

Abstract
Related living kidney donors (LKDs) are at higher risk of end‐stage renal disease (ESRD) compared with unrelated LKDs. A genetic panel was developed to screen 115 genes associated with renal diseases. We used this panel to screen six negative controls, four transplant candidates with presumed genetic renal disease and six related LKDs. After removing common variants, pathogenicity was predicted using six algorithms to score genetic variants based on conservation and function. All variants were evaluated in the context of patient phenotype and clinical data. We identified causal variants in three of the four transplant candidates. Two patients with a family history of autosomal dominant polycystic kidney disease segregated variants in PKD1. These findings excluded genetic risk in three of four relatives accepted as potential LKDs. A third patient with an atypical history for Alport syndrome had a splice site mutation in COL4A5. This pathogenic variant was excluded in a sibling accepted as an LKD. In another patient with a strong family history of ESRD, a negative genetic screen combined with negative comparative genomic hybridization in the recipient facilitated counseling of the related donor. This genetic renal disease panel will allow rapid, efficient and cost‐effective evaluation of related LKDs.
Keywords: clinical research/practice, translational research/science, genetics, kidney transplantation/nephrology, diagnostic techniques and imaging, donors and donation: living, genetics, clinical decision‐making, kidney transplantation: living donor
Short abstract
A pilot study validates the benefit of screening living donors at risk for genetic kidney disease with a comprehensive targeted multigene panel to exclude genetic disease and to facilitate risk assessment and counseling.
Abbreviations
- ADPKD
autosomal dominant polycystic kidney disease
- CAKUT
congenital anomaly of the kidney and urinary tract
- CKD
chronic kidney disease
- ESRD
end‐stage renal disease
- FSGS
focal segmental glomerulosclerosis
- HNF1B
hepatocyte nuclear factor 1β
- LKD
living kidney donor
- MAF
minor allele frequency
- MPS
massively parallel sequencing
- MRI
magnetic resonance imaging
- NGS
next‐generation sequencing
- PCR
polymerase chain reaction
- VUS
variant of unknown significance
- WES
whole‐exome sequencing
Introduction
Kidney transplantation is superior to long‐term dialysis for the management of end‐stage renal disease (ESRD) because it provides greater long‐term survival and better quality of life. Nevertheless, there is an ever‐increasing gap between the need for transplantation and the availability of donor kidneys, with >120 000 patients currently on the deceased donor waitlist in the United States alone. This has resulted in an increasing push to encourage living donation, and today there are almost as many living donors as deceased donors annually in the United States 1. Living kidney donor (LKD) transplants, for those fortunate to receive one, bypass the long waiting time, reduce the likelihood of death while waiting and provide better long‐term allograft and recipient survival compared with deceased donor kidneys 2, 3. In some parts of the world, LKDs are the principal or only source of transplanted organs, and where long‐term dialysis is prohibitively expensive or unavailable, LKD transplants provide the only available therapy for ESRD.
Living donor nephrectomy is generally considered acceptable medical practice, even though there are real risks for the donor, including death, serious injury and failure of the remaining kidney. Recent retrospective studies examining long‐term outcomes of living donation compared with matched nondonor cohorts reported an increased 15‐year and lifetime risk of ESRD for LKDs 4, 5. Although the absolute risk is arguably small, the relative risk is 30 per 10 000 over 15 years and 90 per 10 000 over a lifetime compared with four per 10 000 and 14 per 10 000 in matched controls. Within subpopulations, black men have a 15‐year risk of 90 per 10 000 compared with just nine per 10 000 for white women 4. Although not statistically significant, there is a twofold increased risk of ESRD among biologically related LKDs compared with unrelated LKDs 4. The increased risk may reflect shared inheritance of genetic variants that are deleterious or a common environmental exposure that increases susceptibility to kidney disease.
In the United States, 40% of all LKDs are biologically related to their recipients 1. Many are siblings or adult children of patients with ESRD and are in their third and fourth decades of life, making it difficult to predict future risk of kidney disease. In addition, to guide focused genetic testing of related family members for a specific inherited disease, the transplant recipient's cause of ESRD must be known. Together, diabetes and hypertension are the two most important reported causes of ESRD and account for 60% of the waitlist 1, 6. Most patients with diabetes and/or hypertension and chronic kidney disease (CKD) do not receive a kidney biopsy to verify the diagnosis, and recent studies estimated that as many as 35% of patients with presumed diabetic or hypertensive nephropathy may actually have an alternative diagnosis 7, 8, 9.
Traditionally, establishing and/or confirming the diagnosis of a presumed genetic disease has required Sanger sequencing of the suspected gene for pathogenic variants 10. When candidate genes are large, like COL4A5, sequencing is costly and time consuming. When the disease is heterogeneous, like focal segmental glomerulosclerosis (FSGS), serial gene‐by‐gene screening approaches are inefficient and impractical. These constraints can be largely overcome by using high‐throughput approaches to DNA sequencing (i.e. next‐generation sequencing [NGS] or massively parallel sequencing [MPS]) to sequence a large number of genes simultaneously. Targeted NGS panels have been developed to evaluate patients with a single phenotype, such as steroid‐resistant nephrotic syndrome, FSGS and some ciliopathies 11, 12, 13, 14.
We developed a targeted renal panel that includes 115 genes implicated in a variety of kidney diseases to facilitate a diagnosis in patients with suspected genetic renal disease. We validated this panel for the evaluation of selected LKDs in whom the related transplant recipient's phenotype raised suspicion of or clearly indicated an inherited renal disease. We reported our findings from a pilot study of six controls, four transplant candidates and their six related donors.
Methods
Patient selection
Renal transplant candidates referred to the Organ Transplant Center at the University of Iowa were recruited to the study if they had a known or suspected genetic renal disease and had an asymptomatic younger biological relative who volunteered to be an LKD. Clinical and laboratory data were obtained from the medical record or from patient interviews. Control samples were unrelated persons with no medical or familial history of renal disease. The study was approved by the institutional review board (IRB no. 201301818) for human subject research.
