

Abstract
Natriuretic peptides (NPs) promote diuresis, natriuresis and vasodilation in early chronic heart failure (CHF), countering renin–angiotensin–aldosterone system (RAAS) and sympathetic nervous system (SNS) overstimulation. Despite dramatic increases in circulating NP concentrations as CHF progresses, their effects become blunted. Increases in diuresis, natriuresis, and vasodilation after administration of exogenous atrial (ANP) or brain (BNP) natriuretic peptides are attenuated in patients with advanced CHF compared with controls. Several major factors may account for the reduced effectiveness of the natriuretic peptide system (NPS) in CHF. First, there is reduced availability of active forms of NPs, namely BNP. Second, target organ responsiveness becomes diminished. Third, the counter‐regulatory hormones of the RAAS and SNS, and endothelin‐1 become over‐activated. Therefore, pharmacological approaches to enhance the functional effectiveness of the NPS in CHF have been explored in recent years. In terms of clinical outcomes, studies of synthetic BNP, or of neprilysin inhibitors alone or associated with an angiotensin converting enzyme inhibitor, have been controversial for several reasons. Recently, however, encouraging results have been obtained with the angiotensin receptor neprilysin inhibitor sacubitril/valsartan. The available data show that treatment with sacubitril/valsartan is associated with increased levels of NPs and their intracellular mediator cyclic guanosine monophosphate, suggesting improved functional effectiveness of the NPS, in addition to beneficial effects on mortality and morbidity outcomes. Therefore, combined targeting of the NPS and RAAS with sacubitril/valsartan emerges as the current optimal approach for redressing the neurohormonal imbalance in CHF.
Keywords: Natriuretic peptide, Atrial natriuretic peptide, Brain natriuretic peptide, Heart failure, Neprilysin
Introduction
It has been known for more than 30 years that the natriuretic peptide system (NPS) plays a key compensatory role in chronic heart failure (CHF), counterbalancing overstimulation of the renin–angiotensin–aldosterone system (RAAS) and sympathetic nervous system (SNS). In the early stages of CHF, release of natriuretic peptides (NPs) promotes diuresis, natriuresis and vasodilation, thus reducing both cardiac pre‐load and after‐load. Plasma concentrations of NPs parallel the severity of CHF;1 indeed, NP levels are recommended as a marker for disease severity.2 Despite these profound increases, however, the loss of effectiveness of NPs is a key feature of the CHF syndrome, which significantly impairs the patient's prognosis by aggravating sodium retention, volume overload, vasoconstriction and pressure overload. In this brief review the clinical evidence for reduced effectiveness of the NPS in CHF, as well as the mechanisms involved and the therapeutic implications, will be considered.
The natriuretic peptide system in chronic heart failure
Prolonged RAAS and SNS overstimulation in CHF promotes increased sodium and water retention, with heightened vascular tone, and structural remodelling of the heart, kidney and blood vessels. At the cardiac level, the resulting increases in left ventricular (LV) and atrial pressures stimulate the NPS, augmenting the synthesis and secretion of NPs. In addition to control of extracellular fluid volume and blood pressure, the NPS exerts a broader range of actions, including the control of several metabolic functions.3
There are at least three NP receptors: NPR‐A, NPR‐B, and NPR‐C. Both NPR‐A and NPR‐B are guanylyl cyclase receptors and their activation results in increased cyclic guanosine monophosphate (cGMP) which, in turn, activates downstream kinases.4 A complex array of effects is induced by the interactions of NPs with their receptors, affecting the kidney, blood vessels, heart, endocrine functions, and cell growth and tissue remodelling (Figure 1).3 In the kidney, NP‐related actions have been documented in the glomerulus and all tubular segments.5, 6, 7 Together, these induce afferent arteriole vasodilation with increased glomerular filtration and decreased tubular reabsorption of sodium and water—effects that not only facilitate uropoiesis but also protect the kidney metabolically, promoting oxygen delivery while lowering oxygen consumption. In the vessels, NPs promote vasodilation by enhancing cGMP‐mediated smooth muscle relaxation and increasing capillary permeability. In the heart, all three NPs exert anti‐remodelling effects in the myocardium via local regulation of collagen synthesis and cellular hypertrophy.8 Finally, NPs suppress the RAAS and SNS,7 as well as endothelin and arginine–vasopressin.
Figure 1.
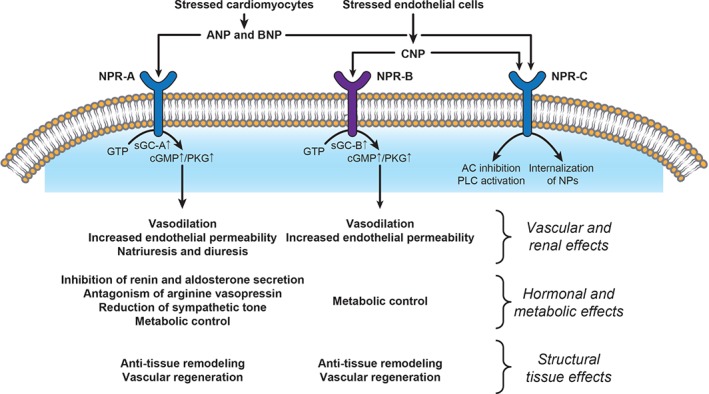
Mechanisms of action and main effects of natriuretic peptides (NPs). AC, adenylyl cyclase; ANP, atrial natriuretic peptide; AVP, arginine–vasopressin; BNP, brain natriuretic peptide; cGMP, cyclic guanosine monophosphate; CNP, C‐type natriuretic peptide; GTP, guanosine triphosphate; NPR, natriuretic peptide receptor; PKG, protein kinase G; PLC, phospholipase C; sGC, soluble guanylate cyclase.
In mild CHF, elevated levels of atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) are believed to play an important role in maintaining sodium balance and systemic haemodynamics. However, as CHF progresses the functional effectiveness of the NPS becomes blunted, impairing the natriuretic, vasodilatory, and hormonal suppressive effects of NPs and contributing to worsening sodium retention and vasoconstriction, with a subsequent detrimental impact on the heart.3 As a consequence, a further increase in cardiac NP production occurs,9 with plasma levels of NPs increasing in direct correlation with the severity of LV dysfunction.10 Deterioration towards more severe CHF is characterized by dominance of the overactivated RAAS and SNS over the counterpoised NPS.
Clinical evidence for a reduced natriuretic peptide system effectiveness in chronic heart failure
Impaired activity of NPs has been demonstrated in both the kidneys and the vasculature, as well as at the level of their endocrine actions, each of which can have profound adverse consequences (Figure 2).
Figure 2.
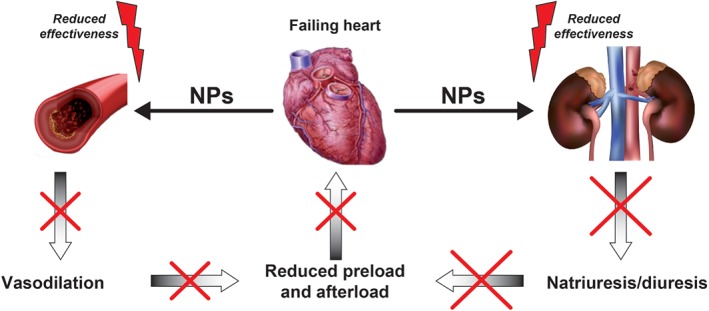
Overview of the main pathophysiological consequences of reduced effectiveness of natriuretic peptides (NPs) in chronic heart failure.
