with a fluorescence stereomicroscope (A1RSi confocal, Nikon).
Pull-down assay.
Soluble recombinant proteins were mixed and incubated in HBSS containing 2 mm CaCl2 and 1 mm MgCl2 for 3 h or overnight at 4°C followed by incubation with anti-Flag antibody (M2)-conjugated agarose (Sigma) for 30 min at 4°C. Subsequently, the agarose suspensions were washed four times with HBSS containing 2 mm CaCl2 and 1 mm MgCl2. Bound proteins were eluted by boiling in SDS sample buffer, separated by SDS-PAGE gel, and analyzed by Western blotting with mouse anti-Myc (DHSB; 1:1000) or rabbit anti-Flag (Sigma; 1:1000) antibodies, respectively. Membranes then were washed with 0.05% TBS-Tween and incubated with fluorescence-labeled secondary antibodies (donkey anti-rabbit IR Dye 800CW, 1:5000; donkey anti-mouse IR Dye 680CW, 1:5000; LI-COR Bioscience) and signals were detected with an Odyssey Infrared Imager and Odyssey software (LI-COR Biosciences). For mass spectrometry analysis, recombinant GluA1-NTD-Fc was mixed with proteins from mice brain homogenate and were separated by 10% SDS-PAGE and silver-stained to visualize the interaction proteins. The selected gel band was cut and sent to protein facility at Peking University for mass spectrometry analysis.
Lentivirus preparations.
Lentiviruses were produced as described previously (Pang et al., 2010). For all lentiviral vectors, viruses were produced in HEK293 cells (ATCC) by cotransfection with three helper plasmids (3.25 μg of pRSV-REV, 8.1 μg of pMDLg/pRRE, and 4.5 μg of vesicular stomatitis virus G-protein expression vector) and 10 μg of lentiviral vector DNA per 75 cm2 culture area using calcium phosphate method (Chen and Okayama, 1987). Lentiviruses were harvested from the medium 48 h after transfection.
Electrophysiological recordings.
Whole-cell voltage-clamp patch recording for neuron and HEK 293 was performed with a MultiClamp 700A amplifier (Molecular Devices). Synaptic responses were evoked through a concentric bipolar electrode (FHC) by 0.5 ms current injection (100 μA) using an Isolated Pulse Stimulator (A-M Systems). Data were digitized at 10 kHz with a 2 kHz low-pass filter. Hippocampal neurons (DIV13-DIV15) were maintained with an external solution containing (in mm) NaCl 150, KCl 4, MgCl2 1, CaCl2 2, HEPES 10, d-glucose 10, with pH adjusted to 7.3–7.4, Osm 315–320 during recording. Whole-cell patches were recorded with 3–5 mΩ borosilicate glass pipettes (World Precision Instruments). The internal solution contained (in mm) KCl 145, NaCl 5, HEPES 10, EGTA 5, MgATP 4, Na2GTP 0.3, with pH adjusted to 7.2, Osm 305. Evoked EPSCs were recorded in bath solution containing 100 μm PTX. Evoked IPSCs were recorded in bath solution containing 10 μm CNQX. The readily releasable vesicle pool was measured by background perfusion of hypertonic sucrose (0.5 m) at a speed of 2 ml/min). HEK293 cells were maintained with an external solution containing (in mm) NaCl 144, KCl 10, MgCl2 1, CaCl2 2, HEPES 10, d-glucose 10, with pH adjusted to 7.3–7.4, Osm 315–320 during recording. The 10 mm glutamate was perfused to the bath solution to record the glutamate-induced currents. The data were analyzed using Clampfit 9.02 (Molecular Devices), Igor 4.0 (WaveMetrics), and Prism 5 (GraphPad Software).
Small hairpin RNAs (shRNAs) and RT-PCR analysis.
The constructs encoding shRNAs against npr were purchased from MISSION shRNA Library (Sigma). The sense sequences of shRNA were as follows: sh472, 5′-GACAGCAACUGGCACCAUAUC-3′; sh473, 5′-CAAGCCACACGGCAUCCUUAU-3′; sh474, 5′-GAUACCUUGGGAGGCCGAUUU-3′; sh475, 5′-CUUGGUCUCUCCCAUCAUAUA-3′. The efficiencies of NPR shRNAs were determined in cortical neurons. At DIV4, the lentiviruses encoding the shRNAs were infected into neurons, and mRNAs were extracted at DIV16. For qPCR analyses of cultured cells, total RNA was isolated from the cultured neurons using RNAqueous Kit (Ambion) following the manufacturer's instructions, treated with DNase (Ambion), and reverse-transcribed and PCR-amplified using one-step reaction (Roche Lightcycler 480 RNA master hydrolysis probes). mRNA levels were quantified by real-time PCR assay using the 7900HT Fast real-time PCR system and RQ analysis software (Applied Biosystems). The real-time PCR PrimeTime assays for npr were performed with purchasing the assay set of Mm.PT.58.11296212.g from Integrated DNA Technologies.
Primary cultures of neurons and exogenous gene delivery.
Mouse primary neurons were prepared as described previously (Maximov and Südhof, 2005). Briefly, the primary cortical or hippocampal neurons were isolated from P0 mice, dissociated by papain digestion, and plated on poly-l-lysine-coated circle glass coverslips. The neurons were cultured for 16 d before experiments. For gene delivery into neurons, they were infected with lentiviruses captured expression vectors encoding Mock, NPR-Flag, or shRNAs (sh472 and sh474) at DIV4. All steps were performed under level II biosafety conditions.
For electrophysiological recordings, hippocampal neurons were cultured from newborn mice pups (Zhang et al., 2010). Neurons were plated on poly-d-lysine-coated glass coverslips and maintained in 5% CO2 at 37°C for 13–15 d before the experiments. Hippocampal neurons were infected with lentivirus at DIV4.
Immunostaining primary neurons.
At DIV16, neurons were fixed with 4% PFA for 15 min at room temperature. For staining surface GluA1 (GluA1surface) or surface GABA(A) receptor γ-2 (GABRG2surface) on neurons, before permeabilization, the cells were blocked with 5% goat serum for 30 min, mouse anti-GluA1 antibody (NeuroMab; 1:500), or mouse anti-GABRG2 antibody (Millipore; 1:250) was added and incubated for 1 h, respectively. Then, the cells were permeabilized with 0.2% Triton X-100 for 10 min. After blocking with 5% goat serum, cells were immunostained with rabbit anti-GluA1 (Millipore; 1:1000), mouse anti-GABRG2 antibody (Millipore; 1:250), chicken anti-MAP2 (EnCor Biotechnology; 1:500), mouse anti-PSD95 (NeuroMab; 1:500), mouse anti-panShank (NeuroMab; 1:500), and rabbit anti-synapsin (EO28; 1:1000) (Hosaka and Südhof, 1999), mouse anti-Gephyrin (Sysy; 1:1000), or guinea pig anti-VGAT (Sysy; 1:500), followed by incubation with AlexaFluor-488-conjugated anti-mouse, anti-rabbit, anti-guinea pig, AlexaFluor-546-conjugated anti-mouse, anti-chicken, and AlexaFluor-633-conjugated anti-chicken antibodies.
Immunoblotting.
At DIV16, neurons were collected with RIPA lysis buffer (10 mm Tris-Cl pH 8.0, 1 mm EDTA, 0.5 mm EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mm NaCl, protease inhibitor mixture). Two days after transfection, HEK293 cells were collected with subcellular fractionation buffer (250 mm sucrose, 20 mm HEPES, pH 7.4, 10 mm KCl, 1.5 mm MgCl2, 1 mm EDTA, 1 mm EGTA, protease inhibitor mixture) and lyzed with a 25 G syringe needle. Supernatant after centrifugation with 720 × g was collected and centrifuged again with 10,000 × g. Obtained supernatants were further centrifuged with 100,000 × g to separate cytosol and membrane proteins. Proteins were separated by 5%–20% gradient Tris-glycine gel and analyzed by Western blotting with rabbit anti-GluA1 (Millipore; 1:1000), mouse anti-GluA2 (Millipore; 1:1000), mouse anti-PSD95 (NeuroMab; 1:500), mouse anti-Gephyrin (Sysy; 1:5000), rabbit anti-NP1 (Thermo Scientific; 1:1000), rabbit anti-NARP (Thermo Scientific; 1:1000), sheep anti-NPR (R&D Systems; 1:250), and mouse ant-Tuj1 (BioLegend; 1:1000) antibodies, followed by incubation with fluorescence-labeled secondary antibodies (donkey anti-mouse or anti-rabbit IR Dye 680CW or IR Dye 800CW, 1:5000; donkey anti-sheep IR Dye 800CW, 1;5000; LI-COR Bioscience), were used and signals were detected with an Odyssey Infrared Imager and Odyssey software (LI-COR Biosciences). The total intensity values for calculated by Odyssey software and each values normalized to Tuj1.
HEK293 cell culture and transfection for electrophysiology.
HEK293 cells were used for both expressing GluA1, stargazin, control, GABRG2 and pentraxins (NPR-ECD, NPR, NP1, NARP) and generating soluble pentraxins. Cells were cultured in incubator supplied with 5% CO2 and maintained at 37°C. HEK 293 cells were cultured in 35 mm dishes before transfection. Total cDNA used for transfection per 35 mm dish was 3 μg with the 2:3 ratio for GluA1 and stargazin. Three hours after transfection, the cells were dissociated with 0.05% trypsin and plated on coverslips pretreated with poly-d-lysine. All HEK293 cell transfections were performed using polyethylenimine (Polysciences). Patch-clamp recordings were performed 24–48 h after transfection.
Immunostaining HEK293 cells.
Surface GluA1 or surface GABRG2 was labeled using nonpermeabilized immunostaining. Total levels of GABRG2 were labeled using permeabilized immunostaining. Transfected HEK293 cells were washed once with PBS, followed by fixation for 10–12 min at room temperature with 4% PFA. After washing 3 times using PBS, cells were blocked using PBS containing 5% milk and 3% goat serum for 30 min at room temperature. The cells were then incubated for 1 h at room temperature with rabbit anti-GluA1 (Millipore; 1:100) or mouse anti-GABRG2 (Sysy; 1:1000) antibodies diluted in the blocking solution. A secondary antibody (Invitrogen) was used after washing 3 times with PBS. Fluoromount-G (Southern Biotechnology) was used to mount the cells on microscope slides. The images were taken with a laser scanning confocal microscope and analyzed using the NIH ImageJ program blindly.