Targeted gene panel
A set of 115 genes implicated in a variety of genetic renal diseases was assembled by enumerating renal phenotypes (e.g. ciliopathy, FSGS, and congenital anomaly of the kidney and urinary tract [CAKUT]) and then assembling a list of known causal genes by literature review. Genes that are implicated in the development of atypical hemolytic‐uremic syndrome and other complement‐mediated glomerular diseases were excluded from this panel. Targeted genomic enrichment and MPS of these 115 genes (hereafter referred to as KidneySeq) was completed as described (genes included in this panel are shown in Tables 1 and S3). Genomic DNA was assessed for quality by gel electrophoresis and spectrophotometry (260/280 ratio of 1.8–2.2; Nanodrop 1000; Thermo Fisher Scientific, Waltham, MA) and quantity by fluorometry (Qubit 2.0 fluorometer; Life Technologies, Carlsbad, CA). Libraries were prepared using a modification of the solution‐based Agilent SureSelect target enrichment system (Agilent Technologies, Santa Clara, CA) using liquid‐handling automation equipment (Perkin Elmer, Waltham, MA). In brief, 3 μg of genomic DNA was randomly fragmented to an average size of 250 bp (Covaris Acoustic Solubilizer; Covaris Inc., Woburn, MA), fragment ends were repaired, A‐tails were added, and sequencing adaptors were ligated before the first amplification. Solid‐phase reverse immobilization purifications were performed between each enzymatic reaction. Hybridization and capture with RNA baits were followed by a second amplification before pooling for sequencing. Minimal amplification was used, typically six cycles for the prehybridization polymerase chain reaction (PCR) and 14 cycles for the posthybridization PCR, using Agilent Herculase II Fusion DNA Polymerase (Agilent Technologies). All samples were bar coded and multiplexed before sequencing on an Illumina MiSeq in pools of five (Illumina Inc, San Diego, CA; performance metrics are shown in Table S1).
Table 1.
Genes implicated in genetic renal diseases and screened by targeted genomic enrichment and massively parallel sequencing
| Gene | Accession number | Locus/alternative name | Exon count |
|---|---|---|---|
| ACTN4 | NM_004924 | 21 | |
| AE1 | NM_000342 | SLC4A1 | 20 |
| AGTR2 | NM_000686 | 3 | |
| AGXT | NM_000030 | 11 | |
| AHI1 | NM_001134830 | JBTS3 | 27 |
| ALMS1 | NM_015120 | 23 | |
| APOL1 | NM_001136540 | FSGS4 | 6 |
| APRT | NM_000485 | 5 | |
| AQP2 | NM_000486 | 4 | |
| ARL13B | NM_001174150 | JBTS8 | 10 |
| ARL6 | NM_001278293 | BBS3 | 8 |
| ATP6V0A4 | NM_020632 | ATP6N1B | 22 |
| AVPR2 | NM_000054 | 3 | |
| BBS1 | NM_024649 | 17 | |
| BBS2 | NM_031885 | 17 | |
| BBS4 | NM_001252678 | 15 | |
| BBS5 | NM_152384 | 12 | |
| BBS7 | NM_018190 | 18 | |
| BMP4 | NM_001202 | 4 | |
| BSND | NM_057176 | 4 | |
| CaSR | NM_000388 | 7 | |
| CC2D2A | NM_001080522 | JBTS9 | 38 |
| CD2AP | NM_012120 | 18 | |
| CEP290 | NM_025114 | JBTS5, MKS4, NPHP6 | 54 |
| CLCN5 | NM_000084 | CLC5 | 12 |
| CLCNKA | NM_004070 | 20 | |
| CLCNKB | NM_000085 | 20 | |
| CLDN16 | NM_006580 | HOMG3 | 5 |
| CLDN19 | NM_001123395 | HOMG5 | 4 |
| CNNM2 | NM_017649 | HOMG6 | 8 |
| COL4A1 | NM_001845 | 52 | |
| COL4A3 | NM_000091 | 52 | |
| COL4A4 | NM_000092 | 48 | |
| COL4A5 | NM_000495 | 51 | |
| COQ2 | NM_015697 | 7 | |
| CREBBP | NM_001079846 | 30 | |
| CTNS | NM_001031681 | 13 | |
| CUL3 | NM_001257197 | 15 | |
| DHCR7 | NM_001163817 | 9 | |
| EGF | NM_001178130 | HOMG4 | 23 |
| EYA1 | NM_000503 | 18 | |
| FGF23 | NM_020638 | 3 | |
| FN1 | NM_002026 | 46 | |
| FRAS1 | NM_001166133 | 42 | |
| FREM2 | NM_207361 | 24 | |
| GATA3 | NM_001002295 | 6 | |
| GLA | NM_000169 | 7 | |
| GLI3 | NM_000168 | 15 | |
| GLIS2 | NM_032575 | NPHP7 | 6 |
| GPC3 | NM_001164617 | 9 | |
| GRHPR | NM_012203 | 9 | |
| HNF1B | NM_000458 | 9 | |
| HOGA1 | NM_138413 | DHDPSL | 7 |
| IFT80 | NM_001190241 | 21 | |
| INF2 | NM_001031714 | FSGS5 | 22 |
| INPP5E | NM_019892 | JBTS1 | 10 |
| INVS | NM_014425 | NPHP2 | 17 |
| IQCB1 | NM_001023570 | NPHP5 | 15 |
| KAL1 | NM_000216 | 14 | |
| KCNJ1 | NM_153766 | ROMK1 | 3 |
| KLHL3 | NM_001257194 | 15 | |
| LAMB2 | NM_002292 | 32 | |
| LMX1B | NM_001174146 | 8 | |
| MKKS | NM_170784 | BBS6 | 6 |
| MKS1 | NM_001165927 | 18 | |
| MYH9 | NM_002473 | 41 | |
| NEK8 | NM_178170 | NPHP9 | 15 |
| NLRP3 | NM_001079821 | 11 | |
| NPHP1 | NM_000272 | JBTS4 | 20 |
| NPHP3 | NM_153240 | 27 | |
| NPHP4 | NM_001291593 | 27 | |
| NPHS1 | NM_004646 | 29 | |
| NPHS2 | NM_001297575 | 7 | |
| NR3C2 | NM_000901 | 9 | |
| OCRL1 | NM_000276 | 24 | |
| OFD1 | NM_003611 | JBTS10 | 23 |
| PAX2 | NM_000278 | 10 | |
| PHEX | NM_000444 | 22 | |
| PKD1 | NM_000296 | ADPKD‐1 | 46 |
| PKD2 | NM_000297 | ADPKD‐2 | 15 |
| PKHD1 | NM_138694 | 67 | |
| PLCE1 | NM_001165979 | NPHS3 | 32 |
| REN | NM_00537 | 10 | |
| RET | NM_020630 | 19 | |
| RPGRIP1L | NM_001127897 | JBTS7, NPHP8, MKS5 | 25 |
| SALL1 | NM_001127892 | 3 | |
| SALL4 | NM_020436 | 4 | |
| SCNN1A | NM_001038 | 13 | |
| SCNN1B | NM_000336 | 13 | |
| SCNN1G | NM_001039 | 13 | |
| SIX1 | NM_005982 | 2 | |
| SIX2 | NM_016932 | 2 | |
| SIX5 | NM_175875 | 3 | |
| SLC12A1 | NM_000338 | NKCC2 | 27 |
| SLC12A3 | NM_000339 | NCCT | 26 |
| SLC26A4 | NM_000441 | 21 | |
| SLC34A1 | NM_001167579 | NPT2a | 9 |
| SLC34A3 | NM_001177316 | NPT2C | 13 |
| SLC3A1 | NM_000341 | 10 | |
| SLC4A4 | NM_001098484 | 26 | |
| SLC7A9 | NM_001126335 | 13 | |
| SMARCAL1 | NM_001127207 | 18 | |
| TCTN1 | NM_001082537 | JBTS13 | 15 |
| TMEM216 | NM_001173990 | JBTS2, MKS2 | 5 |