Reduced renal effectiveness of the natriuretic peptide system in chronic heart failure
An attenuated renal response by patients with CHF to synthetic ANP administered at clinically relevant doses has been demonstrated in two small non‐randomized trials.11, 12 In the first of these, by Cody et al.,11 the response to 60‐min infusions of varying doses of ANP vs. placebo was assessed in seven patients with CHF and in seven healthy controls. The control group showed a dose‐dependent increase in urine volume in response to ANP infusion which reached significance at 0.03 µg/kg.min and 0.10 µg/kg.min. In contrast, CHF patients demonstrated only minimal changes even at the highest dose, with all but one CHF patient remaining within the range of urine volume seen during placebo infusion. A similar difference between the response of CHF patients and controls was seen for sodium excretion (Figure 3). Subsequently, Eiskjaer et al. 12 extended this assessment by examining cGMP level and various renal function parameters following a bolus injection of ANP in a cohort of 12 patients with CHF and 13 healthy control subjects. It was found that ANP induced an increase in urine volume and sodium excretion only in the control group, with no effect in the CHF group (Table 1). The rise in plasma and urinary cGMP levels after ANP injection was significantly diminished in CHF patients (Table 1).12 The same researchers later performed another non‐randomized study that compared the effect of BNP infusion in patients with CHF (n = 9) and healthy controls (n = 10).13 As for ANP, the increase in urinary sodium excretion following BNP infusion was significantly lessened in patients with CHF (60% vs. 71%, P < 0.05). In contrast to ANP, however, there was a smaller difference in distal sodium resorption in CHF patients compared with control subjects (−0.8% vs. –3.7%, P < 0.05). In addition, BNP infusion stimulated a similar increase in cGMP in both CHF patients and control subjects.13 Although limited, these studies point to an impaired renal response to NPs, namely to ANP, in CHF.
Figure 3.
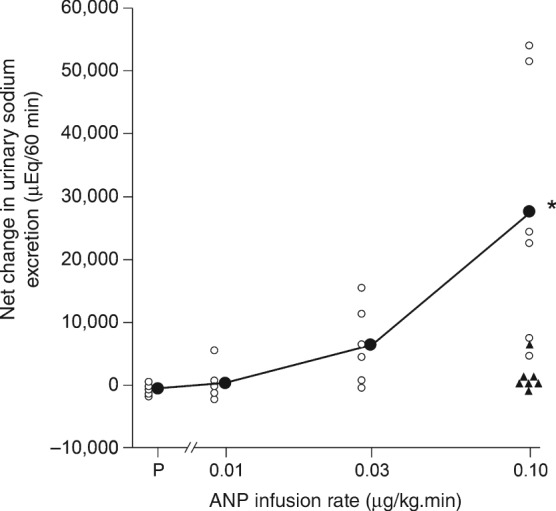
Net change in urinary sodium excretion in response to infusion of atrial natriuretic peptide (ANP) or placebo (P) in seven patients with chronic heart failure (CHF) and seven healthy controls. Open and closed circles indicate individual and mean values, respectively, for controls. Asterisk indicates that the mean value with the highest dose of ANP was higher (P < 0.05) than with P. Triangles indicate individual values for CHF patients with the highest dose of ANP.(Reproduced from Cody et al. 11 with permission.)
Table 1.
Changes in renal parameters after a bolus injection of atrial natriuretic peptide (ANP) (2.0 µg/kg) in 12 patients with chronic heart failure (CHF) and 13 healthy controls
| Patients with CHF (n = 12) | Controls (n = 13) | P‐value vs. placebo | |
|---|---|---|---|
| Diuresis | −3.4 | +62 | <0.01 |
| Natriuresis | +57 | +120 | 0.109 |
| Fractional Na+ excretion | +44 | +99 | 0.121 |
| Proximal fractional Na+ reabsorption | −1.8 | −4.8 | <0.01 |
| Distal fractional Na+ reabsorption | −0.9 | −4.7 | <0.01 |
| Plasma cGMP | +138 | +615 | <0.01 |
| Urinary cGMP | +131 | +729 | <0.01 |
cGMP, cyclic guanosine monophosphate
Results are shown as percentage change from baseline to after ANP injection. Adapted from Eiskjaer et al. 12 with permission.
Reduced vascular effectiveness of natriuretic peptides in chronic heart failure
The first study to examine the local (forearm) vasodilatory response to NPs in CHF, by Nakamura et al.,14 compared the effect of ANP and C‐type natriuretic peptide (CNP) infusions in 11 CHF patients vs. 11 age‐matched healthy control subjects. The vasodilatory response to ANP (maximum change 214% vs. 307%, P < 0.01) and the increase in cGMP (12.0 vs. 31.0 pmol/min.dL tissue, P < 0.02) were lower in CHF patients than in control subjects. The vasodilatory response to CNP was unaffected by the presence of CHF. Nakamura et al. 15 later used the same methodology in a study of ANP and BNP. The forearm vasodilatory response to both peptides was lower (ANP, P < 0.01; BNP, P < 0.05) and the increase in cGMP was smaller (ANP, P < 0.01; BNP, P < 0.05) in CHF patients than in healthy control subjects. Interestingly, in the control group (n = 16), ANP infusion induced greater vasodilation and higher cGMP levels than BNP, whereas in the CHF group (n = 5), both peptides exerted similar effects,15 suggesting that the blunting of NP‐induced vasodilation in CHF is more marked for ANP than for BNP.
The largest study to investigate vasodilatory responses to NPs measured forearm venous and arterial blood flow separately in response to infusions of ANP, BNP, and CNP at successively increasing doses in 53 patients with CHF and 11 control subjects.16 Compared with control subjects, responsiveness of the arterial resistance vasculature to ANP was markedly diminished in CHF patients (Figure 4), whereas the response of venous capacitance vessels was preserved. For BNP and CNP, both the arterial and venous responses were suppressed in CHF patients compared with healthy controls.
Figure 4.
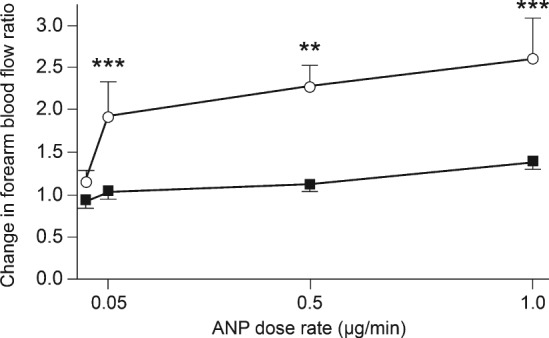
Changes in forearm blood flow response to infusion of atrial natriuretic peptide (ANP) in 53 patients with chronic heart failure (closed squares) and 11 healthy controls (open circles). Changes were assessed in the infused and non‐infused arms and changes in the former were also expressed as a ratio of those in the latter. The first value corresponds to the baseline measurements. **P < 0.01, ***P < 0.001; these refer to within‐group comparison. P < 0.01 refers to area under curve comparison between both groups. (Reproduced from Schmitt et al. 16 with permission.)
