

Abstract
Background
The renal renin-angiotensin system (RAS) is physiologically important for blood pressure regulation. Altered regulation of RAS-related genes has been observed in an animal model of hypertension (spontaneously hypertensive rats – SHRs). The current understanding of certain RAS-related gene expression differences between Wistar-Kyoto rats (WKYs) and SHRs is either limited or has not been compared. The purpose of this study was to compare the regulation of key RAS-related genes in the kidneys of adult WKYs and SHRs.
Material/Methods
Coronal sections were dissected through the hilus of kidneys from 16-week-old male WKYs and SHRs. RT-PCR analysis was performed for Ace, Ace2, Agt, Agtr1a, Agtr1b, Agtr2, Atp6ap2 (PRR), Mas1, Ren, Rnls, and Slc12a3 (NCC).
Results
Increased mRNA expression was observed for Ace, Ace2, Agt, Agtr1a, Agtr1b, and Atp6ap2 in SHRs compared to WKYs. Mas1, Ren, Slc12a3, and Rnls showed no difference in expression between animal types.
Conclusions
This study shows that the upregulation of several key RAS-related genes in the kidney may account for the increased blood pressure of adult SHRs.
MeSH Keywords: Kidney; Rats, Inbred SHR; Rats, Inbred WKY; Receptor, Angiotensin, Type 1; Receptor, Angiotensin, Type 2; Renin-Angiotensin System
Background
The kidney is an important regulator of blood pressure and can be involved in the pathogenesis of hypertension. Renal genes involved in hypertension are responsible for functions such as sodium resorption, renin-angiotensin system (RAS) activity, and catecholamine regulation [1–3]. Hypertension can result from altered gene expression during different time points of development; however, the exact changes that occur during adult life that contribute to hypertension are unclear [2,4].
In this study, renal gene expression was compared between adult hypertensive rats (spontaneously hypertensive rats – SHRs) and normotensive control rats (Wistar-Kyoto rats – WKYs). The genes studied are involved in renal physiology and mechanisms for blood pressure regulation. Most of these genes are related to the RAS and consisted of angiotensin-converting enzymes 1 and 2 (ACE and ACE2; Ace and Ace2), angiotensinogen (AGT; Agt), angiotensin type-1a, type-1b, and type-2 receptors (AT1Ra, AT1Rb, and AT2R; Agtr1a, Agtr1b, and Agtr2), (pro)renin receptor (PRR; Atp6ap2), Mas receptor (Mas; Mas1), and renin (Ren). Two additional genes that were investigated were the Na+/Cl− cotransporter gene (NCC; Slc12a3) and renalase (Rnls). Although altered expression of RAS-related genes has been previously linked to hypertension, information regarding several important genes is still limited and several genes have yet to be studied in the adult SHR kidney [2]. Further, NCC was selected because there are no previous studies that have analyzed its mRNA expression in SHRs. Renalase, an enzyme which lowers the activity of catecholamines (i.e., adrenaline, noradrenaline, and dopamine), was selected because it was shown to be significantly upregulated in the kidneys of critically hypertensive rats (systolic blood pressure >180 mmHg) [5]. This finding also represents the only study to date examining the regulation of renalase in SHRs. Adrenaline and noradrenaline stimulate adrenergic receptors, create the general effect of vasoconstriction throughout the circulatory system, and play a pivotal role in blood pressure homeostasis and hypertension [6]. Because renalase may regulate the activity of catecholamines, it is proposed to be a new candidate gene for hypertension [3]. The purpose of our study was to determine if there are any novel alterations in gene expression that could be responsible for increased blood pressure in SHRs.
Material and Methods
Animals, blood pressure measurements, and tissue collection
Male SHRs and WKYs were purchased from Taconic Farms (Germantown, NY, USA). All protocols regarding the handling of animals have been previously described [7]. Animals were cared for in accordance with the Canadian Council on Animal Care guidelines, and were approved by the Laurentian University Animal Care Committee. Blood Pressure was determined using a non-invasive BP monitor (Coda6, Kent Scientific, CT, USA) as previously described [7]. The kidneys (n=6; 16 weeks of age) were collected and stored at −80°C until use.
