

Abstract
Hepatitis C virus (HCV)–infected patients with cirrhosis are historically a difficult‐to‐treat population and are at risk of hepatic decompensation. In the phase 2 COSMOS study that evaluated simeprevir (HCV NS3/4A protease inhibitor) + sofosbuvir (HCV nucleotide analogue NS5B polymerase inhibitor) ± ribavirin for 12 or 24 weeks in HCV genotype (GT)1–infected patients, high rates of sustained virologic response 12 weeks after planned end of treatment (SVR12) were achieved, including in patients with cirrhosis (METAVIR score F4). This phase 3, open‐label, single‐arm study (OPTIMIST‐2 [NCT02114151]) evaluated the efficacy and safety of 12 weeks of simeprevir + sofosbuvir in HCV GT1–infected treatment‐naive or treatment‐experienced patients with cirrhosis. Patients (aged 18‐70 years) with chronic HCV GT1 infection and documented presence of cirrhosis received oral simeprevir 150 mg once daily + sofosbuvir 400 mg once daily for 12 weeks. The primary efficacy endpoint of the study was the proportion of patients achieving SVR12 versus a composite historical control (SVR12 rate of 70%). Safety and patient‐reported outcomes were assessed. Overall, 103 patients received treatment. SVR12 with simeprevir + sofosbuvir (83%, 95% confidence interval 76%‐91%) met the primary objective of superiority versus the historical control (70%). SVR12 rates for treatment‐naive and treatment‐experienced patients were 88% (44/50) and 79% (42/53), respectively. Adverse events occurred in 72 (70%) patients, with most (64%) being grade 1 or 2. Serious adverse events (none considered related to study treatment) occurred in five (5%) patients, and three (3%) patients discontinued all study treatment due to adverse events. Patient‐reported outcomes improved from baseline to follow‐up week 12. Conclusion: Simeprevir + sofosbuvir for 12 weeks achieved superiority in SVR12 rates versus the historical control in treatment‐naive and treatment‐experienced HCV GT1‐infected patients with cirrhosis and was generally safe and well tolerated. (Hepatology 2016;64:360‐369)
Abbreviations
- AE
adverse event
- BMI
body mass index
- CI
confidence interval
- EOT
end of treatment
- GT
genotype
- HC
historical control
- HCV
hepatitis C virus
- HRQoL
health‐related quality of life
- IFN
interferon
- OPTIMIST‐2
Optimal Treatment With a Simeprevir and Sofosbuvir Therapy
- pegIFN
pegylated interferon‐α‐2a
- PRO
patient‐reported outcome
- QD
once daily
- SVR
sustained virologic response
Chronic hepatitis C viral (HCV) infection is associated with an increased risk of cirrhosis and hepatic decompensation and is a leading cause of liver‐related deaths. Approximately one‐fifth of patients with chronic HCV infection progress to cirrhosis.1 Historically, patients with cirrhosis have achieved lower levels of response to pegylated interferon (pegIFN)–based regimens than patients without cirrhosis, highlighting the difficult‐to‐treat nature of these patients.2
Current treatment guidelines recommend the use of IFN‐free regimens for the treatment of HCV genotype (GT)1–infected patients with cirrhosis.3, 4 Recent approvals of direct‐acting antiviral agents include the simeprevir + sofosbuvir, sofosbuvir + ledipasvir, and paritaprevir/ritonavir/ombitasvir + dasabuvir ± ribavirin combinations. Daclatasvir + sofosbuvir is also approved in the European Union and for the treatment of GT3 in the United States.
Simeprevir, an oral, once‐daily (QD) HCV NS3/4A multigenotypic protease inhibitor, is approved in combination with pegIFN and ribavirin for chronic HCV GT1 and GT4 infections in the United States and European Union. Simeprevir is also approved as part of an IFN‐free combination with sofosbuvir (a QD pangenotypic HCV nucleotide analogue NS5B polymerase inhibitor) for HCV GT1 infection in the United States and GT1 and GT4 infections in the European Union. This combination is also approved for HCV/human immunodeficiency viral coinfection in the European Union.
The efficacy of simeprevir + sofosbuvir for 12 or 24 weeks in HCV GT1‐infected patients with cirrhosis was demonstrated in a phase 2, randomized, open‐label study (COSMOS). The overall rate of sustained virologic response 12 weeks (SVR12) after end of treatment (EOT) was 92% (METAVIR score F0‐F4). In cohort 2 of the study (METAVIR score F3‐F4), SVR12 rates of 93% (38/41) and 96% (44/46) were achieved after 12 and 24 weeks of treatment, respectively; and in patients with compensated cirrhosis (METAVIR score F4), the SVR12 rates following 12 or 24 weeks of treatment were 89% (16/18) and 96% (22/23), respectively, regardless of ribavirin use.
Based on the results from the COSMOS study, the OPTIMIST‐2 (Optimal Treatment With a Simeprevir and Sofosbuvir Therapy) phase 3 trial assessed the efficacy and safety of 12 weeks of simeprevir + sofosbuvir in treatment‐naive and treatment‐experienced patients with chronic HCV GT1 infection and cirrhosis.