Reduced endocrine effectiveness of natriuretic peptides in chronic heart failure
Several studies have assessed the capacity of ANP to inhibit the synthesis and release of renin, aldosterone, noradrenaline, and vasopressin in patients with CHF, administering short‐term infusions (1–2 h) of the peptide to induce a significant increase in its plasma concentration. Cody et al. 11 reported that whereas in 70 healthy controls ANP infusion was followed by significant reductions in plasma renin activity (PRA) and plasma aldosterone concentration, non‐significant reductions in these parameters were observed in 31 CHF patients. In another study performed in 12 CHF patients, ANP infusion was found to reduce plasma aldosterone and noradrenaline concentrations significantly, and PRA concentration non‐significantly.17 Anand et al. 18 did not find effects of ANP infusion on PRA or plasma concentrations of aldosterone, noradrenaline and vasopressin in four patients with CHF. Collectively, these findings suggest an attenuated ability of ANP to counteract the neurohormonal systems activated in CHF. However, additional studies in larger samples of CHF patients are required to definitively confirm that the endocrine effects of the entire NPS are impaired in CHF.
Potential mechanisms of reduced effectiveness of the natriuretic peptide system in chronic heart failure
Various mechanisms have been proposed to account for the diminished effectiveness of the NPS in the setting of CHF (Table 2).3, 19Owing to the differing levels of evidence supporting each proposed mechanism it is not yet possible to ascertain the extent to which each contributes.
Table 2.
Mechanisms of reduced effectiveness of the natriuretic peptide system (NPS) in chronic heart failure
| Decreased availability of biologically active NPs |
|---|
| Reduced production of NPs |
| Increased enzymatic degradation of NPs |
| Increased receptor‐mediated clearance of NPs |
| Diminished target organ responsiveness to NPs |
| Reduced NP receptor expression |
| Desensitization of NP receptors |
| Inhibition of downstream signalling |
| Counter‐regulation of the NPS by antagonistic hormonal systems |
| Over‐activation of the renin‐angiotensin‐aldosterone system |
| Over‐activation of the sympathetic nervous system |
| Over‐activation of endothelin‐1 |
NP, natriuretic peptide.
Decreased availability of biologically active natriuretic peptides
Reduced production of natriuretic peptides
Human BNP is synthesized as a preprohormone of 134 amino acids, containing a signal sequence that is cleaved to yield a 108‐amino acid prohormone. The major circulating form 108‐amino acid precursor, proBNP, is cleaved between amino acids 76 and 77 by the processing enzymes corin and furin to produce the biologically active 32‐amino acid BNP plus the biologically inactive 76‐amino acid N‐terminal peptide [N‐terminal proBNP (NT‐proBNP)]. All three peptides, proBNP, BNP, and NT‐proBNP, are secreted by the heart and circulate in humans together with multiple metabolites of NT‐proBNP and BNP.20 The enzyme dipeptidyl peptidase IV (DPP IV) removes the N‐terminal dipeptide from circulating BNP generating truncated forms that are less potent in terms of biological activity.21
Mass spectroscopy and other sensitive analytical methods have shown that in patients with CHF, mature BNP is only a minor constituent of plasma BNP peptides, with NT‐proBNP comprising a large proportion.22, 23 Commercially available assays do not discriminate between distinct forms of BNP peptides with different levels of biological activity,22 so it is feasible that despite high measured plasma concentrations of these peptides, patients with CHF may be relatively deficient in active BNP. In this regard, certain clinical observations are of interest. First, it has been reported that serum samples from CHF patients show delayed conversion of proBNP to mature BNP, likely because of reduced levels of the soluble form of corin.22 Second, in CHF patients, approximately 70% of circulating proBNP is glycosylated24 and, therefore, resistant to processing by corin25 and/or furin26 to produce NT‐proBNP and BNP. Third, DPP IV activity was found to be increased in the serum of CHF patients.27 Interestingly, inhibition of DPP IV in a porcine model of CHF resulted in improved renal and cardiac function.28
Three forms of ANP can also be detected in human plasma samples: proANP, mid‐regional proANP and the biologically active 28‐amino acid ANP.29 As for BNPs, changes in the cardiomyocyte post‐translational processing of the proANP precursor by corin or in the degradation of the circulating active ANP by DPP IV, may also occur in CHF patients. Thus, combined with the technical limitations of available immunoassay methods, it can be difficult to establish the true plasma concentration of active ANP.
Increased plasma concentrations of both CNP and its precursor NT‐proCNP have been observed in CHF patients compared with controls, with NT‐proCNP levels being 5‐fold and 15‐fold higher than CNP levels in controls and patients, respectively.30, 31
Increased enzymatic degradation of natriuretic peptides
The metallopeptidase neprilysin (NEP) degrades NPs and removes them from the circulation.32 Evidence from rat models of severe CHF has shown a significant increase in renal NEP activity compared with controls, coupled with a threefold increase in renal NEP mRNA expression, suggesting enhanced NP degradation.33 Clinically, Fielitz et al. 34 compared NEP mRNA levels and NEP enzymatic activity in LV samples from patients with CHF and in controls with normal cardiac function (Figure 5). They found both the NEP mRNA content and NEP activity to be approximately fourfold higher in the LV myocardium of the patients with CHF compared with controls. Interestingly, NEP mRNA expression in CHF patients was detected in both cardiomyocytes and non‐cardiomyocytes, and its increase was related to the increase in LV end‐diastolic pressure, suggesting an association with wall stress. On the other hand, several single nucleotide polymorphisms of the NEP gene have been described that alter encoded amino acids.35 In particular, the genetic alteration of just one amino acid, Met73Val, was shown to result in a significant decrease in the quantity and activity of the enzyme.
Figure 5.
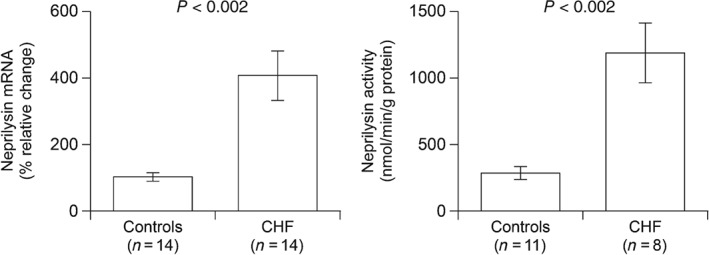
Neprilysin (NEP) messenger RNA (mRNA) and activity in left ventricular myocardial samples from patients with chronic heart failure (CHF) caused by dilated cardiomyopathy and controls. The NEP mRNA was quantified by real‐time polymerase chain reaction assay and NEP activity was measured by an enzymatic assay. Results are expressed as mean ± SEM. (Reproduced from Fielitz et al. 34 with permission.)
