

Abstract
Background
Laminin, a major basement membrane component that has direct contact with pericytes under physiological conditions, actively regulates the proliferation and differentiation/fate determination of pericytes. Recently, two types of pericytes (type I and type II) with different molecular markers and functions have been identified in skeletal muscles. Whether laminin differentially regulates the proliferation and differentiation of these two subpopulations remains unclear.
Methods
Wild-type and pericytic laminin-deficient mice under Nestin-GFP background were used to determine if laminin differentially regulates the proliferation and differentiation of type I and type II pericytes. Specifically, type I and type II pericytes were isolated from these mice, and their proliferation and differentiation were examined in vitro. Moreover, in vivo studies were also performed.
Results
We demonstrate that, although laminin inhibits the proliferation of both type I and type II pericytes in vitro, loss of laminin predominantly induces proliferation of type II pericytes in vivo. In addition, laminin negatively regulates the adipogenic differentiation of type I pericytes and positively regulates the myogenic differentiation of type II pericytes in vitro.
Conclusions
Laminin differentially regulates the proliferation and differentiation of type I and type II pericytes.
Electronic supplementary material
The online version of this article (doi:10.1186/s13287-017-0479-4) contains supplementary material, which is available to authorized users.
Keywords: Laminin, Pericytes, Proliferation, Differentiation
Background
Satellite cells (postnatal muscle progenitor cells) are sandwiched between the basal lamina and myofiber plasma membrane in skeletal muscles [1]. Unlike satellite cells, various other cell populations, including fibro/adipogenic progenitors (FAPs) [2, 3], PW1+/Pax7− interstitial cells (PICs) [4], mesenchymal stem cells (MSCs) [5–7], fibroblasts [8], and pericytes [9, 10], are found in the interstitial space of skeletal muscles. These populations are distinct from each other in their marker expression and differentiation abilities/functions. Specifically, FAPs are α7-integrin−Sca-1+PDGFRα+, and produce both fibroblasts and adipocytes [2, 3]. PICs are Sca-1MedCD34+PW1+Pax7− and have myogenic potential [4]. MSCs express a wide range of stem cell markers and are able to generate adipose, bone, cartilage, and muscle [5–7, 11, 12]. Fibroblasts express ER-TR7, FSP1, and Tcf4, and are involved in tissue fibrosis [8]. Pericytes (perivascular cells covering capillaries in the vascular tree [13]) express a unique array of markers, including PDGFRβ, NG2, CD146, and CD13 [13]. It should be noted, however, that none of these markers are specific for pericytes since they are also expressed by other cell types, and their expression in pericytes is highly dependent on the developmental stages [13]. Accumulating evidence suggests that pericytes are multipotent cells. On one hand, pericytes are able to differentiate into myogenic cells via myogenesis to repair muscle injury [9, 10, 14–16]. On the other hand, pericytes can also differentiate into adipocytes and/or fibroblasts [14, 17–21], exacerbating muscle injury.
Recently, two subtypes (termed type I and type II) of pericytes with different molecular markers and differentiation abilities have been reported in skeletal muscles. Biochemically, both type I and type II pericytes express various classical pericyte markers, including PDGFRβ, NG2, and CD146 [14, 22]. It is the expression of Nestin, an intermediate filament protein, that differentiates these two subtypes of pericytes: type I (PDGFRβ+/NG2+/CD146+ and Nestin−) and type II (PDGFRβ+/NG2+/CD146+ and Nestin+) [14, 22, 23]. It is worth noting that “Nestin” here refers to transgene Nestin-GFP from the Nestin-GFP mice (see Methods), not endogenous Nestin. Since Nestin-GFP cells express low levels of endogenous Nestin in bone marrow [24], caution should be taken when using endogenous Nestin to distinguish type I and type II pericytes. Further studies conducted by Birbrair and colleagues elegantly demonstrate that type I pericytes differentiate into adipogenic and fibrogenic cells, contributing to muscle degeneration, whereas type II pericytes undergo myogenesis, promoting muscle regeneration [14, 23, 25, 26]. These data suggest that type I and type II pericytes are intrinsically different, and have distinct differentiation capability and fate.
The molecular mechanisms underlying pericyte differentiation and fate determination are not fully understood. In a previous study, we identified laminin as a critical regulator in the differentiation and fate determination of PDGFRβ+ pericytes [21]. Specifically, we found that laminin negatively regulates their proliferation, inhibits their adipogenic differentiation, and is indispensable for their myogenic differentiation [21]. Whether laminin differentially regulates the differentiation and fate determination of type I and type II pericytes, however, remains unclear. Using the Nestin-GFP and laminin conditional knockout mice, we report here that laminin inhibits the proliferation of both type I and type II pericytes in vitro. Type II pericytes, however, predominantly proliferate in vivo upon loss of laminin. In addition, laminin prevents adipogenic differentiation of type I pericytes, but is required for myogenic differentiation of type II pericytes. These data suggest that laminin inhibits the proliferation of type II pericytes, and has anti-adipogenic and pro-myogenic effects on type I and type II pericytes, respectively.
Methods
Animals
Pdgfrβ-Cre+ line [27] and laminin γ1 floxed (F/F) transgenic mice [28] were crossed to generate laminin-deficient PKO (F/F:Pdgfrβ-Cre+) mice. These mice were further crossed with Nestin-GFP mice [29, 30] to generate control (F/F:Nestin-GFP) and PKO (F/F:Pdgfrβ-Cre+:Nestin-GFP) mice under Nestin-GFP background. All mice used in this study were in a C57BL/6-FVB-BALB/cBy mixed background. Both males and females were used in this study. All mice were maintained in the animal facility at University of Minnesota with free access to water and food.
Muscle dissection and preparation
Mice were anesthetized and hindlimb muscles were carefully dissected and minced with scissors and blades. The minced muscle fragments were incubated with 0.2% (w/v) type-2 collagenase (Worthington, LS004176) in DMEM at 37 °C for 2 h with shaking, followed by 3 × 15 min incubations with 0.25% trypsin/EDTA at 37 °C. After trituration and centrifugation at 1500 rpm for 6 min, the supernatant was discarded and the pellet resuspended in RBC lysis buffer. After centrifugation, the pellet was resuspended in DMEM + 10% FBS and passed through a 40-μm cell strainer to remove aggregates. The resulting single cell solution was centrifuged again at 1500 rpm for 6 min and resuspended in sorting buffer (20 mM HEPES pH 7.0, 1 mM EDTA, 1% BSA in 1 × Ca/Mg2+-free PBS pH 7.0). The cells were then stained with different antibodies and subjected to FACS sorting.
Cell sorting
Type I and type II pericytes were isolated from wild-type (WT) and PKO mice by FACS as described previously with minor modifications [9, 10, 14]. In brief, the single cell solution was stained with PDGFRβ-PE (eBioscience, 12-1402) on ice for 30 min. After extensive wash, DAPI was added, and PE+GFP− (type I) and PE+GFP+ (type II) cells were sorted using the Sony SH800 sorter. Forward and back scatters (FSC and BSC) and DAPI were used to set a primary gate to exclude dead cells and small debris. Fluorescence minus one (FMO) control for PE was used to set the PDGFRβ-PE gating boundary. Cells dissociated from C57BL/6 mice (not labeled with Nestin-GFP) were used to set the Nestin-GFP gating boundary. The cells were sorted into culturing medium for in vitro experiments.
Cell culture
FACS-isolated type I and type II pericytes were grown in pericyte medium/growth medium (ScienCell, 1201), myogenic differentiation medium (DMEM with 2% horse serum and 1% penicillin/streptomycin), or adipogenic differentiation medium (MesenCult™ Basal Medium supplemented with Adipogenic Stimulatory Supplement, STEMCELL, 05501 and 05503) at 37 °C with 5% CO2. Medium was changed every 2–3 days until analysis.
In vitro proliferation assay
Proliferation was examined using the Click-iT Plus EdU Alexa Fluor 594 Imaging Kit (Life Technologies, C10639). Freshly isolated type I and type II pericytes were plated in uncoated or laminin-111 (Invitrogen, 23017-015, 5 μg/ml)-coated 24-well plates in pericyte medium. Two days after plating, Edu (8 μM) was added to the cells. After 24 h, the cells were fixed and Edu incorporation was analyzed according to the manufacturer’s instructions. For quantification, the percentage of Edu+ nuclei among all nuclei in one field was calculated. At least eight random fields per sample and five samples were used for quantification.
In vitro adipogenic differentiation assay
Adipogenic differentiation was induced using adipogenic differentiation medium. Briefly, freshly isolated type I and type II pericytes were plated in uncoated or laminin-111 (5 μg/ml)-coated 24-well plates in pericyte medium. After 4 days, pericyte medium was replaced with adipogenic differentiation medium containing saline or laminin-111 (5 μg/ml). After 8 or 14 days, the cells were fixed and subjected to perilipin immunostaining, followed by Oil Red O/hematoxylin staining. Mean perilipin intensity per field was used to quantify the extent of adipogenesis. At least eight random fields per sample and four samples were used for quantification.
In vitro myogenic differentiation assay
Myogenic differentiation was induced using myogenic differentiation medium. Briefly, freshly isolated type I and type II pericytes were plated in uncoated or laminin-111 (5 μg/ml)-coated 24-well plates in pericyte medium. After 4 days, pericyte medium was replaced with myogenic differentiation medium containing saline or laminin-111 (5 μg/ml). After 14 days, the cells were fixed and subjected to S-Myosin (DSHB, MF20, 1:100) immunostaining. Mean S-Myosin intensity per field was used to quantify the extent of myogenesis. At least eight random fields per sample and four samples were used for quantification.