Patients and Methods
Patients and Study Design
OPTIMIST‐2 (NCT02114151) was an open‐label, single‐arm, phase 3 study conducted at 35 centers in Canada and the United States and initiated on April 16, 2014 (cutoff for primary analysis January 16, 2015). Eligible patients (age 18‐70 years) had chronic HCV GT1 infection confirmed at screening, plasma HCV RNA concentration >10,000 IU/mL at screening, and presence of cirrhosis determined by any of the following: FibroScan with >12.5 kPa within 6 months of screening, FibroTest score >0.75 and aspartate aminotransferase to platelet ratio >2 at screening, or liver biopsy documenting cirrhosis. Patients had to have undergone hepatic imaging <6 months before screening with no suspicion of hepatocellular carcinoma. No body mass index (BMI) cutoff was applied.
Exclusion criteria included evidence of hepatic decompensation (history or current evidence of ascites, bleeding varices, or hepatic encephalopathy), any liver disease of non‐HCV etiology, and infection/coinfection with HCV non‐GT1, hepatitis B, or human immunodeficiency virus (Supporting Information).
Treatment‐naive and treatment‐experienced patients (prior relapsers, prior nonresponders [partial responders, null responders, and unknown], IFN‐intolerant, and “other” [patients not classified as among any of the aforementioned categories]) were enrolled. Treatment‐experienced patients must have had one (or more) documented course of IFN‐based therapy (Supporting Information).
The study consisted of a screening period of up to 4 weeks, followed by a 12‐week open‐label treatment phase during which patients received oral simeprevir (150 mg QD capsule) and sofosbuvir (400 mg QD tablet) (simeprevir + sofosbuvir). Patients were followed up until 24 weeks after EOT (Supporting Fig. S1; Supporting Information).
The study was approved by the institutional review board or independent ethics committee at each study center and met the ethical principles of the Declaration of Helsinki and good clinical practice guidelines. All patients gave written informed consent.
Outcomes and Procedures
The primary efficacy objective was to demonstrate superiority of simeprevir + sofosbuvir for 12 weeks versus a historical control (HC; Supporting Information), with respect to the proportion of patients achieving SVR12.
Secondary endpoints included proportion of patients achieving SVR 4 and 24 weeks after EOT (SVR4 and SVR24, respectively); proportion of patients with on‐treatment virologic response; rates of on‐treatment failure including viral breakthrough, which was a virologic stopping rule; incidence of viral relapse; evaluation of SVR12 rates in prespecified patient subgroups; assessment of changes from baseline in the HCV NS3/4A and NS5B sequence in patients not achieving SVR; and mean change from baseline for patient‐reported outcome (PRO) assessments at all time points.
SVR12 was evaluated in an exploratory subgroup including patients with baseline NS5A polymorphisms and in a nonprespecified subgroup by baseline serum albumin levels.
Patients were also assessed according to safety endpoints. PRO assessments were completed electronically by patients at specified visits prior to all other study‐related procedures and included the validated Fatigue Severity Scale,5 to evaluate the effect of treatment on fatigue; the Center for Epidemiologic Studies Depression scale,6 to evaluate the frequency of major depressive symptoms; the EuroQoL 5‐Dimensions questionnaire,7 to assess health‐related quality of life (HRQoL); and the Hepatitis C Symptom and Impact Questionnaire version 4, a new tool to assess the severity and frequency of symptoms associated with HCV or its treatment (Supporting Information).
Statistical Analysis
The null hypothesis of this study was that the overall SVR12 rate with simeprevir + sofosbuvir for 12 weeks was not superior to that of an HC (70%). The HC used in this study was a composite of the highest rates of SVR12 for approved direct‐acting antiviral agent regimens that were available at the time of study design, in predefined subpopulations depending on the proportion of treatment‐naive, prior relapser, prior nonresponder, IFN‐intolerant, and other patients enrolled in the study. The SVR12 threshold for each subpopulation was based on the historical data provided in Table 1.8, 9, 10 In this study 95% confidence intervals (CIs) were constructed around the SVR12 rates using a normal approximation with continuity correction. Superiority was concluded if the lower limit of the 95% CI of the SVR12 rate for the simeprevir + sofosbuvir group was greater than the HC SVR12 rate.
Table 1.
Historical SVR12 After EOT Rates in Patients With HCV GT1 Infection and Cirrhosis
| Prior HCV Treatment History | Treatment | Study | Historical SVR12 Rates, % (n/N) | Simeprevir + Sofosbuvir 12 Weeks | |
|---|---|---|---|---|---|
| n (%) | Derived (%) | ||||
| Treatment‐naive | SOF + pegIFN/RBV | NEUTRINO4 | 81 (42/52) | 50 (49) | 39 |
| Relapsers | SMV + pegIFN/RBV | PROMISE5 | 74 (29/39) | 2 (2) | 1 |
| Nonresponder | SMV + pegIFN/RBV | ASPIRE6 | 54 (13/24) | 17 (17) | 9 |
| IFN‐intoleranta | — | — | 5 | 9 (9) | 0 |
| Otherb | — | — | 81 | 25 (24) | 20 |
| Derived HC SVR12 rate | 70% | ||||
SVR rates in IFN‐intolerant patients were set to 5% as no data with approved direct‐acting antiviral/pegIFN/RBV regimens were available for this subgroup at the time of study design.