Increased receptor‐mediated clearance of natriuretic peptides
Beyond its ability to elicit physiological functions, the NPR‐C acts also as a clearance receptor for NPs.36 The finding by Schmitt et al. 16 that identical ANP infusions caused a significantly greater increase in venous effluent plasma ANP levels in healthy controls compared with CHF patients would be in keeping with the notion of upregulation of NPR‐C in CHF. In accord with this, increased expression of the NPR‐C gene has been observed in failing human hearts and is associated with suppression of cGMP production in response to NPs.36 In the same study, reverse remodelling during LV assist device support led to normalization of NPR‐C mRNA levels and to recovery of guanylyl cyclase activity and of the anti‐remodelling effects of BNP.36
Diminished target organ responsiveness to natriuretic peptides
Reduced natriuretic peptide receptor expression
In a series of 97 patients with mild [New York Heart Association (NYHA) class II] or moderate‐to‐severe (NYHA class III–IV) CHF, Tsutamoto et al. 37 determined plasma levels of ANP and cGMP in the femoral artery and vein. In the cohort with mild CHF, plasma cGMP correlated with ANP level (r = 0.75, P < 0.001) but in the moderate‐to‐severe group, the correlation was poor and cGMP level appeared to plateau despite high circulating levels of ANP (Figure 6). The authors concluded that there may be a downregulation of ANP receptors in the peripheral vascular beds of patients with severe CHF. Other authors have reported a similar marked decline in the ratios of plasma cGMP to plasma BNP in the setting of CHF.38 Singh and colleagues39 used a radiolabelled NP analogue to demonstrate a significant downregulation in the density of NPR‐A in cardiomyocytes, and in endothelial cells and smooth muscle cells of intramyocardial vessels, in patients with CHF.
Figure 6.
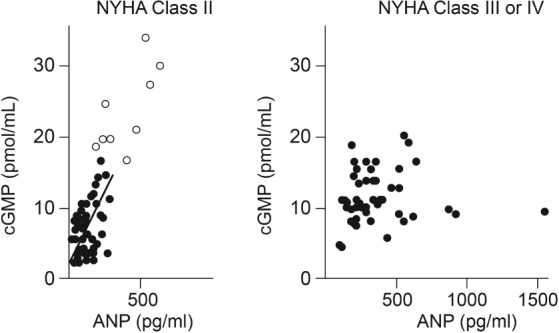
Association of plasma levels of atrial natriuretic peptide (ANP) with cyclic guanosine monophosphate (cGMP) in 43 patients with mild chronic heart failure (closed circles) and acute heart failure (open circles) (left panel), and 45 patients with moderate‐to‐severe CHF (right panel). NYHA, New York Heart Association. (Reproduced from Tsutamoto et al. 37 with permission.)
Desensitization of natriuretic peptide receptors
As suggested by findings from in vitro studies performed with NPR‐A, the diminished effects of the NPS in CHF can also result from NP‐mediated homologous40 and non‐NP‐mediated heterologous41 desensitization of the receptor, resulting in the dephosphorylation of its cytosolic portion. Although the effector molecules involved in homologous desensitization are unclear, the Ca2+‐stimulated phosphatase calcineurin may play an essential role in heterologous desensitization.41
It must also be borne in mind that for guanylyl cyclase receptors, only fully glycosylated and thus dimerized NP receptors are able to crosslink ligand, and bind hormone. In fact, in vitro experiments have shown that glycosylation is crucial for NPR‐A and NPR‐B function.42, 43
Inhibited downstream signalling
It is known that cGMP is degraded by cellular phosphodiesterases (PDEs), such as PDE5. A study in dogs with tachypacing‐induced heart failure has shown that acute administration of a selective PDE5 inhibitor achieved similar haemodynamic responses to treatment with exogenous BNP, and exerted an additive effect to BNP administration.38 In contrast, before CHF induction BNP was associated with the expected cardiovascular effects and PDE5 inhibition had no effect. The reduced ratio of plasma cGMP to plasma BNP seen in the presence of CHF was ameliorated by PDE5 inhibition, but had no effect in non‐failing animals. Enhanced PDE5 activity in CHF may contribute to reduced response to BNP in CHF by impairing its intracellular signal transduction pathways.
Counter‐regulation of the natriuretic peptide system by antagonistic hormonal systems
Over‐activation of the renin–angiotensin–aldosterone system
Although angiotensin II stimulates release of NPs9, 44 chronic RAAS stimulation can overwhelm the effects of the NPS as CHF progresses. Moreover, RAAS activity can impair NP responsiveness. In rats given an infusion of angiotensin II over 12 days, ANP synthesis in the kidneys was increased compared with untreated controls.45 However, the cGMP response was impaired, suggesting reduced response to ANP under conditions of an excess of angiotensin II. Consistent with this, studies in cultured glomerular mesangial cells have demonstrated that the ANP‐induced accumulation of cGMP was significantly inhibited in the presence of angiotensin II.46 Gwathmey et al. 47 have also shown that NEP activity is substantially increased after administration of angiotensin I to isolated proximal tubules from the sheep cortex, an effect that would reduce levels of biologically active NP. Finally, high levels of angiotensin II may also lead to NP receptor downregulation.48
Over‐activation of the sympathetic nervous system
The SNS counteracts the activity of the NPS, particularly at the renal level where both share multiple targets at the arteriolar and tubular level. For example, total NPR density and ANP‐induced cGMP production are higher in denervated kidneys than in non‐denervated kidneys.49 Consistent with this, it has been shown that the blunted natriuretic and diuretic responses to ANP in rats and dogs with CHF is reversed by the alpha 2‐adrenergic agonist clonidine50 and by renal denervation,51 respectively. In contrast, other authors have reported that rats with CHF exhibit blunted natriuretic response to ANP in both intact kidneys and denervated kidneys.52 Thus, the contribution of a rise in sympathetic activity towards renal hyporesponsiveness in CHF is controversial and remains to be evaluated.
Over‐activation of endothelin‐1
Endothelin‐1 also stimulates release of NPs, particularly BNP;44 however, the response to the peptides may be suppressed by high levels of endothelin‐1. A canine model showed that infusion of high‐dose endothelin‐1 inhibited the cardiorenal response to a previous infusion of ANP.53 A randomized, double‐blind study in 142 patients with CHF observed that a 3‐week course of darusentan, an endothelin antagonist, reduced BNP levels but significantly increased the cGMP:BNP ratio, suggesting that endothelin‐1 may reduce the response to BNP.54 Similar findings have been reported from a substudy of a randomized trial of darusentan based on 31 patients with severe CHF.55
Therapeutic implications of the reduced effectiveness of the natriuretic peptide system in chronic heart failure
Recently, several pharmacological approaches have been explored with the aim of enhancing the effectiveness of the NPS in patients with CHF (Table 3).
Table 3.
Summary of the main hormonal effects reported for drugs that aim to increase the effectiveness of the natriuretic peptide system in chronic heart failure
| Effects on plasma NPs | Effects on cGMP | Effects on plasma RAAS | Effects on plasma endothelin‐1 | |
|---|---|---|---|---|
| Nesiritide | ? | ↑Plasma | ↓PRA | ? |
| Candoxatril | ↑ANP, ↑BNP | ↑Plasma and ↑urine | ? | ? |
| Ecadotril | ? | ↑Plasma and ↑urine | No changes in PRA, ANG II or aldosterone | No changes |
| Omapatrilat | ↑ANP, ↑BNP | ↑Plasma | ↓ACE activity | ? |
| Sacubitril/valsartan | ↑ANP, ↑BNP | ↑Plasma and ↑urine | ↑Renin ↑PRA, ↓Aldosterone | ↓ |
ACE, angiotensin‐converting enzyme; ANG II, angiotensin II; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; cGMP, cyclic guanosine monophosphate; NPs, natriuretic peptides; PRA, plasma renin activity; RAAS, renin–angiotensin–aldosterone system.