Immunohistochemistry and immunocytochemistry
Transverse muscle sections (10-μm thick) were cut using a cryostat. Sections of the muscles from one-third and two-thirds along the anterior/posterior axis were collected. Muscle sections or sorted cells were immunostained with anti-laminin γ1 (Abcam, AB3297, 1:200; NeoMarkers, RT-795-P0, 1:100), anti-PDGFRβ (eBioscience, 14-1402, 1:100), anti-S-Myosin (DSHB, MF-20, 1:100), anti-perilipin (Sigma, P1998, 1:500), anti-phospho-histone H3 (Millipore, 06-570, 1:500), or anti-Ki67 (Millipore, AB9260, 1:1000) antibodies overnight at 4 °C, followed by fluorescent secondary antibodies (Invitrogen) for 1 h at room temperature. After mounting, sections were examined and photographed with a LSM 710 confocal microscopy. For immunohistochemistry, five random fields from each section, two sections (one-third and two-thirds along the anterior/posterior axis) per sample, and five animals per condition were used for quantification. For immunocytochemistry, eight random fields from each experiment and four to five independent sorts/experiments were used for quantification.
Histology
Oil Red O staining and hematoxylin staining were performed after immunocytochemistry, according to standard protocols. For Oil Red O staining, after the last step of perilipin immunocytochemistry, sections were washed in PBS and afterwards rinsed in 60% isopropanol. After incubating in Oil Red O (Sigma, O1391) working solution for 15 min, sections were rinsed in 60% isopropanol and water. Then, the sections were either mounted in mounting medium with DAPI (Vector Lab, H-1200) or stained in hematoxylin (Fisher Scientific, SH30-500D) for 2 min and then mounted in mounting medium (Vector Lab, H-1000). The percentage of Oil Red O+ area in each field was used to quantify the extent of adipogenesis. At least eight random fields per sample and four samples were used for quantification.
Quantitative RT-PCR
RNA isolation was performed using Trizol reagent (Invitrogen), followed by RNeasy mini kits (Qiagen). RNA concentration was quantified using a ND1000 spectrophotometer (Nanodrop). Reverse transcription was performed using SuperScript III First-Strand Synthesis System (Invitrogen). Gene expression analysis was performed using SYBR Green PCR Master Mix (Qiagen) on a LightCycler 480 system (Roche). The qRT-PCR data were quantified using the comparative Ct method [31, 32]. Specifically, the Ct values for gene of interest (GOI) and the internal controls (IC) in WT and PKO samples were obtained. The relative expression change of GOI in PKO pericytes compared with WT pericytes is calculated using this equation: fold change = 2–ΔΔCt = [(Ct-GOI – Ct-IC)PKO – (Ct-GOI – Ct-IC)wildtype]. Each sample was performed in duplicate, and four independent/biological replicates were used for quantification. The primers used are listed below. C/EBPα: forward-CCCCTCAGTCCCTGTCTTTAGA, reverse-GGTGGAGGTGCAAAAAGCAA; C/EBPβ: forward-AAGCTGAGCGACGAGTACAAGA, reverse-GTCAGCTCCAGCACCTTGTG; C/EBPδ: forward-GCCCCAAAAGCCAGTAATTGT, reverse-ACAACAGGCCGTGCAGATC; KROX20: forward-CTGGGCAAAGGACCTTGATG, reverse-TCTTTTTGCTCCCCCATTTCT; KLF5: forward-GACATGCCCAGTTCGACAAA, reverse-CATGCCCTGGAACTGTTTCAT; FABP4: forward-CCCAACATGATCATCAGCGTAA, reverse-GTCGTCTGCGGTGATTTCATC; Pax7: forward-GGCCAAACTGCTGTTGATTACC, reverse-GTAGGCTTGTCCCGTTTCCA; MyoD: forward-GACCCAGGAACTGGGATATG, reverse-CGAAACACGGGTCATCATAG; Myf5: forward-TTCCCACCTGCTTCTCTGAAG, reverse-TCAAACTGGTCCCCAAACTCA; Mrf4: forward-CCCCCCTTTCCACCTAATCA, reverse-GACTTTCACTTGAGGTGGTGAGAA; Myog: forward-GACCCTACAGACGCCCACAA, reverse-CAATCTCAGTTGGGCATGGTT; S-Myosin: forward-AACAAGGACCCCCTGAATGAG, reverse-ACCACCGCCACCAGACTCT; GAPDH: forward-GGTGGAGCCAAAAGGGTCAT, reverse-GCATTGCTGACAATCTTGAGTGA; TBP: forward-CCCTTGTACCCTTCACCAAT, reverse-CAGCCAAGATTCACGGTAGA.
Western blotting
Cells were lysed with RIPA buffer (50 mM Tris pH 7.4, 1% NP-40, 0.5% Na-deoxycholate, 1% SDS, 150 mM NaCl, 2 mM EDTA, 1 × protease inhibitor cocktail, and 1 × phosphatase inhibitor cocktail). Total protein levels were determined using the Bio-Rad protein assay kit, and equal amounts of proteins were loaded and separated on SDS-PAGE. After transferring to PVDF membrane (Millipore), proteins were detected using a standard immune-blotting technique. The following primary antibodies were used: anti-Pax7 (Aviva Systems Biology, ARP32742, 1:400), anti-Myf5 (Santa Cruz, sc-302, 1:200), anti-Myogenin (BD Biosciences, 556358, 1:250), anti-phospho-ERK1/2 (Cell Signaling, 4370, 1:1000), anti-ERK1/2 (Cell Signaling, 9107, 1:1000), anti-phospho-p38 (Cell Signaling, 9216, 1:1000), anti-p38 (Cell Signaling, 9212, 1:1000), anti-integrin α7 (Calbiochem, ST1637, 1:500), anti-gpihbp1 (Thermo Scientific, PA1-16976, 1:250), anti-GAPDH (Abcam, ab9484, 1:1000), and anti-β-actin (Sigma, A5441, 1:2000). Target proteins were visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce). The density of target protein bands was quantified using NIH ImageJ software. The expression of phospho-ERK1/2 and phospho-p38 was normalized to ERK1/2 and p38, respectively. The expression of all other proteins was normalized to GAPDH and/or β-actin.
ELISA
The levels of BMP-2 and BMP-4 proteins synthesized by type I pericytes were determined by ELISA. Briefly, WT and PKO type I pericytes were plated in 24-well plates in the absence or presence of exogenous laminin-111 (5 μg/ml). The conditioned medium and cell lysates were collected after adipogenic differentiation for 8 days. BMP-2 and BMP-4 levels were determined by ELISA according to the manufacturer’s instructions (ls-f10947 and ls-f13543, LifeSpan Biosciences). BMP-2 and BMP-4 expression was normalized to total protein level.
Statistics
Results are shown as mean ± SD. Student’s t test performed by SPSS Statistics was used to analyze differences between two groups (genotypes). Two-way ANOVA in SPSS Statistics was used for comparisons involving two factors (genotypes and treatments).
Results
Laminin is abrogated in both type I and type II pericytes in PKO mice
In a previous study [21], we generated a mouse line (PKO) with laminin deficiency in pericytes by crossing the laminin γ1flox/flox (F/F) mice [28] with the Pdgfrβ-Cre+ transgenic line [27]. To investigate if laminin differentially regulates the proliferation and differentiation of type I and type II pericytes, we crossed these mice with Nestin-GFP transgenic line [29, 30] to generate control (WT) and PKO mice under Nestin-GFP background. Consistent with previous reports [14, 22, 23], both type I (PDGFRβ+Nestin−) and type II (PDGFRβ+Nestin+) pericytes were observed in skeletal muscles from WT mice (Fig. 1a). Both cell types were located in the interstitial space and expressed laminin (Fig. 1a). Consistent with our previous finding [21], laminin expression was significantly reduced and PDGFRβ expression increased in PKO muscles (Fig. 1a). Interestingly, GFP+ cells were also increased in PKO muscles, and most, if not all, GFP+ cells expressed PDGFRβ (Fig. 1a). Although no difference in type I pericyte number was observed (Fig. 1b), quantification revealed significantly more type II pericytes in PKO mice, compared to WT controls (Fig. 1c), indicating that loss of laminin preferably enhances type II pericyte number in vivo.
Fig. 1.
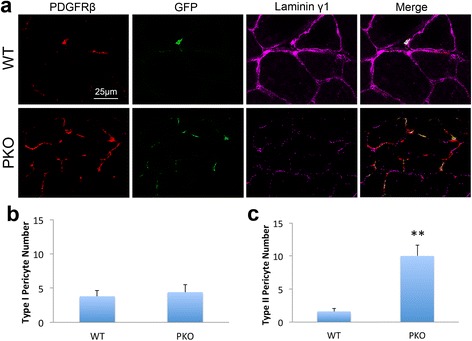
Both type I and type II pericytes are found in skeletal muscles. a Immunohistochemical analysis of PDGFRβ (red), GFP (green), and laminin γ1 (magenta) expression in wild-type (WT) controls and laminin γ1flox/flox:Pdgfrβ-Cre+(PKO) skeletal muscles. b and c Quantification of type I (b) and type II (c) pericyte number in WT and PKO muscles. n = 5. Data are shown as mean ± SD. **p < 0.01 versus WT. Scale bar = 25 μm
In addition, laminin γ1 expression was absent in both PDGFRβ+Nestin− and PDGFRβ+Nestin+ cells in PKO muscles (Fig. 1a), suggesting that laminin expression is diminished in both type I and type II pericytes in PKO mice. To further examine laminin expression in type I and type II pericytes, we isolated these cells from WT and PKO muscles by FACS using a well-established protocol [9, 10, 14, 21] (Fig. 2a). FMO control for PDGFRβ was used to set its gating (Fig. 2b and c). Sorted PDGFRβ+ cells were further separated based on GFP expression, and PDGFRβ+GFP− and PDGFRβ+GFP+ populations were defined as type I and type II pericytes, respectively (Fig. 2d). Representative dot plots for PDGFRβ and GFP are shown in Additional file 1 (Figure S1). The purity of sorted pericytes was examined by immunocytochemistry against PDGFRβ and NG2, and found to be higher than 95%. Consistent with in vivo studies, laminin γ1 expression was detected in both type I and type II pericytes isolated from WT muscles, but was absent in both cell types from PKO muscles (Fig. 2e and f). Quantification showed that almost all WT pericytes (both type I and type II) examined in five independent experiments expressed laminin, while more than 95% of PKO pericytes (both type I and type II) failed to express laminin (Fig. 2g and h). These data again suggest that laminin expression is abrogated in both type I and type II pericytes in PKO mice.