For conservative reasons, the SVR12 rate of the “other” population was set to the same rate as for treatment‐naive patients.
Abbreviations: RBV, ribavirin; SMV, simeprevir; SOF, sofosbuvir.
A sample size of 100 patients was considered sufficient to provide 90% power to show superiority versus the HC using a one‐sided binomial test at a significance level of 0.025 if the SVR12 threshold was no more than 74%.
All analyses were performed on the intent‐to‐treat population (all patients who received at least one dose of study medication). The primary analysis was performed when all patients had completed the SVR12 visit or had discontinued earlier.
Secondary efficacy outcomes were analyzed using descriptive statistics and 95% CIs. All safety data were summarized descriptively. PRO assessments were analyzed using descriptive statistics; mean changes from baseline values were explored for patient subgroups, and cumulative distribution functions were drawn (Supporting Information).
Role of the Funding Source
The study funder designed the trial; was responsible for data collection, analysis, and interpretation; and helped write and review the report. The investigators were also responsible for data interpretation. All authors had final responsibility for the decision to submit for publication. The corresponding author had full access to all study data and had final responsibility for the decision to submit for publication.
Results
Patient Disposition
In total, 147 patients were screened and 103 received at least one dose of treatment and represented the intent‐to‐treat population (Fig. 1). At the time of the primary analysis, four (4%) patients had completed the study and reached the SVR24 (SVR 24 weeks after EOT) time point, 96 (93%) patients were ongoing, and three (3%) patients had discontinued the study (two withdrew consent and one died in a road traffic accident) (Supporting Information).
Figure 1.
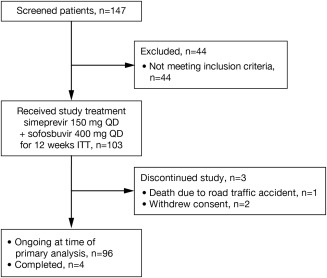
Patient disposition. At the time of the primary analysis, 100% of patients had reached SVR12 after the EOT time point or had discontinued earlier. Abbreviation: ITT, intent to treat.
Table 2 presents baseline demographic and disease characteristics. No NS3 baseline polymorphisms known to impact simeprevir's in vitro activity other than Q80K were observed. The NS5B polymorphism S282T, known to impact sofosbuvir's in vitro activity, was not observed at baseline. Of the patients with sequencing data available, 13 (13%) had baseline NS5A polymorphisms (Supporting Table S1) and six (6%) had a combination of baseline NS5A and Q80K polymorphisms.
Table 2.
Patient Demographics and Baseline Disease Characteristics (Intent‐To‐Treat Population)
| Simeprevir 150 mg QD + Sofosbuvir 400 mg QD (N = 103) | |
|---|---|
| Age, years | 58 (29‐69) |
| Male | 83 (81%) |
| BMI (kg/m2) | 28.8 (18.7‐42.8) |
| ≥30 kg/m2 | 41 (40%) |
| Race | |
| White | 82/101 (81%) |
| Black or African American | 19/101 (19%) |
| Ethnicity | |
| Hispanic or Latino | 16 (16%) |
| HCV GT or subtype and baseline NS3 Q80K polymorphism | |
| 1a | 72 (70%) |
| 1a with baseline Q80Ka | 34 (47%) |
| 1a without baseline Q80Ka | 38 (53%) |
| 1b | 31 (30%) |
| Baseline HCV RNA (log10 IU/mL) | 6.8 (5.0‐7.7) |
| IL28B GTb | |
| CC | 29/102 (28%) |
| CT | 54/102 (53%) |
| TT | 19/102 (19%) |
| Treatment‐naive | 50 (49%) |
| Treatment‐experiencedc | 53 (51%) |
| Platelets <90,000/mm3 d | 19 (18%) |
| Albumin <40 g/Ld | 53 (51%) |
| FibroScan score >20 kPa | 15/26 (58%) |
| Baseline PRO scores | |
| FSSe | 3.4 (0.18) |
| CES‐Df | 13.3 (0.98) |
| EQ‐5D VASg | 70.1 (2.17) |
| HCV‐SIQv4 OBSSg | 17.4 (1.47) |
Data are median (range) or n (%) or n/N (%) except for PRO scores, which are mean (standard error).
Among GT1a‐infected patients.
Single nucleotide polymorphism rs12979860.
Treatment‐experienced patients included prior relapsers, prior nonresponders, IFN‐intolerant, and other patients (n = 2, 17, 9, and 25, respectively).
Most optimal cutoff based on the results of this study.
Normal reference value 2.3, range 1‐7.
Normal reference value 16 (lower threshold for depression), range 0‐60.
Normal reference value not available, range 0‐100.
Abbreviations: CES‐D, Center for Epidemiologic Studies Depression scale; EQ‐5D, EuroQOL‐5 Dimensions questionnaire; FSS, Fatigue Severity Scale; HCV‐SIQv4, Hepatitis C Symptom and Impact Questionnaire version 4; OBSS, Overall Body System Score; VAS, visual analogue scale.
