

Abstract
Derivatives of 3,4-hydroxypyridinones have been extensively studied for in vivo Fe3+ sequestration. Deferiprone, a 1,2-dimethyl-3,4-hydroxypyridinone, is now routinely used for clinical treatment of iron overload disease. Hexadentate tris(3,4-hydroxypyridinone) ligands (THP) complex Fe3+ at very low iron concentrations, and their high affinities for oxophilic trivalent metal ions have led to their development for new applications as bifunctional chelators for the positron emitting radiometal, 68Ga3+, which is clinically used for molecular imaging in positron emission tomography (PET). THP-peptide bioconjugates rapidly and quantitatively complex 68Ga3+ at ambient temperature, neutral pH and micromolar concentrations of ligand, making them amenable to kit-based radiosynthesis of 68Ga PET radiopharmaceuticals. 68Ga-labelled THP-peptides accumulate at target tissue in vivo, and are excreted largely via a renal pathway, providing high quality PET images.
Keywords: hydroxypyridinone, deferiprone, gallium, iron overload, positron emission tomography, molecular imaging, bifunctional chelators
1. Introduction
Positron emission tomography (PET) is a whole body diagnostic three-dimensional molecular imaging modality used in nuclear medicine that detects radiation arising from the decay of unstable positron-emitting radioisotopes. The availability of the positron-emitting isotope, gallium-68 (68Ga) from decay of 68Ge in a bench top 68Ge/68Ga generator is likely to have significant clinical impact on the use of PET for molecular imaging [1]. Clinical use of 68Ga receptor-targeting radiopharmaceuticals for neuroendocrine cancers (68Ga-DOTA-TATE) [2] and prostate cancers (68Ga-HBED-PSMA) [3] has changed patient management in centres that have routine access to such agents. 68Ga molecular imaging agents typically consist of chelators tethered to a receptor-targeted peptide, protein or small molecule. As Ga3+ can incorporate up to six ligands in an octahedral coordination sphere, hexadentate ligands are normally utilised. The Ga3+ ion has a high charge density, and is categorised as a “hard” Lewis acid, and so the majority of hexadentate ligands incorporate oxygen and/or nitrogen donor atoms. Both macrocyclic and acyclic chelators have been explored for 68Ga3+ complexation [1,4,5,6,7].
Clinical radiosyntheses of 68Ga-DOTA-TATE and 68Ga-HBED-PSMA involve heating at 80–100 °C for 5–20 min at pH 3–5 [1,4], followed by post-synthetic purification and work-up to remove impurities, unreacted 68Ga and reaction components that are not physiologically compatible. In the case of 68Ga-DOTA-TATE, heating is required for complexation of 68Ga3+. For 68Ga-HBED-PSMA, three geometric cis/trans isomers are possible, with each possible geometric isomer having a diastereomer. In this case, heating the reaction favours formation of the thermodynamically preferred species.
For 68Ga to be adopted in routine clinical practice, chelators that provide efficient and reproducible kit-based radiolabelling methods—preferably a single manipulation—are required. The chelators that we have developed based on hydroxypyridinones allow one-step quantitative 68Ga3+ radiolabelling at neutral pH and ambient temperature.
Hydroxypyridinone (HP) ligands have high affinities for “hard” metal ions, and originally, the class of HPs described herein was developed extensively for therapeutic in vivo Fe3+ chelation in iron overload disease [8,9,10]. The bidentate HP ligand, deferiprone (1, Figure 1), is used routinely for this treatment [11]. Through chemical modification of substituents, Fe3+ affinity, hydrophilicity, metabolic stability and functionality of HPs can be tailored [10].
Figure 1.

Deferiprone (1): Resonance structures at different protonation states [20].
Ga3+ is an oxophilic metal ion, with a high charge density and ionic radius and coordination preferences similar to Fe3+. The (crystal) ionic radius for Ga3+ is 76 pm, and for high spin Fe3+ is 78.5 pm [12]. Recently, we have investigated the use of HPs for rapid and quantitative chelation of 68Ga3+ (as well as a long-lived PET isotope, 89Zr4+) and adapted a class of HPs to enable functionalisation with peptides and proteins for targeted molecular imaging [13,14,15,16].
Here we describe our research on the development of HPs for Fe3+ chelation, and how the design of such chelators is suitable for direct translation to 68Ga PET radiopharmaceuticals. It is not intended as an exhaustive review of chelators for either treatment of iron overload disease or complexation of radioisotopes of gallium used in nuclear medicine. The literature in both areas has been surveyed in depth in recent years in several excellent reviews and books [4,5,6,7,10,17,18,19].
2. Hydroxypyridinones
HPs consist of a six-membered aromatic N-heterocycle, with a hydroxyl and a ketone functionality. Varying relative positions of the hydroxyl and ketone functional groups within a bidentate HP molecule results in three types of hydroxypyridinones: 1,2-hydroxypyridinone (2); 3,2-hydroxypyridinone (3); and 3,4-hydroxypyridinone (4) (Figure 2). Neutral HPs can be protonated and deprotonated, with the first pKa typically corresponding to protonation/deprotonation at the oxo group, and the second, at the hydroxyl group. Delocalisation of the electrons of the N1 ring atom leads to aromaticity (Figure 1 and Figure 2).
Figure 2.
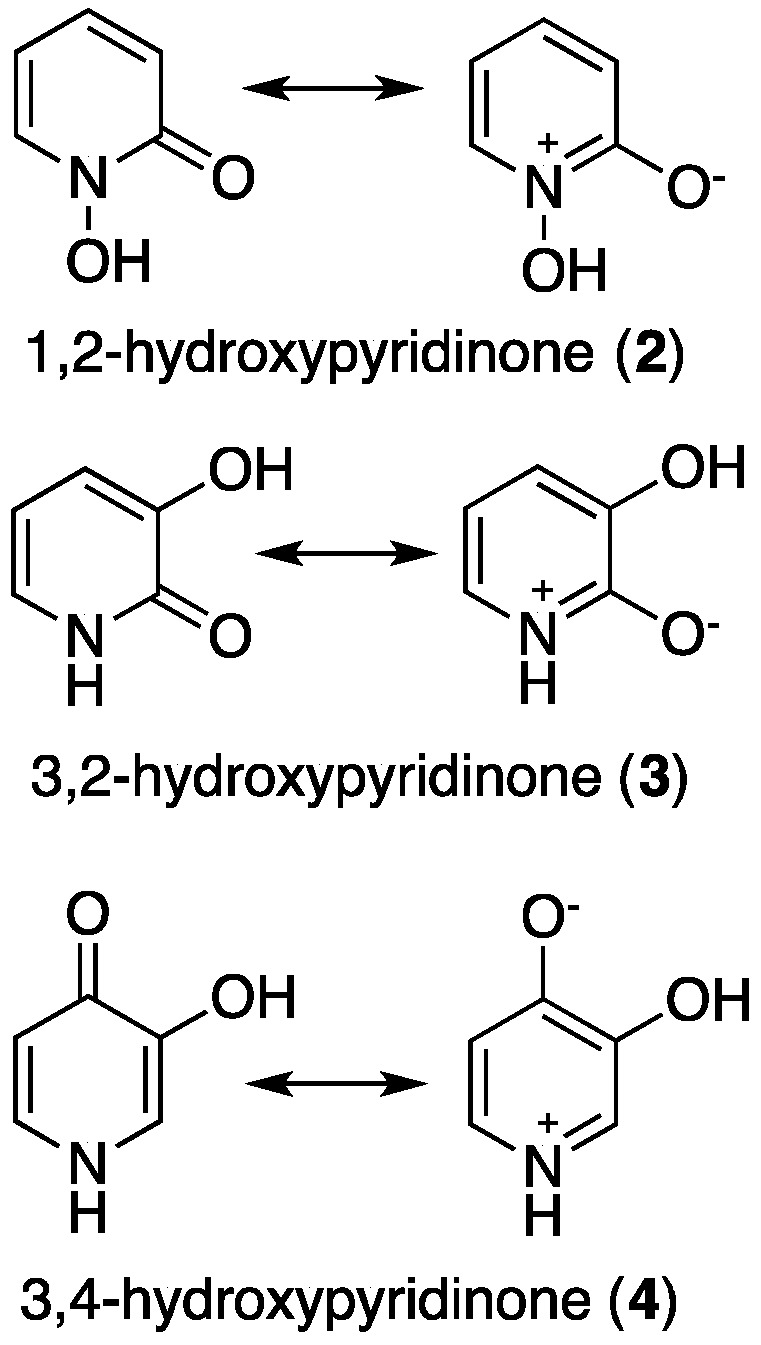
Resonance structures of 1,2-HP, 3,2-HP and 3,4-HP [21].
When HPs are deprotonated at the hydroxyl group, they are capable of complexing metal ions in a bidentate O2 mode, forming five-membered chelate rings. The relative positions of the hydroxyl and ketone groups, as well as ring substituents, influence pKa values and metal binding affinities. In general, pKa (the negative logarithm of the acid dissociation constant) and metal ion affinity values (both stepwise and cumulative stability constants of the metal-chelator complex) follow the order 3,4-HP > 3,2-HP and 1,2-HP (Table 1). This order reflects the relative decrease in delocalisation of the N1 atom lone pairs over the ring, and a corresponding decrease in charge density on the O atoms. The measurement of pM (negative logarithm of free metal concentration) is a more useful comparative measure for chelators than log Ka or affinity constants, as pM accounts for ligand basicity, denticity and protonation. Measurements are typically obtained at pH 7.4, with [total ligand] = 10−5 M and [total metal ion] = 10−6 M. Derivatives of high affinity 3,4-HPs have been extensively studied for iron-overload disease, and generally, 3,4-HPs have higher pM values than homologous 3,2- and 1,2-HPs. Additionally, 3,4-HPs are neutral at physiological pH, and when appropriately functionalised, can cross cell membranes.
Table 1.
Protonation constants, stepwise stability constants (log K) and cumulative Fe3+ stability constants (log β3) for 1,2-HP, 3,2-HP and 3,4-HP.
3,4-HPs have high affinity for both Fe3+ and Ga3+, and form neutral 3:1 bidentate complexes with Fe3+ and Ga3+ (Figure 3, Table 2) (as well as Al3+, In3+, Cr3+, Mn3+ and Gd3+) [24,25,26,27,28,29,30,31]. Both the [Fe(deferiprone)3] [31] and [Ga(deferiprone)3] [29] complexes crystallise in the same octahedral geometries with comparable bond lengths and angles. In the three coordinating deferiprone ligands in both complexes, O3 atoms are fac to each other (see Table 3 for atom numbering scheme). Fe–O3 bond lengths are 1.998 Å, and Fe–O4 bond lengths, 2.038 Å while in the Ga analogue the corresponding Ga–O bond lengths are 1.967 Å, and 1.990 Å, respectively. In both complexes, both C–O bonds are intermediate between single and double bonds, with the C4–O4 bond shorter than the C3–O3 bond. Within each deferiprone ligand, bond angles are 80.91° for O3–Fe–O4, and 83.22° for O3–Ga–O4. Other metal ions (M = Al3+, In3+, Cr3+, Mn3+) form isostructural [M(deferiprone)3] complexes [27,28,29,30], and in all cases, fac geometries are observed in the solid state. However, 1H NMR studies on diamagnetic Ga3+ and Al3+ complexes suggest that conversion between fac and mer geometric isomers occurs in solution [30].
Figure 3.