| TMEM237 | NM_001044385 | JBTS14 | 12 |
| TMEM67 | NM_001142301 | JBTS6, MKS3, NPHP11 | 29 |
| TRPC6 | NM_004621 | FSGS2 | 13 |
| TTC21B | NM_024753 | JBTS11 | 29 |
| TTC8 | NM_144596 | BBS8 | 15 |
| UMOD | NM_001008389 | 11 | |
| UPK3A | NM_001167574 | 4 | |
| WNK1 | NM_001184985 | 28 | |
| WNK4 | NM_032387 | 19 | |
| WNT4 | NM_030761 | 5 | |
| WT1 | NM_000378 | 9 |
Bioinformatic analysis
Data storage and analysis were performed on dedicated computing resources maintained by the Iowa Institute of Human Genetics at the University of Iowa. Sequencing data were archived as fastq files on a secured storage server and then analyzed using locally implemented open source Galaxy software on a high‐performance computing cluster 15. The workflow for variant calling integrated publicly available tools: Reads were mapped using Burrows–Wheeler alignment (BWA–MEM) against human reference genome GRCh37/hg19; duplicates were removed by Picard; realignment, calibration and variant calling were performed with the Genome Analysis Toolkit; and variant annotation was performed with a CLCG annotation and reporting tool developed by our bioinformatics team 16, 17, 18.
Variant prioritization and Sanger validation
The total number of reads per sample varied as a function of the number of samples per run and DNA input per sample. Low‐quality variants (depth <10 or QD <5) were filtered out by quality control. Common variants with minor allele frequency (MAF) >1% in any population were excluded (based on the National Heart, Lung, and Blood Institute GO Exome Sequencing Project [http://evs.gs.washington.edu], the 1000 Genomes Project [http://www.1000genomes.org] and the Exome Aggregation Consortium [http://exac.broadinstitute.org]) unless the variant was a known risk allele. Variants also were filtered based on predicted effect, retaining nonsynonymous single‐nucleotide variants, canonical splicing changes and indels, which were prioritized based on MAF, nucleotide conservation, reported functional and expressive impact, and phenotype correlation. Reference databases that were routinely queried included the Human Gene Mutation Database, ClinVar and our in‐house renal variant database. GERP++ 19, PhyloP 20, MutationTaster 21, PolyPhen‐2 22, SIFT 23 and likelihood ratio tests 24 were used to calculate variant‐specific pathogenicity scores based on the sum of tools predicting a given variant to be deleterious. All reported variants were Sanger validated, as were specific portions of the KidneySeq panel not amenable to targeted genomic enrichment (Table S2).
Variant interpretation
To provide a clinically relevant report, a multidisciplinary board (KidneySeq group meeting) reviewed all genetic data in the context of the available clinical data (Table 3) (case descriptions follow). Standards developed by the American College of Medical Genetics were used to assign variants to one of five categories: pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign and benign 25. Variants with MAF >1% known to be unrelated to disease were classified as “benign.” Variants with an allele frequency greater than expected for the disease and for which computational evidence suggested low likelihood of pathogenicity were classified as “likely benign.” Ultrarare variants reported as pathogenic in the literature and with supporting functional evidence were classified as “pathogenic.” Null variants, such as partial or whole gene deletions, frame‐shift mutations, initiation codon mutations, splice‐site mutations (+1 or −1 or −2) and truncation mutations (if the stop codon was not in the terminal exon) that segregated with disease were classified as “pathogenic” when loss of function was a known mechanism of disease. Novel or rare missense variants that have an unknown impact on protein function were classified as either “likely pathogenic” or “VUS,” a distinction that reflected two considerations: likely pathogenic variants were also (i) missense variants with pathogenicity scores ≥5 (based on GERP++, PhyloP, MutationTaster, PolyPhen‐2, SIFT and LRT), ultrarare (MAF <0.00001%) and found in disease‐related functional domains or loci or (ii) novel and caused loss of function. Based on genotypic findings and the clinical phenotype, additional testing was occasionally recommended.
Table 3.
Transplant candidates tested with KidneySeq
| Case | Clinical diagnosis | Result | Genotype | Genetic diagnosis |
|---|---|---|---|---|
| 1 | ADPKD | Positive | PKD1—NM_000296:c.7866C>G, p.Tyr2622Stop | ADPKD |
| 2 | Alport syndrome/FSGS | Positive | COL4A5—NM_000495:c.3604+1G>A | Alport syndrome |
| 3 | ADPKD | Positive | PKD1—NM_000296:c.11488_11489insGCGACC | ADPKD |
| 4 | CKD | No finding |
This table shows clinical diagnosis and genotype findings for the four transplant candidates tested in this pilot study. ADPKD, autosomal dominant polycystic kidney disease; CKD, chronic kidney disease; FSGS, focal segmental glomerulosclerosis.
Results
Massively parallel sequencing
The targeted regions of 115 candidate genes on KidneySeq covered ≈0.58 Mb of the genome (Table 1). On average, 4.4 million sequence reads per sample were generated for a mean depth of coverage of 586× with >99% of targeted regions covered at ≥10× (Table S1). Approximately 500 variants were detected per sample. These variants were annotated and filtered to identify high‐quality rare and novel variants (Table 2). For each sample, we also identified regions with <10× coverage if they were associated with the disease phenotype (Table S2).
Table 2.