Image acquisition and quantification.
Images were taken with a confocal laser-scanning microscope (A1RSi confocal, Nikon) under constant conditions as to laser power, iris, gain, z-steps, and zoom setting throughout the same set of experiments, and all quantitative measurements were performed with ImageJ 1.50b software. For the quantification of GluA1surface, GluA1total, GABRG2surface, GABRG2total, PSD95, Shank, Synapsin, Homer, VGAT, and Gephyrin puncta along dendrites, 8 z-series optical sections per each observation were first run VAMP2D to get accurate puncta structure (Dumitriu et al., 2012) and projected by the brightest point. After setting “threshold” level for each set of experiments, contiguous puncta were separated from each other by the “segmented particles” tool, and the size and signal intensity of each punctate were read by the “analyze particles” tool. The quantification was performed in a blind manner with respect to the lentiviruses applied to the cultures. Images of HEK293 cells expressing exogenous genes were collected from 3 or 4 separate experiments. The intensities of immunostained signals were measured as the optical mean density within the manually traced area enclosing the HEK293 cells. Statistical significance was evaluated by one-way ANOVA followed by post hoc Tukey's test, or Student's t test.
Results
NPR induces both excitatory and inhibitory heterologous synapses
We examined whether NPR, which differ from the secreted NPs NP1 and NARP, is a membrane protein anchored on the cell surface and might be synaptogenic in the heterologous synapse formation assay. We expressed NPR in HEK293 cells, cocultured the HEK293 cells with dissociated hippocampal neurons from newborn mice, and examined the accumulation of presynaptic or postsynaptic puncta on the surface of the HEK293 cells as a proxy for synapse formation (Scheiffele et al., 2000; Graf et al., 2004). We detected no accumulation of presynaptic bassoon- or synapsin-positive puncta on NPR-expressing HEK293 cells, whereas neuroligin-1-expressing HEK293 cells, used as a positive control, abundantly induced presynaptic specializations in the same experiments (Fig. 1A). When we examined postsynaptic markers, however, we observed massive accumulation of PSD95- or Shank-positive puncta on NPR-expressing HEK293 cells, equivalent to the postsynaptic puncta induced by neurexin-1β, a presynaptic cell adhesion molecule (Fig. 1B) (Ushkaryov et al., 1992). Moreover, we found that NPR-dependent postsynaptic puncta abundantly contained the AMPARs GluA1 and GluA2 (Fig. 1C). Although neurexin-1β-expressing cells also attracted AMPAR-containing puncta, NPR was 2–3 times as effective as neurexin-1β in recruiting AMPARs to synaptic sites.
Figure 1.
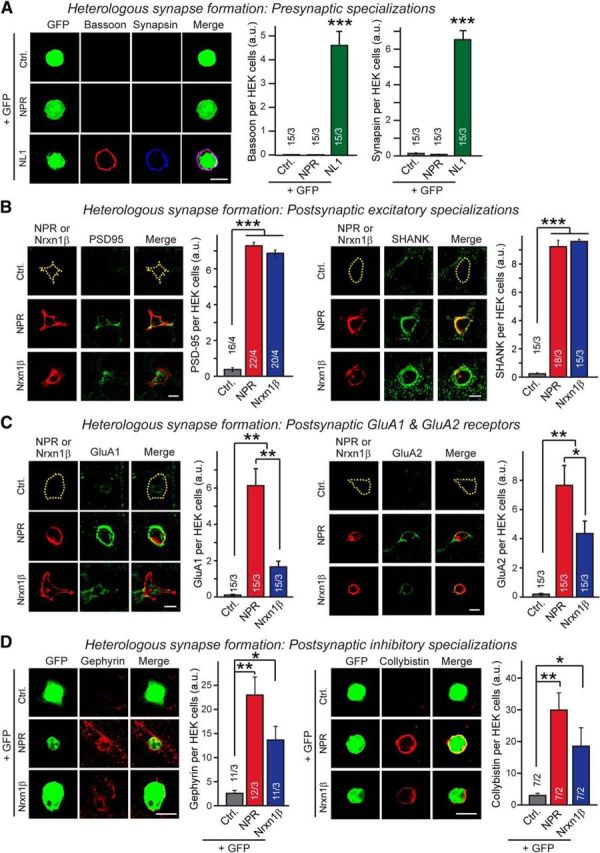
NPR potently induces both excitatory and inhibitory postsynaptic specializations. A, Heterologous synapse formation assay analyzed for presynaptic specializations. Hippocampal mouse neurons were cocultured with HEK293 cells expressing GFP and either no other protein (Ctrl.), or Flag-tagged NPR or HA-tagged neuroligin-1 (NL1; positive control) at DIV16. After 24 h of HEK/neuron cocultures, as shown in the representative images (left), presynaptic terminals formed on HEK293 cells were analyzed by immunocytochemistry for bassoon (red) and synapsin (blue). Right, Bar graphs, Quantifications of the bassoon (left) and synapsin staining intensities (right) from three independent experiments. B, Same as A, except that HEK293 cells were transfected with Flag-tagged neurexin-1β (Nrxn1β, using SS4+ variant) instead of NL1 as a positive control, and that heterologous synapses were analyzed for excitatory postsynaptic specializations using antibodies to PSD95 (left panels) and Shank (right panels). C, Same as B, except that heterologous synapses were analyzed by immunocytochemistry in permeabilized fixed cells for GluA1 and GluA2 proteins. NPR is more efficient in recruiting AMPARs than neurexin-1β. D, Same as B, except that that heterologous synapses were analyzed for inhibitory postsynaptic specializations using antibodies to gephyrin (left panels) and collybistin (right panels). Scale bars, 10 μm. Data are mean ± SEM. Bars represent the number of HEK293 cells/experiments analyzed. *p < 0.05 (one-way ANOVA test with Tukey's post hoc test). **p < 0.01 (one-way ANOVA test with Tukey's post hoc test). ***p < 0.001 (one-way ANOVA test with Tukey's post hoc test).
The data in Figure 1B, C suggest that NPR promotes formation of heterologous excitatory synapses, with an intrinsic activity comparable to that of neurexin-1β, but with a stronger recruitment of AMPARs, which is consistent with the interaction of pentraxins with AMPARs (O'Brien et al., 1999, 2002). When we probed heterologous synapses for the presence of inhibitory synapses, we were surprised to observe robust formation of postsynaptic puncta positive for gephyrin and collybistin (Fig. 1D). This finding was unexpected because it suggests a general synaptogenic activity of NPR that is not specific for excitatory synapses, whereas current concepts view NPs as selective modulators of excitatory synaptic transmission by binding to AMPARs (Chang et al., 2010; Gu et al., 2013; Pelkey et al., 2015).
Neuronal pentraxins increase AMPAR function
Given the induction of both excitatory and inhibitory synapses by NPR, we wanted to confirm that NPs indeed bind to AMPARs. Using cell-surface binding assays and recombinant proteins, we found that NPR, NP1, and NARP specifically interacted with the NTDs of GluA1 and GluA2 (Fig. 2). By assembling into heteropentamers with secreted NP1 and NARP, NPR can potentially nucleate a surface-bound complex containing NP1 or NARP (Kirkpatrick et al., 2000; Xu et al., 2003). Indeed, when we added soluble V5-epitope-tagged NARP and GluA1-NTD to NPR-expressing HEK293 cells, NARP bound together with the GluA1-NTD to NPR (Fig. 2A).
Figure 2.