Fig. 2.
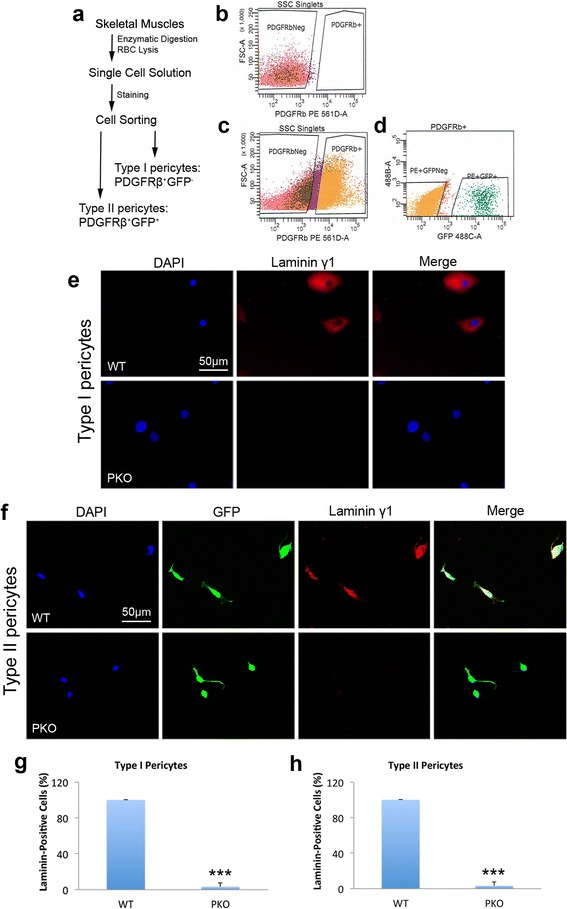
Laminin γ1 expression is abrogated in both type I and type II pericytes isolated from laminin γ1flox/flox:Pdgfrβ-Cre+(PKO) mice. a Diagram of type I and type II pericyte isolation procedure. b Representative plot of PDGFRβ-FMO control. c, d Representative plots showing sorting gates for PDGFRβ (c) and type I and type II pericytes (d). e Immunocytochemical analysis of laminin γ1 (red) expression in type I pericytes isolated from wild-type (WT) and PKO muscles. f Immunocytochemical analysis of laminin γ1 (red) and GFP (green) expression in type II pericytes isolated from WT and PKO muscles. g, h Quantitative data demonstrating the percentage of laminin-positive cells in type I (g) and type II (h) pericytes sorted from WT and PKO mice. n = 5. Data are shown as mean ± SD. ***p < 0.001 versus WT. Scale bars = 50 μm
Loss of laminin preferentially induces type II pericyte proliferation in vivo
In a previous study, we reported increased proliferation of laminin-deficient pericytes [21]. To investigate if the proliferation of type I and/or type II pericytes is regulated by laminin, we first performed in vitro proliferation assay. Type I and type II pericytes were isolated from WT and PKO mice by FACS as described above and their proliferation was examined by Edu incorporation assay 2 days post-isolation (Fig. 3a). Dramatically increased Edu+ cells were observed in PKO type I pericytes, compared to WT controls (Fig. 3b). Statistical analysis showed that there was a significant difference in the percentage of Edu+ cells between WT and PKO type I pericytes (Fig. 3c). Similar result was found for type II pericytes (Fig. 3d and e), suggesting that laminin negatively regulates the proliferation of both type I and type II pericytes in vitro. Additionally, exogenous laminin was able to substantially decrease the proliferation rate of both type I (Fig. 3b and c) and type II (Fig. 3d and e) PKO pericytes without affecting that of WT pericytes, indicating a reversible role of laminin in the proliferation of both type I and type II pericytes. In addition to Edu, an S-phase-specific marker [33], we also examined proliferation using phospho-Histone H3 (pH3), an M-phase-specific marker [34–36]. Similar results were observed; specifically, a higher percentage of pH3+ cells was found in PKO pericytes (both type I and type II), and exogenous laminin substantially reduced their proliferation to baseline levels (Additional file 1: Figure S2), again suggesting that laminin inhibits the proliferation of both type I and type II pericytes in vitro.
Fig. 3.
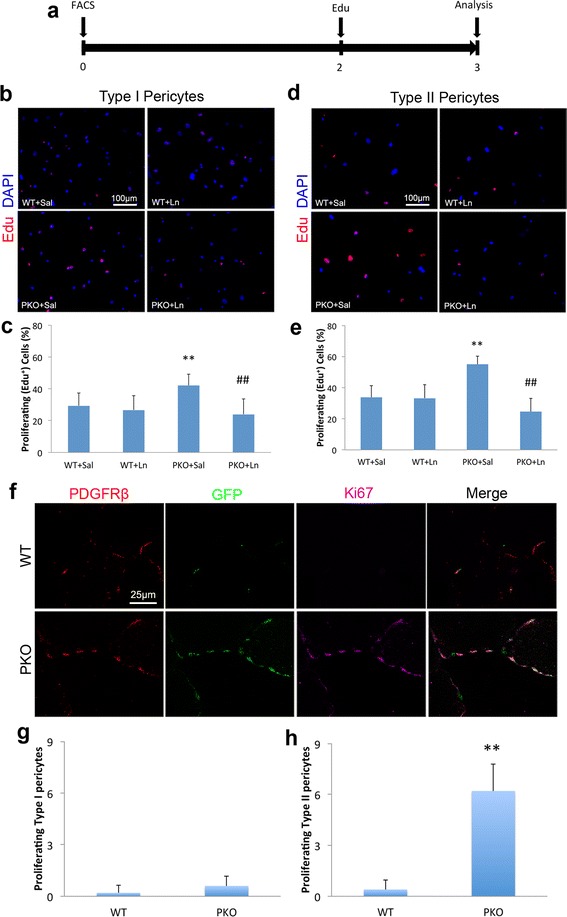
Laminin negatively regulates the proliferation of type I and type II pericytes. a Diagram of experimental design for Edu incorporation assay. b Immunocytochemical analysis of Edu (red) in type I pericytes isolated from wild-type (WT) and laminin γ1flox/flox:Pdgfrβ-Cre+(PKO) mice in the presence of saline (Sal) or laminin-111 (Ln). c Quantification of Edu incorporation in type I pericytes. n = 5. d Immunocytochemical analysis of Edu (red) in type II pericytes isolated from WT and PKO mice in the presence of Sal or Ln. e Quantification of Edu incorporation in type II pericytes. n = 5. f Immunohistochemical analysis of PDGFRβ (red), GFP (green), and Ki67 (magenta) expression in WT and PKO muscles. g, h Quantification of proliferating type I (g) and type II (h) pericyte number in WT and PKO mice. n = 5. Data are shown as mean ± SD. **p < 0.01 versus WT + Sal or WT; ## p < 0.01 versus PKO + Sal. Scale bars = 100 μm in b and d, and 25 μm in f
Next, we further examined the proliferation of pericytes in vivo by immunohistochemistry. Consistent with previous data [21], few proliferating (Ki67+) pericytes were observed in WT muscles (Fig. 3f). In PKO muscles, on the contrary, a large number of Ki67+ cells were identified (Fig. 3f). Surprisingly, these Ki67+ proliferating cells expressed both PDGFRβ and GFP (Fig. 3f). We then quantified the number of proliferating type I (Ki67+PDGFRβ+GFP−) and type II (Ki67+PDGFRβ+GFP+) pericytes in WT and PKO muscles. Although a comparable amount of proliferating type I pericytes was observed in WT and PKO mice (Fig. 3g), significantly more proliferating type II pericytes were detected in PKO muscles (Fig. 3h), suggesting that loss of laminin preferentially induces proliferation of type II pericytes in vivo. These data are consistent with the reported myogenic function of type II pericytes [14] and muscular dystrophic phenotype of the PKO mice [21].
Accumulating evidence suggests that ERK and p38 MAPK pathways play important roles in the proliferation and differentiation of various mammalian cells [37–39]. To determine if these pathways are involved in loss-of-laminin-induced proliferation of pericytes, we examined the activation/phosphorylation of ERK1/2 and p38. Consistent with previous reports demonstrating critical roles of the ERK pathway in pericyte proliferation [40, 41], we found high levels of phosphorylated ERK1/2 in both type I and type II pericytes (Fig. 4), although no significant differences in ERK1/2 activation were observed between WT and PKO pericytes (both type I and type II) (Fig. 4). These data suggest that the ERK pathway regulates pericyte proliferation, but is not responsible for the enhanced proliferation of PKO pericytes. Additionally, we also examined the activation of p38. Negligible levels of phosphorylated p38 were detected in both type I and type II pericytes independent of their genotypes (WT vs. PKO; data not shown), suggesting that p38 may not be involved in the proliferation of type I and type II pericytes.