Administration of exogenous natriuretic peptides
Randomized trials have explored the effects of nesiritide, a recombinant form of human BNP, in CHF. Wang et al. 56 administered intravenous nesiritide (bolus followed by infusion) or placebo for 24 h on consecutive days in a crossover study of 15 patients with CHF and worsening serum creatinine, and found no differences in glomerular filtration rate (GFR) or in effective renal plasma flow, urine output or sodium excretion. A randomized, placebo‐controlled study of 40 patients with CHF and reduced ejection fraction (HFrEF), in which nesiritide was given subcutaneously for 8 weeks, showed a trend to improved estimated GFR compared with placebo.57 Nesiritide administration resulted in an increase in plasma cGMP levels both at the time of the first dose and at the last dose, 8 weeks later, suggesting a maintained response to the peptide.57 Interestingly, a greater and significant decrease in PRA with nesiritide compared with placebo was observed at 8 weeks, suggesting suppression of the RAAS.57 Recently, it was reported that among patients with asymptomatic HFrEF, twice‐daily subcutaneous nesiritide therapy improved the natriuretic and diuretic response to volume expansion at 12 weeks' follow‐up compared with placebo.58 Despite these promising effects, the short bioavailability of nesiritide limits its routine use in clinical practice. Therefore, an NP analogue called M‐ANP has been developed which is less susceptible to degradation by NEP than native NPs. M‐ANP produces greater increases in natriuresis and GFR, and inhibition of aldosterone, compared with endogenous ANP59 and has now entered a clinical development programme for further testing. Another NP analogue that is also less susceptible to degradation by NEP, called cenderitide‐NP (CD‐NP), is currently undergoing phase II clinical trials.
Isolated neprilysin inhibition
Inhibition of NEP impedes the degradation of NPs and increases NP availability. In a large randomized trial, the NEP inhibitor ecadotril demonstrated a dose‐dependent increase in plasma and urinary cGMP levels in patients with CHF NYHA classes II and III compared with placebo, but other neuroendocrine measurements, including PRA, angiotensin II, and endothelin‐1 did not show a meaningful change.60 As a result of its lack of beneficial effects on symptoms, and an unfavourable adverse event profile, the development of ecadotril—like candoxatril, an earlier NEP inhibitor—was discontinued. It appears that NEP inhibition alone is inadequate to counteract the RAAS and other vasoconstrictor hormones in order to restore the neurohormonal balance in CHF.
Simultaneous neprilysin and renin–angiotensin–aldosterone system inhibition
Neprilysin cleaves and degrades several vasoactive peptides in addition to NPs, including angiotensin I and II, bradykinin, endothelin‐1, vasoactive intestinal peptide, and substance P. Thus, although inhibition of NEP increases levels of NPs, the benefits of enhanced NPS activity can be offset by promoting counter‐regulation with angiotensin II and endothelin‐1. Furthermore, as well as inhibiting angiotensin II degradation, NEP inhibition suppresses NEP‐driven cleavage of angiotensin I to generate the biologically active heptapeptide angiotensin (1–7), thus driving the synthetic pathway to produce more angiotensin II.61 These effects have prompted the combination of aminopeptidase inhibiting activities in one molecule, in a drug class known as vasopeptidase inhibitors. The first drug of several drugs with this property was omapatrilat, a single molecule that inhibits both NEP and angiotensin‐converting enzyme (ACE), as well as aminopeptidase P. As expected, omapatrilat was associated with a dose‐dependent increase in plasma ANP, BNP, and cGMP levels, and a decrease in plasma ACE activity in CHF patients treated for 12 weeks.62 Omapatrilat was no more effective than ACE inhibition monotherapy using enalapril63 or lisinopril64 in reducing the risk of death and hospitalization for HF in patients with CHF. However, although statistical significance was not reached, a numerical reduction of these events was observed and the efficacy of this approach was recently suggested by a pooled analysis of these trials and PARADIGM‐HF.65 As omapatrilat inhibits three enzymes that degrade bradykinin (NEP, ACE, and aminopeptidase P), hence increasing bradykinin bioavailability, a high rate of angioedema secondary to increased levels of this molecule occurred in omapatrilat‐treated CHF patients and the drug thus failed to demonstrate an adequate balance between clinical efficacy and safety to justify its approval.
The second drug class with dual action on NEP and the RAAS is the angiotensin receptor‐NEP inhibitors. Sacubitril/valsartan, currently the only available agent of this type, is a salt complex of the angiotensin type 1 receptor blocker valsartan and the NEP inhibitor prodrug sacubitril. In an open‐label, non‐controlled single‐sequence study performed in 30 patients with HFrEF treated with sacubitril/valsartan, significant reductions in plasma NT‐proBNP and increases in plasma cGMP and urinary ANP were observed by days 7 and 21, together with significant increases in plasma renin concentration and PRA, and significant reductions in plasma aldosterone and endothelin–1.66, 67 In a double‐blind randomized trial comparing sacubitril/valsartan with enalapril in 8399 patients with HFrEF, levels of plasma BNP and urinary cGMP were significantly higher during treatment with sacubitril/valsartan than with enalapril.63 The differences between groups were apparent within 4 weeks and were sustained at 8 months.68 Therefore the combination of abrogating angiotensin II‐mediated detrimental cardiovascular actions and potentiation of ANP‐, BNP‐, and CNP‐mediated beneficial cardiovascular actions seems to be involved in the documented ability of sacubitril/valsartan to reduce all‐cause and cardiovascular mortality.69 However, appropriate mechanistic studies are needed for a better insight into the mechanisms of action of sacubitril.70 In favour of this possibility is the observation that plasma levels of NT‐proBNP (a form of BNP that is not degraded by NEP and which reflects haemodynamic cardiac wall stress) is significantly reduced in HFrEF patients treated with sacubitril/valsartan.67 Importantly, non‐serious angioedema was recorded in only 0.5% of HFrEF patients treated with sacubitril/valsartan during a mean period of 27 months, the same incidence as that recorded in enalapril‐treated HFrEF patients.68 The low risk for angioedema under sacubitril/valsartan therapy reflects the fact that it has no effect on ACE or aminopeptidase A, and thus does not significantly increase bradykinin availability.
Conclusions
Reduced effectiveness of the NPS is a central component of the pathophysiology of CHF, aggravating sodium retention, volume overload, vasoconstriction and pressure overload, and impairing patients' prognosis. In CHF, because of the multiple concurrent factors, the kidneys become resistant to the actions of NPs and arterial responsiveness to these peptides is impaired. Increasing the availability of NPs may overcome renal and arterial inefficiency of these peptides but neither exogenous NP administration nor NEP inhibition alone have achieved an adequate clinical response in clinical trials, likely because of overwhelming opposition from the over‐activated RAAS. Therefore, dual therapy inhibiting both the RAAS and NEP seems the most rational approach to restore the neurohormonal balance in CHF, as long as it demonstrates a satisfactory balance between clinical efficacy and safety to justify its implementation. In this conceptual framework, sacubitril/valsartan has emerged as an effective and safe drug for the long‐term treatment of patients with HFrEF.
Acknowledgements
The author acknowledges the contribution of Caroline Dunstall from Novartis who helped in the writing process.