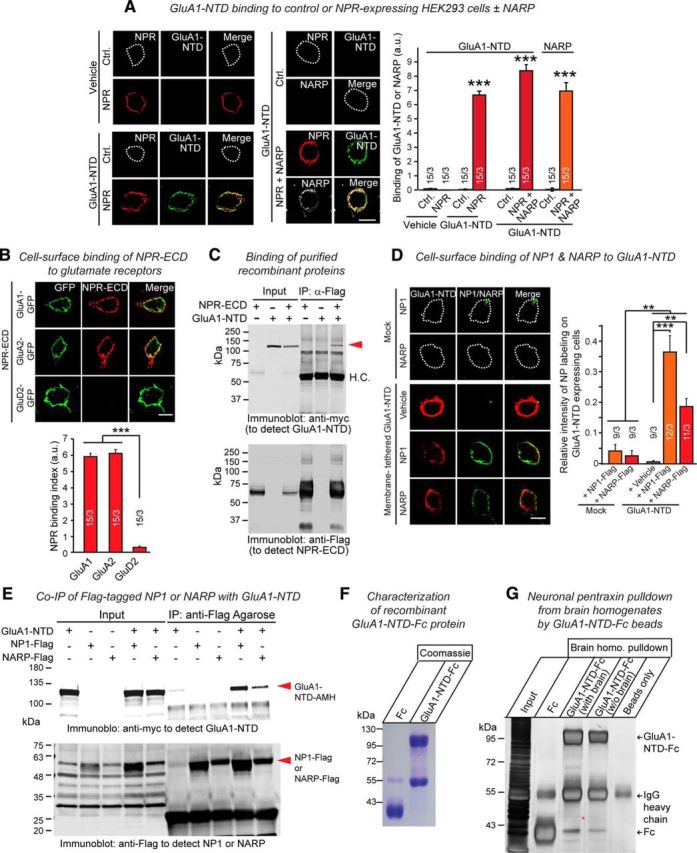
The extracellular domains of neuronal pentraxin receptor (NPR-ECD) and the secreted neuronal pentraxins NP1 and NARP (NP2) directly bind to GluA1-NTD. A, Recombinant GluA1-N-terminal domain (GluA1-NTD) binds to either NPR alone or NPR-NARP complexes expressed in HEK293 cells. Control HEK293 cells (Ctrl.) or HEK293 cells expressing Flag-tagged NPR-Flag or together with V5-tagged NARP were incubated with control medium or medium containing recombinant GluA1-NTD fused to the human Fc-domain for 2 h, and immunostained with anti-Flag (red), anti-Fc (green), and anti-V5 antibodies (white) without permeabilization. Bar graphs represent staining intensities, demonstrating that GluA1-NTD binding is similarly observed in the absence or presence of NARP. B, Recombinant NPR extracelluar domain (NPR-ECD) binds to GluA1 and GluA2, but not GluD2 expressed in HEK293 cells. HEK293 cells transfected with GluA1-GFP, GluA2-GFP, or GluD2-GFP (all together with stargazin to facilitate GluA receptor function) (Chen et al., 2000) were incubated for 2 h at room temperature with vehicle or recombinant NPR extracellular domains (NPR-ECD, composed of the extracellular sequences NPR with a C-terminal Flag- and His-tag). Cells were immunostained with anti-Flag antibody. NPR-ECD (red) binds to both HEK293 cells transfected GluA1-GFP or GluA2-GFP but not GluD2-GFP. Bar graphs represent intensities of staining signals for NPR-ECD on the surface of HEK293 cells expressing GluA1-GFP, GluA2-GFP, or GluD2-GFP as labeled. C, Recombinant NPR-ECD binds to GluA1-NTD. The purified recombinant proteins with indicated combinations using NPR-ECD with C-terminal Flag-tag (NPR-ECD-Flag) and GluA1-NTD with C-terminal alkaline phosphatase-, Myc-, and His-tag (GluA1-NTD-AMH) were mixed and incubated for 3 h or overnight at 4°C with rotating. NPR-ECD-Flag was pulled down with anti-Flag agarose, and the precipitates were analyzed by immunoblotting with anti-Flag and anti-Myc antibodies. D, Cell surface binding assay reveals direct binding of recombinant NP1 and NARP to HEK293 cells expressing membrane-tethered GluA1-NTD on their surface. Control-transfected HEK293 cells or HEK293 cells transfected with a plasmid encoding the HA-tagged GluA1-NTD fused to the transmembrane region of the PDGF receptor as a membrane tether were incubated with vehicle or NP1 or NARP (both C-terminally tagged with a Flag-epitope and a hexa-histidine sequence for purification; 50 μg/ml each) for 2 h at room temperature. Cells were immunostained with anti-Flag (NP1 or NARP; green) or anti-HA antibodies (GluA1-NTD; red), and the staining intensity was quantified. Bar graphs represent intensities of staining signals for the vehicle NP1-Flag-His or NARP-Flag-His on the surface of HEK293 cells expressing membrane-tethered GluA1-NTD-HA. E, Recombinant NP1 or NARP with GluA1-NTD. The purified recombinant proteins with indicated combinations using NP1 or NARP with both C-terminal Flag- and His-tag (NP1-Flag-His or NARP-Flag-His, respectively) and GluA1-NTD-AMH were mixed and incubated for 3 h or overnight at 4°C with rotating. NP1-Flag-His or NARP-Flag-His was pulled down with anti-Flag agarose, and the precipitates were analyzed by immunoblotting with anti-Flag and anti-Myc antibodies. F, Purification of recombinant GluA1-NTD-Fc and Fc from medium of transfected HEK293T cells. GluA1-NTD-Fc encodes the extracellular NTD domain of GluA1 (ending at … DRYEGY) following Fc domain. G, Brain proteins pulled down with GluA1-NTD-Fc and Fc were separated by 10% SDS-PAGE and silver-stained. Asterisk indicates sequenced band identified as NP1 and NPR. Scale bars, 10 μm. Data are mean ± SEM. Numbers in bars indicate number of HEK293 cells/experiments analyzed. **p < 0.01 (one-way ANOVA test with Tukey's post hoc test). ***p < 0.001 (one-way ANOVA test with Tukey's post hoc test).
We next asked whether NPs might directly influence AMPAR function. We coexpressed GluA1 with stargazin in HEK293T cells to ensure efficient surface-transport of GluA1 (Chen et al., 2000), and additionally coexpressed EGFP (control), NARP, NP1, or NPR. For NPR, we used both the secreted extracellular NPR domains (NPR-ECD) and full-length membranous NPR. We then measured the AMPAR response to bath-applied glutamate. In control cells, glutamate induced an initial fast peak current followed by a plateau current (Fig. 3A). Surprisingly, all NPs significantly increased the glutamate-induced peak and plateau currents, suggesting that NPs enhance AMPAR function in this experimental setup (Fig. 3A). To test whether NPs increase glutamate-induced GluA1 currents by directly binding to surface-exposed GluA1, we incubated HEK293T cells expressing GluA1 and stargazin with soluble recombinant NPs (Fig. 3B). When we measured glutamate-induced currents, we found again that addition of NPs to the medium increased AMPAR currents similar to coexpressed NPs. These results suggest that NPs directly activate surface AMPARs, either by increasing the intrinsic activity of AMPARs or by stabilizing them on the cell surface. In support of the second hypothesis, we found that coexpression of soluble NPs with GluA1 significantly increased GluA1 surface levels in HEK293 cells without affecting total GluA1 protein levels (Fig. 4A–C). However, we did not observe a similar effect of NPs on the membrane localization of GABAR, at least as far tested with one particular isoform GABA(A) receptor γ-2 (GABRG2). Coexpression of NPs with GABRG2 showed no significant changes of GABRG2 surface levels in HEK293 cells (Fig. 4D–F).
Figure 3.

NPR, NP1, and NARP binding enhances GluA1 function. A, Glutamate-induced currents in HEK293T cells expressing GluA1 without or with neuronal pentraxins. Left, Sample traces. Right, Summary graphs of the peak and plateau amplitudes. Currents were induced by a 2 s application of 10 mm K-glutamate (GluK) to patched HEK293T cells cotransfected with GluA1 and stargazin expression vectors, and with a control plasmid (Ctrl.) or plasmids encoding Flag-tagged neuronal pentaxins. Both NPR and extracellular domains of NPR (NPR-ECD) were used because NPR physiologically binds to GluA1 in trans. B, Same as A, except that HEK cells were transfected only with GluA1 and stargazin expression plasmids, and secreted neuronal pentraxins were added to the medium for 24 h before recordings. Recombinant neuronal pentraxins were obtained in the medium of separately transfected HEK293T cells that secrete the proteins into the medium, using medium from control transfected HEK293T cells as a control. Data are mean ± SEM. Numbers in bars indicate number of HEK293 cells/experiments analyzed. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test).
Figure 4.
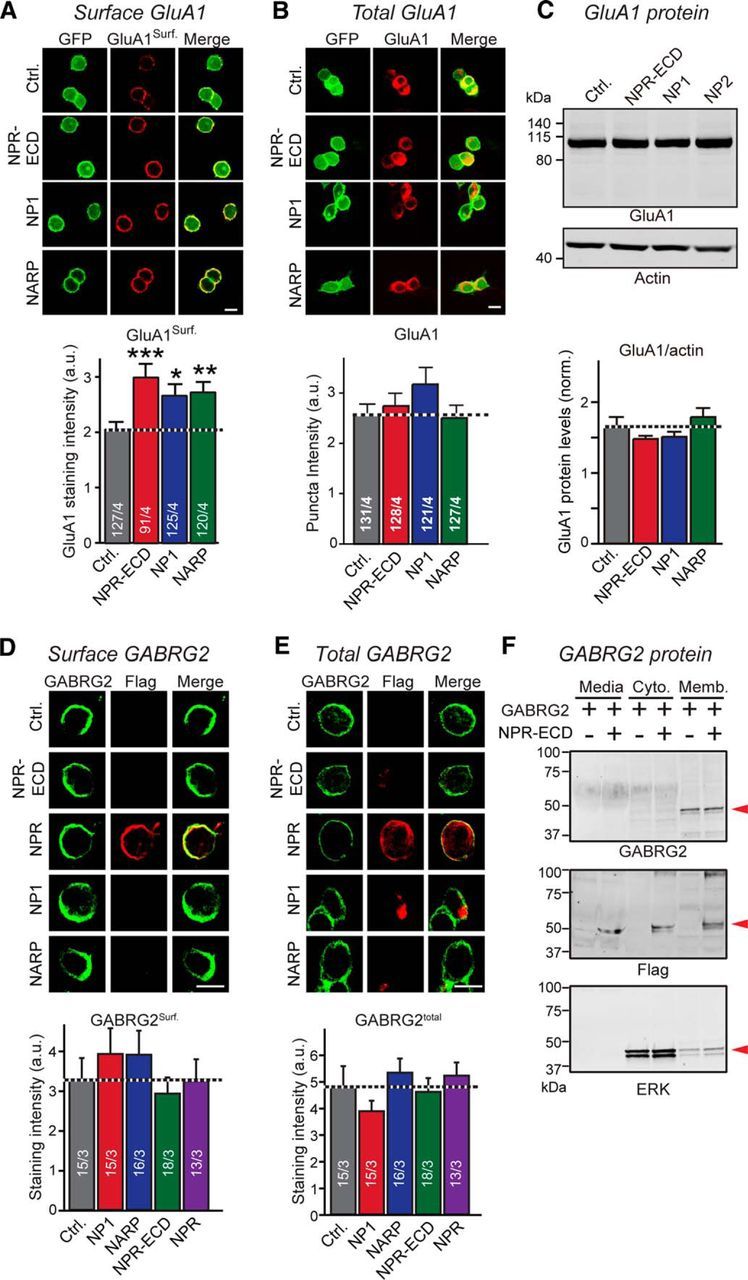
NPR, NP1, and NARP binding increases surface levels of GluA1, but not of GABRG2 in HEK293T cells. A, Neuronal pentraxins stabilize surface levels of GluA1 in HEK293T cells. Surface GluA1 levels were measured in HEK293T cells expressing NPR-ECD-Flag, NP1-Flag-His, NARP-Flag-His, or control plasmid together with GluA1-IRES GFP and stargazin-YFP. Nonpermeabilized cells were stained with an antibody that recognizes the GluA1-NTD. Top, Representative images. Bottom, Summary graph. B, Same as A, except measuring total level of GluA1 in HEK293T cells transfected. Transfected cells were stained with permeabilization using a GluA1 antibody. Top, Representative images. Bottom, Summary graph. C, Total GluA1 levels are unchanged by neuronal pentraxins. Total GluA1 protein levels were measured in HEK293T cells transfected as in A and B by quantitative immunoblotting, using actin as an internal control. Top, Representative blots. Bottom, Summary graph. D, Neuronal pentraxins do not affect surface stabilization of GABRG2 in HEK293 cells. Surface GABRG2 levels were measured in HEK293 cells expressing NP1-Flag-His, NARP-Flag-His, NPR-ECD-Flag, NPR-Flag, or control plasmid together with GABRG2. Nonpermeabilized cells were stained with an antibody recognizes the GABRG2 (green) and Flag (red). Top, Representative images. Bottom, Summary graph. E, Same as D, except measuring total level of GABRG2 in HEK293 cells transfected. Transfected cells were stained with permeabilization using a GABRG2 antibody. Top, Representative images. Bottom, Summary graph. F, Extracellular (Media), membranous (Memb.), and cytosolic (Cyto.) protein levels of GABRG2 were measured. Lysates from HEK293 cells expressing GABRG2 together with NPR-ECD-Flag or control plasmid were collected after subcellular fractionation. GABRG2, NPR-ECD, and ERK (subcellular fraction marker) were observed by immunoblotting (red arrowheads). GABRG2 mainly localize cell membrane and were hardly affected by NPR-ECD expression. Scale bars, 10 μm. Data are mean ± SEM. Numbers in bars indicate number of HEK293 cells/experiments analyzed. *p < 0.05 (Student's t test). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test).