Fig. 4.
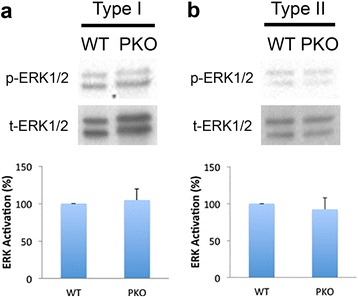
ERK1/2 activation is not responsible for the enhanced proliferation of PKO pericytes. a Western blots and quantification of phosphorylated ERK1/2 (p-ERK1/2) expression in wild-type (WT) and laminin γ1flox/flox:Pdgfrβ-Cre+(PKO) type I pericytes. Total ERK1/2 (t-ERK1/2) was used as a loading control. n = 3. b Western blots and quantification of p-ERK1/2 expression in WT and PKO type II pericytes. t-ERK1/2 was used as a loading control. n = 3. Data are shown as mean ± SD
Laminin inhibits adipogenesis of type I pericytes
Accumulating evidence suggests that type I rather than type II pericytes are adipogenic [14, 23, 25]. To investigate the role of laminin in pericyte adipogenesis, we isolated type I and type II pericytes from WT and PKO muscles and examined their adipogenic differentiation in vitro (Fig. 5a). After 8 days in adipogenic medium, few WT type I pericytes expressed the adipocyte marker perilipin or were stained by Oil Red O, a dye that specifically labels lipids in adipocytes (Fig. 5b). PKO type I pericytes, however, showed features of mature adipocytes at both the morphological and biochemical levels. Cells with typical adipocyte morphology (large cells with multiple vacuoles) that were positive for perilipin and Oil Red O were observed (Fig. 5b). Quantifications revealed significantly higher levels of perilipin intensity (Fig. 5c) and Oil Red O+ area (Fig. 5d) in PKO type I pericytes, suggesting that laminin inhibits the adipogenic differentiation of type I pericytes. Furthermore, we found that the adipogenic differentiation of PKO type I pericytes was dramatically diminished in the presence of exogenous laminin (Fig. 5b–d), suggesting an anti-adipogenic potential of laminin. These data are consistent with previous studies demonstrating anti-adipogenic and beneficial roles of exogenous laminin in muscular dystrophy [21, 42–45]. To explore the molecular mechanism underlying the inhibitory effect of laminin in the adipogenesis of type I pericytes, we first examined the expression of gpihbp1, which has been shown to be involved in pericyte adipogenesis [21]. A significantly lower level of gpihbp1 was found in PKO type I pericytes, and exogenous laminin was able to increase gpihbp1 expression in type I PKO pericytes (Fig. 6a), suggesting that gpihbp1 may regulate the adipogenesis of type I pericytes. Next, we further examined the expression of various adipogenic factors, including C/EBPα, C/EBPβ, C/EBPδ, KROX20, KLF5, and FABP4, in type I pericytes by quantitative RT-PCR. Compared to WT type I pericytes, PKO type I pericytes demonstrated a dramatic (four- to six-fold) increase in the expression of C/EBPβ, KROX20, and KLF5, and a slight decrease of C/EBPα 4 days after adipogenic differentiation (Fig. 5e). These data suggest that laminin negatively regulates adipogenesis of type I pericytes via inhibiting the expression of C/EBPβ, KROX20, and KLF5.
Fig. 5.
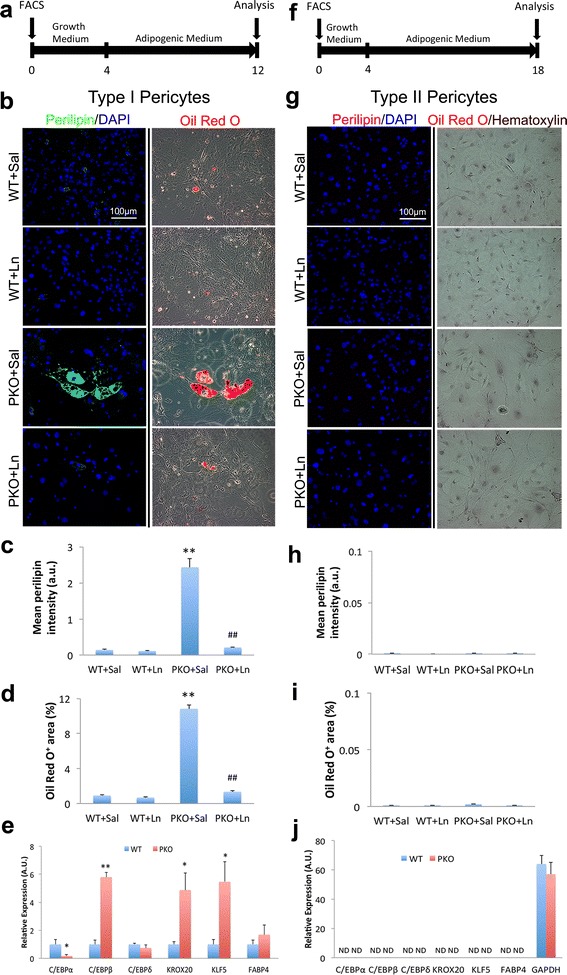
Laminin inhibits adipogenic differentiation of type I pericytes. a Diagram of experimental design for adipogenic differentiation of type I pericytes. b Immunocytochemical analysis of perilipin (green) and Oil red O (red) staining in wild-type (WT) and laminin γ1flox/flox:Pdgfrβ-Cre+(PKO) type I pericytes in the presence of saline (Sal) or laminin-111 (Ln). c Quantification of perilipin (green) expression in type I pericytes after adipogenic differentiation. n = 4. d Percentage of Oil Red O+ (red) area in type I pericytes after adipogenic differentiation. n = 4. e Quantitative RT-PCR analyses of C/EBPα, C/EBPβ, C/EBPδ, KROX20, KLF5, and FABP4 expression in type I pericytes 4 days after adipogenic differentiation. n = 4. f Diagram of experimental design for long-term adipogenic differentiation of type II pericytes. g Immunocytochemical analysis of perilipin (magenta) and Oil red O (red)/hematoxylin (brown) staining in WT and PKO type II pericytes in the presence of sal or Ln. h Quantification of perilipin (magenta) expression in type II pericytes after adipogenic differentiation. n = 4. i Percentage of Oil Red O+ (red) area in type II pericytes. n = 4. j Quantitative RT-PCR analyses of C/EBPα, C/EBPβ, C/EBPδ, KROX20, KLF5, and FABP4 expression in type II pericytes 4 days after adipogenic differentiation. n = 4. Data are shown as mean ± SD. *p < 0.05, **p < 0.01 versus WT or WT + Sal; ## p < 0.01 versus PKO + Sal. Scale bars = 100 μm
Fig. 6.
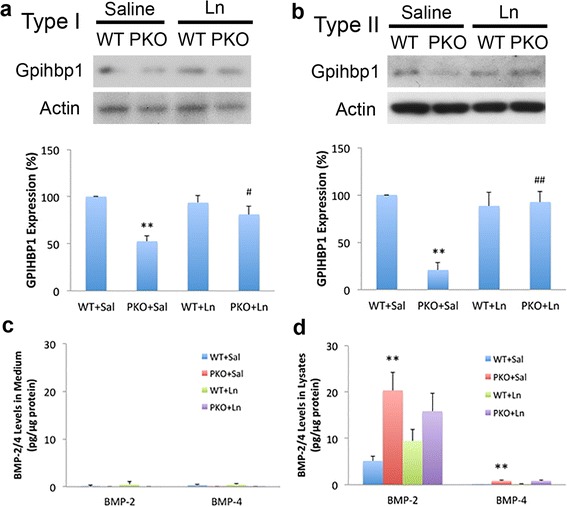
Gpihbp1 and BMP-2/4 expression in subpopulations of pericytes. a Western blots and quantification of gpihbp1 expression in wild-type (WT) and laminin γ1flox/flox:Pdgfrβ-Cre+(PKO) type I pericytes in the presence of saline (Sal) or laminin-111 (Ln). Actin was used as a loading control. n = 4. b Western blots and quantification of gpihbp1 expression in WT and PKO type II pericytes in the presence of Sal or Ln. Actin was used as a loading control. n = 4. c ELISA analyses of BMP-2 and BMP-4 levels in conditioned medium from type I pericytes after adipogenic differentiation for 8 days. n = 3. d ELISA analyses of BMP-2 and BMP-4 levels in cell lysates from type I pericytes after adipogenic differentiation for 8 days. n = 3. Data are shown as mean ± SD. **p < 0.01 versus WT + Sal; # p < 0.05, ## p < 0.01 versus PKO + Sal
Accumulating evidence shows that BMP-2 and BMP-4 promote adipogenic differentiation of various types of progenitor/stem cells, including mesenchymal precursor cells [46–50]. To investigate if loss-of-laminin-induced adipogenic differentiation of type I pericytes is driven by BMP proteins, we examined BMP-2 and BMP-4 levels from type I pericytes 8 days after adipogenic differentiation. Minimal levels of BMP-2 and BMP-4 were detected in conditioned medium independent of genotypes (WT vs. PKO) and environment (with vs. without exogenous laminin) (Fig. 6c), suggesting that either these proteins were not secreted by type I pericytes or were mobilized to cell surface after secretion. Thus, we further examined BMP-2 and BMP-4 levels in cell lysates from type I pericytes. Compared to WT cells, PKO type I pericytes expressed significantly higher levels of BMP-2 and BMP-4 (Fig. 6d). It should be noted, however, that the absolute level of BMP-2 is substantially higher than that of BMP-4 (Fig. 6d), suggesting that BMP-2 may play a more important role than BMP-4. In addition, exogenous laminin slightly reduced BMP-2 expression in type I PKO, although this change was not statistically significant (Fig. 6d). Laminin treatment also failed to affect BMP-4 expression in type I PKO pericytes (Fig. 6d). Together, these results suggest that both BMP proteins, especially BMP-2, may mediate loss-of-laminin-induced adipogenic differentiation of type I pericytes.