Representations of the molecular structures of [Fe(deferiprone)3] [31] (CCDC number 1183332) (left) and [Ga(deferiprone)3] (CCDC number 1164388) [29] (right). Only one stereoisomer is depicted for each complex. Orange—iron, pink—gallium, blue—ni trogen, grey—carbon, red—oxygen, and white—hydrogen. These diagrams (generated in Mercury software courtesy of the Cambridge Crystal Database) represent crystallographically determined structures. Hydrogen atoms of methyl groups are omitted for clarity.
Table 2.
Protonation constants and Fe3+ and Ga3+ stability constants for deferiprone (1) (measured at 22.5–25 °C, ionic strength 0.1–0.2 M; for pM values, pH 7.4, with [total ligand] = 10−5 M and [total metal ion] = 10−6 M).
| Metal Ion | pKa1 | pKa2 | log K1 | log K2 | log K3 | log β3 | pM3+ | Ref. |
|---|---|---|---|---|---|---|---|---|
| Fe3+ | 3.56 | 9.64 | 14.92 | 12.23 | 9.79 | 37.2 | – | [20] |
| 3.68 | 9.77 | 14.56 | 12.19 | 9.69 | 36.4 | 19.4 | [34] | |
| 3.62 | 9.76 | 15.14 | 11.54 | 9.24 | 35.92 | 18.3 | [35] | |
| – | – | 15.10 | 11.51 | 9.27 | 35.88 | – | [25] | |
| 3.61 | 9.78 | 15.03 | 27.42 | – | 37.35 | 20.74 | [36] | |
| Ga3+ | – | – | 13.17 | 12.26 | 10.33 | 35.76 | – | [25] |
| 3.70 | 9.86 | 17.07 | 12.19 | 9.16 | 38.42 | – | [37] |
Table 3.
Protonation constants and Fe3+ stability constants for 3,4-HP derivatives.
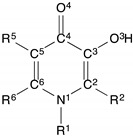 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R5 | R6 | pKa1 | pKa2 | log β3 (Fe3+) | pFe3+ | Ref. |
| 1 | CH3 | CH3 | H | H | 3.68 | 9.77 | 36.4 | 19.4 | [34] |
| 3.61 | 9.78 | 37.35 | 20.74 | [36] | |||||
| 4 | H | H | H | H | 3.34 | 9.01 | 35.1 | – | [20,23] |
| 5 | H | CH3 | H | H | 3.70 | 9.76 | 37.2 | – | [20] |
| 3.64 | 9.73 | 36.63 | 20.17 | [36] | |||||
| 6 | CH3 | CH2OH | H | H | 2.92 | 9.11 | 35.3 | 20.9 | [34] |
| 7 | CH2CH3 | CH2CH3 | H | H | 3.81 | 9.93 | 36.8 | 19.7 | [34] |
| 7 | CH2CH3 | CH2OH | H | H | 2.80 | 9.27 | 35.3 | 21.0 | [34] |
| 9 | CH2CH3 | CH(OH)CH3 | H | H | 3.03 | 8.77 | 35.1 | 21.4 | [34] |
| 10 | H | CON(CH3)2 | H | CH3 | 2.53 | 8.20 | 33.2 | 20.4 | [38] |
| 11 | H | CONHCH3 | H | CH3 | 6.66 | 2.32 | 32.5 | 22.8 | [38] |
| 12 | CH2CH3 | CH3 | H | H | 3.65 | 9.88 | 37.7 | – | [20] |
| 13 | CH3 | CH3 | CH3 | H | 3.37 | 10.32 | 37.93 | 19.71 | [36] |
| 14 | H | CH3 | CH3 | H | 3.43 | 10.27 | 37.28 | 19.22 | [36] |
In living organisms, iron plays an essential role in transport of oxygen, electron transfer, and activation and functioning of enzymes. Iron transport and metabolism are tightly controlled in living organisms. In patients with iron overload disease, a disruption to iron homeostasis results in “free” iron, largely present as a citrate-albumin complex [32]. This iron pool gives rise to Fenton redox cycling between Fe2+ and Fe3+, producing free radicals that result in toxic tissue damage [33]:
| Fe2+ + H2O2 → Fe3+ + HO· + OH− | (1) |
| Fe3+ + H2O2 → Fe2+ + HOO· + H+ | (2) |
Therapeutic chelators, including 3,4-HPs, are clinically used for sequestration of Fe3+ in vivo [7,8,9,10]. 3,4-HPs can be functionalised at the N1, C2, C5 or C6 positions of the ring (Table 3). An understanding of the effects of substitution is critical in optimizing 3,4-HP derivatives for either therapeutic Fe3+ chelation, or radiopharmaceutical Ga3+ chelation.
3. 3,4-Hydroxypyridinones for Fe3+ Complexation
3.1. Tailoring 3,4-HP Properties by Ring Substitution
Substitution at the C2 position results in pronounced effects on metal ion affinity. Alkylation increases pyridinone ring electron density, and increases pKa values and log K and log β3(Fe3+) constants (log β3 = 35.1 when R2 = H (4); log β3 = 37.2 when R2 = CH3 (5)) [20], although pFe3+ values are not markedly affected [26].
Substitution of C2 alkyl groups for 1′-hydroxyalkyl groups decreases pKa and log β3(Fe3+) constants (compare 1 with 6; and 7, 8 and 9) [34]. This decrease in affinity is a result of: (i) a decrease in ring electron density due to a negative induction effect of the C2 1′-hydroxylalkyl group; and (ii) intramolecular hydrogen bonding between the C2 1′-hydroxyalkyl group and the deprotonated C3 hydroxyl group (Figure 4). This hydrogen bonding stabilises the ionised C3 hydroxyl group. However, although Fe3+ affinity is decreased, the concurrent lowering of pKa2 actually results in an increase in pFe3+. The decrease in proton affinity favours the negative form that coordinates to Fe3+. In a side-by-side comparison at pH 7.4, pFe3+ values of the C2 1′-hydroxyalkyl derivatives are higher than C2 alkyl derivatives [34].
Figure 4.

Intramolecular hydrogen bonding can stabilise ionised hydroxyl groups of 3,4-HPs.
Similar effects are also observed upon introduction of a C2 amido group (Figure 4) [38]. A combination of a negative induction effect and hydrogen bonding from the C2 amido NH to the C3 hydroxyl group decreases Fe3+ stability constants, but the concurrent decrease in pKa serves to increase pFe3+ values at physiological pH relative to C2 alkyl derivatives. The C2 doubly alkylated N(CH3)2 amido group of 10 cannot form a hydrogen bond with the C3 hydroxyl group (Figure 4), and so its pFe3+ value is lower when compared to 11 bearing a singly alkylated NH(CH3) that can form a hydrogen bond (Table 3) [38].
In contrast to C2, varying alkyl substituents at the N1 position (H, methyl, ethyl) of 3,4-HP does not markedly affect the proton or metal ion affinity of 3,4-HPs (compounds 1, 5, 12 in Table 3) [20]. The exception to this is the C2 amido derivatives discussed above, where N1 alkylation sterically inhibits coplanarity of the C2 amido group with the HP ring, disrupting hydrogen bonding and thus decreasing pFe3+ values [38]. As such, N1 alkyl substitution can be a useful strategy for tailoring chelators’ lipophilicity, cell permeability [20], in vivo biodistribution [34], and rates of metabolism [39] without deleterious effects on metal ion affinity. Substitution at N1 sites has also been utilised to functionalise 3,4-HPs with fluorescent tags [40] or biological vectors [41].
Alkyl substitution at the C5 position increases 3,4-HP affinity for Fe3+, however as there is a concurrent increase in pKa2, pFe3+ does not increase relative to derivatives that do not contain a C5 alkyl group (compounds 13 and 14) [36]. There are no studies directly comparing and quantifying the effect of C6 substitution, but existing data of C6 methylated derivatives suggest that alkylation has little influence on 3,4-HP metal affinity [36].
Increasing the lipophilicity of 3,4-HPs increases their cell membrane permeability. Higher intracellular 3,4-HP accumulation results in greater intracellular Fe3+ scavenging, however excessive cellular uptake of 3,4-HPs results in toxicity [20]. A prodrug strategy that involves oral administration of an N1-substituted hydrophobic ester 3,4-HP (15, Figure 5) has proved particularly successful in scavenging iron in a preclinical iron overloaded rat model [34]. In iron overload disease, excess iron is stored in the liver, and the rat model mimics this. The ester compound 15 is absorbed effectively in the gastrointestinal tract, and delivered to the liver, where it enters target iron-overloaded hepatocyte cells. The compound is proposed to undergo ester hydrolysis and metabolism intracellularly (16, 17) prior to complexing Fe3+ [20]. The complex is then excreted, recovering excess iron from diseased animals efficiently. In contrast, when the more hydrophilic N1 propyl alcohol derivative 16 (Figure 5) is directly administered to rats, absorption is less effective, and Fe3+ recovery is lower.
Figure 5.