Binding of the pentraxin domain of NPR to the NTD of GluA1 mediates cell adhesion and induces heterologous synapse formation
NPR thus induces excitatory heterologous synapse formation with preferential accumulation of AMPARs, binds to AMPARs, and stabilizes AMPARs on the cell surface. NPs share three sequence regions, an N-terminal domain of relatively low complexity, a central short coiled-coil domain, and a large C-terminal pentraxin domain that is homologous to other pentraxin domains, but exhibits only modest identity with these domains (Fig. 5A). NPR additionally contains an N-terminal transmembrane region (TMR) preceded by a short cytoplasmic sequence. Using HEK293 cells expressing various domains of NPR tethered to its TMR, we measured binding of recombinant soluble GluA1-NTD, produced as an IgG-Fc-domain fusion protein of various NPR domains (Fig. 5B). We observed efficient binding of GluA1-NTD to the pentraxin-domain of NPR, but not to its other domains.
Figure 5.
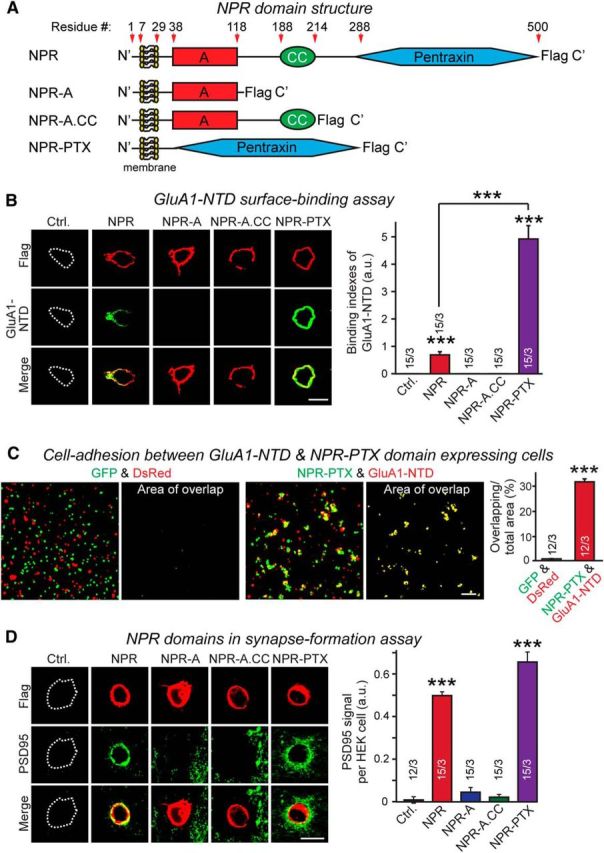
The pentraxin domain of NPR mediates binding to GluA1-NTD and trans-cellular adhesion between cells expressing NPR and GluA1, and induces postsynaptic specializations during heterologous synapse formation. A, Schematic structures of NPR and of various NPR truncation mutants. NPR comprises an N-terminal TMR, an A domain containing low-complexity sequences, a predicted coiled-coil domain (CC), and a C-terminal pentraxin domain that accounts for nearly half of the entire protein. All NPR constructs contained a C-terminal Flag epitope. B, Recombinant GluA1-NTD (expressed as a soluble Fc-domain fusion protein) efficiently binds to HEK293 cells expressing full-length NPR or the pentraxin domain of NPR, but not to NPR proteins lacking the pentraxin domain. Left, Representative images. Right, Summary graph. The apparently stronger binding of GluA1-NTD to the NPR pentraxin domain as opposed to full-length NPR is likely due to increased expression levels between the two constructs. C, Cells expressing the membrane-tethered NPR pentraxin domain (NPR-PTX) readily form cell adhesion complexes with cells expressing the membrane-anchored GluA1-NTD. Separate sets of HEK293 cells were transfected with EGFP or DsRed (control), or cotransfected with EGFP and NPR-PTX or DsRed and GluA1-NTD, cells were mixed, and cell adhesion was measured by imaging the overlap between EGFP and DsRed signals at low magnifications. Left, Representative images. Right, Summary graph. D, NPR pentraxin domain is sufficient to induce excitatory postsynaptic specialization during heterologous synapse formation. Hippocampal mouse neurons were cocultured with control HEK293 cells (Ctrl.), or HEK293 cells expressing Flag-tagged NPR proteins shown in A. Postsynaptic specializations formed on the HEK293 cells were analyzed by immunocytochemistry for the Flag epitope (red, to visualize the NPR proteins) and PSD95 (green, to visualize postsynaptic specializations). Left, Representative images. Right, Summary graphs of the PSD95 staining intensity. Scale bars: B, D, 10 μm; C, 100 μm. Graphs represent mean ± SEM. Numbers in bars indicate number of HEK293 cells/experiments analyzed. ***p < 0.001 (one-way ANOVA test with Tukey's post hoc test).
To test whether the interaction of NPR with GluA1 could induce an intercellular junction (i.e., whether GluA1 and NPR could function as heterophilic cell adhesion molecules), we mixed EGFP- and DsRed-labeled HEK293 cells that expressed the NPR pentraxin domain and the GluA1-NTD on the surface. We observed strong aggregation, confirming a trans-cellular interaction (Fig. 5C). Finally, we asked whether the membrane-tethered NPR pentraxin domain alone suffices to induce synapse formation when expressed in HEK293 cells, and observed abundant induction of postsynaptic specializations, equivalent to that obtained with full-length NPR (Fig. 5D).
Thus, the pentraxin-domain of NPR binds to AMPARs and mediates its synapse induction activity. Is it the actual binding of NPR to the AMPAR-NTDs that induces synapse formation? To address this question, we first tested whether addition of purified GluA1-NTD inhibits heterologous excitatory synapse formation induced by NPR. We observed strong inhibition of synapse formation; in those areas of HEK293 cells in which synapses still formed, no bound GluA1-NTD was detected, suggesting that the NTD-caused inhibition was incomplete in these experiments because GluA1-NTD binding was incomplete (Fig. 6A,B). Importantly, addition of GluA1-NTD had no effect on inhibitory synapse formation, suggesting that GluA1-NTD did not inhibit synapse formation by a steric mechanism because it still binds to the NPR on the HEK293 cells under these conditions (Fig. 6C).
Figure 6.
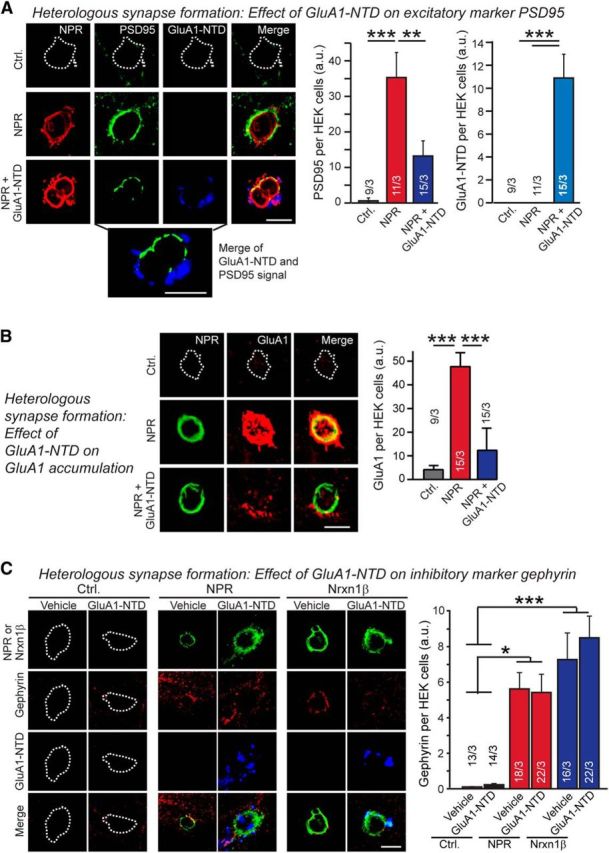
Recombinant GluA1-NTD inhibits NPR-mediated induction of postsynaptic excitatory, but not inhibitory, specializations during heterologous synapse formation. A, Addition of recombinant GluA1-NTD (added as a purified Fc-fusion protein) inhibits NPR-induced heterologous synapse formation as visualized by staining for the postsynaptic density marker PSD95. Control or NPR-expressing HEK293 cells were cocultured with hippocampal neurons (Fig. 1). HEK/neuron cocultures were performed in the absence and presence of 100 μg/ml GluA1-NTD for 24 h. The recombinant GluA1-NTD was added immediately before plating HEK cell on neurons at DIV16. Induced postsynaptic specializations on the HEK293 cells were assessed by staining for PSD95 and binding of the GluA1-NTD to the cells by staining for the Fc-domain that was fused to GluA1-NTD. Left, Representative images. Right, Quantifications of the PSD95 (left) and GluA1-NTD staining intensities (right). Expanded picture below representative images shows merged PSD95 and GluA1-NTD labeling under the NPR + GluA1-NTD condition to illustrate that two molecules are mutually exclusive, suggesting that the remaining PSD95 induction in the presence of GluA1-NTD is due to incomplete occupation of all NPRs on the cells. B, Same as A, except measuring the concentration of GluA1 receptors induced by NPR during heterologous synapse formation. C, Same as A, except measuring the induction of inhibitory synapses by staining for gephyrin. In addition, HEK293 cells expressing neurexin-1β were included as a positive control. Scale bars, 10 μm. Graphs represent mean ± SEM. Numbers in bars indicate number of HEK293 cells/experiments analyzed. *p < 0.05 (one-way ANOVA test with Tukey's post hoc test). **p < 0.01 (one-way ANOVA test with Tukey's post hoc test). ***p < 0.001 (one-way ANOVA test with Tukey's post hoc test).