Funding
The author acknowledges the contribution of a freelance medical writer (C. Dunstall), funded by Novartis Pharma AG, Basel, Switzerland.
Conflict of interest: the author has served as an advisor and/or as a speaker for Novartis, Bayer, Merck, Sharp and Dohme, and Abbvie.
References
- 1. Abassi Z, Karram T, Ellaham S, Winaver J, Hoffman A. Implications of the natriuretic peptide system in the pathogenesis of heart failure: diagnostic and therapeutic importance. Pharmacol Ther 2004;102:223–241. [DOI] [PubMed] [Google Scholar]
- 2. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, González‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) . Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016;18:891–975. [DOI] [PubMed] [Google Scholar]
- 3. Volpe M, Carnovali M, Mastromarino V. The natriuretic peptides system in the pathophysiology of heart failure: from molecular basis to treatment. Clin Sci (Lond) 2016;130:57–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levin ER, Gardner DG, Samson WK. Natriuretic peptides. N Engl J Med 1998;339:321–328. [DOI] [PubMed] [Google Scholar]
- 5. Volpe M, Rubattu S, Burnett J Jr. Natriuretic peptides in cardiovascular diseases: current use and perspectives. Eur Heart J 2014;35:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gupta DK, Wang TJ. Natriuretic peptides and cardiometabolic health. Circ J 2015;79:1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kerkelä R, Ulvila J, Magga J. Natriuretic peptides in the regulation of cardiovascular physiology and metabolic events. J Am Heart Assoc 2015;4:e002423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Calvieri C, Rubattu S, Volpe M. Molecular mechanisms underlying cardiac antihypertrophic and antifibrotic effects of natriuretic peptides. J Mol Med (Berl) 2012;90:5–13. [DOI] [PubMed] [Google Scholar]
- 9. Focaccio V, Volpe M, Ambrosio G, Lembo G, Pannain S, Rubattu S, Enea I, Pignalosa S, Chiariello M. Angiotensin II directly stimulates release of atrial natriuretic factor in isolated rabbit hearts. Circulation 1993;87:192–198. [DOI] [PubMed] [Google Scholar]
- 10. Raine AE, Erne P, Bürgisser E, Müller FB, Bolli P, Burkart F, Bühler FR. Atrial natriuretic peptide and atrial pressure in patients with congestive heart failure. N Engl J Med 1986;315:533–537. [DOI] [PubMed] [Google Scholar]
- 11. Cody RJ, Atlas SA, Laragh JH, Kubo SH, Covit AB, Ryman KS, Shaknovich A, Pondolfino K, Clark M, Camargo MJ. Atrial natriuretic factor in normal subjects and heart failure patients. Plasma levels and renal, hormonal, and hemodynamic responses to peptide infusion. J Clin Invest 1986;78:1362–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eiskjaer H, Bagger JP, Danielsen H, Jensen JD, Jespersen B, Thomsen K, Pedersen EB. Attenuated renal excretory response to atrial natriuretic peptide in congestive heart failure in man. Int J Cardiol 1991;33:61–74. [DOI] [PubMed] [Google Scholar]
- 13. Jensen KT, Eiskjaer H, Carstens J, Pedersen EB. Renal effects of brain natriuretic peptide in patients with congestive heart failure. Clin Sci 1999;96:5–15. [PubMed] [Google Scholar]
- 14. Nakamua M, Arakawa N, Yoshida H, Makita S, Hiramori K. Vasodilatory effects of C‐type natriuretic peptide on forearm resistance vessels are distinct from those of atrial natriuretic peptide in chronic heart failure. Circulation 1994;90:1210–1214. [DOI] [PubMed] [Google Scholar]
- 15. Nakamura M, Arakawa N, Yoshida H, Makita S, Niinuma H, Hiramori K. Vasodilatory effects of B‐type natriuretic peptide are impaired in patients with chronic heart failure. Am Heart J 1998;135 : 414–420. [DOI] [PubMed] [Google Scholar]
- 16. Schmitt M, Gunaruwan P, Payne N, Taylor J, Lee L, Broadley AJ, Nightingale AK, Cockcroft JR, Struthers AD, Tyberg JV, Frenneaux MP. Effects of exogenous and endogenous natriuretic peptides on forearm vascular function in chronic heart failure. Arterioscler Thromb Vasc Biol 2004;24:911–917. [DOI] [PubMed] [Google Scholar]
- 17. Molina CR, Fowler MB, McCrory S, Peterson C, Myers BD, Schroeder JS, Murad F. Hemodynamic, renal and endocrine effects of atrial natriuretic peptide infusion in severe heart failure. J Am Coll Cardiol 1988;12:175–186. [DOI] [PubMed] [Google Scholar]
- 18. Anand IS, Kalra GS, Ferrari R, Harris P, Poole‐Wilson PA. Hemodynamic, hormonal, and renal effects of atrial natriuretic peptide in untreated congestive cardiac failure. Am Heart J 1989;118:500–505. [DOI] [PubMed] [Google Scholar]
- 19. Egom EE, Feridooni T, Hotchkiss A, Kruzliak P, Pasumarthi KB. Mechanisms of renal hyporesponsiveness to BNP in heart failure. Can J Physiol Pharmacol 2014;93:399–403. [DOI] [PubMed] [Google Scholar]
- 20. D'Alessandro R, Masarone D, Buono A, Gravino R, Rea A, Salerno G, Golia E, Ammendola E, Del Giorno G, Santangelo L, Russo MG, Calabrò R, Bossone E, Pacileo G, Limongelli G. Natriuretic peptides: molecular biology, pathophysiology and clinical implications for the cardiologist. Future Cardiol 2013;9:519–534. [DOI] [PubMed] [Google Scholar]
- 21. Liang F, O'Rear J, Schellenberger U, Tai L, Lasecki M, Schreiner GF, Apple FS, Maisel AS, Pollitt NS, Protter AA. Evidence for functional heterogeneity of circulating B‐type natriuretic peptide. J Am Coll Cardiol 2007;49:1071–1078. [DOI] [PubMed] [Google Scholar]
- 22. Huntley BK, Sandberg SM, Heublein DM, Sangaralingham SJ, Burnett JC Jr, Ichiki T. Pro‐B‐type natriuretic peptide‐1–108 processing and degradation in human heart failure. Circ Heart Fail 2015;8:89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miller WL, Phelps MA, Wood CM, Schellenberger U, Van Le A, Perichon R, Jaffe AS. Comparison of mass spectrometry and clinical assay measurements of circulating fragments of B‐type natriuretic peptide in patients with chronic heart failure. Circ Heart Fail 2011;4:355–360. [DOI] [PubMed] [Google Scholar]
- 24. Semenov AG, Tamm NN, Seferian KR, Postnikov AB, Karpova NS, Serebryanaya DV, Koshkina EV, Krasnoselsky MI, Katrukha AG. Processing of pro‐B‐type natriuretic peptide: furin and corin as candidate convertases. Clin Chem 2010;56:1166–1176. [DOI] [PubMed] [Google Scholar]