Homogenization of tissue, RNA extraction, and reverse transcription PCR (RT-PCR)
Coronal sections approximately 2 mm thick were cut along the hilus of each kidney. Samples were placed in TRIZOL reagent (Sigma-Aldrich Corp., MO, USA), homogenized with the QIAGEN TissueLyser (Newtown, PA, USA), and RNA was extracted by following the manufacturer’s instructions. RNA concentrations were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE, USA). RNA samples were treated with DNase as per the manufacturer’s instructions (Sigma-Aldrich), and reverse transcribed using the M-MLV reverse transcriptase kit (Promega, Madison, WI, USA). PCRs were performed using the GoTaq flexi DNA polymerase (Promega), appropriate primers, and amplification conditions (Table 1). PCR products were resolved on a 2% TAE gel and imaged with the BioRad UV ChemiDoc UV analyzer. Densitometry was performed using Quantity One software (BioRad). Specific gene expression was normalized to rat 28s rRNA (Rn28s1) and fold changes in the SHR were calculated with respect to WKYs. Statistical analyses were performed using GraphPad Prism software (La Jolla, CA, USA). Differences between SHRs and WKYs were analyzed using an unpaired t test.
Table 1.
Primer specifications and conditions used for conventional reverse transcription PCR.
| Gene name and NCBI accession number | Forward primer sequence (5′-3′) | Reverse primer sequence (5′-3′) | Amplicon size (bp) | Annealing temperature (°C) |
|---|---|---|---|---|
| Rn28s1 NR_046246 | AGGGATAACTGGCTTGTGGC | TAAACCCAGCTCACGTTCCC | 143 | 58 |
| Ace NM_012544.1 | GCCCCCTGTACAAGTGTGAT* | TAGGAAGAGCAGCACCCACT* | 347 | 61 |
| Ace2 NM_001012006.1 | CAGGAAGCTGAAGACCTGTCT | TTCAACTGTTTGTTCTTGTCTG | 251 | 56 |
| Agtr1a NM_030985.4 | CAAAGCTTGCTGGCAATGTA | TCCAGCTCCTGACTTGTCCT | 228 | 59 |
| AT1Ra | CTGCCACATTCCCTGAGTTAAC | ATCACCACCAAGCTGTTTCC | 302 | ?? |
| Agtr1b NM_031009.2 | GAGTGACAGAGACCAGACCAGAC | ATCACCACCAAGCTGTTTCC | 307 | 63 |
| Agtr2 NM_012494.3 | TAATCTCAACGCAACTGGCACC | GCCAAAAGGAGTAAGTCAGCCA | 222 | 59 |
| Mas1 NM_012757.2 | GGCGGTCATCATCTTCATAGC | CTTCTTCTTACTGCTGCCCAC | 313 | 59 |
| Slc12a3 NM_019345.3 | ATGTTCCTGCTTACCTGGTGG | GTGCTCACAAAGTCCACAAG | 251 | 57 |
| Atp6ap2 NM_001007091.1 | CCGTGGCACCATGGCTGTGCT | GCAAGCCCTGGCCAAGACAG | 204 | 65 |
| Agt NM_134432.2 | TTCAGGCCAAGACCTCCC* | CCAGCCGGGAGGTGCAGT* | 309 | 63 |
| Rnls NM_001014167.1 | GATAACAAGTGGGAAGTCTC | CATAAAAGAGGCCCAGAGC | 187 | 54 |
| Ren NM_012642.4 | CACTCTTGTTGCTCTGGACCT | GGGGTACCAATGCCGATCTC | 250 | 63 |
Results
At 16 weeks of age, average systolic, diastolic, and mean arterial blood pressures were higher in SHRs compared to WKYs, as previously described [7]. Renal gene expression analysis revealed that mRNA levels of several genes related to the RAS were significantly higher in SHRs when compared to WKYs. Figure 1 shows that mRNA levels in SHRs were significantly increased for AT1Ra (1.71-fold; P<0.0001), ACE (1.27-fold; P<0.0001), PRR (1.12-fold; P=0.0003), ACE2 (1.16-fold; P=0.0017), AT1Rb (1.97-fold; P=0.0044), and AGT (1.21-fold; P=0.022). AT2R expression could not be detected in either WKYs or SHRs. Further, 2 RAS genes (Mas and renin) showed no difference in expression between WKYs and SHRs (Figure 2). Neither renalase nor NCC showed significant differences (Figure 3).