In sharp contrast to type I pericytes, type II pericytes failed to show signs of adipogenesis after 8 days in adipogenic medium (data not shown). To determine if this is due to a relatively short differentiation time, we allowed type II pericytes to differentiate in adipogenic condition for up to 14 days (Fig. 5f). Neither perilipin expression (Fig. 5g and h) nor Oil Red O signal (Fig. 5g and i) was observed in type II pericytes, independent of their genotypes (WT vs. PKO). In addition, the same results were observed in the presence of exogenous laminin (Fig. 5g–i). Consistent with these results, quantitative RT-PCR failed to detect any adipogenic factors described above in type II pericytes (Fig. 5j). Altogether, these data strongly suggest that type II pericytes are not adipogenic.
Laminin is indispensable for myogenesis of type II pericytes
It has been shown that type II, but not type I, pericytes have myogenic potential and are able to differentiate into myogenic cells to repair muscle injury [14, 23, 25]. To determine the role of laminin in pericyte myogenesis, we isolated type I and type II pericytes from WT and PKO muscles using FACS, as described above, and examined their myogenic ability in vitro (Fig. 7a). Consistent with previous reports that type II rather than type I pericytes are myogenic [14, 23, 25], we failed to detect sarcomere myosin (S-Myosin), a marker for mature myotubes/myofibers, in type I pericytes after 14 days in myogenic medium regardless of their genotypes (WT vs. PKO) or environment (with vs. without exogenous laminin) (Fig. 7b). Quantification from four independent experiments revealed negligible levels of S-Myosin expression in type I pericytes (Fig. 7c). Similarly, quantitative RT-PCR failed to detect any myogenic factors, including Pax7, MyoD, Myf5, Mrf4, Myog, and S-Myosin, in type I pericytes 4 days after myogenic differentiation (Fig. 7d). These data strongly indicate that type I pericytes are unable to undergo myogenic differentiation.
Fig. 7.
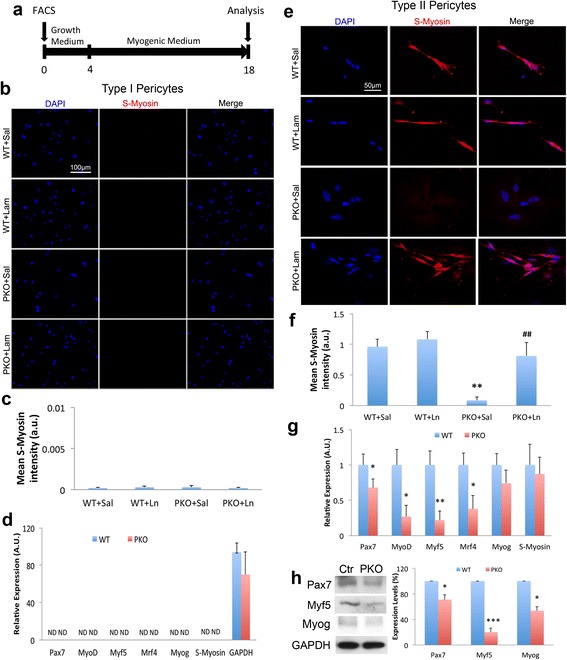
Laminin promotes myogenic differentiation of type II pericytes. a Diagram of experimental design for myogenic differentiation. b Immunocytochemical analysis of S-Myosin (red) in wild-type (WT) and laminin γ1flox/flox:Pdgfrβ-Cre+(PKO) type I pericytes in the presence of saline (Sal) or laminin-111 (Ln). c Quantification of S-Myosin (red) expression in type I pericytes after myogenic differentiation. n = 4. d Quantitative RT-PCR analyses of Pax7, MyoD, Myf5, Mrf4, myogenin (Myog), and S-Myosin expression in type I pericytes 4 days after myogenic differentiation. n = 4. e Immunocytochemical analysis of S-Myosin (red) in WT and PKO type II pericytes in the presence of sal or Ln. f Quantification of S-Myosin (red) expression in type II pericytes after myogenic differentiation. n = 4. **p < 0.01 versus WT + Sal. g Quantitative RT-PCR analyses of Pax7, MyoD, Myf5, Mrf4, Myog, and S-Myosin expression in type II pericytes 4 days after myogenic differentiation. n = 4. h Western blots and quantification of Pax7, Myf5, and Myog expression in WT and PKO type II pericytes 7 days after myogenic differentiation. GAPDH was used as a loading control. n = 4–5. Data are shown as mean ± SD. # p < 0.05 versus PKO + Sal; *p < 0.05, **p < 0.01, ***p < 0.001 versus WT. Scale bars = 100 μm in b and 50 μm in d
Next, we performed in vitro myogenic assay using type II pericytes isolated from WT and PKO muscles. After 14 days in myogenic medium, type II pericytes isolated from WT mice aggregated together, forming myotubes that were S-Myosin-positive (Fig. 7e). Type II pericytes isolated from PKO mice, on the other hand, failed to express S-Myosin or form myotube-like structures after differentiation for 14 days (Fig. 7e). Interestingly, exogenous laminin successfully induced the formation of S-Myosin+ myotubes in PKO type II pericytes without affecting WT type II pericytes (Fig. 7e). Quantification revealed a significant reduction of S-Myosin expression in PKO type II pericytes compared to WT cells, and a substantial increase of S-Myosin expression in PKO type II pericytes treated with exogenous laminin compared to those treated with saline (Fig. 7f). In addition, we also examined the expression of Myogenin (Myog), a key transcription factor indispensable for myogenesis. Compared to WT type II pericytes, PKO type II pericytes demonstrated significantly lower levels of Myog (Additional file 1: Figure S3). Additionally, exogenous laminin successfully enhanced the expression of Myog in PKO type II pericytes without affecting that in WT cells (Additional file 1: Figure S3). These results suggest that laminin is indispensable for the myogenic differentiation of type II pericytes.
To explore the molecular mechanism underlying the role of laminin in the myogenesis of type II pericytes, we first examined the expression of gpihbp1, which has been shown to regulate pericyte myogenesis [21]. Compared to WT type II pericytes, PKO type II pericytes showed reduced expression of gpihbp1 (Fig. 6b). Additionally, exogenous laminin was able to increase gpihbp1 expression in PKO type II pericytes (Fig. 6b), suggesting that myogenesis of type II pericytes may be mediated by gpihbp1. Next, we examined the expression of various myogenic factors in type II pericytes by quantitative RT-PCR. Compared to WT controls, PKO type II pericytes demonstrated a significant reduction in the expression of Pax7, MyoD, Myf5, and Mrf4 4 days after myogenic differentiation (Fig. 7g). In addition, we further examined the expression of Pax7, Myf5, and Myog in type II pericytes at protein level 7 days after myogenic differentiation. Western blot analyses revealed a 30% and 80% reduction of Pax7 and Myf5 in PKO type II pericytes, respectively, compared to WT controls (Fig. 7h). Although Myog mRNA was not altered at 4 days after myogenic differentiation (Fig. 7g), its protein level was dramatically reduced in PKO type II pericytes by 7 (Fig. 7h) and 14 (Additional file 1: Figure S3) days after myogenic differentiation. Furthermore, we also examined the activation of the p38 MAPK pathway, which plays a crucial role in the myogenesis of satellite cells and/or myoblasts [51–55]. Negligible levels of phosphorylated p38 were detected in type II pericytes independent of their genotypes (WT vs. PKO; data not shown), suggesting that the p38 MAPK pathway plays a minimal role in myogenic differentiation of type II pericytes. Together, these data suggest that: (1) laminin induces myogenesis of type II pericytes via activating various myogenic transcription factors; and (2) laminin is involved in the differentiation of pericytes into Pax7+, Myf5+, and Myog+ myogenic cells during myogenesis, which is consistent with previous studies showing that muscle regeneration fails when Pax7 expression is genetically ablated [8, 56, 57].
It has been shown that integrin α7 (ITGA7), a molecular marker for myoblasts, functions as a laminin receptor. To examine whether the differences in myogenic differentiation between type I and type II pericytes in the presence or absence of exogenous laminin are due to abnormal expression of ITGA7, we performed Western blot analysis. Consistent with a previous study reporting ITGA7 mRNA transcript in pericytes/MSCs [58], ITGA7 protein (120 kDa) was detected in both type I and type II pericytes (Fig. 8). Compared to WT controls, substantially lower levels of ITGA7 were found in PKO pericytes (both type I and type II) (Fig. 8), suggesting that laminin level positively correlates with ITGA7 expression in pericytes. In addition, exogenous laminin dramatically increased ITGA7 expression in PKO pericytes (both type I and type II) without affecting that in WT pericytes (Fig. 8), again suggesting that laminin may regulate ITGA7 expression in pericytes. Since ITGA7 is expressed in both type I and type II pericytes, it is less likely to be responsible for their different myogenic potential.
Fig. 8.