- 25. Dong N, Chen S, Yang J, He L, Liu P, Zheng D, Li L, Zhou Y, Ruan C, Plow E, Wu Q. Plasma soluble corin in patients with heart failure. Circ Heart Fail 2010;3:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vidricaire G, Denault JB, Leduc R. Characterization of a secreted form of human furin endoprotease. Biochem Biophys Res Commun 1993;195:1011–1018. [DOI] [PubMed] [Google Scholar]
- 27. dos Santos L, Salles TA, Arruda‐Junior DF, Campos LC, Pereira AC, Barreto AL, Antonio EL, Mansur AJ, Tucci PJ, Krieger JE, Girardi AC. Circulating dipeptidyl peptidase IV activity correlates with cardiac dysfunction in human and experimental heart failure. Circ Heart Fail 2013;6:1029–1038. [DOI] [PubMed] [Google Scholar]
- 28. Gomez N, Touihri K, Matheeussen V, Mendes Da Costa A, Mahmoudabady M, Mathieu M, Baerts L, Peace A, Lybaert P, Scharpé S, De Meester I, Bartunek J,Vanderheyden M, Mc Entee K. Dipetidyl peptidase IV inhibition improves cardiorenal function in overpacing‐induced heart failure. Eur J Heart Fail 2012;14:14–21. [DOI] [PubMed] [Google Scholar]
- 29. Goetze JP, Hansen LH, Terzic D, Zois NE, Albrethsen J, Timm A, Smith J, Soltysinska E, Lippert SK, Hunter I. Atrial natriuretic peptides in plasma. Clin Chim Acta 2015;443:25–28. [DOI] [PubMed] [Google Scholar]
- 30. Prickett TC, Yandle TG, Nicholls MG, Espiner EA, Richards AM. Identification of amino‐terminal pro‐C‐type natriuretic peptide in human plasma. Biochem Biophys Res Commun 2001;286:513–517. [DOI] [PubMed] [Google Scholar]
- 31. Wright SP, Prickett TC, Doughty RN, Frampton C, Gamble GD, Yandle TG, Sharpe N, Richards M. Amino‐terminal pro‐C‐type natriuretic peptide in heart failure. Hypertension 2004;43:94–100. [DOI] [PubMed] [Google Scholar]
- 32. Potter LR. Natriuretic peptide metabolism, clearance and degradation. FEBS J 2011;278:1808–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Knecht M, Pagel I, Langenickel T, Philipp S, Scheuermann‐Freestone M, Willnow T, Bruemmer D, Graf K, Dietz R, Willenbrock R. Increased expression of renal neutral endopeptidase in severe heart failure. Life Sci 2002;71:2701–2712. [DOI] [PubMed] [Google Scholar]
- 34. Fielitz J, Dendorfer A, Pregla R, Ehler E, Zurbrügg HR, Bartunek J, Hetzer R, Regitz‐Zagrosek V. Neutral endopeptidase is activated in cardiomyocytes in human aortic valve stenosis and heart failure. Circulation 2002;105:286–289. [DOI] [PubMed] [Google Scholar]
- 35. Pereira NL, Aksoy P, Moon I, Peng Y, Redfield MM, Burnett JC Jr, Wieben ED, Yee VC, Weinshilboum RM. Natriuretic peptide pharmacogenetics: membrane metallo‐endopeptidase (MME): common gene sequence variation, functional characterization and degradation. J Mol Cell Cardiol 2010;49:864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuhn M, Voss M, Mitko D, Stypmann J, Schmid C, Kawaguchi N, Grabellus F, Baba HA. Left ventricular assist device support reverses altered cardiac expression and function of natriuretic peptides and receptors in end‐stage heart failure. Cardiovasc Res 2004;64:308–314. [DOI] [PubMed] [Google Scholar]
- 37. Tsutamoto T, Kanamori T, Morigami N, Sugimoto Y, Yamaoka O, Kinoshita M. Possibility of downregulation of atrial natriuretic peptide receptor coupled to guanylate cyclase in peripheral vascular beds of patients with chronic severe heart failure. Circulation 1993;87:70–75. [DOI] [PubMed] [Google Scholar]
- 38. Forfia PR, Lee M, Tunin RS, Mahmud M, Champion HC, Kass DA. Acute phosphodiesterase 5 inhibition mimics hemodynamic effects of B‐type natriuretic peptide and potentiates B‐type natriuretic peptide effects in failing but not normal canine heart. J Am Coll Cardiol 2007;49:1079–1088. [DOI] [PubMed] [Google Scholar]
- 39. Singh G, Kuc RE, Maguire JJ, Fidock M, Davenport AP. Novel snake venom ligand dendroaspis natriuretic peptide is selective for natriuretic peptide receptor‐A in human heart: downregulation of natriuretic peptide receptor‐A in heart failure. Circ Res 2006;99:183–190. [DOI] [PubMed] [Google Scholar]
- 40. Schröter J, Zahedi RP, Hartmann M, Gassner B, Gazinski A, Waschke J, Sickmann A, Kuhn M. Homologous desensitization of guanylyl cyclase A, the receptor for atrial natriuretic peptide, is associated with a complex phosphorylation pattern. FEBS J 2010;277:2440–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fortin Y, De Léan A. Role of cyclic GMP and calcineurin in homologous and heterologous desensitization of natriuretic peptide receptor‐A. Can J Physiol Pharmacol 2006;84:539–546. [DOI] [PubMed] [Google Scholar]
- 42. Koller K, Lipari T, Goeddel DV. Proper glycosylation and phosphorylation of the type A natriuretic peptide receptor are required for hormone‐stimulated guanylyl cyelase activity. J Biol Chem 1993;268:5997–6003. [PubMed] [Google Scholar]
- 43. Fenrick R, McNicoll N, De Léan A. Glycosylation is critical for natriuretic peptide receptor‐B function. Mol Cell Biochem 1996;165:103–109. [DOI] [PubMed] [Google Scholar]
- 44. Hensen J, Abraham WT, Lesnefsky EJ, Levenson B, Groves BM, Schröder K, Schrier RW, Dürr J. Atrial natriuretic peptide kinetic studies in patients with cardiac dysfunction. Kidney Int 1992;41:1333–1339. [DOI] [PubMed] [Google Scholar]
- 45. Bae EH, Ma SK, Lee J, Kim SW. Altered regulation of renal nitric oxide and atrial natriuretic peptide systems in angiotensin II‐induced hypertension. Regul Pept 2011;170:31–37. [DOI] [PubMed] [Google Scholar]
- 46. Haneda M, Kikkawa R, Maeda S, Togawa M, Koya D, Horide N, Kajiwara N, Shigeta Y. Dual mechanism of angiotensin II inhibits ANP‐induced mesangial cGMP accumulation. Kidney Int 1991;40:188–194. [DOI] [PubMed] [Google Scholar]
- 47. Gwathmey TM, Westwood BM, Pirro NT, Tang L, Rose JC, Diz DI, Chappell MC. Nuclear angiotensin‐(1–7) receptor is functionally coupled to the formation of nitric oxide. Am J Physiol Renal Physiol 2010;299:F983–F990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Charloux A, Piquard F, Doutreleau S, Brandenberger G, Geny B. Mechanisms of renal hyporesponsiveness to ANP in heart failure. Eur J Clin Invest 2003;33:769–778. [DOI] [PubMed] [Google Scholar]
- 49. Awazu M, Kon V, Harris RC, Imada T, Inagami T, Ichikawa I. Renal sympathetic nerves modulate glomerular ANP receptors and filtration. Am J Physiol 1991;261:F29–F35. [DOI] [PubMed] [Google Scholar]