Cirrhosis was determined by FibroScan in 26/103 (25%) patients, FibroTest score in 23/103 (22%) patients, and liver biopsy in 54/103 (52%) patients. In total, 19 (18%) patients had baseline platelets <90,000 mm3, 53 (51%) patients had albumin <40 g/L, and 15/26 (58%) patients had a FibroScan score of >20 kPa.
Efficacy
The primary efficacy end point, SVR12, was met by 83% (86/103, 95% CI 76%‐91%) of HCV GT1‐infected patients with cirrhosis receiving simeprevir + sofosbuvir for 12 weeks, and the primary objective of superiority to the HC was achieved (lower limit of the 95% CI of the SVR12 rate [76%] > HC rate [70%]).
Table 3 presents results for other secondary efficacy outcomes. SVR12 rates were higher (88% [44/50]) in treatment‐naive patients than in treatment‐experienced patients (79% [42/53]). HCV RNA <25 IU/mL undetectable at week 4 was achieved by 85/102 (83%) patients, 73 (86%) of whom achieved SVR12.
Table 3.
Primary and Key Secondary Efficacy End Points (Intent‐To‐Treat Population)
| Response | Simeprevir 150 mg QD + Sofosbuvir 400 mg QD (N = 103) | 95% CI |
|---|---|---|
| SVR12a | 86/103 (83%) | 76‐91 |
| Treatment‐naive | 44/50 (88%) | 78‐98 |
| Treatment‐experienced | 42/53 (79%) | 67‐91 |
| Week 4 HCV RNA <25 IU/mL (undetectable) | 73/85 (86%) | 77‐93 |
| Week 4 HCV RNA <25 IU/mL (detectable) | 13/16 (81%) | 54‐96 |
| Week 4 HCV RNA ≥25 IU/mL | 0/1 | — |
| SVR4 | 89/103 (86%) | 79‐94 |
| On‐treatment failureb | 3/103 (3%) | 1‐8 |
| Viral breakthroughc | 2/103 (2%) | 0‐7 |
| Viral relapsed | 13/99 (13%) | 7‐21 |
Data are n/N (%) or 95% CI.
Defined as HCV RNA <25 IU/mL detectable or undetectable 12 weeks after EOT.
Confirmed HCV RNA <25 IU/mL detectable or ≥25 IU/mL at EOT.
For both patients multiple missed doses were reported prior to breakthrough; confirmed >1.0 log10 increase in HCV RNA from nadir or confirmed HCV RNA >100 IU/mL in patients who had previously achieved HCV RNA <25 IU/mL.
Failure to achieve SVR12 with HCV RNA <25 IU/mL undetectable (or unconfirmed detectable) at EOT and HCV RNA ≥25 IU/mL during follow‐up.
The SVR12 rates were similar for patients with HCV GT1a and 1b infection (83% and 84%, respectively). Patients with IL28B CC or CT GTs had higher rates of SVR12 than patients with the TT GT: 86%, 85%, and 79%, respectively. Furthermore, patients with baseline albumin levels ≥40 g/L had higher SVR12 rates (94%) than patients with levels <40 g/L (74%) (Table 4).
Table 4.
SVR12 and Viral Relapse According to Baseline Demographics and Disease Characteristics (Prespecified and Nonprespecified Patient Subgroups; Intent‐To‐Treat Population)
| Response | Simeprevir 150 mg QD + Sofosbuvir 400 mg QD (N = 103) | 95% CI | Relapse Ratea | 95% CI |
|---|---|---|---|---|
| Sex | ||||
| Male | 68/83 (82%) | 73‐91 | 12/80 (15%) | 8‐25 |
| Female | 18/20 (90%) | 74‐100 | 1/19 (5%) | 0‐26 |
| BMI (kg/m2) | ||||
| <25 | 15/19 (79%) | 58‐100 | 4/19 (21%) | 6‐46 |
| ≥25‐<30 | 40/43 (93%) | 84‐100 | 1/41 (2%) | 0‐13 |
| ≥30 | 31/41 (76%) | 61‐90 | 8/39 (21%) | 9‐37 |
| Raceb | ||||
| White | 68/82 (83%) | 73‐90 | 11/79 (14%) | 7‐24 |
| Black or African American | 17/19 (89%) | 67‐99 | 1/18 (6%) | 0‐27 |
| Ethnicity | ||||
| Hispanic or Latino | 13/16 (81%) | 54‐96 | 2/15 (13%) | 2‐41 |
| HCV GT or subtype and baseline NS3 Q80K polymorphism | ||||
| 1a | 60/72 (83%) | 74‐93 | 8/68 (12%) | 5‐22 |
| 1a with baseline Q80K | 25/34 (74%) | 57‐90 | 6/31 (19%) | 8‐38 |
| 1a without baseline Q80K | 35/38 (92%) | 82‐100 | 2/37 (5%) | 1‐18 |
| 1b | 26/31 (84%) | 69‐98 | 5/31 (16%) | 6‐34 |
| Baseline HCV RNA | ||||
| <6,000,000 log10 IU/mL | 43/51 (84%) | 71‐93 | 6/49 (12%) | 5‐25 |
| ≥6,000,000 log10 IU/mL | 43/52 (83%) | 70‐92 | 7/50 (14%) | 6‐27 |
| Baseline platelet count | ||||
| <90,000/mm3 | 13/19 (68%) | 45‐92 | 4/17 (24%) | 7‐50 |