Figure 1.

Elevated expression of renal RAS genes in SHRs compared to WKYs. RT-PCR analysis of transcripts for ACE, ACE2, AGT, AT1Ra, AT1Rb, and PRR (n=6). Data are normalized to the 28s gene and fold changes between WKYs and SHRs are expressed as mean ±SEM. Upper panels show representative gel bands. Significant differences between strains are denoted by * p≤0.05; ** p≤0.01, and *** p≤0.001.
Figure 2.
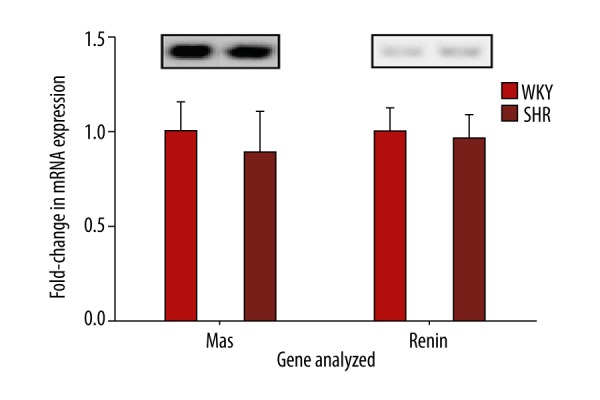
Renal RAS genes that remain unchanged between WKYs and SHRs. RT-PCR analysis of transcripts for Mas and renin (n=6). Data are normalized to the 28s gene and fold changes between WKYs and SHRs are expressed as mean ±SEM. Upper panels show representative gel bands.
Figure 3.
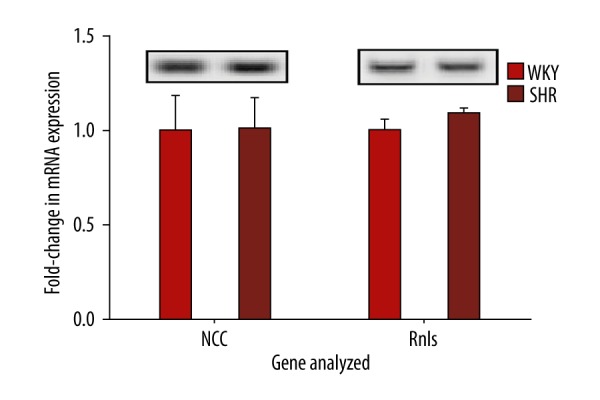
Transcript analysis of renal NCC and renalase in WKYs and SHRs. RT-PCR analysis of transcripts for renal NCC and renalase (n=6). Data are normalized to the 28s gene and fold changes between WKYs and SHRs are expressed as mean ±SEM. Upper panels show representative gel bands.
Discussion
All genes that were found to be upregulated in the SHR belonged to the RAS (Figure 1). Previous studies have shown similar results for ACE, PRR, Mas, renin, and renalase [5,8–11]. Our data for the expression of AGT, ACE2, AT1Ra, and AT2R are inconsistent with several previous reports suggesting that the regulation of these genes may depend on factors such as age, sex, stress hormones, epigenetics, sodium intake, and/or complex feedback mechanisms [4,9,10,12].