- 50. Feng QP, Hedner T, Hedner J, Pettersson A. Blunted renal response to atrial natriuretic peptide in congestive heart failure rats is reversed by the alpha 2‐adrenergic agonist clonidine. J Cardiovasc Pharmacol 1990;16:776–782. [DOI] [PubMed] [Google Scholar]
- 51. Koepke JP, DiBona GF. Blunted natriuresis to atrial natriuretic peptide in chronic sodium‐retaining disorders. Am J Physiol 1987;252:F865–F871. [DOI] [PubMed] [Google Scholar]
- 52. Patel KP, Zhang PL, Carmines PK. Neural influences on renal responses to acute volume expansion in rats with heart failure. Am J Physiol 1996;271:H1441–H1448. [DOI] [PubMed] [Google Scholar]
- 53. Ota K, Kimura T, Shoji M, Inoue M, Sato K, Ohta M, Yamamoto T, Tsunoda K, Abe K, Yoshinaga K. Interaction of ANP with endothelin on cardiovascular, renal, and endocrine function. Am J Physiol 1992;262:E135–E141. [DOI] [PubMed] [Google Scholar]
- 54. Philipp S, Monti J, Pagel I, Langenickel T, Notter T, Ruschitzka F, Lüscher T, Dietz R, Willenbrock R. Treatment with darusentan over 21 days improved cGMP generation in patients with chronic heart failure. Clin Sci (Lond) 2002;103(Suppl 48):249S–253S. [DOI] [PubMed] [Google Scholar]
- 55. Bergler‐Klein J, Pacher R, Berger R, Bojic A, Stanek B. Neurohormonal and hemodynamic effects of the selective endothelin antagonist darusentan in advanced chronic heart failure. J Heart Lung Transplant 2004;23:20–27. [DOI] [PubMed] [Google Scholar]
- 56. Wang DJ, Dowling TC, Meadows D, Ayala T, Marshall J, Minshall S, Greenberg N, Thattassery E, Fisher ML, Rao K, Gottlieb SS. Nesiritide does not improve renal function in patients with chronic heart failure and worsening serum creatinine. Circulation 2004;110:1620–1625. [DOI] [PubMed] [Google Scholar]
- 57. Chen HH, Glockner JF, Schirger JA, Cataliotti A, Redfield MM, Burnett JC Jr. Novel protein therapeutics for systolic heart failure: chronic subcutaneous B‐type natriuretic peptide. J Am Coll Cardiol 2012;60:2305–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McKie PM, Schirger, JA , Benike SL, Harstad LK, Slusser JP, Hodge DO, Redfield MM, Burnett JC Jr, Chen HH. Chronic subcutaneous brain natriuretic peptide therapy in asymptomatic systolic heart failure. Eur J Heart Fail 2016;18:433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McKie PM, Cataliotti A, Huntley BK, Martin FL, Olson TM, Burnett JC Jr. A human atrial natriuretic peptide gene mutation reveals a novel peptide with enhanced blood pressure‐lowering, renal‐enhancing, and aldosterone‐suppressing actions. J Am Coll Cardiol 2009;54:1024–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cleland JG, Swedberg K. Lack of efficacy of neutral endopeptidase inhibitor ecadotril in heart failure. The International Ecadotril Multi‐centre Dose‐ranging Study Investigators. Lancet 1998;351:1657–1658. [DOI] [PubMed] [Google Scholar]
- 61. Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin‐converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J 2004;383:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McClean DR, Ikram H, Mehta S, Heywood JT, Rousseau MF, Niederman AL, Sequeira RF, Fleck E, Singh SN, Coutu B, Hanrath P, Komajda M, Bryson CC,Qian C, Hanyok JJ; Group Omapatrilat Hemodynamic Study. Vasopeptidase inhibition with omapatrilat in chronic heart failure: acute and long‐term hemodynamic and neurohumoral effects. J Am Coll Cardiol 2002;39:2034–2041. [DOI] [PubMed] [Google Scholar]
- 63. Packer M, Califf RM, Konstam MA, Krum H, McMurray JJ, Rouleau JL, Swedberg K. Comparison of omapatrilat and enalapril in patients with chronic heart failure: the Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE). Circulation 2002;106:920–926. [DOI] [PubMed] [Google Scholar]
- 64. Rouleau JL, Pfeffer MA, Stewart DJ, Isaac D, Sestier F, Kerut EK, Porter CB, Proulx G, Qian C, Block AJ. Comparison of vasopeptidase inhibitor, omapatrilat, and lisinopril on exercise tolerance and morbidity in patients with heart failure: IMPRESS randomised trial. Lancet 2000;356:615–620. [DOI] [PubMed] [Google Scholar]
- 65. Solomon SD, Claggett B, McMurray JJ, Hernandez AF, Fonarow GC. Combined neprilysin and renin–angiotensin system inhibition in heart failure with reduced ejection fraction: a meta‐analysis. Eur J Heart Fail 2016;18:1238–1243. [DOI] [PubMed] [Google Scholar]
- 66. Kobalava C, Kotovskaya Y, Averkov O, Pavlikova E, Moiseev V, Albrecht D, Chandra P, Ayalasomayajula S, Prescott MF, Pal P, Langenickel TH, Jordaan P, Rajman I. Pharmacodynamic and pharmacokinetic profiles of sacubitril/valsartan (LCZ696) in patients with heart failure and reduced ejection fraction. Cardiovasc Ther 2016;34:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kobalava Z, Averkov O, Meray I, Alexandriya L, Moiseev V, Albrecht D, Feng A, Cahndra P, Jordaan PJ. Natriuretic peptide degradation inhibition in the presence of angiotensin receptor blockade following short‐term treatment with LCZ696 in heart failure patients: effect on ANP, BNP, NT‐proBNP and cGMP. Eur Heart J 2011;32(Suppl 1):784–785.21169282 [Google Scholar]
- 68. Packer M, McMurray JJ, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile M, Andersen K, Arango JL, Arnold JM, Bělohlávek J, Böhm M, Boytsov S, Burgess LJ, Cabrera W, Calvo C, Chen CH, Dukat A, Duarte YC, Erglis A, Fu M, Gomez E, Gonzàlez‐Medina A, Hagège AA, Huang J, Katova T, Kiatchoosakun S, Kim KS, Kozan Ö, Llamas EB, Martinez F, Merkely B, Mendoza I, Mosterd A, Negrusz‐Kawecka M, Peuhkurinen K,Ramires FJ, Refsgaard J, Rosenthal A, Senni M, Sibulo AS Jr, Silva‐Cardoso J, Squire IB, Starling RC, Teerlink JR, Vanhaecke J, Vinereanu D, Wong RC; PARADIGM‐HF Investigators and Coordinators . Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation 2015;131:54–61 [DOI] [PubMed] [Google Scholar]
- 69. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR; Investigators PARADIGM‐HF and Committees. Angio‐tensin neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014;371:993–1004. [DOI] [PubMed] [Google Scholar]
- 70. McMurray JJ. Neprilysin inhibition to treat heart failure: a tale of science, serendipity, and second chances. Eur J Heart Fail 2015;17:242–247. [DOI] [PubMed] [Google Scholar]