| ≥90,000/mm3 | 73/84 (87%) | 79‐95 | 9/82 (11%) | 5‐20 |
| Baseline albumin levels | ||||
| <40 g/L | 39/53 (74%) | 61‐86 | 11/50 (22%) | 12‐36 |
| ≥40 g/L | 47/50 (94%) | 86‐100 | 2/49 (4%) | 1‐14 |
| HCV GT1a | 30/31 (97%) | 83‐100 | 0/30 | 0‐12 |
| 1a with baseline Q80K | 12/12 (100%) | 74‐100 | 0/12 | 0‐27 |
| 1a without baseline Q80K | 18/19 (95%) | 74‐100 | 0/18 | 0‐19 |
| HCV GT1b | 17/19 (89%) | 67‐99 | 2/19 (11%) | 1‐33 |
| FibroScan scorec | ||||
| >12.5‐≤20 kPa | 11/11 (100%) | 96‐100 | 0/11 | 0‐29 |
| >20 kPa | 12/15 (80%) | 56‐100 | 2/14 (14%) | 2‐43 |
| IL28B GTd | ||||
| CC | 25/29 (86%) | 72‐100 | 3/28 (11%) | 2‐28 |
| CT | 46/54 (85%) | 75‐96 | 7/53 (13%) | 6‐25 |
| TT | 15/19 (79%) | 58‐100 | 2/17 (12%) | 2‐36 |
Data are n/N (%) or 95% CI.
The incidence of viral relapse was only calculated for patients with undetectable HCV RNA levels at EOT and with at least one follow‐up HCV RNA measurement.
N = 101.
N = 26.
N = 102.
HCV GT1a–infected patients without baseline Q80K polymorphisms had a higher SVR12 rate (92%) than those with baseline Q80K polymorphisms (74%) (Table 4). All patients with baseline NS5A polymorphisms achieved SVR12 (100%), as did 82% of patients without baseline NS5A polymorphisms (Fig. 2). Of the patients with both NS3 Q80K and NS5A polymorphisms at baseline, 100% achieved SVR12.
Figure 2.
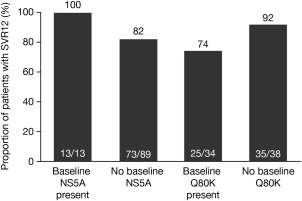
SVR12 by baseline NS5A and Q80K polymorphisms (intent‐to‐treat population).
In total, 17/103 (17%) patients did not achieve SVR12 (including one patient who had undetectable HCV RNA at EOT but missing data at the SVR12 time point). On‐treatment failure occurred in three of 103 (3%) patients; of these, one (1%) discontinued treatment early due to an adverse event (AE), with HCV RNA detectable at the time of discontinuation, and two (2%) experienced viral breakthrough at weeks 4 and 8. For both patients who experienced viral breakthrough, multiple missed doses were reported prior to breakthrough. Thirteen (13%) patients experienced viral relapse, which most commonly occurred at follow‐up week 4. A lower proportion of treatment‐naive patients experienced viral relapse (6% [3/47]) versus treatment‐experienced patients (19% [10/52]). For patients with week 4 HCV RNA <25 IU/mL undetectable, <25 IU/mL detectable, and ≥25 IU/mL, 10/83 (12%), 3/16 (19%), and 0/0 patients experienced viral relapse, respectively. Relapse according to subgroup is shown in Table 4. Viral relapse occurred in a higher proportion of patients with baseline Q80K polymorphisms (19% [6/31]), platelets <90,000/mm3 (24% [4/17]), albumin <40 g/L (22% [11/50]), or FibroScan score >20 kPa (14% [2/14]) versus patients without baseline Q80K polymorphisms (5% [2/37]), platelets ≥90,000/mm3 (11% [9/82]), albumin ≥40 g/L (4% [2/49]), or FibroScan score >12.5≤20 kPa (0% [0/11]). Patients with IL28B CC, CT, and TT GTs had similar viral relapse rates: 11% (3/28), 13% (7/53), and 12% (2/17), respectively.
Most (79% [11/14]) patients not achieving SVR12 with sequencing data available had emerging NS3 mutations associated with resistance to simeprevir at time of failure at position 168 alone (n = 7), R155K alone (n = 1), or combined with mutations D168E, I170T, or N174G (n = 1 each). No emerging NS5B S282T or other mutations were observed (Supporting Information).
Safety
In total, 72 (70%) patients reported an AE during simeprevir + sofosbuvir treatment (Table 5); most AEs were grade 1 or 2 (64%). AEs leading to permanent discontinuation of at least one study drug occurred in three (3%) patients: an infected bite, rash, and death (road traffic accident). The most common AEs reported (≥10% patients) were headache (20%), fatigue (20%), and nausea (11%); these were grade 1 or 2, were transient, and did not lead to permanent treatment discontinuation (Table 5).