AMPA and GABAR antagonists selectively inhibit excitatory and inhibitory heterologous synapse formation, respectively
Thus far, our results suggest that NPR promotes formation of both excitatory and inhibitory heterologous synapses, and that the former are induced by direct binding of NPR to AMPAR-NTDs. These results raise the possibility that simply recruiting AMPARs to a particular area of the neuronal plasma membrane is synaptogenic, and may mediate signaling via AMPAR-mediated ion fluxes. In support of this hypothesis, the AMPAR antagonist NBQX strongly inhibited NPR-induced heterologous excitatory synapse formation (Fig. 7A,B). NBQX had no effect on NPR-induced inhibitory synapse formation and did not alter heterologous excitatory or inhibitory synapse formation by neurexin-1β (Fig. 7A,B). Moreover, NBQX did not impair NPR binding to AMPARs, suggesting that its inhibition of heterologous excitatory synapse formation was due to its inhibition of AMPAR function (Fig. 7C). The GABAR antagonist PTX, conversely, selectively impaired heterologous inhibitory synapse formation by NPR but not by neurexin-1β (Fig. 8A,B). Thus, activation of neurotransmitter receptors appears to be essential for full heterologous synapse formation by NPR. However, heterologous synapse formation did not depend on neuronal network activity because silencing neurons with TTX did not impair NPR-induced heterologous excitatory synapse formation (Fig. 8C).
Figure 7.

AMPAR antagonist NBQX selectively inhibits NPR-mediated induction of excitatory and postsynaptic specializations during heterologous synapse formation but has no effect on neurexin-1β-induced heterologous synapse formation. A, NBQX impairs heterologous synapse formation induced by NPR, but not by neurexin-1β. Control HEK293 cells or HEK293 cells transfected with expression plasmids encoding Flag-tagged NPR or neurexin-1β (Nrxn1β, SS4+ variant) were cocultured with hippocampal neurons at DIV16. In the absence or presence of 20 μm NBQX added right before seeding the HEK cells onto neurons, heterologous synapses were analyzed by immunolabeling for GluA1 levels (blue) and for the postsynaptic density protein PSD95 (red) at DIV17, after 24 h of coculture. Left, Representative images. Right, Summary graphs of the PSD95 and GluA1 staining intensity. B, NBQX impairs excitatory, but not inhibitory, heterologous synapse formation. Experiments were performed as described for A with inclusion of neurexin-1β as a positive control, but heterologous synapses on a HEK293 cell were simultaneously analyzed for the excitatory postsynaptic marker Homer and the inhibitory postsynaptic marker gephyrin. HEK293 cells contain a mixture for both excitatory and inhibitory postsynaptic specializations. C, NBQX does not change binding levels of NPR-ECD to GluA1 and GluA2 expressed in HEK293 cells. HEK293 cells transfected with GluA1-GFP, GluA2-GFP, or GluD2-GFP (all together with stargazin to facilitate GluA receptor function) (Chen et al., 2000) were incubated for 2 h at room temperature with recombinant NPR extracellular domains (NPR-ECD, composed of the extracellular sequences NPR with a C-terminal Flag- and His-tag) in the presence or the absence of 20 μm NBQX. Cells were immunostained with anti-Flag antibody. NPR-ECD (red) binds to both HEK293 cells transfected GluA1-GFP or GluA2-GFP but not GluD2-GFP. Bar graphs represent intensities of staining signals for NPR-ECD on the surface of HEK293 cells expressing GluA1-GFP, GluA2-GFP, or GluD2-GFP as labeled. Scale bars, 10 μm. Graphs represent mean ± SEM. Numbers in bars indicate number of HEK293 cells/experiments analyzed. **p < 0.01 (one-way ANOVA test with Tukey's post hoc test). ***p < 0.001 (one-way ANOVA test with Tukey's post hoc test).
Figure 8.

GABAR antagonist PTX selectively inhibits NPR-mediated induction of inhibitory postsynaptic specializations during heterologous synapse formation but has no effect on neurexin-1β induced heterologous synapse formation. A, B, PTX impairs inhibitory heterologous synapse formation induced by NPR but not by neurexin-1β, as analyzed by staining heterologous synapses for gephyrin (A) or collybistin (B). Experiments were performed as described for A, except that the HEK293 cell/neuron cocultures were incubated with 50 μm PTX for 24 h and analyzed by staining for the indicated postsynaptic markers. C, Neuronal silencing with TTX does not impair NPR-induced heterologous excitatory synapse formation. Cocultures of neurons and HEK293 cells expressing GFP with Mock or NPR-Flag were incubated with or without 500 nm TTX. Top, Cells were immunostained with antibodies against GluA1 (red) or PSD95 (blue). Bar graphs represent intensities of staining signals for the GluA1 or PSD95 on the surface of HEK293 cells expressing Mock or NPR-Flag (bottom). Scale bars, 10 μm. Graphs represent mean ± SEM. Numbers in bars indicate number of HEK293 cells/experiments analyzed. **p < 0.01 (one-way ANOVA test with Tukey's post hoc test). ***p < 0.001 (one-way ANOVA test with Tukey's post hoc test).
NPR knockdown decreases excitatory synapse organization in cultured neurons
Although our results suggest that NPR functions generally to shape synapses, these results are entirely based on the heterologous synapse formation assay. This assay has been shown to be an invaluable tool for identifying synaptic functions in general but has proven unreliable in interpreting the nature of these functions. For example, neuroligins and neurexins are powerfully synaptogenic in this assay (Scheiffele et al., 2000; Graf et al., 2004; Chubykin et al., 2005; Nam and Chen, 2005) but are not required for synapse formation in vivo (Missler et al., 2003; Varoqueaux et al., 2006). Instead, neuroligins and neurexins in vivo primarily shape the properties of synapses without influencing their numbers. Thus, to relate heterologous synapse formation induced by NPR to its function in a physiological synapse, we probed the effects of knocking down NPR expression in hippocampal neurons cultured from newborn mice. We designed four different shRNAs to knock down NPR, and identified two shRNAs that were highly effective (>80% knockdown; Fig. 9A). Because knockdowns with shRNAs often produce off-target effect, we also examined the effect of NPR rescues on the knockdown phenotype, and furthermore tested the effect of the “rescue” (which is always overexpression) on neurons without knockdowns (Figs. 9, 10, 11–12).
Figure 9.

Membrane-bound NPR stabilizes secreted NPs. A, qRT-PCR of neurons expressing the indicated shRNAs targeting NPR identifies highly effective NPR knockdown shRNAs. Cortical neurons were infected with lentiviruses expressing the indicated shRNAs (shCtrl = control shRNA; sh472 to sh475 = four different NPR-targeted shRNAs) at DIV4, and harvested at DIV16. Total RNA was isolated from the neuronal cell lysate and used for qPCR of NPR and β-actin mRNAs. β-actin was used as an endogenous control to normalize for experimental variation, and measured mRNA levels were normalized to those of β-actin and the control. Dotted lines indicate control values at 1.0, and the cutoff of shRNAs considered to be effective at 0.2. Data are results from two independent experiments, each performed in duplicate (samples a–d) to assess variability. Two shRNAs (sh472 and sh474) were chosen as the most effective ones and used in mixed viruses for NPR knockdown experiments. B, Quantification of NPs and synaptic protein levels in cultured hippocampal neurons infected lentiviruses expressing control, NPR shRNAs, NPR shRNAs plus shRNA-resistant cDNA, or only the NPR cDNA. Primary hippocampal neurons were infected viruses encoding indicated genes at DIV4. Neurons were collected at DIV16 in separate experiments and protein levels for synapsin, GluA1, GluA2, PSD95, Gephyrin, NP1, NARP, NPR, or Tuj1 were determined by quantitative immunoblotting using fluorescently labeled secondary antibodies. Tuj1 was used as a loading control. Left, Representative images. Right, Summary graph. Numbers in bars indicate number of independent experiments analyzed. Graphs represent mean ± SEM. Numbers in bars indicate number of neurons/experiments analyzed. *p < 0.05 (one-way ANOVA test with Tukey's post hoc test). **p < 0.01 (one-way ANOVA test with Tukey's post hoc test). ***p < 0.001 (one-way ANOVA test with Tukey's post hoc test).
Figure 10.

NPR knockdown impairs postsynaptic organization of excitatory synapses without decreasing excitatory synapse numbers. A, B, NPR knockdown does not alter the density of synapsin- or VgluT1-positive synaptic puncta but decreases the synaptic content of the excitatory postsynaptic density proteins PSD95 and Shank. Hippocampal neurons were infected control lentiviruses or lentiviruses expressing NPR shRNAs, NPR shRNAs plus shRNA-resistant cDNA, or only the NPR cDNA at DIV4. Neurons were immunolabeled at DIV16 in separate experiments for synapsin, VGluT1, PSD95, or Shank (all green) and MAP2 (red or blue), and the density, area, and staining intensity of puncta were quantified: A, Representative images. B, Summary graphs. Scale bars, 10 μm. Data are mean ± SEM. Numbers in bars indicate number of neurons/experiments analyzed. *p < 0.05 (one-way ANOVA test with Tukey's post hoc test). **p < 0.01 (one-way ANOVA test with Tukey's post hoc test). ***p < 0.001 (one-way ANOVA test with Tukey's post hoc test).
Figure 11.

NPR knockdown impairs GluA1 content and transmission strength of excitatory synapses. A, B, NPR knockdown decreases the overall surface levels of GluA1, but not total GluA1 levels. Hippocampal neurons were infected control lentiviruses or lentiviruses expressing NPR shRNAs, NPR shRNAs plus shRNA-resistant cDNA, or only the NPR cDNA at DIV4. Neurons were immunolabeled at DIV16 in separate experiments for surface GluA1 receptors before permeabilization (red), and then permeabilized and stained for total GluA1 receptors using a different secondary antibody (green), and additionally stained for MAP2 (blue). A, Representative images. B, Summary graphs of the surface GluA1 levels and total GluA1 levels normalized for MAP2 as an internal standard as well as of the ratio of surface to total GluA1 levels. C, D, NPR KD reduces the density of GluA1-positive synaptic puncta but has no apparent effect on the GluA1 content of such puncta. Hippocampal neurons obtained as described for A and B, and synaptic puncta were imaged in neurons obtained. The density, area, and staining intensity of puncta were quantified. C, Representative images. D, Summary graphs. E, NPR knockdown decreases the amplitude of action potential-evoked EPSCs. Control neurons and neurons with the NPR knockdown without or with rescue were produced as described for A and B, and EPSCs were monitored in whole-cell patch-clamp recordings after stimulation with a multicentric extracellular electrode in the presence of 0.1 mm PTX. Left, Representative traces. Right, Summary graphs of the peak EPSC amplitudes. F, NPR knockdown decreases the total synaptic charge transfer at excitatory synapses after stimulation of neurotransmitter release with hypertonic sucrose, which triggers release of all transmitters in readily-releasable vesicles (Rosenmund and Stevens, 1996). Control and NPR knockdown neurons were analyzed by whole-cell recordings as described for E, except that recordings were performed in the presence of 0.1 mm PTX and 1 μm TTX, and that presynaptic neurotransmitter release was induced by application of 0.5 m sucrose for 30 s. EPSCs were quantified by integrating the total charge transfer during the sucrose application. Left, Representative traces. Right, Summary graph of the total charge transfer. Scale bars: A, 50 μm; C, 10 μm. Graphs represent mean ± SEM. Numbers in bars indicate number of neurons/experiments analyzed. Statistical significance: B, D, One-way ANOVA test with Tukey's post hoc test; E, F, Student's t test. *p < 0.05. ***p < 0.001.