Variant filtering for the samples and controls included in this study
| Case 1 | Case 2 | Case 3 | Case 4 | Control 1 | Control 2 | Control 3 | Control 4 | Control 5 | Control 6 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Total number of variants | 421 | 546 | 471 | 515 | 561 | 566 | 509 | 523 | 523 | 466 |
| Quality filter (Q_VAR >50, QD >5 and observed % >30) | 385 | 522 | 433 | 489 | 527 | 532 | 490 | 499 | 500 | 445 |
| Rarity filter MAF <1% | 8 | 30 | 11 | 14 | 44 | 19 | 42 | 23 | 12 | 16 |
| Functional filters (exonic, nonsynonymous, splice) | 2 | 7 | 4 | 5 | 5 | 5 | 6 | 5 | 5 | 5 |
Q_VAR, quality of the variant (quality of the identification of the nucleotide generated by automated DNA sequencing); QD, Phred‐like quality score divided by depth; MAF, minor allele frequency.
Sanger sequencing
For confirmation purposes, exons carrying a variant determined to be pathogenic were Sanger sequenced (Table 3). Primers for PCR and for sequencing were designed using Primer 3 and are available upon request 26. In addition, the duplicated regions of the PKD1 gene (exons 1–34) were Sanger sequenced using published primers in those patients with suspected polycystic kidney disease 27.
Patients and KidneySeq multidisciplinary group meetings
Four transplant candidates with their six related LKDs participated in this study. The cohort included two patients with autosomal dominant polycystic kidney disease (ADPKD), one patient with suspected Alport syndrome and one patient with presumed hypertensive nephropathy who had a sibling with ESRD, raising suspicion of a genetically undefined inherited kidney disease (Figure 1). All patients and donors were white; the patients ranged in age from 40 to 63 years, and the donor candidates ranged in age from 20 to 36 years.
Figure 1.
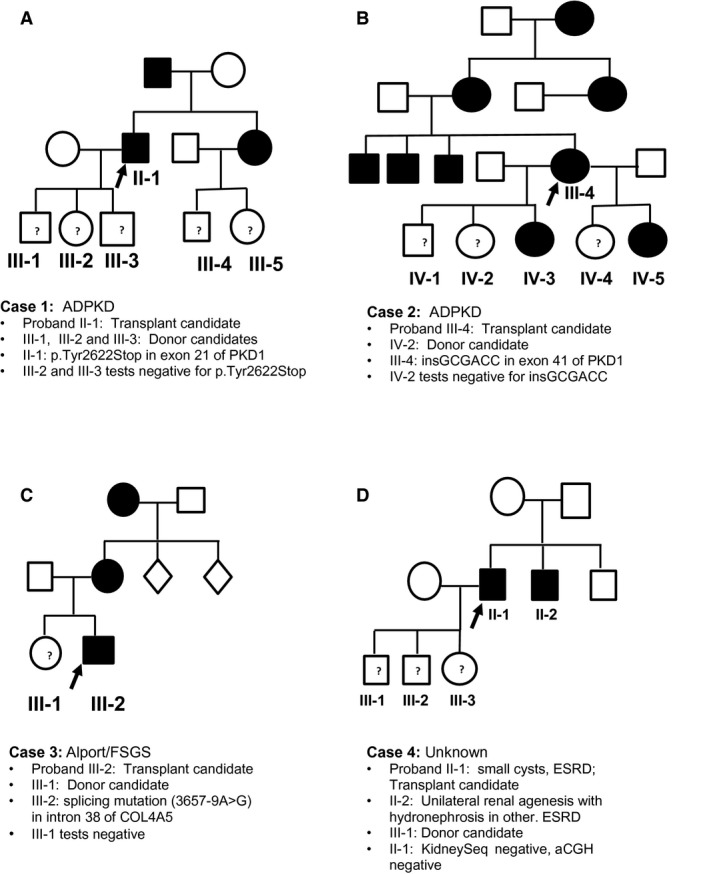
Pedigree chart of candidates and donors tested. Transplant candidates are shown as the probands. ADPKD, autosomal dominant polycystic kidney disease; ESRD, end‐stage renal disease; FSGS, focal segmental glomerulosclerosis.
Case 1
The first patient was diagnosed with ADPKD in her early 50s when workup for a urinary tract infection in the setting of family history of ADPKD revealed multiple cysts in bilaterally enlarged kidneys (Figure 1A). She presented for transplant evaluation at age 63 years, and a daughter aged 25 years wished to be evaluated as a living donor. Genetic testing of the transplant candidate revealed a heterozygous 6‐bp insertion in exon 41 of PKD1, which resulted in the in‐frame insertion of Ala‐Thr. This insertion has not been reported in the ADPKD Mutation Database (http://pkdb.mayo.edu) or in population databases. Segregation analysis identified this insertion in the patient's affected brother and in two other affected daughters. Based on the change in protein length, absence of controls, cosegregation with disease and close proximity of this in‐frame insertion to another in‐frame insertion classified as pathogenic in the ADPKD Mutation Database, this variant was classified as “likely pathogenic.” The donor candidate was negative for the insertion and was accepted to continue her donor evaluation. Unfortunately, the transplant candidate developed major complications from peripheral vascular disease, and that has precluded her transplant.
Case 2
The second patient was diagnosed with ADPKD in his late 30s when workup for severe hypertension in the setting of a positive family history of ADPKD revealed bilateral enlarged cystic kidneys (Figure 1B). He presented for a transplant evaluation at age 51 years, and his three children, aged 20, 22, and 25 years, wished to be evaluated as living donors. Genetic testing of the transplant candidate revealed a nonsense mutation in exon 21 of PKD1 (p.Tyr2622X) that has been reported to be pathogenic 28. Pre‐ and posttest genetic counseling was provided to the candidate's three unaffected children. The mutation segregated in the family, and two of the three children were negative for the mutation. The 25‐year‐old son completed his evaluation and had normal urinalysis, normal kidney function, and no kidney cysts on computed tomography angiography. He underwent donor nephrectomy, and both recipient and donor are doing well.