Table 5.
Summary of AEs (Intent‐To‐Treat Population)
| AE | Simeprevir 150 mg QD + Sofosbuvir 400 mg QD (N = 103) |
|---|---|
| Any AE | 72 (70%) |
| Worst grade 1 or 2 | 66 (64%) |
| Worst grade 3 | 5 (5%) |
| Worst grade 4a | 1 (1%) |
| Serious AE | 5 (5%) |
| Deathsb | 1 (1%) |
| AE leading to permanent discontinuationc | 3 (3%) |
| Most common AEs (>10% patients) | |
| Headache | 21 (20%) |
| Fatigue | 21 (20%) |
| Nausea | 11 (11%) |
| AE of special interest | |
| Increased bilirubin | 2 (2%) |
| Grade 3 or 4 | 0 |
| AEs of clinical interest | |
| Rash (any type) | 16 (16%) |
| Photosensitivity conditions | 5 (5%) |
| Pruritus | 14 (14%) |
| Dyspnea | 3 (3%) |
Data are n (%).
Grade 4 AE was an infected bite.
Death due to road traffic accident.
AEs leading to the permanent stop of at least one study drug were an infected bite, rash, and death due to a road traffic accident.
Grade 3 and 4 AEs occurred in five (5%) patients and one (1%) patient, respectively (Supporting Information). Serious AEs were reported by five (5%) patients and were cellulitis, an infected bite, limb injury, a road traffic accident, anemia, and noncardiac chest pain; no serious AEs were considered possibly related to treatment.
Increased bilirubin was reported as an AE by two (2%) patients. No grade 3/4 AEs, serious AEs, or treatment discontinuations related to increased bilirubin were reported. All rash or photosensitivity AEs were grade 1/2 except for one grade 3 rash.
Of the laboratory abnormalities, treatment‐emergent bilirubin increases were isolated, asymptomatic, transient, and not associated with an increase in liver transaminases. One patient experienced a grade 3 bilirubin increase (not considered an AE). Asymptomatic, transient increases in amylase and lipase were seen; these were mostly mild to moderate in severity and were mostly not reported as AEs. Pancreatitis was not reported (Supporting Table S2).
Patient‐Reported Outcomes
At baseline, patients had more fatigue and depressive symptoms, greater impairment in functioning, and worse HRQoL than are commonly reported for the general population (Table 2).
All PRO scores improved from baseline to week 12 of follow‐up, with clinically important improvements seen in Fatigue Severity Scale and HRQoL (EuroQoL 5‐Dimensions visual analogue scale). HCV‐related symptom severity significantly reduced from baseline after 4 weeks of simeprevir + sofosbuvir treatment and continued to improve (Hepatitis C Symptom and Impact Questionnaire version 4 overall body system score). Depressive symptoms (Center for Epidemiologic Studies Depression scale) decreased during treatment, but the score improvement did not reach levels considered clinically important. For patients who achieved SVR12, improvements in fatigue severity and HRQoL were clinically important (Supporting Fig. S2 and Table S3; Supporting Information).
Discussion
Findings from the phase 3, open‐label, OPTIMIST‐2 study demonstrated that simeprevir + sofosbuvir for 12 weeks was efficacious and well tolerated in treatment‐naive and treatment‐experienced patients with HCV GT1 infection and cirrhosis. The primary objective of the study was met as simeprevir + sofosbuvir demonstrated superiority in SVR12 rates (83%) compared with the HC (70%).
The SVR12 data in OPTIMIST‐2 provide further evidence of the clinical effectiveness of simeprevir + sofosbuvir in HCV GT1–infected patients with cirrhosis. The SVR12 rate of 83% reported for patients with cirrhosis in this study is similar to the SVR12 rate of 86% (6/7) reported for 12 weeks of simeprevir + sofosbuvir without ribavirin in HCV GT1‐infected patients with cirrhosis in the COSMOS study.11 Furthermore, a prospective study in HCV GT1a–infected patients with cirrhosis (Child‐Pugh grade A) receiving simeprevir + sofosbuvir without ribavirin for 12 weeks reported SVR12 rates of 93%.12 Also, preliminary findings from real‐world evidence studies of simeprevir + sofosbuvir in HCV GT1–infected patients, including those with decompensated cirrhosis, demonstrated overall SVR12 rates of 75% (TRIO study)13 and 74% (HCV‐TARGET study).14
The OPTIMIST‐2 study used an SVR12 composite HC, an approach used in other IFN‐free studies involving treatment‐naive and treatment‐experienced patients with cirrhosis (ION‐1,15 ION‐2,16 and TURQUOISE II17).