Figure 12.

NPR knockdown impairs organization and strength of inhibitory synapses without changing inhibitory synapse numbers. A, B, NPR knockdown decreases gephyrin content of inhibitory postsynaptic specializations without altering the density of VGAT- or gephyrin-positive synaptic puncta. Hippocampal neurons infected control lentiviruses or lentiviruses expressing NPR shRNAs, NPR shRNAs plus shRNA-resistant cDNA, or only the NPR cDNA were immunolabeled for VGAT (green), gephyrin (red), and MAP2 (blue). The density, area, and staining intensity of puncta were quantified. A, Representative images. B, Summary graphs. C–F, NPR knockdown does not change levels of puncta for both surface and total GABRG2. Hippocampal neurons were immunolabeled at DIV16 in separate experiments for surface GABRG2 before permeabilization (C), or for total GABRG2 after permeabilization (E) costaining MAP2. The density, area, and staining intensity of puncta were quantified. C, E, Representative images. D, F, Summary graphs. G, NPR knockdown decreases the amplitude of action potential-evoked IPSCs. Control neurons and neurons with the NPR knockdown without or with rescue were produced as described for A and B, and IPSCs were monitored in whole-cell patch-clamp recordings after stimulation with a multicentric extracellular electrode in the presence of 10 μm CNQX. Left, Representative traces. Right, Summary graphs of the peak IPSC amplitudes. H, NPR knockdown decreases the total synaptic charge transfer at inhibitory synapses after stimulation of neurotransmitter release with hypertonic sucrose. Neurons were obtained and analyzed by whole-cell recordings as described for C, except that recordings were performed in the presence of 10 μm CNQX and 1 μm TTX, and that presynaptic neurotransmitter release was induced by application of 0.5 m sucrose for 30 s, which induces exocytosis of all release-ready vesicles. IPSCs were quantified by integrating the total charge transfer during the sucrose application. Left, Representative traces. Right, Summary graph of the total charge transfer. Scale bars, 10 μm. Graphs represent mean ± SEM. Numbers in bars indicate number of neurons/independent experiments analyzed. Statistical significance: B, D, F, One-way ANOVA test with Tukey's post hoc test; G, H, Student's t test. *p < 0.05. **p < 0.01. ***p < 0.001.
We first examined how the NPR knockdown and/or overexpression affected the levels of NPR, of other synaptic proteins and of NP1 and NARP. Although the NPR knockdown suppressed NPR mRNA levels ∼80%, it decreased NPR protein levels only ∼40% (Fig. 9B). NPR rescue/overexpression, conversely, increased NPR protein levels 8- to 10-fold. Total levels of selected synaptic proteins (e.g., synapsin, GluA1, GluA2, PSD95, and gephyrin) were not significantly changed by NPR knockdown or rescue/overexpression. Interestingly, the NPR knockdown decreased the levels for NARP to the same extent as the NPR levels (∼40%) but had no effect on NP1 levels (Fig. 9B). The NPR rescue/overexpression, conversely, elevated NARP levels 2- to 5-fold and NP1 levels 1.5- to 2-fold (Fig. 9B). These results suggest that secreted NPs are produced in excess of NPR and that the recruitment of secreted NPs by membrane-bound NPR stabilizes secreted NPs.
We then examined the effect of manipulating NPR levels by knockdown or rescue/overexpression on overall synapse density, measured as the density of synapsin- and vGlut1-positive puncta on a dendritic segment (Fig. 10A,B). We observed no significant change. However, when we examined excitatory postsynaptic specializations using two different markers (PSD95 and Shanks), we detected a 25%–35% decrease in the density of synaptic puncta and an additional 40%–60% decrease in the strength of the PSD95 or Shank signal per synaptic punctum. All of these changes were reversed by the NPR rescue/overexpression; however, NPR overexpression did not produce significant increase any of these parameters compared with controls (Fig. 10A,B). Thus, NPR seems to be rate-limiting for the development of excitatory postsynaptic specializations in cultured neurons, but overexpression of NPR does not in itself induce formation of excitatory postsynaptic specializations or synapses.
The observation that the NPR knockdown decreased the apparent levels of postsynaptic density markers per remaining synapse more than the density of postsynaptic specialization without altering total synapse numbers suggests that the NPR knockdown leads to a loss of components of postsynaptic specializations, but not of synapses, and that the apparent decrease in the number of synapses as measured by postsynaptic density markers is due to this decrease. To test this interpretation, we studied the effects of the NPR knockdown on the surface concentration and the total levels of GluA1 (Fig. 11A). We found that the NPR knockdown lowered surface GluA1 levels ∼60% but had no effect on total GluA1 levels. This phenotype was completely rescued by NPR overexpression, which on its own had no significant effect on GluA1 surface levels. As a result, we observed a large decrease in the ratio of surface to total GluA1 receptors (Fig. 11B). This decrease is consistent with the observation in transfected HEK293 cells in which NPs increased the surface levels of GluA1 by interacting with GluA1 directly on the cell surface and without affecting total GluA1 levels (Fig. 4).
We next examined by immunocytochemistry the synaptic levels of GluA1 as a function of the NPR knockdown. The number of synaptic surface GluA1 puncta was decreased ∼60% by the NPR knockdown; again, this phenotype was rescued by NPR overexpression, whereas NPR overexpression in control neurons had no effect (Fig. 11C,D). The NPR knockdown did not alter total GluA1 levels but dramatically decreased the ratio of surface/total GluA1. To probe for changes in synaptic transmission induced by the NPR knockdown, we patched the cultured neurons and measured action potential-evoked EPSCs (Fig. 11E). The NPR knockdown caused an ∼60% decrease in EPSC amplitude that was fully reversed by NPR overexpression, whereas NPR overexpression in control neurons by itself had no significant effect. Surprised by this dramatic effect, we examined it further by measuring EPSCs evoked by hypertonic sucrose, which triggers exocytosis of the entire readily releasable pool of vesicles at a synapse (Rosenmund and Stevens, 1996). We observed a similar ∼60% decrease in sucrose-evoked EPSCs induced by the NPR knockdown; again, this phenotype was rescued by NPR overexpression, which on its own did not alter EPSCs in control neurons (Fig. 11F).
The data are consistent with the mechanistic studies shown in Figure 4 that NPR “traps” GluA1 and other AMPARs on the neuronal surface in synaptic specializations, whereas the heterologous synapse formation data suggest that this trapping activates a signaling cascade, which organizes postsynaptic specializations. The knockdown results indicate that NPR is rate-limiting for excitatory synapses in cultured neurons, and thus NPR serves as a mediator of synapse formation in a broad sense. It is puzzling, however, that the relatively small decrease in NPR protein levels of ∼40% (Fig. 9B) causes such dramatic effects on excitatory synapses. One reason for this unexpectedly large effect of a relatively small decrease in NPR levels may be that NPR always functions in a complex with secreted NPs as suggested by the effects of manipulating NPR levels on secreted NPs (Xu et al., 2003). In this manner, small changes in NPR levels may produce bigger effects because of these small changes cause equivalent changes in secreted NPs.
NPR knockdown equally impairs inhibitory synapses in cultured neurons
Do NPR levels also influence inhibitory synapses? We detected no change in the density of inhibitory synapses as a function of the NPR knockdown, as measured either by gephyrin or VGAT immunoreactivity (Fig. 12A,B). However, we did observe a decrease in gephyrin staining per synaptic punctum, both in terms of the size of gephyrin-positive clusters and in terms of its staining intensity. Both decreases were reversed by NPR rescue/overexpression, which had no effect on these parameters in control neurons. No change was detected for the presynaptic inhibitory marker VGAT, suggesting that the NPR knockdown impedes the maturation of inhibitory postsynaptic specializations without affecting inhibitory synapse numbers (Fig. 12A,B).
We next examined by immunocytochemistry the levels of GABRG2 as a function of the NPR knockdown and rescue/overexpression but detected no significant change in either surface or total GABRG2 levels (Fig. 12C–F). Consistent with the decrease in inhibitory postsynaptic specializations by the NPR knockdown, however, the NPR knockdown dramatically decreased IPSCs evoked by action potentials (Fig. 12G) or by application of hypertonic sucrose (Fig. 12H). Both the ∼30% decrease in action potential-evoked IPSCs and the ∼70% decrease in sucrose-evoked IPSCs were rescued by NPR rescue/overexpression, which again had no effect on these parameters compared with naive neurons (Fig. 12G,H). Thus, as for excitatory synapses, a relatively small decrease in NPR protein levels appears to cause a profound change in inhibitory synapses, whereas NPR overexpression on its own does not alter these synapses.
Discussion
Ever since their identification two decades ago, NPs have been studied intensely. Pioneering work from Paul Worley and colleagues documented that NPs are presynaptic molecules that bind to postsynaptic AMPARs in a trans-synaptic interaction and thereby regulate excitatory synapses (O'Brien et al., 1999, 2002; Xu et al., 2003; Sia et al., 2007). Subsequent studies focused on the function of NPs in specific circuits, in particular on the role of NARP as an immediate-early gene in behavioral plasticity (Bjartmar et al., 2006; Chang et al., 2010; Johnson et al., 2010; Knackstedt et al., 2010; Koch and Ullian, 2010; Blouin et al., 2013; Gu et al., 2013; Pelkey et al., 2015). Overall, these studies established that NPs act in the activity-dependent regulation of AMPARs in specific neural circuits but raised new basic cell-biology questions of synapse organization. For example, what activity-dependent versus activity-independent functions do NPs precisely have in synapses because most NPs are not activity-dependent, what are the functional effects of clustering AMPARs by NPs, and do NPs only function in excitatory synapses or perform a more general role in all synapses, including inhibitory synapses? Furthermore, how does NPR, which is the least studied although arguably the most interesting NP since it can recruit the other NPs into a membrane-associated complex, function in synapse?