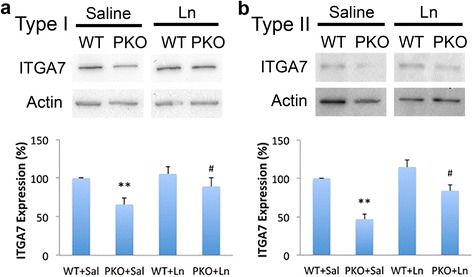
Laminin regulates ITGA7 expression in both type I and type II pericytes. a Western blots and quantification of integrin α7 (ITGA7; 120 kDa) expression in wild-type (WT) and laminin γ1flox/flox:Pdgfrβ-Cre+(PKO) type I pericytes in the presence of saline (Sal) or laminin-111 (Ln). Actin was used as a loading control. n = 3. b Western blots and quantification of ITGA7 (120 kDa) expression in WT and PKO type II pericytes in the presence of Sal or Ln. Actin was used as a loading control. n = 3. Data are shown as mean ± SD. **p < 0.01 versus WT + Sal; # p < 0.05 versus PKO + Sal
Discussion
Consistent with previous reports [14, 23, 25], we showed that type I pericytes are adipogenic and type II pericytes are myogenic under in vitro conditions. We also demonstrated that loss of laminin increases the proliferation of both type I and type II pericytes in vitro, but preferentially induces the proliferation of type II pericytes in vivo. In addition, lack of laminin also promotes adipogenic differentiation of type I pericytes and inhibits myogenic differentiation of type II pericytes in vitro. These results are consistent with: (1) the observation that both type I and type II pericytes have direct contact with laminin-rich basement membrane under physiological conditions; (2) the “inhibitory” effect of laminin on pericyte differentiation in the brain [59]; and (3) our previous report [21] that loss of laminin in PDGFRβ+ pericytes (both type I and type II) increases their proliferation, promotes their adipogenic differentiation, and diminishes their myogenic differentiation. Together, our data suggest that laminin actively but differentially regulates the proliferation and differentiation of type I and type II pericytes. The mechanism linking laminin and different responses of type I and type II pericytes, however, remains not fully understood. Our mechanistic studies showed that: (1) loss of laminin activates the adipogenic program in type I pericytes by enhancing the expression of various adipogenic factors, including C/EBPβ, KROX20, and KLF5; and (2) loss of laminin inactivates the myogenic program in type II pericytes by decreasing the expression of almost all myogenic transcription factors, including Pax7, MyoD, Myf5, Mrf4, and Myog. These data suggest that the intrinsic difference between type I and type II pericytes may be responsible for their distinct behaviors to loss of laminin. Consistent with previous studies [14, 60], adipogenic factors were detected in type I but not type II pericytes after adipogenic differentiation, whereas myogenic factors were found in type II but not type I pericytes after myogenic differentiation. These results support that type I and type II pericytes are intrinsically different and have distinct differentiation programs. Altogether, our data suggest that laminin differentially regulates the differentiation and fate determination of type I and type II pericytes by acting on distinct transcription factors in these cells.
It should be noted that Nestin-GFP+ myofibers are not detected in Nestin-GFP transgenic mice in vivo, although type II pericytes show myogenic activity in vitro. This may be due to the following three possibilities. First, type II pericytes may lose Nestin-GFP expression after myogenic differentiation, generating GFP-negative myofibers. Second, the contribution of pericytes to myogenesis in vivo may be below the detection level under physiological conditions. Third, pericytes do not have myogenic capability in vivo, but obtain myogenic potential in vitro. Recently, it has been shown that transplantation of DsRed-expressing type II pericytes into injured muscles leads to DsRed-positive myofibers after 2 weeks [14], suggesting that type II pericytes are able to undergo myogenic differentiation in vivo, at least under pathological conditions. Therefore, we favor the first two possibilities.
In the in vitro proliferation assay, we showed that exogenous laminin failed to decrease the proliferation of WT pericytes. This could be due to the fact that endogenous laminin produced by WT pericytes is sufficient to inhibit and maintain the proliferation at baseline. An alternative explanation is that endogenous pericytic laminin and exogenous laminin may be different in their compositions and functions. The exogenous laminin we used in this study is mouse laminin-111, which is the only commercially available recombinant laminin of mouse origin. Although expressed at the embryonic stage, this laminin isoform is absent in adult skeletal muscles [61–67]. In addition, although the exact laminin isoforms produced by pericytes are not clear, many other laminin chains, including laminin α4 and β2, are detected [68]. Ongoing projects in our laboratory focus on identifying laminin isoforms synthesized by pericytes and investigating their biological functions.
The fact that exogenous laminin successfully reversed the proliferation and differentiation phenotypes of PKO pericytes suggests that laminin-111 could at least partially compensate for the loss of pericyte-derived laminin isoforms. This is consistent with previous reports on the compensatory effects of laminin-111. For example, expressing laminin α1 is able to improve the general health (body weight and lifespan) and muscle deficit in laminin α2-deficient mice [69–74], a widely used congenital muscular dystrophy model [75–77]. Furthermore, recombinant laminin-111 has been shown to improve muscle pathology and function in various muscular dystrophy models, including those caused by lack of laminin α2 [42, 44] and laminin γ1 [21]. Additionally, laminin α1, especially its globular domains, has been shown to partially compensate for the developmental defect caused by loss of laminin α5 [78, 79]. Given the diverse biological functions and strong therapeutic potential of laminin-111, it is not surprising to see that the deficits in proliferation and differentiation of PKO pericytes can be rescued by exogenous laminin-111.
In muscle injury induced by glycerol or BaCl2, both type I and type II pericytes were reported to expand in vivo [14]. In contrast to that study, we found that type II pericytes predominantly proliferate in pericytic laminin-deficient mice. Given that type II pericytes have myogenic capability [14] and that the pericytic laminin-deficient mutants develop severe muscular dystrophy [21], this finding suggests that laminin is indispensable for the myogenesis of type II pericytes. The discrepancy may be explained by either pericytic laminin only regulates the proliferation of type II pericytes, or different injury models create different microenvironments that lead to distinct outcomes. Given that loss of pericytic laminin enhances the proliferation of both type I and type II pericytes in vitro, we favor the second possibility. The exact microenvironment created in PKO mice and how it induces predominant proliferation of type II pericytes in vivo, however, remain unclear. Answers to these questions would allow us to identify subtype-specific pathways, separate subtype-specific functions, and hopefully achieve therapeutic potential by manipulating different subtypes of pericytes.
Conclusions
Laminin predominantly inhibits the proliferation of type II pericytes in vivo, and negatively and positively regulates the adipogenic and myogenic differentiation of type I and type II pericytes, respectively.
Acknowledgements
We thank Drs. Volkhard Lindner (Maine Medical Center Research Institute) and Grigori Enikolopov (Cold Spring Harbor Laboratory) for the Pdgfrβ-Cre and Nestin-GFP mice, respectively.
Funding
This work was supported by the Fund-A-Fellow grant from the Myotonic Dystrophy Foundation (MDF-FF-2014-0013) and Scientist Development Grant from American Heart Association (16SDG29320001).
Availability of data and materials
Three supplementary figures are available. Material used in this study is available from the corresponding author on reasonable request.
Authors’ contributions
YY designed the experiments. JG, AN, and YY performed the experiments, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The use of mice in this study was approved by the Institutional Animal Care and Use Committee at the University of Minnesota (1502-32293A). All experimental procedures were performed in compliance with the Animal Welfare Act and the NIH Guide for the Care and Use of Laboratory Animals. Euthanasia was performed according to the standards of the American Veterinary Medical Association.
Abbreviations
- F/F
Laminin γ1flox/flox
- FAP
Fibro/adipogenic progenitor
- FMO
Fluorescence minus one
- ITGA7
Integrin α7
- MSC
Mesenchymal stem cell
- Myog
Myogenin
- pH3
Phospho-Histone H3
- PIC
PW1+/Pax7– interstitial cell
- PKO
Laminin γ1flox/flox:Pdgfrβ-Cre+
- WT
Wild-type
Additional file
Figure S1. Representative dot plots for PDGFRβ-PE and Nestin-GFP in FACS analysis. (a) Representative dot plot for PDGFRβ-FMO control. (b) Representative dot plot showing sorting gates. Figure S2. Laminin inhibits the proliferation of type I and type II pericytes in vitro. (a) Quantification of pH3+ cell percentage in type I pericytes in the presence of sal or ln. n = 4. (b) Quantification of pH3+ cell percentage in type II pericytes in the presence of Sal or Ln. n = 4. Ln laminin-111, Sal saline, WT wildtype. Data are shown as mean ± SD. **p < 0.01 versus WT + Sal; ## p < 0.01 versus PKO + Sal. Figure S3. Laminin induces Myogenin expression in type II pericytes after myogenic differentiation. Western blots and quantification of Myog expression in WT and PKO type II pericytes after myogenic differentiation. Actin was used as a loading control. n = 4. Ln laminin-111, Sal saline, WT wildtype. Data are shown as mean ± SD. **p < 0.01 versus WT + Sal; # p < 0.05 versus PKO + Sal. (DOCX 17413 kb)
Contributor Information
Jyoti Gautam, Email: fjyoti@d.umn.edu.
Abhijit Nirwane, Email: anirwane@d.umn.edu.
Yao Yao, Phone: 218-726-6082, Email: yyao@umn.edu.