In previous studies, the expression of AGT mRNA has been observed to be either similar, upregulated, or downregulated in SHRs compared to WKYs [4,9,11]. These findings suggest that RAS activation, which is initiated by AGT, is likely regulated by a complex mechanism. Factors that may play a role in this mechanism include age, sex hormones, and cortisol [4,11,13]. However, the increase in AGT mRNA reported here is consistent with the evidence that it is commonly upregulated in the SHR kidney during adulthood.
At 16 weeks of age, ACE and PRR showed 1.27- and 1.12-fold increases in the SHR, respectively. Previous studies have reported much higher expression of these 2 genes in 18-week-old SHRs, which may be explained by different experimental methods (e.g., the use of qPCR and a different sample area for tissue collection) or differences in epigenetic modifications (e.g., H3Ac and H3K4me3 histone modifications) [8,9]. Upregulation of ACE, demonstrated in this and other studies of hypertension, support the functional importance of its known vasoconstrictive effects [2]. Regarding PRR, this receptor has been previously associated with an increase in blood pressure when binding renin or the renin precursor, (pro)renin [14]. Although renin expression is unchanged between WKYs and SHRs, upregulation of PRR may still independently increase the activity of this receptor [11]. However, when PRR is overexpressed in mice, hypertensive effects are not observed [14]. Although it has been reported that PRR mRNA is increased in the kidneys of SHRs and diabetic rats, this increase may be caused by its regulation by AT1R activity [9,15]. The increase in PRR expression suggests that this receptor may play a physiological role in blood pressure regulation in SHRs, but its cellular mechanism remains to be elucidated.
Increased AT1R expression may be another mechanism that contributes to hypertension. Previous studies have reported upregulation of renal AT1Ra in a transgenerational animal model of hypertension [16]. Our study supports this finding in SHRs, while other studies with 18-week-old SHRs did not [9]. AT1Rb, which is not present in humans, was also upregulated in our study. Although studies using AT1Rb knockout mice suggest the redundancy of this receptor subtype, both AT1Rs appear to be responsible for kidney structure and function, and AT1Rb may also compensate for lowered AT1Ra [17]. Both AT1Ra and AT1Rb were elevated in our study, suggesting that AT1Rb is not solely expressed as a compensatory mechanism. Although the hypertensive effects of AT1Ra are evident, the effects of upregulated AT1Rb have not been reported [2].
Interestingly, our study shows that AT2R was not expressed in either SHR or WKY kidneys. Binding of angiotensin (Ang) II to AT2R stimulates vasodilatory effects that are antagonistic to the effects of AT1Rs [12]. Previously, research on renal AT2R expression in rats and humans has shown that it is highly expressed throughout the kidney during fetal development [2,18]. However, it is well known that both humans and rats undergo a dramatic reduction in AT2R expression postnatally [2,18]. AT2R expression in humans and rats may still exist in the glomeruli, proximal tubules, or collecting ducts during adulthood, but it appears to be only activated by sodium depletion [12,18]. Controlled sodium intake in our study may be the reason for a lack of overall AT2R expression, and comparison of with the increased expression of AT1Rs suggests that the most available Ang II receptors in the adult kidney may be AT1Rs. Protein levels should be assessed to determine their ratio of AT1R/AT2R in areas of renal Ang II production and to determine if this ratio contributes to increased blood pressure.
The current study also provides evidence of a complex regulatory mechanism between ACE2 and the Mas receptor. The physiological function of ACE2 is to produce Ang (1–7) from Ang II, which then binds to Mas and stimulates vasodilation [10]. Previously, in studies analyzing 16-week-old SHRs, renal Mas expression was unchanged but ACE2 expression was 1.5-fold lower than in WKYs; higher levels of Ang (1–7) might be responsible for this decrease [10]. In contrast, the 1.16-fold significant increase in ACE2 mRNA shown in our study presents a new observation for this gene. However, if the ACE2 upregulation found in our study was stimulated by lower levels of Ang (1–7), Mas would likely have been upregulated by this same feedback mechanism. Because Mas was unchanged between SHRs and WKYs in our study and previous studies, Mas may be tightly controlled by another mechanism in addition to Ang (1–7) regulation [10]. Further, it is likely that increased ACE2 increases Ang (1–7) production, thereby compensating for increased Ang II. Nevertheless, their overall effects on blood pressure are largely dependent on the expression of Mas, AT2R, and AT1Rs. Results from our study show that the only receptors upregulated were AT1Rs; therefore, the effects of increased Ang II production in the adult kidney may still be largely vasoconstrictive.