SVR12 rates in prespecified subgroups of patients were explored to evaluate the potential benefit of treatment in these patients. SVR12 rates were higher for treatment‐naive patients (88%) than treatment‐experienced patients (79%). This is in contrast to secondary analyses in the phase 2 COSMOS study, which demonstrated high SVR12 rates for patients with cirrhosis regardless of prior treatment history, possibly due to smaller patient numbers in the COSMOS study.11 SVR12 rates were also higher for HCV GT1–infected patients without baseline Q80K polymorphisms than those with Q80K polymorphisms. This finding is in contrast with those from a prospective study in patients with cirrhosis which noted no differences due to baseline NS3 Q80K polymorphisms.12
Notably, in the current study, an SVR12 rate of 94% was observed for patients with albumin ≥40 g/L, and SVR12 rates ≥85% were observed for patients with IL28B CC or CT GTs and patients with platelet count ≥90,000/mm3. Similar SVR12 rates were observed for patients in this study with albumin ≥40 g/L regardless of the presence or absence of baseline Q80K polymorphisms. These high SVR12 rates suggest that successful treatment outcomes in these patient subgroups may be possible with the combination of simeprevir + sofosbuvir for 12 weeks, although the numbers in some of these subgroups were small.
The viral relapse rate in this study (13% overall) was higher in treatment‐experienced than treatment‐naive patients and was the primary reason that patients did not achieve SVR12. No predisposing baseline characteristics were identified. In the COSMOS study, two of six relapsers were patients with METAVIR scores of F4, both treated for 12 weeks; no patients with cirrhosis treated for 24 weeks relapsed, which suggests that some patients with cirrhosis may benefit from 24‐week treatment.11 The albumin cutoff level of 40 g/L could be useful in identifying these patients as patients with cirrhosis and albumin <40 g/L treated with simeprevir + sofosbuvir for 12 weeks in this study experienced higher rates of viral relapse than those with ≥40 g/L. In the United States, simeprevir + sofosbuvir is recommended for 24 weeks.18 In contrast, in the European Union, 12‐week treatment is recommended and patients are assessed on an individual case basis for extension to 24‐week treatment (with or without ribavirin).19 For patients with cirrhosis, European Union treatment guidelines recommend 12 weeks with ribavirin or extension to 24‐week treatment in patients for whom ribavirin is contraindicated.3
In this study, all patients with cirrhosis with baseline NS5A polymorphisms achieved SVR12, indicating that these polymorphisms had no impact on the efficacy of the simeprevir + sofosbuvir combination. Consistent with previous reports,8, 11, 20, 21 most patients with treatment failure had emerging NS3 mutations, while no NS5B mutations were observed.
The safety results in the OPTIMIST‐2 study demonstrate that simeprevir + sofosbuvir was well tolerated in the patient population evaluated. Safety findings were consistent with those observed in the OPTIMIST‐1 study22 and the COSMOS study,11 and no new safety issues were identified.
Patients enrolled in this study were experiencing more fatigue and depressive symptoms, greater impairment in functioning, and worse HRQoL at baseline than are commonly reported for the general population. PRO scores significantly improved from baseline as seen at the week 12 follow‐up visit coincident with the SVR12 assessment.
A strength of this study was the inclusion of patients with cirrhosis exclusively, as was the inclusion of a patient population that reflected clinical practice. The lack of a BMI eligibility cutoff and the platelet count eligibility cutoff of 50,000/mm3 were further strengths. Limitations of this study included the lack of a comparator arm, the open‐label nature of the trial, and the low patient numbers in some subgroups, which limited the conclusions that could be drawn.
In conclusion, simeprevir + sofosbuvir for 12 weeks was efficacious and well tolerated by treatment‐naive and treatment‐experienced patients with chronic HCV GT1 infection and cirrhosis.
Supporting information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28422/suppinfo.
Supporting Information
Acknowledgment
We thank all patients and their families for participating in this trial. We also thank the study site personnel and Quintiles (Clinical Research Organization, Clinical Trial Services) for their contributions to the conduct of this study. Medical writing assistance was provided by Kimberley Haines of Complete Medical Communications, funded by Janssen.
Potential conflict of interest: Dr. DeJesus consults, advises, and is on the speakers' bureau for Gilead. He consults and advises Janssen. Dr. Felizarta consults, is on the speakers' bureau, and received grants from Merck. He is on the speakers' bureau and received grants from AbbVie and Janssen. Dr. Godofsky advises and received grants from Janssen. He received grants from Gilead. Dr. Lawitz consults, advises, is on the speakers' bureau, and received grants from AbbVie, Bristol‐Myers Squibb, Gilead, and Janssen. He consults, advises, and received grants from Achillion, Enanta, Idenix, Merck, Novartis, Santaris, and Theravance. He consults and advises Regulus. He received grants from Boehringer Ingelheim, Eisai, Exelixis, Galactin, GenFit, GlaxoSmithKline, Hologic, Intercept, Nitto Denko, Romark, Roche, Salix, Synageva, Tacere, and Tobira. Dr. Kugelmas consults, advises, is on the speakers' bureau, and received grants from Janssen and Gilead. Dr. Poleynard is on the speakers' bureau for Gilead. Dr. Sheikh advises, is on the speakers' bureau, and received grants from Gilead and Bristol‐Myers Squibb. He advises and received grants from Intercept. He is on the speakers' bureau and received grants from AbbVie. He received grants from Johnson and Johnson and Merck. Dr. Tobias is on the speakers' bureau for Gilead, Bristol‐Myers Squibb, and AbbVie. Dr. Yoshida received grants from Janssen, Gilead, AbbVie, Hoffman LaRoche, Merck, Vertex, Boehringer Ingelheim, and Novartis. Dr. Ghalib received grants from AbbVie, Achillion, Anadys, Biolex, Boehringer Ingelheim, Bristol‐Myers Squibb, Evoke, Gilead, Idenix, Idera, Inhibitex, Janssen, Merck, Novartis, Pfizer, Pharmasett, Salix, Takeda, Vertex, Virochem, and Zymogenetics. Dr. De La Rosa is employed by and owns stock in Johnson & Johnson. Dr. Fevery is employed by and owns stock in Johnson & Johnson. Dr. Witek is employed by and owns stock in Johnson & Johnson. Dr. Scott is employed by and owns stock in Johnson & Johnson. Dr. Lenz is employed by and owns stock in Johnson & Johnson and Janssen. Dr. Kalmeijer is employed by and owns stock in Johnson & Johnson. Dr. Sinha is employed by Janssen.