Schematic representation of the use of hydrophobic esters to enhance both the absorption from the gastrointestinal tract (GIT) and the hepatic extraction of the chelator. Subsequent intracellular hydrolysis occurs yielding a hydrophilic chelator which can undergo further metabolism to the extremely hydrophilic negatively charged 1-carboxyalkyl metabolite in the liver. Reprinted with permission from Liu, Z.D.; et al. Synthesis, physicochemical characterisation, and biological evaluation of 2-(1′-hydroxyalkyl)-3-hydroxypyridin-4-ones: Novel iron chelators with enhanced pFe3+ values. J. Med. Chem. 1999, 42, 4814–4823. Copyright American Chemical Society.
3.2. Deferiprone, Desferrioxamine and Deferasirox
Deferiprone (or Ferriprox) (1), developed by Hider and colleagues, has been clinically approved (in Europe in 1999, and the USA, in 2011) for treatment of iron overload diseases, including hemochromatosis and transfusion-dependant thalassemia [42]. The advantage of deferiprone is that it is both orally active and effective at sequestering Fe3+ from the blood stream, heart (where iron overload toxicity can be fatal) and liver (where excess iron is stored).
Two other approved treatments for iron overload are available. The first is the hexadentate hydroxamate chelator, desferrioxamine-B (DFO, 18, Figure 6), which forms a complex with Fe3+ with a metal to ligand stoichiometry of 1:1 and an overall charge of +1 under physiological conditions. DFO requires parenteral infusion over 8–12 h, several times a week, as it is not orally absorbed [42]. Clinical studies have demonstrated that either deferiprone alone, or a combination of deferiprone and DFO, are more effective therapies for myocardial iron overload than DFO alone. Additionally, a combination of oral deferiprone and parenteral DFO therapies is more viable for patients than parenteral DFO therapy alone, as combination therapy results in fewer parenteral infusions.
Figure 6.

Structure of desferrioxamine B (DFO) and deferasirox.
Like deferiprone, deferasirox [43] (19, Figure 6) is an effective orally active treatment for iron overload with clinical approval (in Europe in 2006 and the USA in 2005). It is a tridentate chelator, with Fe3+ complexes bearing a 3-charge at physiological pH. Data from clinical comparisons of the efficacy of deferiprone and deferasirox are conflicting and inconclusive, possibly due to variations in doses of the two treatments [44,45,46,47]. Some clinical data suggest that deferiprone is more effective at reducing iron levels in cardiac tissue [46,47].
3.3. Synthesis of 3,4-HPs
The structural diversity of 3,4-HPs is a result of extensive synthetic research that spans several decades. It is beyond the scope of this review to describe all of these synthetic routes in great detail, but it is worth highlighting common starting routes, and routes that give rise to N1- and C2-substituted 3,4-HPs that are important precursors for new, bioactive compounds.
Pyranones such as maltol (20) containing a benzyl (Bn) protecting group can simply be converted to pyridinones by reaction with primary amines (Scheme 1) [20]. This allows diverse substitution at N1, including incorporation of reactive groups such as carboxylates or primary amines that lead to further functionalization [20,40].
Scheme 1.

Synthetic route for N1-substituted 3,4-HPs.
Compound 22 is a pyranone containing a Bn protecting group, and a reactive alcohol. It is a key synthetic precursor enabling versatile manipulation of functionality at the C2 position of 3,4-HPs (Scheme 2). It is synthesised in high yields from the commercially available and inexpensive precursor, kojic acid (21) in four steps [48,49]. As discussed above, the Bn-protected 22 can be converted to Bn-protected pyridinone 23 by reaction with methylamine [49]. Protection of the ethyl alcohol group (and subsequent deprotection) increases yields in this reaction. A Mitsunobu reaction of 23 with phthalamide gives a Bn-protected pyridinone (24) that contains a phthalamide at the C2 position, that can be simply converted to a primary amine (25) [49]. Bn-protected 25 is a very useful precursor for preparation of tris(hydroxypyridinones) such as 26 (see below).
Scheme 2.

Synthesis of a useful 3,4-HP with a reactive amine (25) that is used for synthesis of THP-Ac (26).
Alternatively, 22 can be converted at the alcohol to a carboxylic acid in two steps to give pyranone 27 (Scheme 3) [48]. Compound 27 can then be coupled with 2-mercaptothiazoline using appropriate reagents to give pyranone 28 that contains an active amide. Compound 28 can be reacted with primary or secondary amines, resulting in pyranone amide derivatives that can be converted to Bn-protected pyridinones by reaction with methylamine or ammonia [38,48]. For example, Bn-protected precursors to compounds 10 and 11 are prepared in this fashion.
Scheme 3.

Synthesis of 3,4-HP precursors containing primary and secondary amides.
Reactive carboxylates are synthetically accessible from Bn-protected pyridinones (Scheme 4) [50]. Bn-protected 29 can be further protected, and the methyl group substituted to ultimately yield 30. Compound 30 can be converted to a carboxylate, yielding 31. The carboxylate group of 31 can be further activated with an N-hydroxysuccinimide if required (32). Such derivatives have been used to prepare tris(hydroxypyridinones) such as 33 and 34 as well as other amide derivatives (35) [50].
Scheme 4.

Synthesis of a 3,4-HP containing a reactive carboxylate that allows synthesis of THPs 33 and 34.
Deprotection of Bn-protected hydroxyl groups proceeds via either hydrogenation reactions (catalysed by palladium on carbon) followed by acidification, or treatment with dissolved boron trichloride in an aprotic solvent, followed by addition of an alcohol (Scheme 1, Scheme 2 and Scheme 4).
4. Hexadentate Tris(hydroxypyridinone) Ligands
4.1. Topology and Fe3+ Affinity
Incorporation of three bidentate 3,4-HP ligands into a tripodal construct provides hexadentate tris(hydroxypyridinone) ligands (THPs) that, in a suitably designed scaffold, saturate the coordination sphere of octahedral metal ions. THPs have been designed for applications in gastrointestinal scavenging of Fe3+ [51], antimicrobial activity (via deprivation of microbes’ Fe3+ pool) [52,53], and fluorescence imaging of cellular Fe3+ distribution [54]. The topology of tripodal THPs is critically important to formation of octahedral complexes with 1:1 stoichiometry. The backbone of the tripod should be connected ortho to a coordinating O atom [55].
The synthesis of the first generation of THP ligands utilised 3,4-HP groups with a C2 carboxylate substitution (32), allowing reaction with tripodal polyamines to yield compounds such as 33 and 34 (Scheme 4). In 33 and 34, 3,4-HP units are attached via a C2 amide group, and fulfil the topology requirements outlined above for formation of hexadentate compounds with 1:1 stoichiometry. For compound 34, log K1 = 30.7, whereas for the bidentate homologue 35, log β3 = 31.4 [50]. In this case, the affinity of the bidentate 3,4-HP for Fe3+ is already optimal, and incorporation into a hexadentate form does not result in an increase in thermodynamic stability, as might normally be expected for an increase in ligand denticity and accompanying lower entropic costs. The pFe3+ of 34 (30.5 at pH 7.4) is higher than the pFe3+ of 35 (22.0) [50]. This arises because the formation constant of a hexadentate complex of 34 has only a first order dependence on ligand concentration (1:1 ligand to metal stoichiometry), whereas that of 35 necessarily has third order dependence on free ligand concentration (3:1 ligand to metal stoichiometry). In solutions of 34 where the hexadentate ligand concentration = 10 µM, the total concentration of single 3,4-HP units is three times greater than in solutions of 35 where the bidentate ligand concentration = 10 µM.
Instead of polyamine tripods, the next generation of THP ligands derivatised tripodal carboxylate groups, using 3,4-HP units with a C2 aminomethyl substituent (25, Scheme 2) [51]. In the resulting compounds, for example THP-Ac (26, Scheme 2), each 3,4-HP group is tethered to the tripod via an amidomethyl linker at the C2 position. The N1 and C6 positions are methylated. With the exception of C6-methylation, THP-Ac’s single 3,4-HP unit is structurally similar to deferiprone. For THP-Ac, log K1 = 32.52 and pFe3+ = 28.47 [51].
The incorporation of three 3,4-HP groups into a tripodal ligand also has implications for the lability of hexadentate complexes. For example, in [Fe(THP-Ac)], dissociation of a single 3,4-HP unit is likely to be followed by its rapid recoordination to the metal centre. During dissociation, this 3,4-HP unit will remain spatially close to the metal centre, as the other two 3,4-HP groups, to which it is covalently tethered, are likely to still be bound to Fe3+. On the other hand, in [Fe(deferiprone)3], if a deferiprone ligand dissociates, it has a lower probability of recoordinating to the same metal centre, as it is not anchored to any other coordinating ligands. The activation energy barrier to dissociation of [Fe(THP-Ac)] is higher than that of [Fe(deferiprone)3], and [Fe(THP-Ac)] is more kinetically inert than [Fe(deferiprone)3].
4.2. Dendrimers Based on THP Units
Dendritic THP molecules, for example 36, have incorporated between three and six THP groups, allowing coordination of between three and six equivalents of coordinatively saturated Fe3+ per molecule [51,56]. Dendrimers have been prepared from both 25 and 32. Affinity constants as well as pFe3+ values of dendrimer 36 (Figure 7) (log K1 = 32.74, pFe3+ = 28.69) and other derivatives do not meaningfully deviate from values for the single THP-Ac homologue 26, indicating that maximum Fe3+ binding efficiency is achieved using 3,4-HP motifs in a tripodal THP topology.
Figure 7.