References
- 1.Mauro A. Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–5. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uezumi A, Fukada S, Yamamoto N, Takeda S, Tsuchida K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat Cell Biol. 2010;12:143–52. doi: 10.1038/ncb2014. [DOI] [PubMed] [Google Scholar]
- 3.Joe AW, Yi L, Natarajan A, Le Grand F, So L, Wang J, Rudnicki MA, Rossi FM. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat Cell Biol. 2010;12:153–63. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitchell KJ, Pannerec A, Cadot B, Parlakian A, Besson V, Gomes ER, Marazzi G, Sassoon DA. Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat Cell Biol. 2010;12:257–66. doi: 10.1038/ncb2025. [DOI] [PubMed] [Google Scholar]
- 5.Zheng B, Cao B, Crisan M, Sun B, Li G, Logar A, Yap S, Pollett JB, Drowley L, Cassino T, et al. Prospective identification of myogenic endothelial cells in human skeletal muscle. Nat Biotechnol. 2007;25:1025–34. doi: 10.1038/nbt1334. [DOI] [PubMed] [Google Scholar]
- 6.Nesti LJ, Jackson WM, Shanti RM, Koehler SM, Aragon AB, Bailey JR, Sracic MK, Freedman BA, Giuliani JR, Tuan RS. Differentiation potential of multipotent progenitor cells derived from war-traumatized muscle tissue. J Bone Joint Surg Am. 2008;90:2390–8. doi: 10.2106/JBJS.H.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jackson WM, Nesti LJ, Tuan RS. Potential therapeutic applications of muscle-derived mesenchymal stem and progenitor cells. Expert Opin Biol Ther. 2010;10:505–17. doi: 10.1517/14712591003610606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murphy MM, Lawson JA, Mathew SJ, Hutcheson DA, Kardon G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development. 2011;138:3625–37. doi: 10.1242/dev.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dellavalle A, Maroli G, Covarello D, Azzoni E, Innocenzi A, Perani L, Antonini S, Sambasivan R, Brunelli S, Tajbakhsh S, Cossu G. Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat Commun. 2011;2:499. doi: 10.1038/ncomms1508. [DOI] [PubMed] [Google Scholar]
- 10.Dellavalle A, Sampaolesi M, Tonlorenzi R, Tagliafico E, Sacchetti B, Perani L, Innocenzi A, Galvez BG, Messina G, Morosetti R, et al. Pericytes of human skeletal muscle are myogenic precursors distinct from satellite cells. Nat Cell Biol. 2007;9:255–67. doi: 10.1038/ncb1542. [DOI] [PubMed] [Google Scholar]
- 11.Granero-Molto F, Weis JA, Longobardi L, Spagnoli A. Role of mesenchymal stem cells in regenerative medicine: application to bone and cartilage repair. Expert Opin Biol Ther. 2008;8:255–68. doi: 10.1517/14712598.8.3.255. [DOI] [PubMed] [Google Scholar]
- 12.Salem HK, Thiemermann C. Mesenchymal stromal cells: current understanding and clinical status. Stem Cells. 2010;28:585–96. doi: 10.1002/stem.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 14.Birbrair A, Zhang T, Wang ZM, Messi ML, Enikolopov GN, Mintz A, Delbono O. Role of pericytes in skeletal muscle regeneration and fat accumulation. Stem Cells Dev. 2013;22:2298–314. doi: 10.1089/scd.2012.0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sampaolesi M, Blot S, D’Antona G, Granger N, Tonlorenzi R, Innocenzi A, Mognol P, Thibaud JL, Galvez BG, Barthelemy I, et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature. 2006;444:574–9. doi: 10.1038/nature05282. [DOI] [PubMed] [Google Scholar]
- 16.Sampaolesi M, Torrente Y, Innocenzi A, Tonlorenzi R, D’Antona G, Pellegrino MA, Barresi R, Bresolin N, De Angelis MG, Campbell KP, et al. Cell therapy of alpha-sarcoglycan null dystrophic mice through intra-arterial delivery of mesoangioblasts. Science. 2003;301:487–92. doi: 10.1126/science.1082254. [DOI] [PubMed] [Google Scholar]
- 17.Sims DE. Recent advances in pericyte biology—implications for health and disease. Can J Cardiol. 1991;7:431–43. [PubMed] [Google Scholar]
- 18.Diaz-Flores L, Gutierrez R, Varela H, Rancel N, Valladares F. Microvascular pericytes: a review of their morphological and functional characteristics. Histol Histopathol. 1991;6:269–86. [PubMed] [Google Scholar]
- 19.Doherty MJ, Canfield AE. Gene expression during vascular pericyte differentiation. Crit Rev Eukaryot Gene Expr. 1999;9:1–17. [PubMed] [Google Scholar]
- 20.Farrington-Rock C, Crofts NJ, Doherty MJ, Ashton BA, Griffin-Jones C, Canfield AE. Chondrogenic and adipogenic potential of microvascular pericytes. Circulation. 2004;110:2226–32. doi: 10.1161/01.CIR.0000144457.55518.E5. [DOI] [PubMed] [Google Scholar]
- 21.Yao Y, Norris EH, Mason CE, Strickland S. Laminin regulates PDGFRβ + cell stemness and muscle development. Nat Commun. 2016;7:11415. doi: 10.1038/ncomms11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birbrair A, Zhang T, Wang ZM, Messi ML, Enikolopov GN, Mintz A, Delbono O. Skeletal muscle pericyte subtypes differ in their differentiation potential. Stem Cell Res. 2013;10:67–84. doi: 10.1016/j.scr.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birbrair A, Zhang T, Files DC, Mannava S, Smith T, Wang ZM, Messi ML, Mintz A, Delbono O. Type-1 pericytes accumulate after tissue injury and produce collagen in an organ-dependent manner. Stem Cell Res Ther. 2014;5:122. doi: 10.1186/scrt512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–62. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O. Type 1 pericytes participate in fibrous tissue deposition in aged skeletal muscle. Am J Physiol Cell Physiol. 2013;305:C1098–113. doi: 10.1152/ajpcell.00171.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Birbrair A, Zhang T, Wang ZM, Messi ML, Olson JD, Mintz A, Delbono O. Type 2 pericytes participate in normal and tumoral angiogenesis. Am J Physiol Cell Physiol. 2014;307:C25–38. doi: 10.1152/ajpcell.00084.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cuttler AS, LeClair RJ, Stohn JP, Wang Q, Sorenson CM, Liaw L, Lindner V. Characterization of Pdgfrb-Cre transgenic mice reveals reduction of ROSA26 reporter activity in remodeling arteries. Genesis. 2011;49:673–80. doi: 10.1002/dvg.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen ZL, Strickland S. Laminin gamma1 is critical for Schwann cell differentiation, axon myelination, and regeneration in the peripheral nerve. J Cell Biol. 2003;163:889–99. doi: 10.1083/jcb.200307068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enikolopov G, Overstreet-Wadiche L, Ge S. Viral and transgenic reporters and genetic analysis of adult neurogenesis. Cold Spring Harb Perspect Biol. 2015;7:a018804. doi: 10.1101/cshperspect.a018804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mignone JL, Kukekov V, Chiang AS, Steindler D, Enikolopov G. Neural stem and progenitor cells in nestin-GFP transgenic mice. J Comp Neurol. 2004;469:311–24. doi: 10.1002/cne.10964. [DOI] [PubMed] [Google Scholar]
- 31.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Solovjeva LV, Demin SJ, Pleskach NM, Kuznetsova MO, Svetlova MP. Characterization of telomeric repeats in metaphase chromosomes and interphase nuclei of Syrian hamster fibroblasts. Mol Cytogenet. 2012;5:37. doi: 10.1186/1755-8166-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu D, Bai J, Duan Q, Costa M, Dai W. Covalent modifications of histones during mitosis and meiosis. Cell Cycle. 2009;8:3688–94. doi: 10.4161/cc.8.22.9908. [DOI] [PubMed] [Google Scholar]
- 35.Zhu W, Lee CY, Johnson RL, Wichterman J, Huang R, DePamphilis ML. An image-based, high-throughput screening assay for molecules that induce excess DNA replication in human cancer cells. Mol Cancer Res. 2011;9:294–310. doi: 10.1158/1541-7786.MCR-10-0570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang B, Zhang T, Wang G, Wang G, Chi W, Jiang Q, Zhang C. GSK3beta-Dzip1-Rab8 cascade regulates ciliogenesis after mitosis. PLoS Biol. 2015;13:e1002129. doi: 10.1371/journal.pbio.1002129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9–18. doi: 10.1038/sj.cr.7290105. [DOI] [PubMed] [Google Scholar]
- 38.Cheng P, Alberts I, Li X. The role of ERK1/2 in the regulation of proliferation and differentiation of astrocytes in developing brain. Int J Dev Neurosci. 2013;31:783–9. doi: 10.1016/j.ijdevneu.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 39.Sun Y, Liu WZ, Liu T, Feng X, Yang N, Zhou HF. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res. 2015;35:600–4. doi: 10.3109/10799893.2015.1030412. [DOI] [PubMed] [Google Scholar]
- 40.Tomkowicz B, Rybinski K, Sebeck D, Sass P, Nicolaides NC, Grasso L, Zhou Y. Endosialin/TEM-1/CD248 regulates pericyte proliferation through PDGF receptor signaling. Cancer Biol Ther. 2010;9:908–15. doi: 10.4161/cbt.9.11.11731. [DOI] [PubMed] [Google Scholar]