In trying to address these questions, we made eight principal observations (illustrated in Fig. 13). First, NPR is highly synaptogenic during heterologous synapse formation, such that NPR expressed in HEK293 cells induces postsynaptic, but not presynaptic, specializations in cocultured neurons as potently as neurexin-1β (Fig. 1). Second, NPR stimulates formation of both excitatory and inhibitory heterologous synapses (Figs. 1, 567-8), consistent with the expression of NPR in both excitatory and inhibitory neurons. Third, binding of NPs to the NTDs of AMPARs stabilizes AMPARs on the cell surface and enhances overall AMPAR function (Figs. 3, 4). This suggests that cell-surface clustering of AMPARs by NPs increases AMPAR activity. Direct surface stabilization of AMPARs by NPs is consistent with the much larger recruitment of GluA1 and GluA2 to heterologous synapses by NPR than by neurexin-1β (Fig. 1C). Fourth, the NPR pentraxin domain directly binds to the GluA1-NTD and mediates trans-synaptic interactions; strikingly, the NPR pentraxin domain on its own induces heterologous synapse formation (Fig. 5). Thus, trans-synaptic engagement of postsynaptic AMPARs by presynaptic NPR appears to be sufficient to activate a signaling cascade that causes assembly of postsynaptic specializations (Fig. 6). Fifth, NPR-induced heterologous synapse formation is inhibited by antagonists of AMPARs or GABARs. NBQX selectively inhibited NPR-induced excitatory heterologous synapse formation ∼50% (Fig. 7A,B), while PTX selectively inhibited NPR-induced inhibitory heterologous synapse formation ∼40% (Fig. 8A,B). Thus, NPR does not induce heterologous synapse formation by clustering active receptors. Sixth, changes in NPR protein levels in cultured hippocampal neurons by knockdowns or rescue/overexpression cause precisely corresponding changes in NARP protein levels and, to a lesser extent, in NP1 protein levels. These observations suggest that NPR levels determine the levels of secreted NPs (Fig. 9B), probably because membrane recruitment of secreted NPs by NPR stabilizes secreted NPs. Seventh, NPR is rate-limiting for assembly of functional excitatory synapses. Knockdown of NPR did not alter total excitatory synapse numbers but decreased the levels of the postsynaptic density proteins, suppressed surface GluA1 levels, and impaired excitatory synaptic transmission (Figs. 10, 11). Eighth and last, NPR is also essential for normal organization of inhibitory synapses, again without affecting synapse numbers. Knockdown of NPR caused a decrease in postsynaptic gephyrin levels; in parallel, the NPR knockdown produced a large decrease in IPSC amplitudes (Fig. 12).
Figure 13.
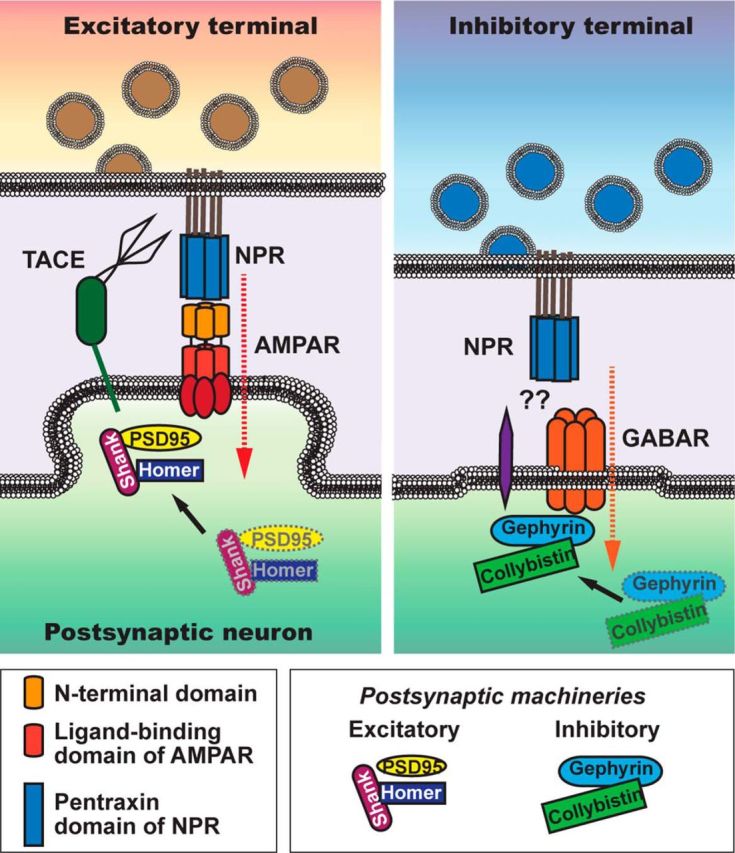
Diagram model of the synaptic function of NPR. Diagram illustrates the trans-synaptic signaling role proposed for NPR at both excitatory (left) and inhibitory synapses (right). Presynaptic NPR, either as a homomeric pentamer or as a heteropentameric complex with NP1 and/or NARP, binds to postsynaptic ligands in both excitatory and inhibitory synapses and thereby activates trans-synaptic signaling. In excitatory synapses, NPR binds via its pentraxin domain to the NTD of AMPARs and activates AMPARs by stabilizing them on the surface, whereas in inhibitory synapses NPR binds to an as yet unknown postsynaptic ligand, but its trans-synaptic signaling function also requires postsynaptic receptor activity.
These eight observations suggest a general function for NPs as trans-synaptic organizers whose demonstrated role in regulating circuits is mediated by their biochemical reorganization of specific synapses (Fig. 13). We envision that, in this function, NPs act more as modulators of synaptic strength and synapse properties than as essential building blocks of synapses because NPs are not actually essential for synapse formation or for overall nervous system function (Bjartmar et al., 2006). Importantly, our results imply that NPR determines overall NP function because it recruits secreted NPs to the presynaptic plasma membrane and stabilizes them in that manner.
Our results also raise new questions. First, why were the general, rather robust activities of NPs in synapses not observed in previous studies? We believe that the most likely reason for this oversight is that previous studies focused on the role of AMPAR binding by NPs in neural circuits. To the best of our knowledge, no previous study probed the role of NPs in inhibitory synapses. Moreover, although triple NP KO mice were shown to be viable and double NP1/NARP KO mice were found to exhibit reduced AMPAR-mediated synaptic transmission in the developing visual system (Bjartmar et al., 2006; Koch and Ullian, 2010), no basic analyses of synaptic transmission were performed.
Second, why do heterologous synapse formation assays and cultured hippocampal mouse neurons appear to provide different results? The heterologous synapse formation data seem to show a function for NPR in synapse formation, whereas the cultured neuron data appear to demonstrate a role, not in the actual formation of synapses, but in the assembly of the postsynaptic machinery. When assessed on the background of the field, however, it appears that the heterologous synapse formation assay monitors synapse organization rather than synapse formation. For example, neurexins and neuroligins are powerful agents in heterologous synapse formation assays (Scheiffele et al., 2000; Graf et al., 2004; Chubykin et al., 2005; Nam and Chen, 2005) (Fig. 1), but deletions of neurexins or neuroligins in cultured neurons or in vivo in brain primarily impair synapse function but not synapse numbers (Missler et al., 2003; Varoqueaux et al., 2006; Chubykin et al., 2007). It seems likely that heterologous synapse formation is induced whenever trans-synaptic activation of a presynaptic or postsynaptic compartment occurs, independent of the physiological function of this trans-synaptic activation.
Third, how does NPR induce organization of postsynaptic excitatory specializations by clustering AMPARs, and how does NPR act in inhibitory synapses? We show that the NPR pentraxin domain, when tethered to the membrane, was sufficient for this activity (Fig. 5), and that inhibiting AMPAR or GABAR functions decreased excitatory or inhibitory synapse formation, respectively (Figs. 7, 8). Thus, local neuronal excitation cannot simply mediate synapse formation because the receptor activation mechanisms also appear to apply to inhibitory synapses. Identifying a target of NPR in inhibitory synapse will lead to a better understanding of how it regulates inhibitory postsynaptic specializations. Preliminary studies failed to identify a direct interaction of NPR with GABARs (S.-J.L. and T.C.S., unpublished data), and the mechanisms underlying our somewhat surprising observations remain to be examined.
Finally, what are the relative functions of the secreted versus membrane-bound forms of NPs, and how is their ratio regulated? Our data show that, in cultured neurons, NPR levels determine those of secreted NPs, presumably because secreted NP1 and NARP are produced in excess and assembled into membrane-bound presynaptic complexes via NPR. However, NPR can be proteolytically cleaved by TACE in an activity-dependent manner, effectively converting NPR into a secreted NP (Cho et al., 2008). Moreover, because NARP, but not NPR, expression is activity-dependent, increased NARP expression would presumably lead to increased NARP degradation without increased NPR expression. Thus, one possible explanation for the changes in neural circuit function that have been observed in an activity- and NARP-dependent manner is that these changes are due to a switch in the relation of secreted versus membrane-bound NPs, and that secreted NPs and cleaved NPR are rapidly degraded because they induce endocytosis of postsynaptic binding partners, thereby controlling synaptic strength. Indeed, Cho et al. (2008) suggested a model whereby LTD is induced because it stimulates cleavage of NPR, thereby causing endocytosis of AMPARs that were clustered by NPs (Cho et al., 2008). An alternative interpretation of these experiments, however, might be that the NPR KO decreased the starting synaptic strength due to a decrease in surface AMPARs, and that LTD was thus occluded without any direct role of NPR in LTD itself. Because TACE has many neuronal substrates and its cleavage efficiency of NPR is unknown, it is unclear whether TACE's role in LTD is related to its cleavage of NPR. Thus, more data are needed to address this question.