Dendritic 3,4-HPs such as (36) can coordinate multiple Fe3+ ions.
4.3. Derivatising THP Ligands
THP compounds of the same topology and substitution as THP-Ac (26) are synthetically accessible from the β-alanine derivative, THP-NH2 (37, Figure 8) using similar reaction routes to that described in Scheme 2 [51]. THP derivatives such as 33 or 34 are less amenable to derivatisation in this fashion. The presence of an apical primary amine in THP-NH2 allows attachment to dendritic scaffolds [51], biomolecules [13,14,15,16], polymer units [57], and fluorophores [54]. This ability to functionalise THP compounds makes them attractive for technological applications where trivalent metal ion complexes of high affinity and kinetic stability are required. We have explored THP derivatives for PET imaging with Ga3+.
Figure 8.

THP derivatives.
5. Tris(hydroxypyridinone) Ligands for Radiolabelling with 68Ga3+
5.1. Radiolabelling Peptides with 68Ga for PET Imaging: The Case for Tris(hydroxypyridinone) Derivatives
Peptide-based molecular imaging agents can rapidly accumulate at target tissue and clear from circulation within 1–2 h. The 68 min half-life and positron emission properties of 68Ga (β+ 90%, Emax = 1880 KeV) match these requirements for PET imaging of peptide receptor expression. Moreover, 68Ga is conveniently available by elution of the 68Ge/68Ga generator to produce no-carrier-added solutions of 68Ga3+ in hydrochloric acid [58].
Hydrated Ga3+ species such as [Ga(H2O)6]3+ exist in aqueous solution below pH 4. As the pH is raised above 4, the poorly soluble hydroxide species Ga(OH)3 predominates in solution, until pH > 6.3, where tetradentate [Ga(OH)4]− predominates [59,60]. For efficient 68Ga3+ radiolabelling of chelate-peptide conjugates at neutral or near neutral pH, chelate complex formation must effectively compete with unreactive 68Ga-colloid formation. Preferably, the rate of chelation will be diffusion-controlled, so that complex formation outcompetes 68Ga3+ colloid formation. Additionally, the amounts of 68Ga eluted from clinical generators are in the range of 200–2000 MBq, approximately equivalent to 2–20 pmol of 68Ga3+ in 1–5 mL of solution. Highly efficient chelators are required to quantitatively complex such low concentrations of metal ion without using excessively high chelator concentration. In clinical formulations of chelator bioconjugates, low concentrations of chelator-peptide conjugate are important. Clinical formulations do not usually separate unlabelled bioconjugate from labelled conjugate, and large amounts of unlabelled conjugate can lead to saturation of target receptors in vivo.
As THP ligands have extraordinarily high pFe3+ values, and Ga3+ has similar coordination preferences to Fe3+, it was reasoned that such chelators could be very efficient at quantitatively coordinating 68Ga3+ at low chelator concentrations [13]. The acyclic nature of THP, and hence flexibility compared to macrocyclic ligands, results in low activation barriers to complexation, resulting in rapid rates of reaction at room temperature.
It is also critical that the 68Ga3+ complex is sufficiently kinetically stable over the period of time required for imaging (1–2 h) to withstand transchelation by competing endogenous proteins, such as transferrin, and other ligands that compete for Ga3+ in vivo [4]. The iron transport protein transferrin is abundant in serum and its two metal binding sites have high affinity for Fe3+ (log β1 = 22.8, log β2 = 44.3) and Ga3+ (log β1 = 20.3, log β2 = 39.6) [61]. Transchelation of 68Ga3+ to endogenous ligands results in increased non-target tissue uptake and lower tumour/diseased tissue uptake, decreasing PET image quality.
As 3,4-HP and THP derivatives can bind Fe3+ under physiological conditions, with the resulting complexes excreted, it was hypothesised that a THP Ga3+ complex could be sufficiently stable in vivo. Mouse biodistribution studies with the γ-emitting isotope, 67Ga3+ (half-life = 78 h), comparing the biodistribution of [67Ga(deferiprone)3] with [67Ga(citrate)3] show that mice administered the deferiprone complex intravenously have lower 67Ga blood activity one day post-administration than animals administered the citrate complex [37]. Subsequent studies demonstrate that 67Ga: (i) clears more rapidly from animals administered the deferiprone complex compared to animals administered the citrate complex; and (ii) clears predominantly via a renal pathway in animals administered the deferiprone complex [37,62]. N1-functionalised derivatives of 3,4-HP are also efficacious at sequestering 67Ga3+ in vivo, with biodistribution of radioactivity modified by N1 substituents [62]. Such results suggest that [67Ga(deferiprone)3] has appreciable stability in vivo, and that 3,4-HPs can effectively compete with endogenous protein ligands for Ga3+ [37,62].
5.2. Tris(hydroxypyridinone) Bioconjugates
We first reported the utility of THP-Ac (26, Figure 8) as a basis for highly efficient 68Ga labelling under very mild conditions after undertaking side-by-side comparisons of THP-Ac with chelators already commonly used to complex 68Ga3+: macrocyclic derivatives DOTA (41) (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) and NOTA (42) (1,4,7-triazacyclononane-1,4,7-triacetic acid), and the acyclic chelator HBED (43) (bis(2-hydroxybenzyl)ethylenediaminediacetic acid) (Figure 9) [13]. Each chelator was reacted with generator-produced 68Ga3+ at progressively lower chelator concentrations (100 nM–1 mM, each 100 µL corresponding to amounts of 10 pmol–100 nmol) (Figure 10), with the reasoning that the most efficient, rapidly complexing chelators would maintain high labelling efficiency at the lowest ligand concentrations. Optimised reaction conditions (as reported in the radiochemical literature) for each chelator were employed. At a concentration of 10 µM, THP-Ac complexes 68Ga3+ in 5 min in >98% radiochemical yield at pH 6.5 at room temperature. Radiochemical yields for DOTA are >95% at the same chelator concentration, but this requires heating at 100 °C at pH 4.4. NOTA has been reported to coordinate 68Ga3+ efficiently at room temperature, but at 10 µM concentration at room temperature, pH 4.4, radiochemical yields only average 80%. At 10 µM, HBED is able to complex 68Ga3+ in >96% radiochemical yield at pH 4.6. Whilst HBED radiochemical yields under acidic conditions are comparable to those of THP-Ac at near neutral pH, HBED forms geometric isomers when complexed to Ga3+ [4]. From a regulatory perspective, the presence of different geometric isomers is undesirable, as it is possible that the different isomers have different pharmacological profiles.
Figure 9.

Chelators for 68Ga3+.
Figure 10.

Radiolabelling yield versus ligand concentration for 68Ga-DOTA (pH 4.4, 30 min, 100 °C), 68Ga-NOTA (pH 3.6, 10 min, room temperature), 68Ga-HBED (pH 4.6, 10 min, room temperature) and 68Ga-THP, (pH 6.5, 5 min, room temperature). All experiments were conducted with the same batch of 68Ga eluate. All radiolabelling buffers were 0.2 M acetic acid/sodium acetate. Reprinted with permission from Berry, D.J.; et al. Efficient bifunctional gallium-68 chelators for positron emission tomography: tris(hydroxypyridinone) ligands. Chem. Commun. 2011, 47, 7068–7070. Copyright Royal Society of Chemistry.
The β-alanine-derived compound, THP-NH2 (37) [51], can be functionalised for bioconjugation via a maleimide [13,16] and isothiocyanates [14,15] (Figure 8). The bifunctional chelators THP-mal (38), THP-Ph-NCS (39) and THP-NCS (40) have all been conjugated to biomolecules. Amide conjugation of THP-NH2 (37) to activated carboxylates of peptides and proteins is also a viable conjugation strategy.
The protein C2A, containing an engineered cysteine residue, has been conjugated to THP-mal (38), resulting in a single equivalent of THP-mal attached per protein molecule [13]. The conjugate can be radiolabelled with 68Ga3+ at pH 5.5 (6 nmol of protein in 100 µL, at a concentration of 60 µM), giving quantitative labelling after 5 min. In vivo PET imaging in healthy mice demonstrates that 68Ga-THP-mal-C2A clears to the kidneys, and does not release any 68Ga3+ over a 90 min period. In contrast, in mice administered solutions of unchelated 68Ga3+, radioactivity is distributed throughout the body 90 min post-injection. In competition studies where 68Ga-THP-Ac is incubated with transferrin, [68Ga(THP-Ac)] remains intact. On the other hand, when 68Ga-transferrin is incubated with THP-Ac, 68Ga is quickly transchelated to form a complex with THP-Ac [13].
THP-Ph-NCS and THP-NCS have both been attached to the cyclic pentapeptide, c(RGDfK) via lysine sidechains [14]. The “RGD” peptide motif targets αvβ3 integrin receptors that are expressed on the surface of many metastatic tumour cells, as well as inflamed tissue and blood vessels undergoing angiogenesis. Both THP-Ph-NCS-RGD (41, Figure 11) and THP-NCS-RGD (42, Figure 11) can be radiolabelled with generator-produced 68Ga3+ eluate at conjugate concentrations of 4–5 µM, or total amounts of 10–12 nmol, giving radiochemical yields of 95%–99%. These reactions proceed in aqueous solution, pH 5.5–6.5 in less than 5 min at ambient temperature to give a single product corresponding to either [68Ga(THP-Ph-NCS-RGD)] or [68Ga(THP-NCS-RGD)].
Figure 11.