Case 3
The third transplant candidate presented at age 40 years for an evaluation together with his sister, who wished to be considered as a donor (Figure 1C). The patient had had an earlier renal transplant that lasted 17 years. He first presented at age 18 years when hematuria and proteinuria were noted on an athletic physical examination. A renal biopsy at the time showed FSGS on light microscopy with segmental mesangial and glomerular capillary loop staining for IgM and C3 and glomerular basement membrane lamellations with segmental thickening and thinning on electron microscopy. Ophthalmology examination showed anterior lenticonus and mild retinal pigmentary epithelial clumping, but an audiogram showed no deafness. His mother has proteinuria and hematuria, and his maternal grandmother had “Bright's disease.” The clinical picture with laboratory data was consistent with an X‐linked or autosomal dominant hereditary nephritis suggestive of Alport syndrome, although hereditary FSGS was also a possibility. Genetic testing identified a splice site mutation in intron 38 of COL4A5 (3657‐9A>G). This variant has been reported as pathogenic, confirming X‐linked Alport syndrome 29. The 35‐year‐old sister had negative urinalysis and a negative slit lamp examination and was negative for the splicing mutation. She was accepted as a donor but was blood type incompatible so is awaiting a match in the paired kidney donor program.
Case 4
The fourth case was a man aged 59 years who presented for a transplant evaluation with his 30‐year‐old son, who wished to be his living donor (Figure 1D). The patient had hypertension and advanced CKD with hematuria and proteinuria on dipstick testing. An ultrasound at first presentation several years earlier was noted to show a few small scattered cysts in both kidneys, consistent with hypertensive nephrosclerosis with acquired cysts, although other tubulointerstitial kidney diseases could not be ruled out. The patient's younger sibling had presented at age 37 years with advanced CKD, an absent left kidney and right‐sided hydronephrosis on ultrasound. On retrograde pyelography, this sibling had moderate right‐sided caliectasis with a possible filling defect in the ureter and narrowing consistent with obstructive right‐sided urolithiasis or congenital ureteropelvic junction obstruction or unilateral vesicoureteric reflux. The left ureteric orifice was cannulated and appeared to have a blind end within 1 cm, consistent with an involuted multicystic dysplastic kidney or left‐sided renal agenesis.
In these two siblings, we considered disease associated with hepatocyte nuclear factor 1β (HNF1B) presenting as interstitial kidney disease in one and as a CAKUT in the other. Comprehensive renal gene panel testing in the transplant candidate did not identify any likely pathogenic variants in any of the genes on KidneySeq, including HNF1B. Of note, copy number variant analysis of HNF1B was normal, a relevant finding because about half of HNF1B‐associated disease arises from gene or chromosomal microdeletions on 17q12 30, 31. We confirmed this finding using array chromosomal gene hybridization as an orthogonal technology. Having found no likely pathogenic variants, the son was counseled and completed his donor evaluation with no detectable abnormalities on functional testing and proceeded to donor nephrectomy. Both recipient and donor are doing well.
Discussion
LKDs have a greater lifetime risk of ESRD than otherwise matched controls 4, 5. Whether this increase reflects unrecognized risk factors that are not affected by the donation process or whether the loss of one kidney increases the risk of kidney disease in a subset of donors is not known. In either case, genetic susceptibility may contribute to the risk, with nephrectomy either promoting progressive CKD or simply shortening the time to reach ESRD once CKD begins.
Because ≈40% of LKDs are close biological relatives of the transplant recipient, it is imperative, if appropriate, to exclude presymptomatic genetic disease prior to accepting a donor candidate for nephrectomy. There are published instances in which this precaution was not taken and the genetic risk to a sibling LKD was unrecognized, only to have the donor develop the same kidney disease years later 32, 33. Assessing this risk is difficult because recipient candidates who progress to ESRD are often not appropriately phenotyped with a renal biopsy and are seldom genotyped for possible genetic causes of disease.
We designed, developed and validated a targeted gene panel to provide comprehensive genetic testing for 115 genes implicated in a wide variety of renal diseases (Table S3). Although this gene panel was developed to facilitate the genetic diagnosis in patients with hereditary kidney diseases, in this publication, we described its utility for the evaluation of asymptomatic LKDs without evident kidney disease who nevertheless have a family history of kidney disease.
There are many reasons to consider comprehensive gene panel testing in this setting. First, although >60% of transplant‐eligible patients have diabetes or hypertension as the stated cause of their renal disease, this diagnosis is often based on association rather than probable causality. If biopsy correlation is available, up to one‐third of patients with diabetes or hypertension may have an alternative diagnosis to explain their ESRD 7, 8, 9. In another 20% of transplant candidates, the cause of ESRD is unknown, preventing a focused genetic evaluation of related family members 1, 6.
Second, some diseases such as HNF1B‐associated kidney disease (also known as renal cysts and diabetes) have limited penetrance and variable expression, which makes clinical diagnosis challenging. Although heterozygosity for pathogenic variants in HNF1B represents the most common monogenic cause of developmental kidney disease 30, 34, the disease is a multisystem disorder. Renal cysts are the most frequently presenting feature, but the spectrum of possible renal structural abnormalities includes renal hypodysplasia, pelvic–ureteric junction obstruction, horseshoe kidney, unilateral renal agenesis, single kidneys and renal hypoplasia 35. Extrarenal phenotypes also occur, and other affected family members might present with early onset diabetes (maturity onset diabetes of the young type 5) or genital abnormalities 36, 37. This complexity and the often apparently limited number of affected relatives can reduce suspicion of a genetic disease.
Third, some types of kidney diseases (e.g. FSGS) are genetically heterogeneous, with at least 15 known loci that cause dominant or recessive disease, and this list is growing, making traditional gene testing impractical 38, 39. Furthermore, classically distinct genetic diseases can phenocopy other diseases, blurring the difference between phenotypes. Variants in, for example, other syndromic glomerular disease genes; the Alport genes, COL4A3/COL4A4; and the gene for nail–patella syndrome, LMX1B, can be identified in a number of patients without extrarenal features who have histological FSGS 12, 40, 41, 42. Variants in ciliary disease genes TTC21B and NPHP4 that typically cause juvenile nephronophthisis have been recently reported as causing inherited FSGS 43, 44, 45. Phenotypic similarities mean that often a large number of candidate genes are associated with a given renal disease, making gene‐by‐gene screening prohibitive in terms of cost and time.
Fourth, genetic diseases that present in adult life, with the exception of ADPKD, do not have accepted diagnostic tests—short of genetic testing—that have been validated for presymptomatic screening to exclude disease in a living donor at risk. Even with ADPKD, although age‐dependent ultrasound and magnetic resonance imaging (MRI) criteria for the exclusion of disease have been developed, there are many scenarios in which diagnostic certainty is insufficient, making genetic screening requisite to establish or exclude a diagnosis 46, 47.