Previous studies analyzing NCC in younger SHRs reported that although protein levels of NCC were unchanged, ACE activity may redistribute the localization of several sodium transporters, including the NCC [1]. NCC was chosen for our study to determine if older SHRs had an alternate regulation of this gene. Although, the level of NCC transcripts were unchanged, the localization of NCC may still be influenced by an increased expression of ACE, leading to differential sodium absorption and blood pressure regulation.
Renalase expression was also unchanged in the SHRs. Previous studies show that renalase expression is increased in SHR kidneys with systolic blood pressures >180 mmHg [5]. The SHRs of our study had an average systolic blood pressure of 188.3 mmHg [7]. This suggests that there may be unknown factors responsible for increased renalase expression, in addition to the severity of the blood pressure increase. The currently unknown regulatory mechanism for renalase may not decrease catecholamines in adult SHRs sufficiently to effectively reduce the hypertensive effects of the RAS.
Conclusions
The present study adds new information and better characterizes which genes are important contributors to hypertension. While the expression of certain genes in the adult SHR kidney may be more variable, and affected by age (e.g., ACE2, AGT, AT1Ra, AT1Rb, and renalase), others may be more stable in their regulation patterns (e.g., ACE, Mas, PRR, and renin). Our analysis of multiple genes has led to the conclusion that the complex mechanisms involved in gene regulation result in an overall alteration of RAS physiology. Since RAS is a potent regulator of blood pressure, understanding all potential alterations in its physiology remains one of the most important steps in understanding the etiology of hypertension.
Acknowledgements
We thank Dr. Anne Monique Nuyt for the AT1Rb primer sequences used for RT-PCR.
Footnotes
Source of support: This study was supported by funds from the Canadian Institutes for Health Research (CIHR) and the Northern Ontario School of Medicine (NOSM) Research Development Fund. CRW was supported by the Ontario Graduate Scholarship
References
- 1.Yang LE, Leong PKK, McDonough AA. Reducing blood pressure in SHR with enalapril provokes redistribution of NHE3, NaPi2, and NCC and decreases NaPi2 and ACE abundance. Am J Physiol Renal Physiol. 2007;293:F1197–208. doi: 10.1152/ajprenal.00040.2007. [DOI] [PubMed] [Google Scholar]
- 2.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: From physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev. 2007;59:251–87. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 3.Wu Y, Xu J, Velazquez H, et al. Renalase deficiency aggravates ischemic myocardial damage. Kidney Int. 2011;79:853–60. doi: 10.1038/ki.2010.488. [DOI] [PubMed] [Google Scholar]
- 4.Tamura K, Umemura S, Nyui N, et al. Tissue-specific regulation of angiotensinogen gene expression in spontaneously hypertensive rats. Hypertension. 1996;27:1216–23. doi: 10.1161/01.hyp.27.6.1216. [DOI] [PubMed] [Google Scholar]