Representative PET maximum intensity projection of Balb/c nu/nu mice bearing U87MG tumours on right flank at 1 h post-injection of: (a) [68Ga(THP-Ph-NCS-RGD)]; and (b) [68Ga(THP-NCS-RGD)]. Black arrow, tumour; grey arrow, bladder; dashed arrow, kidneys and liver. Reprinted with permission under a Creative Commons Attribution (CC-BY) License from Ma, M.T.; et al. New tris(hydroxypyridinone) bifunctional chelators containing isothiocyanate groups provide a versatile platform for rapid one-step labelling and PET imaging with 68Ga3+. Bioconjugate Chem. 2016, 27, 309–318. Copyright American Chemical Society.
Both [68Ga(THP-Ph-NCS-RGD)] and [68Ga(THP-NCS-RGD)] retain affinity for αvβ3 integrin receptors in vitro and in vivo [14]. PET imaging and biodistribution studies of mice bearing U87MG tumours demonstrate that both [68Ga(THP-Ph-NCS-RGD)] and [68Ga(THP-NCS-RGD)]: (i) selectively target αvβ3 integrin receptors; and (ii) clear from the body within 1–2 h post-injection, predominantly via a renal route (Figure 11).
The molecular imaging agent, [68Ga(DOTA-TATE)] is routinely used for PET imaging of neuroendocrine tumours. DOTA-TATE (43, Figure 12) is a conjugate of the macrocyclic chelator DOTA (27, Figure 9) and Tyr3-octreotate, an eight amino acid cyclic peptide that targets somatostatin 2 receptors (SSTR) overexpressed on the surface of neuroendocrine tumours. Using THP-NCS, THP-TATE (44, Figure 12) has been synthesised [15]. Similar to RGD conjugates, THP-TATE can be radiolabelled at room temperature at concentrations of 5 µM (10 nmol of conjugate) at near neutral pH in less than 2 min. In contrast, DOTA-TATE requires temperatures of 80–90 °C at pH 3–5, with reaction times of 5–10 min, and in clinical radiolabelling protocols, post-synthetic purifications procedures are invariably employed [15]. PET imaging and biodistribution experiments show that [68Ga(THP-TATE)] has similar uptake in SSTR-positive tumours to the clinical standard [68Ga(DOTA-TATE)]. [68Ga(THP-TATE)] clears via a renal pathway, but significantly higher kidney and liver retention of [68Ga(THP-TATE)] is observed (Figure 12). In PET images of mice bearing SSTR2-positive tumours administered [68Ga(THP-TATE)], tumours can be clearly delineated.
Figure 12.

Representative PET maximum intensity projections of Balb/c nu/nu mice bearing AR47J tumours on the right flank 1 h PI of: (a) [68Ga(DOTA-TATE)]; and (b) [68Ga(THP-TATE)]. Black arrow, tumour; grey arrow, bladder; dashed arrow, kidneys. Reprinted with permission under a Creative Commons Attribution (CC-BY) License from Ma, M.T.; et al. Rapid kit-based 68Ga-labelling and PET imaging with THP-Tyr3-octreotate: a preliminary comparison with DOTA-Tyr3-octreotate. EJNMMI Res. 2015, 5, 52. Copyright.
In all of these radiolabelled derivatives, the radiotracer demonstrates high serum stability and in vivo stability, with no evidence of dissociation of 68Ga3+ from the THP chelator.
5.3. Preparation, Radiolabelling and In Vitro Uptake of a Trastuzumab Immunoconjugate
New protein constructs that target receptors with high affinity and specificity have similar utility to peptides in molecular imaging. For proteins with short circulation times and rapid accumulation at target tissue, 68Ga PET imaging will be clinically viable, provided that appropriate radiolabelling protocols are available. The sensitivity of proteins’ tertiary structures to acidic pH and extremes of temperature requires mild radiolabelling conditions, raising problems when using conventional Ga3+ chelators. HBED, NOTA and DFO are capable of radiolabelling Ga3+ isotopes at room temperature, but HBED [63] and NOTA [64] require low pH for reactions to proceed quantitatively, and DFO complexes of Ga3+ are unstable [65].
Trastuzumab is a therapeutic monoclonal antibody used for treatment of breast cancer. It targets the human epidermal growth factor receptor 2 (HER2). In vivo, antibodies such as trastuzumab require extended periods of time (6 to 48 h) to clear circulation and accumulate at HER2-positive target tissue, and given the short half-life of 68Ga, it is likely impractical to use 68Ga-labelled trastuzumab to image HER2 expression. Nonetheless, it is instructive to radiolabel THP-PhNCS-trastuzumab to assess whether a THP protein conjugate that is sensitive to acidic pH (less than pH 5) can be radiolabelled rapidly under mild conditions (pH 6–7) to provide a formulation suitable for injection without further purification.
The bifunctional chelator THP-PhNCS has been conjugated to the monoclonal antibody (mAb), trastuzumab by incubating a HEPES buffered solution containing both reagents under mild conditions [66,67]. The immunoconjugate, THP-PhNCS-trastuzumab has been isolated using solid phase size exclusion chromatography [66,67]. Addition of generator-produced 68Ga3+ (~10 MBq, 50 µL, aqueous 0.1 M HCl) to solutions of THP-PhNCS-trastuzumab (50 µL, 0.65 mg·mL−1, 0.2 M ammonium acetate, pH 6–7) results in formation of [68Ga(THP-PhNCS-trastuzumab)], with specific activities of up to 50 MBq·nmol−1 and radiochemical yields of 99% as measured by size exclusion HPLC (Figure 13a, red trace). The major signal at 7.98 min in the radiochromatogram matches the retention time of native trastuzumab, and the minor signal at 6.83 min is typical of formation of radiolabelled immunoconjugate aggregates [66]. Addition of 68Ga3+ solutions to samples containing native, unconjugated trastuzumab do not provide labelled antibody (Figure 13a, black trace)—the mobile phase in these experiments contains ethylenediaminetetraacetate (EDTA), and the single signal in the radiochromatogram with a retention time of 12.08 min corresponds to [68Ga(EDTA)]−.
Figure 13.

(a) Size exclusion radio-HPLC traces of [68Ga(THP-PhNCS-trastuzumab)] (red) and [68Ga(EDTA)]− (black); and (b) In vitro uptake of [68Ga(THP-PhNCS-trastuzumab)] and [68Ga(THP-PhNCS-trastuzumab)] in the presence of an inhibitory concentration of trastuzumab, and 68Ga3+ in HER2-positive HCC1954 cells, and [68Ga(THP-PhNCS-trastuzumab)] in HER2-negative A375 cells, after 30 min incubation. Uptake is expressed as a percentage of added radioactivity (AR)/1 × 106 cells (n = 3, error bars correspond to standard error of the mean).
To establish that the labelled immunoconjugate, [68Ga(THP-PhNCS-trastuzumab)], retains affinity for HER2 receptors, [68Ga(THP-PhNCS-trastuzumab)] has been incubated with HER2-positive HCC1954 cells and HER2-negative A375 cells. Additionally, a blockade experiment where [68Ga(THP-PhNCS-trastuzumab)] is incubated with HCC1954 cells in the presence of a large excess of native trastuzumab has been undertaken. Uptake of [68Ga(THP-PhNCS-trastuzumab)] in HCC1954 cells measures 35.12 ± 0.53 percentage of added radioactivity per one million cells (%AR/million cells), whereas uptake in A375 cells measures 1.98% ± 0.02 %AR/million cells and uptake in the trastuzumab blockade measures 0.30% ± 0.03 %AR/million, indicating that uptake of [68Ga(THP-PhNCS-trastuzumab)] is receptor-mediated, and that in vitro [68Ga(THP-PhNCS-trastuzumab)] retains affinity for HER2-expressing cells (Figure 13b). Addition of a solution containing 68Ga3+ to HCC1954 cells does not result in significant uptake of activity (1.09% ± 0.05 %AR/million cells).
Thus, THP enables efficient and rapid 68Ga3+ radiolabelling of proteins under mild, aqueous conditions. This radiolabelling strategy will have utility for 68Ga PET imaging of proteins such as fusion proteins and antibody fragments that have shorter clearance times than full length antibodies.
5.4. Other THP Derivatives
Alternative THP chelators such as NTP(PrHP)3 (45) have been reported for complexation of the SPECT isotope, 67Ga [68,69]. In these derivatives (Figure 14), the 3,4-HP groups are attached to the tripodal scaffold via the N1 ring atoms, and the chelating O atoms are meta and para to the linker. Such a topology can lead to formation of either dinuclear structures or structures where one 3,4-HP unit has dissociated [55]. Both species are generally more kinetically labile than species of a 1:1 stoichiometry and are not ideal for in vivo applications. The presence of more than one complex structure is also undesirable, as these species can have different biological behaviours. NTP(PrHP)3, with its tripodal topology centred on a tertiary amine rather than carbon, is not readily adapted to use as a bifunctional chelator. SPECT imaging and biodistribution studies with 67Ga3+ indicate that most [67Ga-NTP(PrHP)3] rapidly clears circulation within 1 h, and remaining [67Ga-NTP(PrHP)3] does not release 67Ga3+ over 24 h [69].
Figure 14.

Structure of NTP(PrHP)3.
6. Hydroxypyridinones for Radiolabelling 89Zr4+
We have also investigated THP-Ac and a THP-mal-trastuzumab conjugate for radiolabelling with the long-lived PET isotope, zirconium-89 (89Zr4+) (half-life = 78 h) [16]. The Zr4+ ion is oxophilic, and bifunctional hexadentate DFO derivatives are most commonly utilised for incorporating 89Zr4+ into antibodies. There is evidence that [89Zr(DFO)]+ derivatives are unstable in vivo, with observations of 89Zr accumulation in the skeleton over the course of a week [16]. Here, skeletal uptake is assumed to be indicative of dissociation of 89Zr4+ from a chelator-conjugate.
Both THP-Ac and THP-mal-trastuzumab can be radiolabelled with 89Zr4+ at room temperature and near neutral pH in quantitative yield. For THP-Ac, this was achieved at 1 mM concentration and 10 µL volume, corresponding to 10 nmol of ligand. Competition experiments indicated a thermodynamic preference for Zr4+ coordination to THP-Ac over DFO. This is consistent with higher metal ion affinities of 3,4-HPs compared to hydroxamates. However, experiments in which 89Zr-THP-mal-trastuzumab was administered to healthy animals indicate that in vivo, 89Zr4+ dissociates from THP-mal-trastuzumab within 48 h, with activity accumulating in the skeleton (Figure 15). In vivo stability of the [89Zr(THP)]+ complex is inferior to that of the [89Zr(DFO)]+ complex, possibly a result of the different chelator topologies.
Figure 15.