Finally, comprehensive genetic testing takes on even greater importance for specific ethnic groups. A prime example is the contribution of West African ancestry to the risk of FSGS and CKD associated with two common alleles in the gene apolipoprotein L1 (APOL1), referred to as G1 and G2 48, 49. The G1 allele is composed of two missense variants in linkage disequilibrium, Ser342Gly and Ile384Met, and the G2 allele is an in‐frame deletion of two amino acids, delN388/Y389. In the Yoruba people of Nigeria, the prevalence of these alleles is 40% and 8%, respectively, reflecting the heterozygous protection they afford to carriers from infection with Trypanosoma brucei rhodesiense. In African Americans, G1 is found in 52% of those with and 18–23% of those without FSGS; for the G2 allele, the percentages are 23% and 15%, respectively. Under a recessive model (i.e. carriers of two risk alleles: G1/G1, G1/G2 or G2/G2), there is a seven‐ to 10‐fold increased risk of hypertension‐associated renal disease and a 10‐ to 17‐fold increased risk of FSGS. These two APOL1 risk alleles also affect allograft outcomes of the donor kidney. Kidneys from deceased African American donors with two APOL1 risk variants fail more rapidly after transplantation than kidneys from donors with no or one risk allele; however, the APOL1 allele status of the transplant recipient does not affect outcome 50, 51, 52. Taken together, some have suggested that all African American kidney donors should be screened for these APOL1 risk alleles 10, 53, 54.
In this pilot series, we tested four transplant candidates to determine the genetic basis of disease (Table 3). In two candidates, the clinical diagnosis of ADPKD was easily made on the basis of strong family history of enlarged cystic kidneys and autosomal dominant inheritance; however, their children were all aged <30 years, limiting the utility of imaging‐based screening. In a third candidate, although there was a high suspicion of Alport disease based on the clinical features of childhood‐onset hematuria and proteinuria and glomerular‐basement membrane lamellations with segmental thinning on ultrastructural examination of a renal biopsy, there were some inconsistencies; for example, there was no hearing deficit, and the light microscopy and immunofluorescence suggested FSGS. The fourth case was the most problematic because there was no unifying diagnosis for the two affected siblings in the pedigree. Nevertheless, negative screening in this case reduced concern about a common genetic disease and was valuable in providing counseling to the donor candidate.
The KidneySeq panel includes many genes not associated with ESRD or CKD but with other distinct renal phenotypes. The clinical utility of their inclusion is multifold. First, the added sequencing cost of additional genes is trivial. Second, by including all known causes of genetic renal disease, it becomes possible to restrict the bioinformatic analysis, if necessary, to the genes associated with the phenotype of interest. As more genes are discovered to be causes of renal diseases, updating a single targeted panel also becomes more practical than updating multiple phenotype‐defined panels (e.g. a panel limited to FSGS). Third, phenotypes are often blurry with the absence of pathognomonic clinical, imaging or biopsy information, making it unclear whether the focus should be on a glomerular disease or a tubulointerstitial disease. Moreover, as stated earlier, even when the phenotype is clear, there is significant variability in the phenotypic expression of some genes.
Who are candidates for genetic screening? For living donors, we recommend genetic testing in all persons with a clear family history of CKD or ESRD or when two or more family members have kidney disease of unknown or uncertain etiology, unless an alternative screening test with a negative predictive value close to 100% is available. Genetic testing should also be considered for living donors with just one first‐degree relative with CKD or ESRD unless that renal disease is clearly diabetic, immunologic (e.g. lupus nephritis), vascular, obstructive, or drug or toxin related. About 40% of the 5000 annual living donors in the United States are biologically related to their recipients; 8–10% of recipients have a known genetic diagnosis like polycystic kidney disease and 18–20% have an unknown cause of ESRD 1, 6. At a conservative estimate, 5–10% of these unknown causes may have gene variants that confer a Mendelian risk of future disease. We suggest that 9–12% of LKDs may benefit from formal testing to exclude monogenic kidney disease. Such testing could include imaging studies with high negative predictive value (e.g. MRI for ADPKD), focused genetic testing for diseases like Alport (COL3A3, COL3A4 and COL3A5) or comprehensive screening using targeted gene panels. Expanded genetic testing may also increase the living donor pool by excluding genetic disease in susceptible persons who are currently not being accepted because of clinical uncertainty.
Whole‐exome sequencing (WES) is increasingly used for the diagnosis of monogenic disorders in a research setting and has been proposed by some as the preferred clinical genetic diagnostic test when locus heterogeneity is extreme or when the phenotype is indistinct 55, 56. When applied to clinical diagnostics, however, the bioinformatic analysis of WES data must be restricted to genes known to be clinically implicated in the disease under consideration. Compared with targeted panels like KidneySeq, the aggregate sequencing and analysis costs of WES are far higher, the depth of sequencing is lower, the bioinformatic throughput is slower, and the type of analysis is more restricted—all points that favor the use of targeted panels in the clinical arena.
Diagnostic laboratories offering genetic panels must be certified (College of American Pathologists or Clinical Laboratory Improvement Amendments program). In addition, we strongly advocate that sequencing and bioinformatic data be reviewed by a multidisciplinary group in the context of the clinical data. This group should include, at a minimum, research scientists with expertise in targeted genomic enrichment and MPS, bioinformaticians, clinical geneticists and physicians with expertise in genetic renal diseases. We also recommend that biological relatives who are considering becoming LKDs be offered pre‐ and posttest genetic counseling. Genetic counselors can assist in the evaluation of an appropriate family history in addition to providing counseling and interpretation of test results. Last, both donor candidates and clinicians should understand the benefits and limitations of genetic testing.
There are several limitations to genetic testing for LKDs. First, the majority of kidney disease is polygenic or secondary to diabetes, hypertension or autoimmune conditions or from infections or toxins. Second, not all genetic variants are identified by targeted NGS panels (or WES), including variants in 5′ regulatory regions, introns or untranslated exonic regions. Third, a negative screen may falsely reduce perceived risk and thus provide misleading reassurance to the transplant center and the donor. Fourth, some identified VUSs may be exceedingly difficult to interpret, leading the transplant center and/or the donor to unwarranted dissuation from donation. Finally, significant variants unrelated to the phenotype (unsolicited but nevertheless medically significant discoveries) may be identified that are actionable and that need to be addressed.