The overall perspective that emerges from our experiments is that NPs are synaptic organizer proteins that are anchored on the presynaptic membrane by NPR and trans-synaptically regulate synapse assembly. According to this perspective, NPR's role is to organize trans-synaptic complexes; if there is at least one NPR molecule for every four secreted NP1 and NARP molecules, the latter would enhance the NPR signal by assembling into complexes with NPR. If there is less than one NPR molecule for every four NP1 and NARP molecules, however, NP1 and NARP would bind to postsynaptic partners without constraining them on the surface, maybe inducing their endocytosis. In this case, secreted NPs would suppress NPR function by blocking a trans-synaptic signal, or decrease the function of the postsynaptic binding partners by inducing their endocytosis. It is thus unclear whether NARP as an immediate-early gene primarily acts to enhance synapse formation by amplifying NPR function or to inhibit NPR function by serving as a dominant negative, an exciting question that has broad implications for the regulation of synaptic strength.
Footnotes
This work was supported by National Institute of Mental Health Grant MH052804 to T.C.S. We thank Dr. L. Chen for providing the GluA1-GFP, GluA2-GFP, GluD2-GFP, stargazin, and GluA1-NTD-Fc constructs.
The authors declare no competing financial interests.
References
- Bjartmar L, Huberman AD, Ullian EM, Rentería RC, Liu X, Xu W, Prezioso J, Susman MW, Stellwagen D, Stokes CC, Cho R, Worley P, Malenka RC, Ball S, Peachey NS, Copenhagen D, Chapman B, Nakamoto M, Barres BA, Perin MS (2006) Neuronal pentraxins mediate synaptic refinement in the developing visual system. J Neurosci 26:6269–6281. 10.1523/JNEUROSCI.4212-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin AM, Han S, Pearce AM, Cheng K, Lee JJ, Johnson AW, Wang C, During MJ, Holland PC, Shaham Y, Baraban JM, Reti IM (2013) Role of medial prefrontal cortex Narp in the extinction of morphine conditioned place preference. Learn Mem 20:75–79. 10.1101/lm.028621.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MC, Park JM, Pelkey KA, Grabenstatter HL, Xu D, Linden DJ, Sutula TP, McBain CJ, Worley PF (2010) Narp regulates homeostatic scaling of excitatory synapses on parvalbumin-expressing interneurons. Nat Neurosci 13:1090–1097. 10.1038/nn.2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Okayama H (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7:2745–2752. 10.1128/MCB.7.8.2745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA (2000) Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 408:936–943. 10.1038/35050030 [DOI] [PubMed] [Google Scholar]
- Cho RW, Park JM, Wolff SB, Xu D, Hopf C, Kim JA, Reddy RC, Petralia RS, Perin MS, Linden DJ, Worley PF (2008) mGluR1/5-dependent long-term depression requires the regulated ectodomain cleavage of neuronal pentraxin NPR by TACE. Neuron 57:858–871. 10.1016/j.neuron.2008.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chubykin AA, Liu X, Comoletti D, Tsigelny I, Taylor P, Südhof TC (2005) Dissection of synaptic induction by neuroligins: effect of neuroligin mutation associated with autism. J Biol Chem 280:22365–22374. 10.1074/jbc.M410723200 [DOI] [PubMed] [Google Scholar]
- Chubykin AA, Atasoy D, Etherton MR, Brose N, Kavalali ET, Gibson JR, Südhof TC (2007) Activity-dependent validation of excitatory versus inhibitory synapses by neuroligin-1 versus neuroligin-2. Neuron 54:919–931. 10.1016/j.neuron.2007.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitriu D, Berger SI, Hamo C, Hara Y, Bailey M, Hamo A, Grossman YS, Janssen WG, Morrison JH (2012) Vamping: stereology-based automated quantification of fluorescent puncta size and density. J Neurosci Methods 209:97–105. 10.1016/j.jneumeth.2012.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz I, Lerer-Goldshtein T, Okamoto H, Appelbaum L (2015) Reduced synaptic density and deficient locomotor response in neuronal activity-regulated pentraxin 2a mutant zebrafish. FASEB J 29:1220–1234. 10.1096/fj.14-258350 [DOI] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM (2004) Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119:1013–1026. 10.1016/j.cell.2004.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Huang S, Chang MC, Worley P, Kirkwood A, Quinlan EM (2013) Obligatory role of the immediate early gene NARP in critical period plasticity. Neuron 79:335–346. 10.1016/j.neuron.2013.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka M, Südhof TC (1999) Homo- and heterodimerization of synapsins. J Biol Chem 274:16747–16753. 10.1074/jbc.274.24.16747 [DOI] [PubMed] [Google Scholar]
- Johnson AW, Han S, Blouin AM, Saini J, Worley PF, During MJ, Holland PC, Baraban JM, Reti IM (2010) Localized disruption of Narp in medial prefrontal cortex blocks reinforce devaluation performance. Learn Mem 17:620–626. 10.1101/lm.1937210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick LL, Matzuk MM, Dodds DC, Perin MS (2000) Biochemical interactions of the neuronal pentraxins: neuronal pentraxin (NP) receptor binds to taipoxin and taipoxin-associated calcium binding protein 49 via NP1 and NP2. J Biol Chem 275:17786–17792. 10.1074/jbc.M002254200 [DOI] [PubMed] [Google Scholar]
- Knackstedt LA, Moussawi K, Lalumiere R, Schwendt M, Klugmann M, Kalivas PW (2010) Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J Neurosci 30:7984–7992. 10.1523/JNEUROSCI.1244-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch SM, Ullian EM (2010) Neuronal pentraxins mediate silent synapse conversion in the developing visual system. J Neurosci 30:5404–5414. 10.1523/JNEUROSCI.4893-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Südhof TC (2005) Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron 48:547–554. 10.1016/j.neuron.2005.09.006 [DOI] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC (2003) α-Neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423:939–948. 10.1038/nature01755 [DOI] [PubMed] [Google Scholar]
- Nam CI, Chen L (2005) Postsynaptic assembly induced by neurexin-neuroligin interaction and neurotransmitter. Proc Natl Acad Sci U S A 102:9137–9142. 10.1073/pnas.0502038102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Xu D, Petralia RS, Steward O, Huganir RL, Worley P (1999) Synaptic clustering of AMOA receptors by the extracellular immediate-early gene product Narp. Neuron 23:309–323. 10.1016/S0896-6273(00)80782-5 [DOI] [PubMed] [Google Scholar]
- O'Brien R, Xu D, Mi R, Tang X, Hopf C, Worley P (2002) Synaptically targeted narp plays an essential role in the aggregation of AMPA receptors at excitatory synapses in cultures spinal neurons. J Neurosci 22:4487–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Cao P, Xu W, Südhof TC (2010) Calmodulin controls synaptic strength via presynaptic activation of calmodulin kinase II. J Neurosci 30:4132–4142. 10.1523/JNEUROSCI.3129-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkey KA, Barksdale E, Craig MT, Yuan X, Sukumaran M, Vargish GA, Mitchell RM, Wyeth MS, Petralia RS, Chittajallu R, Karlsson RM, Cameron HA, Murata Y, Colonnese MT, Worley PF, McBain CJ (2015) Pentraxins coordinate excitatory synapse maturation and circuit integration of the parvalbumin interneurons. Neuron 85:1257–1272. 10.1016/j.neuron.2015.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF (1996) Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16:1197–1207. 10.1016/S0896-6273(00)80146-4 [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T (2000) Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101:657–669. 10.1016/S0092-8674(00)80877-6 [DOI] [PubMed] [Google Scholar]
- Schlimgen AK, Helms JA, Vogel H, Perin MS (1995) Neuronal pentraxin, a secreted protein with homology to acute phase proteins of the immune system. Neuron 14:519–526. 10.1016/0896-6273(95)90308-9 [DOI] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Brechet A, Zolles G, Berkefeld H, Müller CS, Bildl W, Baehrens D, Hüber B, Kulik A, Klöcker N, Schulte U, Fakler B (2012) High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron 74:621–633. 10.1016/j.neuron.2012.03.034 [DOI] [PubMed] [Google Scholar]
- Schwenk J, Baehrens D, Haupt A, Bildl W, Boudkkazi S, Roeper J, Fakler B, Schulte U (2014) Regional diversity and developmental dynamics of the AMPA receptor proteome in the mammalian brain. Neuron 84:41–54. 10.1016/j.neuron.2014.08.044 [DOI] [PubMed] [Google Scholar]
- Sia GM, Béïque JC, Rumbaugh G, Cho R, Worley PF, Huganir RL (2007) Interaction of the N-terminal domain of the AMPA receptor GluR4 subunit with the neuronal pentraxin NP1 mediates GluR4 synaptic recruitment. Neuron 55:87–102. 10.1016/j.neuron.2007.06.020 [DOI] [PubMed] [Google Scholar]
- Tsui CC, Copeland NG, Gilbert DJ, Jenkins NA, Barnes C, Worley PF (1996) Narp, a novel member of the pentraxin family, promotes neurite outgrowth and is dynamically regulated by neuronal activity. J Neurosci 16:2463–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Mishina M (2008) The amino-terminal domain of glutamate receptor δ2 triggers presynaptic differentiation. Biochem Biophys Res Commun 377:1315–1319. 10.1016/j.bbrc.2008.10.170 [DOI] [PubMed] [Google Scholar]
- Ushkaryov YA, Petrenko AG, Geppert M, Südhof TC (1992) Neurexins: synaptic cell surface proteins related to the alpha-latrotoxin receptor and laminin. Science 257:50–56. 10.1126/science.1621094 [DOI] [PubMed] [Google Scholar]
- Varoqueaux F, Aramuni G, Rawson RL, Mohrmann R, Missler M, Gottmann K, Zhang W, Südhof TC, Brose N (2006) Neuroligins determine synapse maturation and function. Neuron 51:741–754. 10.1016/j.neuron.2006.09.003 [DOI] [PubMed] [Google Scholar]
- Xu D, Hopf C, Reddy R, Cho RW, Guo L, Lanahan A, Petralia RS, Wenthold RJ, O'Brien RJ, Worley P (2003) Narp and NP1 form heterocomplexes that function in developmental and activity-dependent synaptic plasticity. Neuron 39:513–528. 10.1016/S0896-6273(03)00463-X [DOI] [PubMed] [Google Scholar]
- Zhang C, Atasoy D, Araç D, Yang X, Fucillo MV, Robinson AJ, Ko J, Brunger AT, Südhof TC (2010) Neurexins physically and functionally interact with GABAA receptors. Neuron 66:403–416. 10.1016/j.neuron.2010.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]