PET scans of C57Bl/6j mice administered: (a) [89Zr(oxalate)4]4−; (b) 89Zr-THP-mal-trastuzumab; and (c) 89Zr-DFO-mal-trastuzumab. For the animal administered [89Zr(ox)4]4− 98% of the injected dose remains 4 h post-injection, and is associated predominantly with the skeleton. For the animal administered 89Zr-THP-mal-trastuzumab, the skeleton is clearly visible from three days post-injection, indicative of dissociation of 89Zr4+ from THP-mal-trastuzumab over time. This is in marked contrast to the animal administered 89Zr-DFO-trastuzumab, where the blood pool remains visible out to seven days post-injection. Reprinted with permission under a Creative Commons Attribution (CC-BY) License from Ma, M.T.; et al. Tripodal tris(hydroxypyridinone) ligands for immunoconjugate PET imaging with 89Zr4+: comparison with desferrioxamine-B. Dalton Trans. 2015, 44, 4884–4900. Copyright Royal Society of Chemistry.
Zr4+ can accommodate up to eight donor atoms in its coordination sphere, and these coordination requirements are likely to be a factor in the instability of [89Zr(THP)]+ in vivo. The presence of two coordination sites unoccupied by THP chelator allows coordination of endogenous ligands, and provides pathways to transmetallation/ligand exchange.
It is possible that a tetrakis(3,4-hydroxypyridinone) ligand could impart greater in vivo stability, but there are no reports of such ligands to date. Other researchers have studied octadentate tetrakis(hydroxypyridinone) derivatives for 89Zr4+ incorporating 1,2-HPs and 3,2-HPs. The bifunctional open chain (1,2-HP)4 chelator (46, Figure 16) can: (i) coordinate Zr4+ in an octadentate environment; and (ii) be conjugated to trastuzumab. Immunoconjugate (1,2-HP)4-trastuzumab can be radiolabelled with 89Zr4+ at room temperature, and PET imaging and biodistribution studies demonstrate high in vivo stability of the complex, with concomitant high tumour uptake [70].
Figure 16.