In summary, the reasons to include comprehensive genetic testing in the evaluation of prospective renal transplant recipients and donors are compelling. We showed that a targeted sequencing approach works well and detects single‐nucleotide changes and more complex indels and copy number variants. Areas that are not adequately captured must be clearly defined so that complementary sequencing methods can be included in the analytical pipeline to ensure comprehensive coverage, and all likely pathogenic or pathogenic variants should be Sanger confirmed on a new DNA sample extracted from the originally received blood samples (Figures S1 and S2). Finally, to ensure a clinically meaningful report, a multidisciplinary review of all variants in the context of the phenotypic data is essential.
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Supporting information
Figure S1: KidneySeq test workflow. The diagram in this figure shows the test workflow. Samples received in the laboratory were entered into a database. Quality of samples was assessed after several steps (DNA extraction, library preparation, and hybridization and capture). Successful samples were then pooled in batches of five samples and sequenced in the MiSeq. Sequencing data were analyzed through an in‐house–developed pipeline (Figure S2), and an internal report was generated. Variants in this report were evaluated for interpretation at the multidisciplinary board meeting, those variants interpreted as etiologic were Sanger sequenced and a final results letter was generated.
Figure S2: Analysis pipeline for processing massively parallel sequencing data. The pipeline shows processing of raw sequencing reads to variant detection and report generation, which includes FastQC to monitor quality, Burrows–Wheeler alignment to map reads to thereference genome, Picard to remove read duplicates, the Genome Analysis Toolkit for variant detection across the KidneySeq target regions, Freebayes to call variants in the PKD1 gene, and an in‐house–developed tool to annotate and filter variants and generate a final complete report.
Table S1: Total sequence reads and percentage of the target region covered.
Table S2: Target regions covered with <10×.
Table S3: Broad disease phenotypes, genes tested, and modes of inheritance.
Thomas CP, Mansilla MA, Sompallae R, Mason SO, Nishimura CJ, Kimble MJ, Campbell CA, Kwitek AE, Darbro BW, Stewart ZA & Smith RJH. Screening of Living Kidney Donors for Genetic Diseases Using a Comprehensive Genetic Testing Strategy. Am J Transplant 2017; 17: 401–410
Contributor Information
C. P. Thomas, Email: christie-thomas@uiowa.edu
R. J. H. Smith, Email: richard-smith@uiowa.edu
References
- 1. Hart A, Smith JM, Skeans MA, et al. Kidney. Am J Transplant 2016; 16: 11–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lamb KE, Lodhi S, Meier‐Kriesche HU. Long‐term renal allograft survival in the United States: A critical reappraisal. Am J Transplant 2011; 11: 450–462. [DOI] [PubMed] [Google Scholar]
- 3. Hariharan S, Johnson CP, Bresnahan BA, Taranto SE, McIntosh MJ, Stablein D. Improved graft survival after renal transplantation in the United States, 1988 to 1996. N Engl J Med 2000; 342: 605–612. [DOI] [PubMed] [Google Scholar]
- 4. Muzaale AD, Massie AB, Wang M, et al. Risk of end‐stage renal disease following live kidney donation. JAMA 2014; 311: 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mjøen G, Hallan S, Hartmann A, et al. Long‐term risks for kidney donors. Kidney Int 2014; 86: 162–167. [DOI] [PubMed] [Google Scholar]
- 6. 2015 USRDS annual data report: Epidemiology of kidney disease in the United States. United States Renal Data System NIoH, National Institute of Diabetes and Digestive and Kidney Diseases. Bethesda, MD, 2015.
- 7. Christensen PK, Larsen S, Horn T, Olsen S, Parving H‐H. Causes of albuminuria in patients with type 2 diabetes without diabetic retinopathy. Kidney Int 2000; 58: 1719–1731. [DOI] [PubMed] [Google Scholar]
- 8. Sharma SG, Bomback AS, Radhakrishnan J, et al. The modern spectrum of renal biopsy findings in patients with diabetes. Clin J Am Soc Nephrol 2013; 8: 1718–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Caetano ERSP, Zatz R, Saldanha LB, Praxedes JN. Hypertensive nephrosclerosis as a relevant cause of chronic renal failure. Hypertension 2001; 38: 171–176. [DOI] [PubMed] [Google Scholar]
- 10. Kuppachi S, Smith RJH, Thomas CP. Evaluation of genetic renal diseases in potential living kidney donors. Curr Transplant Rep 2015; 2: 1–14. [Google Scholar]
- 11. Sadowski CE, Lovric S, Ashraf S, et al. A single‐gene cause in 29.5% of cases of steroid‐resistant nephrotic syndrome. J Am Soc Nephrol 2015; 26: 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gast C, Pengelly RJ, Lyon M, et al. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dial Transplant 2016; 31: 961–970. [DOI] [PubMed] [Google Scholar]
- 13. McCarthy HJ, Bierzynska A, Wherlock M, et al. Simultaneous sequencing of 24 genes associated with steroid‐resistant nephrotic syndrome. Clin J Am Soc Nephrol 2013; 8: 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Redin C, Le Gras S, Mhamdi O, et al. Targeted high‐throughput sequencing for diagnosis of genetically heterogeneous diseases: Efficient mutation detection in Bardet‐Biedl and Alström Syndromes. J Med Genet 2012; 49: 502–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goecks J, Nekrutenko A, Taylor J. Galaxy: A comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 2010; 11: R86–R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. Cornell University Library, 2013: arXiv:1303.3997v1301 [q‐bio.GN].
- 17. DePristo MA, Banks E, Poplin RE, et al. A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet 2011; 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garrison E, Marth G. Haplotype‐based variant detection from short‐read sequencing. Cornell University Library, 2012: arXiv:1207.3907 [q‐bio.GN].