- 41.Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci. 2016;19:771–83. doi: 10.1038/nn.4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rooney JE, Knapp JR, Hodges BL, Wuebbles RD, Burkin DJ. Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy. Am J Pathol. 2012;180:1593–602. doi: 10.1016/j.ajpath.2011.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rooney JE, Gurpur PB, Burkin DJ. Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2009;106:7991–6. doi: 10.1073/pnas.0811599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Van Ry PM, Minogue P, Hodges BL, Burkin DJ. Laminin-111 improves muscle repair in a mouse model of merosin-deficient congenital muscular dystrophy. Hum Mol Genet. 2014;23:383–96. doi: 10.1093/hmg/ddt428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goudenege S, Lamarre Y, Dumont N, Rousseau J, Frenette J, Skuk D, Tremblay JP. Laminin-111: a potential therapeutic agent for Duchenne muscular dystrophy. Mol Ther. 2010;18:2155–63. doi: 10.1038/mt.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guan J, Zhang J, Zhu Z, Niu X, Guo S, Wang Y, Zhang C. Bone morphogenetic protein 2 gene transduction enhances the osteogenic potential of human urine-derived stem cells. Stem Cell Res Ther. 2015;6:5. doi: 10.1186/scrt539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin S, Seong JK, Bae YS. Ahnak stimulates BMP2-mediated adipocyte differentiation through Smad1 activation. Obesity (Silver Spring) 2016;24:398–407. doi: 10.1002/oby.21367. [DOI] [PubMed] [Google Scholar]
- 48.Sottile V, Seuwen K. Bone morphogenetic protein-2 stimulates adipogenic differentiation of mesenchymal precursor cells in synergy with BRL 49653 (rosiglitazone) FEBS Lett. 2000;475:201–4. doi: 10.1016/S0014-5793(00)01655-0. [DOI] [PubMed] [Google Scholar]
- 49.Huang H, Song TJ, Li X, Hu L, He Q, Liu M, Lane MD, Tang QQ. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci U S A. 2009;106:12670–5. doi: 10.1073/pnas.0906266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gustafson B, Hammarstedt A, Hedjazifar S, Hoffmann JM, Svensson PA, Grimsby J, Rondinone C, Smith U. BMP4 and BMP antagonists regulate human white and beige adipogenesis. Diabetes. 2015;64:1670–81. doi: 10.2337/db14-1127. [DOI] [PubMed] [Google Scholar]
- 51.Gillespie MA, Le Grand F, Scime A, Kuang S, von Maltzahn J, Seale V, Cuenda A, Ranish JA, Rudnicki MA. p38-{gamma}-dependent gene silencing restricts entry into the myogenic differentiation program. J Cell Biol. 2009;187:991–1005. doi: 10.1083/jcb.200907037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Segales J, Perdiguero E, Munoz-Canoves P. Regulation of muscle stem cell functions: a focus on the p38 MAPK signaling pathway. Front Cell Dev Biol. 2016;4:91. doi: 10.3389/fcell.2016.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perdiguero E, Ruiz-Bonilla V, Gresh L, Hui L, Ballestar E, Sousa-Victor P, Baeza-Raja B, Jardi M, Bosch-Comas A, Esteller M, et al. Genetic analysis of p38 MAP kinases in myogenesis: fundamental role of p38alpha in abrogating myoblast proliferation. EMBO J. 2007;26:1245–56. doi: 10.1038/sj.emboj.7601587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zetser A, Gredinger E, Bengal E. p38 mitogen-activated protein kinase pathway promotes skeletal muscle differentiation. Participation of the Mef2c transcription factor. J Biol Chem. 1999;274:5193–200. doi: 10.1074/jbc.274.8.5193. [DOI] [PubMed] [Google Scholar]
- 55.Yi P, Chew LL, Zhang Z, Ren H, Wang F, Cong X, Zheng L, Luo Y, Ouyang H, Low BC, Zhou YT. KIF5B transports BNIP-2 to regulate p38 mitogen-activated protein kinase activation and myoblast differentiation. Mol Biol Cell. 2015;26:29–42. doi: 10.1091/mbc.E14-03-0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lepper C, Partridge TA, Fan CM. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 2011;138:3639–46. doi: 10.1242/dev.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-Morel B, Guenou H, Malissen B, Tajbakhsh S, Galy A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–56. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- 58.Valero MC, Huntsman HD, Liu J, Zou K, Boppart MD. Eccentric exercise facilitates mesenchymal stem cell appearance in skeletal muscle. PLoS One. 2012;7:e29760. doi: 10.1371/journal.pone.0029760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yao Y, Chen ZL, Norris EH, Strickland S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun. 2014;5:3413. doi: 10.1038/ncomms4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Birbrair A, Zhang T, Wang ZM, Messi ML, Enikolopov GN, Mintz A, Delbono O. Skeletal muscle neural progenitor cells exhibit properties of NG2-glia. Exp Cell Res. 2013;319:45–63. doi: 10.1016/j.yexcr.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patton BL, Miner JH, Chiu AY, Sanes JR. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J Cell Biol. 1997;139:1507–21. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gullberg D, Tiger CF, Velling T. Laminins during muscle development and in muscular dystrophies. Cell Mol Life Sci. 1999;56:442–60. doi: 10.1007/PL00000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pedrosa-Domellof F, Tiger CF, Virtanen I, Thornell LE, Gullberg D. Laminin chains in developing and adult human myotendinous junctions. J Histochem Cytochem. 2000;48:201–10. doi: 10.1177/002215540004800205. [DOI] [PubMed] [Google Scholar]
- 64.Ringelmann B, Roder C, Hallmann R, Maley M, Davies M, Grounds M, Sorokin L. Expression of laminin alpha1, alpha2, alpha4, and alpha5 chains, fibronectin, and tenascin-C in skeletal muscle of dystrophic 129ReJ dy/dy mice. Exp Cell Res. 1999;246:165–82. doi: 10.1006/excr.1998.4244. [DOI] [PubMed] [Google Scholar]
- 65.Schuler F, Sorokin LM. Expression of laminin isoforms in mouse myogenic cells in vitro and in vivo. J Cell Sci. 1995;108(Pt 12):3795–805. doi: 10.1242/jcs.108.12.3795. [DOI] [PubMed] [Google Scholar]
- 66.Sasaki T, Giltay R, Talts U, Timpl R, Talts JF. Expression and distribution of laminin alpha1 and alpha2 chains in embryonic and adult mouse tissues: an immunochemical approach. Exp Cell Res. 2002;275:185–99. doi: 10.1006/excr.2002.5499. [DOI] [PubMed] [Google Scholar]
- 67.Aumailley M, Smyth N. The role of laminins in basement membrane function. J Anat. 1998;193(Pt 1):1–21. doi: 10.1046/j.1469-7580.1998.19310001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–101. doi: 10.1182/blood-2009-05-222364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gawlik K, Miyagoe-Suzuki Y, Ekblom P, Takeda S, Durbeej M. Laminin alpha1 chain reduces muscular dystrophy in laminin alpha2 chain deficient mice. Hum Mol Genet. 2004;13:1775–84. doi: 10.1093/hmg/ddh190. [DOI] [PubMed] [Google Scholar]
- 70.Hager M, Gawlik K, Nystrom A, Sasaki T, Durbeej M. Laminin {alpha}1 chain corrects male infertility caused by absence of laminin {alpha}2 chain. Am J Pathol. 2005;167:823–33. doi: 10.1016/S0002-9440(10)62054-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gawlik KI, Mayer U, Blomberg K, Sonnenberg A, Ekblom P, Durbeej M. Laminin alpha1 chain mediated reduction of laminin alpha2 chain deficient muscular dystrophy involves integrin alpha7beta1 and dystroglycan. FEBS Lett. 2006;580:1759–65. doi: 10.1016/j.febslet.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 72.Gawlik KI, Durbeej M. Transgenic overexpression of laminin alpha1 chain in laminin alpha2 chain-deficient mice rescues the disease throughout the lifespan. Muscle Nerve. 2010;42:30–7. doi: 10.1002/mus.21616. [DOI] [PubMed] [Google Scholar]
- 73.Gawlik KI, Li JY, Petersen A, Durbeej M. Laminin alpha1 chain improves laminin alpha2 chain deficient peripheral neuropathy. Hum Mol Genet. 2006;15:2690–700. doi: 10.1093/hmg/ddl201. [DOI] [PubMed] [Google Scholar]
- 74.Gawlik KI, Akerlund M, Carmignac V, Elamaa H, Durbeej M. Distinct roles for laminin globular domains in laminin alpha1 chain mediated rescue of murine laminin alpha2 chain deficiency. PLoS One. 2010;5:e11549. doi: 10.1371/journal.pone.0011549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Miyagoe Y, Hanaoka K, Nonaka I, Hayasaka M, Nabeshima Y, Arahata K, Nabeshima Y, Takeda S. Laminin alpha2 chain-null mutant mice by targeted disruption of the Lama2 gene: a new model of merosin (laminin 2)-deficient congenital muscular dystrophy. FEBS Lett. 1997;415:33–9. doi: 10.1016/S0014-5793(97)01007-7. [DOI] [PubMed] [Google Scholar]
- 76.Kuang W, Xu H, Vachon PH, Liu L, Loechel F, Wewer UM, Engvall E. Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. J Clin Invest. 1998;102:844–52. doi: 10.1172/JCI3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo LT, Zhang XU, Kuang W, Xu H, Liu LA, Vilquin JT, Miyagoe-Suzuki Y, Takeda S, Ruegg MA, Wewer UM, Engvall E. Laminin alpha2 deficiency and muscular dystrophy; genotype-phenotype correlation in mutant mice. Neuromuscul Disord. 2003;13:207–15. doi: 10.1016/s0960-8966(02)00266-3. [DOI] [PubMed] [Google Scholar]
- 78.Kikkawa Y, Miner JH. Molecular dissection of laminin alpha 5 in vivo reveals separable domain-specific roles in embryonic development and kidney function. Dev Biol. 2006;296:265–77. doi: 10.1016/j.ydbio.2006.04.463. [DOI] [PubMed] [Google Scholar]
- 79.Nishimune H, Valdez G, Jarad G, Moulson CL, Muller U, Miner JH, Sanes JR. Laminins promote postsynaptic maturation by an autocrine mechanism at the neuromuscular junction. J Cell Biol. 2008;182:1201–15. doi: 10.1083/jcb.200805095. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Three supplementary figures are available. Material used in this study is available from the corresponding author on reasonable request.