- 5.Fedchenko V, Globa A, Buneeva O, Medvedev A. Renalase mRNA levels in the brain, heart, and kidneys of spontaneously hypertensive rats with moderate and high hypertension. Med Sci Monit Basic Res. 2013;19:267–70. doi: 10.12659/MSMBR.889540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tank AW, Lee Wong D. Peripheral and central effects of circulating catecholamines. Compr Physiol. 2015;5:1–15. doi: 10.1002/cphy.c140007. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen P, Peltsch H, de Wit J, et al. Regulation of the phenylethanolamine N-methyltransferase gene in the adrenal gland of the spontaneous hypertensive rat. Neurosci Lett. 2009;461:280–84. doi: 10.1016/j.neulet.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 8.Lee H-A, Cho H-M, Lee D-Y, et al. Tissue-specific upregulation of angiotensin-converting enzyme 1 in spontaneously hypertensive rats through histone code modifications. Hypertension. 2012;59:621–26. doi: 10.1161/HYPERTENSIONAHA.111.182428. [DOI] [PubMed] [Google Scholar]
- 9.Lee H-A, Lee D-Y, Lee H-J, et al. Enrichment of (pro)renin receptor promoter with activating histone codes in the kidneys of spontaneously hypertensive rats. J Renin Angiotensin Aldosterone Syst. 2012;13:11–18. doi: 10.1177/1470320311415738. [DOI] [PubMed] [Google Scholar]
- 10.Tan Z, Wu J, Ma H. Regulation of angiotensin-converting enzyme 2 and Mas receptor by Ang-(1–7) in heart and kidney of spontaneously hypertensive rats. J Renin Angiotensin Aldosterone Syst. 2011;12:413–19. doi: 10.1177/1470320311402109. [DOI] [PubMed] [Google Scholar]
- 11.Kobori H, Ozawa Y, Suzaki Y, Nishiyama A. Enhanced intrarenal angiotensinogen contributes to early renal injury in spontaneously hypertensive rats. J Am Soc Nephrol. 2005;16:2073–80. doi: 10.1681/ASN.2004080676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyata N, Park F, Li XF, Cowley AW. Distribution of angiotensin AT1 and AT2 receptor subtypes in the rat kidney. Am J Physiol. 1999;277:F437–46. doi: 10.1152/ajprenal.1999.277.3.F437. [DOI] [PubMed] [Google Scholar]
- 13.Klett C, Ganten D, Hellmann W, et al. Regulation of hepatic angiotensinogen synthesis and secretion by steroid hormones. Endocrinology. 1992;130:3660–68. doi: 10.1210/endo.130.6.1597163. [DOI] [PubMed] [Google Scholar]
- 14.Rosendahl A, Niemann G, Lange S, et al. Increased expression of (pro)renin receptor does not cause hypertension or cardiac and renal fibrosis in mice. Lab Invest. 2014;94:863–72. doi: 10.1038/labinvest.2014.83. [DOI] [PubMed] [Google Scholar]
- 15.Siragy HM, Huang J. Renal (pro)renin receptor upregulation in diabetic rats through enhanced angiotensin AT1 receptor and NADPH oxidase activity. Exp Physiol. 2008;93:709–14. doi: 10.1113/expphysiol.2007.040550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collett JA, Hart AK, Patterson E, et al. Renal angiotensin II type 1 receptor expression and associated hypertension in rats with minimal SHR nuclear genome. Physiol Rep. 2013;1:e00104. doi: 10.1002/phy2.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oliverio MI, Kim HS, Ito M, et al. Reduced growth, abnormal kidney structure, and type 2 (AT2) angiotensin receptor-mediated blood pressure regulation in mice lacking both AT1A and AT1B receptors for angiotensin II. Proc Natl Acad Sci USA. 1998;95:15496–501. doi: 10.1073/pnas.95.26.15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ozono R, Wang ZQ, Moore AF, et al. Expression of the subtype 2 angiotensin (AT2) receptor protein in rat kidney. Hypertension. 1997;30:1238–46. doi: 10.1161/01.hyp.30.5.1238. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz-Ortega M, Lorenzo O, Egido J. Angiotensin III up-regulates genes involved in kidney damage in mesangial cells and renal interstitial fibroblasts. Kidney Int. 1998;54:S41–45. doi: 10.1046/j.1523-1755.1998.06811.x. [DOI] [PubMed] [Google Scholar]