Structures of octadentate (HP)4 derivatives evaluated for 89Zr4+ immunoconjugate radiolabelling.
Bifunctional macrobicyclic octadentate (3,2-HP)4 (47, Figure 16) has also been derivatised for conjugation to trastuzumab [71]. The coordination environment of the Zr4+ complex is not well defined, and chromatographic analysis indicates that multiple Zr-bound (3,2-HP)4 species form under the mild reaction conditions described (room temperature, 15 min incubation). PET imaging and biodistribution studies in mice using 89Zr-labelled (3,2-HP)4-antibody conjugates suggest that at least one form of 89Zr4+-(3,2-HP)4 is unstable in vivo, as significantly higher bone uptake is observed for (3,2-HP)4 conjugates compared to DFO conjugates. This highlights the difficulty in interpreting in vivo results based on radiolabelled compounds that contain metal complexes in more than one conformation.
7. Concluding Remarks
Derivatisation of 3,4-HPs via substitution of ring protons allows for tailoring of chelator properties including the proton and Fe3+ affinities, and in vivo distribution and reactivity towards endogenous enzymes. 3,4-HPs can be further functionalised with fluorescent tags and biologically active motifs, and can be incorporated into chelators of higher denticity. Hexadentate THP chelators based on 3,4-HPs possess extraordinarily high pFe3+ values, and are very potent Fe3+ scavengers. As Fe3+ and Ga3+ have similar coordination preferences, THP chelators are ideal candidates for development of PET radiopharmaceuticals based on 68Ga3+, and direct translation of this chemistry has enabled rapid development of bioconjugates of THP chelators for 68Ga PET imaging.
Simplicity of radiolabelling with minimal need for complex equipment and radiochemical expertise is likely to be a key to the wider availability of 68Ga PET, and this is afforded by appropriate design of a 68Ga chelator. THP derivatives fulfil these requirements whereas other established chelator designs do not. Current clinical radiosynthetic protocols based on DOTA and HBED derivatives require heating (>80 °C), low pH (3–5) and post-synthetic purification/formulation. In contrast, THP compounds can be radiolabelled and formulated by treatment with generator-produced 68Ga3+ in over 95% radiochemical yield under ambient conditions in less than 5 min, at low chelator concentrations, neutral pH and in aqueous solution. There is no requirement for post-synthetic purification or reformulation, as reactions are quantitative and components (solvents and buffers) are physiologically compatible. Bifunctional THP derivatives enable a means of attachment to peptides and proteins, and biological studies have demonstrated that the peptide radiotracers are stable in vivo with respect to dissociation of the 68Ga-THP complex, retain affinity for target receptors and clear rapidly from circulation. All of these characteristics make THP superlative for one-step kit-based radiosynthesis.
Acknowledgments
Ruslan Cusnir acknowledges the Swiss National Science Foundation for an Early Postdoc Mobility Fellowship (P2LAP3-168441). Cinzia Imberti acknowledges the National Institute for Health Research Biomedical Research Centre for a PhD studentship. This research was supported by the Centre of Excellence in Medical Engineering Centre funded by the Wellcome Trust and the Engineering and Physical Sciences Research Council (WT088641/Z/09/Z), the King’s College London and University College London Comprehensive Cancer Imaging Centre funded by Cancer Research UK and the Engineering and Physical Sciences Research Council in association with the Medical Research Council and Department of Health (England), and by the National Institute for Health Research Biomedical Research Centre at Guy’s and St Thomas’ NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the National Health Service, the National Institute for Health Research or the Department of Health.
Abbreviations
| Bn | Benzyl |
| DFO | Deferrioxamine B |
| EDTA | Ethylenediaminetetraacetate |
| GIT | Gastrointestinal tract |
| HER2 | Human epidermal growth factor receptor 2 |
| HP | Hydroxypyridinone |
| HPLC | High performance liquid chromatography |
| PET | Positron Emission Tomography |
| RGD | Cyclic RGDfK peptide |
| TATE | Octreotate |
| THP | Tris(hydroxypyridinone) |
| SSTR2 | Somatostatin 2 receptor |
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Blower P.J. A nuclear chocolate box: The periodic table of nuclear medicine. Dalton Trans. 2015;44:4819–4844. doi: 10.1039/C4DT02846E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hofman M.S., Kong G., Neels O.C., Eu P., Hong E., Hicks R.J. High management impact of Ga-68 DOTATATE (Ga Tate) PET/CT for imaging neuroendocrine and other somatostatin expressing tumours. J. Med. Imaging Radiat. Oncol. 2012;56:40–47. doi: 10.1111/j.1754-9485.2011.02327.x. [DOI] [PubMed] [Google Scholar]
- 3.Afshar-Oromieh A., Zechmann C.M., Malcher A., Eder M., Eisenhut M., Linhart H.G., Holland-Letz T., Hadaschik B.A., Giesel F.L., Debus J., et al. Comparison of PET imaging with a 68Ga-labelled PSMA ligand and 18F-choline-based PET/CT for the diagnosis of recurrent prostate cancer. Eur. J. Nucl. Med. Mol. Imaging. 2014;41:11–20. doi: 10.1007/s00259-013-2525-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma M.T., Blower P.J. Chelators for diagnostic molecular imaging with radioisotopes of copper, gallium and zirconium. In: Crichton R.R., Ward R.J., Hider R.C., editors. Metal Chelation in Medicine. The Royal Society of Chemistry; Cambridge, UK: 2017. pp. 260–312. [Google Scholar]
- 5.Price E.W., Orvig C. Matching chelators to radiometals for radiopharmaceuticals. Chem. Soc. Rev. 2014;43:260–290. doi: 10.1039/C3CS60304K. [DOI] [PubMed] [Google Scholar]
- 6.Price T.W., Greenman J., Stasiuk G.J. Current advances in ligand design for inorganic positron emission tomography tracers 68Ga, 64Cu, 89Zr and 44Sc. Dalton Trans. 2016;45:15702–15724. doi: 10.1039/C5DT04706D. [DOI] [PubMed] [Google Scholar]
- 7.Spang P., Herrmann C., Roesch F. Bifunctional gallium-68 chelators: Past, present, and future. Semin. Nucl. Med. 2016;46:373–394. doi: 10.1053/j.semnuclmed.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Liu Z.D., Hider R.C. Design of iron chelators with therapeutic application. Coord. Chem. Rev. 2002;232:151–171. doi: 10.1016/S0010-8545(02)00050-4. [DOI] [Google Scholar]
- 9.Liu Z.D., Hider R.C. Design of clinically useful iron(III)-selective chelators. Med. Res. Rev. 2002;22:26–64. doi: 10.1002/med.1027. [DOI] [PubMed] [Google Scholar]
- 10.Zhou T., Ma Y., Kong X., Hider R.C. Design of iron chelators with therapeutic application. Dalton Trans. 2012;41:6371–6389. doi: 10.1039/c2dt12159j. [DOI] [PubMed] [Google Scholar]
- 11.Hider R.C., Kontoghiorghes G., Silver J. Pharmaceutically Active 3-Hydroxypyrid-2-and-4-ones. GB 2118176A. UK Patent Application. 1982
- 12.Shannon R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A. 1976;A32:751–767. doi: 10.1107/S0567739476001551. [DOI] [Google Scholar]
- 13.Berry D.J., Ma Y., Ballinger J.R., Tavare R., Koers A., Sunassee K., Zhou T., Nawaz S., Mullen G.E.D., Hider R.C., et al. Efficient bifunctional gallium-68 chelators for positron emission tomography: Tris(hydroxypyridinone) ligands. Chem. Commun. 2011;47:7068–7070. doi: 10.1039/c1cc12123e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma M.T., Cullinane C., Imberti C., Baguna Torres J., Terry S.Y.A., Roselt P., Hicks R.J., Blower P.J. New tris(hydroxypyridinone) bifunctional chelators containing isothiocyanate groups provide a versatile platform for rapid one-step labeling and pet imaging with 68Ga3+ Bioconjug. Chem. 2016;27:309–318. doi: 10.1021/acs.bioconjchem.5b00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma M.T., Cullinane C., Waldeck K., Roselt P., Hicks R.J., Blower P.J. Rapid kit-based 68Ga-labelling and PET imaging with THP-Tyr3-octreotate: A preliminary comparison with DOTA-Tyr3-octreotate. EJNMMI Res. 2015;5:52. doi: 10.1186/s13550-015-0131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma M.T., Meszaros L.K., Paterson B.M., Berry D.J., Cooper M.S., Ma Y., Hider R.C., Blower P.J. Tripodal tris(hydroxypyridinone) ligands for immunoconjugate PET imaging with 89Zr4+: Comparison with desferrioxamine-B. Dalton Trans. 2015;44:4884–4900. doi: 10.1039/C4DT02978J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gateau C., Mintz E., Delangle P. Rational design of copper and iron chelators to treat Wilson’s disease and hemochromatosis. In: Storr T., editor. Ligand Design in Medicinal Inorganic Chemistry. John Wiley & Sons, Ltd.; Chichester, UK: 2014. pp. 287–319. [Google Scholar]
- 18.Gumienna-Kontecka E., Pyrkosz-Bulska M., Szebesczyk A., Ostrowska M. Iron chelating strategies in systemic metal overload, neurodegeneration and cancer. Curr. Med. Chem. 2014;21:3741–3767. doi: 10.2174/0929867321666140706143402. [DOI] [PubMed] [Google Scholar]
- 19.Price E.W., Orvig C. The Chemistry of Inorganic Nuclides (86Y 68Ga 64Cu 89Zr 124I) In: Long N., Wong W.-T., editors. The Chemistry of Molecular Imaging. John Wiley & Sons; Hoboken, NJ, USA: 2015. pp. 105–136. [Google Scholar]
- 20.Dobbin P.S., Hider R.C., Hall A.D., Taylor P.D., Sarpong P., Porter J.B., Xiao G., van der Helm D. Synthesis, physicochemical properties, and biological evaluation of N-substituted 2-alkyl-3-hydroxy-4(1H)-pyridinones: Orally active iron chelators with clinical potential. J. Med. Chem. 1993;36:2448–2458. doi: 10.1021/jm00069a002. [DOI] [PubMed] [Google Scholar]
- 21.Hider R.C., Hall A.D. Clinically useful chelators of tripositive elements. Prog. Med. Chem. 1991;28:41–173. doi: 10.1016/s0079-6468(08)70363-1. [DOI] [PubMed] [Google Scholar]
- 22.Li Y.J., Martell A.E. Potentiometric and spectrophotometric determination of stabilities of the 1-hydroxy-2-pyridinone complexes of trivalent and divalent metal ions. Inorg. Chim. Acta. 1993;214:103–111. [Google Scholar]
- 23.Scarrow R.C., Riley P.E., Abu-Dari K., White D.L., Raymond K.N. Ferric ion sequestering agents. 13. Synthesis, structures, and thermodynamics of complexation of cobalt(III) and iron(III) tris complexes of several chelating hydroxypyridinones. Inorg. Chem. 1985;24:954–967. doi: 10.1021/ic00200a030. [DOI] [Google Scholar]
- 24.Clarke E.T., Martell A.E. 1-Methyl-3-hydroxy-2-pyridinone and 1,4-dihydroxy-2-pyridinone complexes of the trivalent metal ions of iron(III), gallium(III), aluminum(III), indium(III) and gadolinium(III): Potentiometric and spectrophotometric determination of stabilities. Inorg. Chim. Acta. 1992;196:185–194. doi: 10.1016/S0020-1693(00)86122-8. [DOI] [Google Scholar]
- 25.Clarke E.T., Martell A.E. Stabilities of 1,2-dimethyl-3-hydroxy-4-pyridinone chelates of divalent and trivalent metal ions. Inorg. Chim. Acta. 1992;191:56–63. doi: 10.1016/S0020-1693(00)80327-8. [DOI] [Google Scholar]
- 26.Amelia Santos M. Hydroxypyridinone complexes with aluminum. In vitro/vivo studies and perspectives. Coord. Chem. Rev. 2002;228:187–203. doi: 10.1016/S0010-8545(02)00035-8. [DOI] [Google Scholar]
- 27.Hsieh W.-Y., Liu S. Synthesis and characterization of Cr(III) complexes with 3-hydroxy-4-pyrones and 1,2-dimethyl-3-hydroxy-4-pyridinone (DMHP): X-ray crystal structures of Cr(DMHP)3·12H2O and Cr(ma)3. Synth. React. Inorg. Met.-Org. Nano-Met. Chem. 2005;35:61–70. doi: 10.1081/SIM-200047549. [DOI] [Google Scholar]
- 28.Hsieh W.-Y., Liu S. Synthesis, characterization, and structures of Mn(DMHP)3·12H2O and Mn(DMHP)2Cl·0.5H2O. Inorg. Chem. 2005;44:2031–2038. doi: 10.1021/ic0490627. [DOI] [PubMed] [Google Scholar]
- 29.Nelson W.O., Karpishin T.B., Rettig S.J., Orvig C. Aluminum and gallium compounds of 3-hydroxy-4-pyridinones: Synthesis, characterization, and crystallography of biologically active complexes with unusual hydrogen bonding. Inorg. Chem. 1988;27:1045–1051. doi: 10.1021/ic00279a022. [DOI] [Google Scholar]