- 19. Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comput Biol 2010; 6: e1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005; 15: 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease‐causing potential of sequence alterations. Nat Methods 2010; 7: 575–576. [DOI] [PubMed] [Google Scholar]
- 22. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010; 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009; 4: 1073–1081. [DOI] [PubMed] [Google Scholar]
- 24. Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res 2009; 19: 1553–1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007; 23: 1289–1291. [DOI] [PubMed] [Google Scholar]
- 27. Rossetti S, Hopp K, Sikkink RA, et al. Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J Am Soc Nephrol 2012; 23: 915–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Audrézet M‐P, Cornec‐Le Gall E, Chen J‐M, et al. Autosomal dominant polycystic kidney disease: Comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat 2012; 33: 1239–1250. [DOI] [PubMed] [Google Scholar]
- 29. Martin P, Heiskari N, Pajari H, et al. Spectrum of COL4A5 mutations in Finnish Alport syndrome patients. Hum Mutat 2000; 15: 579–579. [DOI] [PubMed] [Google Scholar]
- 30. Heidet L, Decramer S, Pawtowski A, et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin J Am Soc Nephrol 2010; 5: 1079–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Faguer S, Decramer S, Chassaing N, et al. Diagnosis, management, and prognosis of HNF1B nephropathy in adulthood. Kidney Int 2011; 80: 768–776. [DOI] [PubMed] [Google Scholar]
- 32. Winn MP, Alkhunaizi AM, Bennett WM, et al. Focal segmental glomerulosclerosis: A need for caution in live‐related renal transplantation. Am J Kidney Dis 1999; 33: 970–974. [DOI] [PubMed] [Google Scholar]
- 33. Kofman T, Audard V, Narjoz C, et al. APOL1 polymorphisms and development of CKD in an identical twin donor and recipient pair. Am J Kidney Dis 2014; 63: 816–819. [DOI] [PubMed] [Google Scholar]
- 34. Thomas R, Sanna‐Cherchi S, Warady BA, Furth SL, Kaskel FJ, Gharavi AG. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr Nephrol 2011; 26: 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bockenhauer D, Jaureguiberry G. HNF1B‐associated clinical phenotypes: The kidney and beyond. Pediatr Nephrol 2015; 31: 707–714. [DOI] [PubMed] [Google Scholar]
- 36. Clissold RL, Hamilton AJ, Hattersley AT, Ellard S, Bingham C. HNF1B‐associated renal and extra‐renal disease—an expanding clinical spectrum. Nat Rev Nephrol 2015; 11: 102–112. [DOI] [PubMed] [Google Scholar]
- 37. Oram RA, Edghill EL, Blackman J, et al. Mutations in the hepatocyte nuclear factor‐1β (HNF1B) gene are common with combined uterine and renal malformations but are not found with isolated uterine malformations. Am J Obstet Gynecol 2010; 203: e1–e5. [DOI] [PubMed] [Google Scholar]
- 38. Pollak MR. Familial FSGS. Adv Chronic Kidney Dis 2014; 21: 422–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lovric S, Ashraf S, Tan W, Hildebrandt F. Genetic testing in steroid‐resistant nephrotic syndrome: When and how? Nephrol Dial Transplant 2015; DOI: 10.1093/ndt/gfv355 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 40. Malone AF, Phelan PJ, Hall G, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int 2014; 86: 1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pierides A, Voskarides K, Athanasiou Y, et al. Clinico‐pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol Dial Transplant 2009; 24: 2721–2729. [DOI] [PubMed] [Google Scholar]
- 42. Boyer O, Woerner S, Yang F, et al. LMX1B mutations cause hereditary FSGS without extrarenal involvement. J Am Soc Nephrol 2013; 24: 1216–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cong EH, Bizet AA, Boyer O, et al. A homozygous missense mutation in the ciliary gene TTC21B causes familial FSGS. J Am Soc Nephrol 2014; 25: 2435–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bullich G, Vargas I, Trujillano D, et al. Contribution of the TTC21B gene to glomerular and cystic kidney diseases. Nephrol Dial Transplant 2016; DOI: 10.1093/ndt/gfv453 [Epub ahead of print]. [DOI] [PubMed]
- 45. Mistry K, Ireland JHE, Ng RCK, Henderson JM, Pollak MR. Novel mutations in NPHP4 in a consanguineous family with histological findings of focal segmental glomerulosclerosis. Am J Kidney Dis 2007; 50: 855–864. [DOI] [PubMed] [Google Scholar]
- 46. Pei Y, Hwang Y‐H, Conklin J, et al. Imaging‐based diagnosis of autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2015; 26: 746–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pei Y, Obaji J, Dupuis A, et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol 2009; 20: 205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Friedman DJ, Pollak MR. Apolipoprotein L1 and kidney disease in African Americans. Trends Endocrinol Metab 2016; 27: 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kruzel‐Davila E, Wasser WG, Aviram S, Skorecki K. APOL1 nephropathy: From gene to mechanisms of kidney injury. Nephrol Dial Transplant 2016; 31: 349–358. [DOI] [PubMed] [Google Scholar]
- 50. Freedman BI, Pastan SO, Israni AK, et al. APOL1 genotype and kidney transplantation outcomes from deceased African American donors. Transplantation 2016; 100: 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Freedman BI, Julian BA, Pastan SO, et al. Apolipoprotein L1 gene variants in deceased organ donors are associated with renal allograft failure. Am J Transplant 2015; 15: 1615–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee BT, Kumar V, Williams TA, et al. The APOL1 genotype of African American Kidney transplant recipients does not impact 5‐year allograft survival. Am J Transplant 2012; 12: 1924–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Riella LV, Sheridan AM. Testing for high‐risk APOL1 alleles in potential living kidney donors. Am J Kidney Dis 2015; 66: 396–401. [DOI] [PubMed] [Google Scholar]
- 54. Freedman BI, Julian BA. Should kidney donors be genotyped for APOL1 risk alleles? Kidney Int 2015; 87: 671–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang Y, Muzny DM, Reid JG, et al. Clinical whole‐exome sequencing for the diagnosis of mendelian disorders. N Engl J Med 2013; 369: 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Xue Y, Ankala A, Wilcox WR, Hegde MR. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next‐generation sequencing: Single‐gene, gene panel, or exome/genome sequencing. Genet Med 2015; 17: 444–451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: KidneySeq test workflow. The diagram in this figure shows the test workflow. Samples received in the laboratory were entered into a database. Quality of samples was assessed after several steps (DNA extraction, library preparation, and hybridization and capture). Successful samples were then pooled in batches of five samples and sequenced in the MiSeq. Sequencing data were analyzed through an in‐house–developed pipeline (Figure S2), and an internal report was generated. Variants in this report were evaluated for interpretation at the multidisciplinary board meeting, those variants interpreted as etiologic were Sanger sequenced and a final results letter was generated.
Figure S2: Analysis pipeline for processing massively parallel sequencing data. The pipeline shows processing of raw sequencing reads to variant detection and report generation, which includes FastQC to monitor quality, Burrows–Wheeler alignment to map reads to thereference genome, Picard to remove read duplicates, the Genome Analysis Toolkit for variant detection across the KidneySeq target regions, Freebayes to call variants in the PKD1 gene, and an in‐house–developed tool to annotate and filter variants and generate a final complete report.
Table S1: Total sequence reads and percentage of the target region covered.
Table S2: Target regions covered with <10×.
Table S3: Broad disease phenotypes, genes tested, and modes of inheritance.