- 30.Matsuba C.A., Nelson W.O., Rettig S.J., Orvig C. Neutral water-soluble indium complexes of 3-hydroxy-4-pyrones and 3-hydroxy-4-pyridinones. Inorg. Chem. 1988;27:3935–3939. doi: 10.1021/ic00295a012. [DOI] [Google Scholar]
- 31.Charalambous J., Dodd A., McPartlin M., Matondo S.O.C., Pathirana N.D., Powell H.R. Synthesis and X-ray crystal structure of tris(1,2-dimethyl-3-hydroxypyrid-4-onato)iron(III) Polyhedron. 1988;7:2235–2237. doi: 10.1016/S0277-5387(00)81813-6. [DOI] [Google Scholar]
- 32.Evans R.W., Rafique R., Zarea A., Rapisarda C., Cammack R., Evans P.J., Porter J.B., Hider R.C. Nature of non-transferrin-bound iron: Studies on iron citrate complexes and thalassemic sera. J. Biol. Inorg. Chem. 2008;13:57–74. doi: 10.1007/s00775-007-0297-8. [DOI] [PubMed] [Google Scholar]
- 33.Fenton H.J.H. LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894;65:899–910. doi: 10.1039/CT8946500899. [DOI] [Google Scholar]
- 34.Liu Z.D., Khodr H.H., Liu D.Y., Lu S.L., Hider R.C. Synthesis, physicochemical characterization, and biological evaluation of 2-(1′-hydroxyalkyl)-3-hydroxypyridin-4-ones: Novel iron chelators with enhanced pFe3+ values. J. Med. Chem. 1999;42:4814–4823. doi: 10.1021/jm991080o. [DOI] [PubMed] [Google Scholar]
- 35.Motekaitis R.J., Martell A.E. Stabilities of the iron(III) chelates of 1,2-dimethyl-3-hydroxy-4-pyridinone and related ligands. Inorg. Chim. Acta. 1991;183:71–80. doi: 10.1016/S0020-1693(00)82997-7. [DOI] [Google Scholar]
- 36.Xie Y.-Y., Lu Z., Kong X.-L., Zhou T., Bansal S., Hider R. Systematic comparison of the mono-, dimethyl- and trimethyl 3-hydroxy-4(1H)-pyridones—Attempted optimization of the orally active iron chelator, deferiprone. Eur. J. Med. Chem. 2016;115:132–140. doi: 10.1016/j.ejmech.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 37.Clevette D.J., Lyster D.M., Nelson W.O., Rihela T., Webb G.A., Orvig C. Solution chemistry of gallium and indium 3-hydroxy-4-pyridinone complexes in vitro and in vivo. Inorg. Chem. 1990;29:667–672. doi: 10.1021/ic00329a021. [DOI] [Google Scholar]
- 38.Piyamongkol S., Ma Y.M., Kong X.L., Liu Z.D., Aytemir M.D., van der Helm D., Hider R.C. Amido-3-hydroxypyridin-4-ones as iron(III) ligands. Chem. Eur. J. 2010;16:6374–6381. doi: 10.1002/chem.200902455. [DOI] [PubMed] [Google Scholar]
- 39.Li J., Lu Z., Kong X., Ma Y., Zhang X., Bansal S.S., Abbate V., Hider R.C. Design and synthesis of novel pegylated iron chelators with decreased metabolic rate. Future Med. Chem. 2015;7:2439–2449. doi: 10.4155/fmc.15.154. [DOI] [PubMed] [Google Scholar]
- 40.Ma Y., Luo W., Quinn P.J., Liu Z., Hider R.C. Design, synthesis, physicochemical properties, and evaluation of novel iron chelators with fluorescent sensors. J. Med. Chem. 2004;47:6349–6362. doi: 10.1021/jm049751s. [DOI] [PubMed] [Google Scholar]
- 41.Abbate V., Reelfs O., Kong X., Pourzand C., Hider R.C. Dual selective iron chelating probes with a potential to monitor mitochondrial labile iron pools. Chem. Commun. 2016;52:784–787. doi: 10.1039/C5CC06170A. [DOI] [PubMed] [Google Scholar]
- 42.Galanello R. Deferiprone in the treatment of transfusion-dependent thalassemia: A review and perspective. Ther. Clin. Risk Manag. 2007;3:795–805. [PMC free article] [PubMed] [Google Scholar]
- 43.Heinz U., Hegetschweiler K., Acklin P., Faller B., Lattmann R., Schnebli H.P. 4-[3,5-bis(2-hydroxyphenyl)-1,2,4-triazol-1-yl]-benzoic acid: A novel efficient and eelective iron(III) complexing agent. Angew. Chem. Int. Ed. 1999;38:2568–2570. doi: 10.1002/(SICI)1521-3773(19990903)38:17<2568::AID-ANIE2568>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 44.Xia S., Zhang W., Huang L., Jiang H. Comparative efficacy and safety of deferoxamine, deferiprone and deferasirox on severe thalassemia: A meta-analysis of 16 randomized controlled trials. PLoS ONE. 2013;8:e82662. doi: 10.1371/journal.pone.0082662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cermak J., Jonasova A., Vondrakova J., Cervinek L., Belohlavkova P., Neuwirtova R. A comparative study of deferasirox and deferiprone in the treatment of iron overload in patients with myelodysplastic syndromes. Leuk. Res. 2013;37:1612–1615. doi: 10.1016/j.leukres.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 46.Zachariah M., Tony S., Bashir W., Al Rawas A., Wali Y., Pathare A. Comparative assessment of deferiprone and deferasirox in thalassemia major patients in the first two decades-single centre experience. J. Pediatr. Hematol. Oncol. 2013;30:104–112. doi: 10.3109/08880018.2012.762568. [DOI] [PubMed] [Google Scholar]
- 47.Pepe A., Meloni A., Capra M., Cianciulli P., Prossomariti L., Malaventura C., Putti M.C., Lippi A., Romeo M.A., Bisconte M.G., et al. Deferasirox, deferiprone and desferrioxamine treatment in thalassemia major patients: Cardiac iron and function comparison determined by quantitative magnetic resonance imaging. Haematologica. 2010;96:41. doi: 10.3324/haematol.2009.019042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Z.D., Piyamongkol S., Liu D.Y., Khodr H.H., Lu S.L., Hider R.C. Synthesis of 2-amido-3-hydroxypyridin-4(1H)-ones: Novel iron chelators with enhanced pFe3+ values. Bioorg. Med. Chem. 2001;9:563–573. doi: 10.1016/S0968-0896(00)00273-X. [DOI] [PubMed] [Google Scholar]
- 49.Liu Z.D., Kayyali R., Hider R.C., Porter J.B., Theobald A.E. Design, synthesis, and evaluation of novel 2-substituted 3-hydroxypyridin-4-ones: Structure-activity investigation of metalloenzyme inhibition by iron chelators. J. Med. Chem. 2002;45:631–639. doi: 10.1021/jm010817i. [DOI] [PubMed] [Google Scholar]
- 50.Piyamongkol S., Zhou T., Liu Z.D., Khodr H.H., Hider R.C. Design and characterization of novel hexadentate 3-hydroxypyridin-4-one ligands. Tetrahedron Lett. 2005;46:1333–1336. doi: 10.1016/j.tetlet.2004.12.115. [DOI] [Google Scholar]
- 51.Zhou T., Neubert H., Liu D.Y., Liu Z.D., Ma Y.M., Kong X.L., Luo W., Mark S., Hider R.C. Iron binding dendrimers: A novel approach for the treatment of hemochromatosis. J. Med. Chem. 2006;49:4171–4182. doi: 10.1021/jm0600949. [DOI] [PubMed] [Google Scholar]
- 52.Xie Y.-Y., Liu M.-S., Hu P.-P., Kong X.-L., Qiu D.-H., Xu J.-L., Hider R.C., Zhou T. Synthesis, physico-chemical properties, and antimicrobial evaluation of a new series of iron(III) hexadentate chelators. Med. Chem. Res. 2013;22:2351–2359. doi: 10.1007/s00044-012-0229-1. [DOI] [Google Scholar]
- 53.Zhou Y.-J., Liu M.-S., Osamah A.R., Kong X.-L., Alsam S., Battah S., Xie Y.-Y., Hider R.C., Zhou T. Hexadentate 3-hydroxypyridin-4-ones with high iron(III) affinity: Design, synthesis and inhibition on methicillin resistant staphylococcus aureus and pseudomonas strains. Eur. J. Med. Chem. 2015;94:8–21. doi: 10.1016/j.ejmech.2015.02.050. [DOI] [PubMed] [Google Scholar]
- 54.Nunes A., Podinovskaia M., Leite A., Gameiro P., Zhou T., Ma Y., Kong X., Schaible U.E., Hider R.C., Rangel M. Fluorescent 3-hydroxy-4-pyridinone hexadentate iron chelators: Intracellular distribution and the relevance to antimycobacterial properties. J. Biol. Inorg. Chem. 2010;15:861–877. doi: 10.1007/s00775-010-0650-1. [DOI] [PubMed] [Google Scholar]
- 55.Zhou T., Hider R.C., Kong X. Mode of iron(III) chelation by hexadentate hydroxypyridinones. Chem. Commun. 2015;51:5614–5617. doi: 10.1039/C4CC10339D. [DOI] [PubMed] [Google Scholar]
- 56.Zhou T., Liu Z.D., Neubert H., Kong X.L., Ma Y.M., Hider R.C. High affinity iron(III) scavenging by a novel hexadentate 3-hydroxypyridin-4-one-based dendrimer: Synthesis and characterization. Bioorg. Med. Chem. Lett. 2005;15:5007–5011. doi: 10.1016/j.bmcl.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 57.Zhou Y.-J., Kong X.-L., Li J.-P., Ma Y.-M., Hider R.C., Zhou T. Novel 3-hydroxypyridin-4-one hexadentate ligand-based polymeric iron chelator: Synthesis, characterization and antimicrobial evaluation. Med. Chem. Commun. 2015;6:1620–1625. doi: 10.1039/C5MD00264H. [DOI] [Google Scholar]
- 58.Velikyan I. Prospective of 68Ga-radiopharmaceutical development. Theranostics. 2014;4:47–80. doi: 10.7150/thno.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jackson G.E., Byrne M.J. Metal ion speciation in blood plasma: Gallium-67-citrate and MRI contrast agents. J. Nucl. Med. 1996;37:379–386. [PubMed] [Google Scholar]
- 60.Moerlein S.M., Welch M.J. The chemistry of gallium and indium as related to radiopharmaceutical production. Int. J. Nucl. Med. Biol. 1981;8:277–287. doi: 10.1016/0047-0740(81)90034-6. [DOI] [PubMed] [Google Scholar]
- 61.Harris W.R., Pecoraro V.L. Thermodynamic binding constants for gallium transferrin. Biochemistry. 1983;22:292–299. doi: 10.1021/bi00271a010. [DOI] [PubMed] [Google Scholar]
- 62.Santos M.A., Gil M., Gano L., Chaves S. Bifunctional 3-hydroxy-4-pyridinone derivatives as potential pharmaceuticals: Synthesis, complexation with Fe(III), Al(III) and Ga(III) and in vivo evaluation with 67Ga. J. Biol. Inorg. Chem. 2005;10:564–580. doi: 10.1007/s00775-005-0003-7. [DOI] [PubMed] [Google Scholar]
- 63.Eder M., Krivoshein A.V., Backer M., Backer J.M., Haberkorn U., Eisenhut M. ScVEGF-PEG-HBED-CC and scVEGF-PEG-NOTA conjugates: Comparison of easy-to-label recombinant proteins for [68Ga] PET imaging of VEGF receptors in angiogenic vasculature. Nucl. Med. Biol. 2010;37:405–412. doi: 10.1016/j.nucmedbio.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 64.Velikyan I., Maecke H., Langstrom B. Convenient preparation of 68Ga-based PET-radiopharmaceuticals at room temperature. Bioconjug. Chem. 2008;19:569–573. doi: 10.1021/bc700341x. [DOI] [PubMed] [Google Scholar]
- 65.Govindan S.V., Michel R.B., Griffiths G.L., Goldenberg D.M., Mattes M.J. Deferoxamine as a chelator for 67Ga in the preparation of antibody conjugates. Nucl. Med. Biol. 2005;32:513–519. doi: 10.1016/j.nucmedbio.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 66.Cooper M.S., Ma M.T., Sunassee K., Shaw K.P., Williams J.D., Paul R.L., Donnelly P.S., Blower P.J. Comparison of 64Cu-complexing bifunctional chelators for radioimmunoconjugation: Labeling efficiency, specific activity, and in vitro/in vivo stability. Bioconjug. Chem. 2012;23:1029–1039. doi: 10.1021/bc300037w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cooper M.S., Sabbah E., Mather S.J. Conjugation of chelating agents to proteins and radiolabeling with trivalent metallic isotopes. Nat. Protoc. 2006;1:314–317. doi: 10.1038/nprot.2006.49. [DOI] [PubMed] [Google Scholar]
- 68.Chaves S., Marques S.M., Matos A.M.F., Nunes A., Gano L., Tuccinardi T., Martinelli A., Santos M.A. New tris(hydroxypyridinones) as iron and aluminium sequestering agents: Synthesis, complexation and in vivo studies. Chem. Eur. J. 2010;16:10535–10545. doi: 10.1002/chem.201001335. [DOI] [PubMed] [Google Scholar]
- 69.Chaves S., Mendonca A.C., Marques S.M., Prata M.I., Santos A.C., Martins A.F., Geraldes C.F.G.C., Santos M.A. A gallium complex with a new tripodal tris-hydroxypyridinone for potential nuclear diagnostic imaging: Solution and in vivo studies of 67Ga-labeled species. J. Inorg. Biochem. 2011;105:31–38. doi: 10.1016/j.jinorgbio.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 70.Deri M.A., Ponnala S., Kozlowski P., Burton-Pye B.P., Cicek H.T., Hu C., Lewis J.S., Francesconi L.C. p-SCN-Bn-HOPO: A superior bifunctional chelator for 89Zr immunoPET. Bioconjug. Chem. 2015;26:2579–2591. doi: 10.1021/acs.bioconjchem.5b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tinianow J.N., Pandya D.N., Pailloux S.L., Ogasawara A., Vanderbilt A.N., Gill H.S., Williams S.-P., Wadas T.J., Magda D., Marik J. Evaluation of a 3-hydroxypyridin-2-one (2,3-HOPO) based macrocyclic chelator for 89Zr4+ and its use for immunopet imaging of HER2 positive model of ovarian carcinoma in mice. Theranostics. 2016;6:511–521. doi: 10.7150/thno.14261. [DOI] [PMC free article] [PubMed] [Google Scholar]