Supported by Janssen.
[The copyright for this article was changed on January 30, 2017, after original online publication.]
REFERENCES
- 1. Westbrook RH, Dusheiko G. Natural history of hepatitis C. J Hepatol 2014;61:S58‐S68. [DOI] [PubMed] [Google Scholar]
- 2. Ferenci P. Treatment of hepatitis C in difficult‐to‐treat patients. Nat Rev Gastroenterol Hepatol 2015;12:284‐292. [DOI] [PubMed] [Google Scholar]
- 3. European Association for the Study of the Liver . Clinical practice guidelines: recommendations on treatment of hepatitis C 2015. http://www.easl.eu/research/our‐contributions/clinical‐practice‐guidelines/detail/recommendations‐on‐treatment‐of‐hepatitis‐c‐2015. Accessed May 5, 2015. [DOI] [PubMed]
- 4. American Association for the Study of Liver Diseases . HCV guidance: recommendations for testing, managing, and treating hepatitis C. www.hcvguidelines.org/full‐report‐view. Accessed May 5, 2015.
- 5. Kleinman L, Mannix S, Yuan Y, Kummer S, L'Italien G, Revicki D. Review of patient‐reported outcome measures in chronic hepatitis C. Health Qual Life Outcomes 2012;10:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Radloff LS. A self‐report depression scale for research in the general population. Appl Psychol Meas 1977;1:385‐401. [Google Scholar]
- 7. EuroQol Group . EuroQol—a new facility for the measurement of health‐related quality of life. Health Policy 1990;16:199‐208. [DOI] [PubMed] [Google Scholar]
- 8. Forns X, Lawitz E, Zeuzem S, Gane E, Bronowicki JP, Andreone P, et al. Simeprevir with peginterferon and ribavirin leads to high rates of SVR in patients with HCV genotype 1 who relapsed after previous therapy: a phase 3 trial. Gastroenterology 2014;146:1669‐1679. [DOI] [PubMed] [Google Scholar]
- 9. Lawitz E, Mangia A, Wyles D, Rodriguez‐Torres M, Hassanein T, Gordon SC, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013;368:1878‐1887. [DOI] [PubMed] [Google Scholar]
- 10. Zeuzem S, Berg T, Gane E, Ferenci P, Foster GR, Fried MW, et al. Simeprevir (TMC435) in treatment‐experienced HCV genotype 1 patients; the phase IIb, randomized controlled ASPIRE trial. Gastroenterology 2014;146:430‐441. [DOI] [PubMed] [Google Scholar]
- 11. Lawitz E, Sulkowski MS, Ghalib R, Rodriguez‐Torres M, Younossi ZM, Corregidor A, et al. Simeprevir plus sofosbuvir, with or without ribavirin, to treat chronic infection with hepatitis C virus genotype 1 in non‐responders to pegylated interferon and ribavirin and treatment‐naive patients: the COSMOS randomised study. Lancet 2014;384:1756‐1765. [DOI] [PubMed] [Google Scholar]
- 12. Pearlman BL, Ehleben C, Perrys M. The combination of simeprevir and sofosbuvir is more effective than that of peginterferon, ribavirin, and sofosbuvir for patients with hepatitis C–related Child's class A cirrhosis. Gastroenterology 2015;148:762‐770. [DOI] [PubMed] [Google Scholar]
- 13. Flamm S, Bacon B, Dieterich D, Kowdley K, Lawitz E, Milligan S, et al. Evaluation of efficacy of sofosbuvir and simeprevir‐based regimens in a real‐life population of HCV patients with cirrhosis; data from the TRIO network. Poster 983 presented at the American Association for the Study of Liver Disease (AASLD), Boston, MA, November 7‐11, 2014.
- 14. Reddy KR, Lim JK, Kuo A, Di Bisceglie AM, Vargas HE, Galati JS, et al. All oral HCV therapy is safe and effective in patients with decompensated cirrhosis: report from HCV‐TARGET. Oral presentation O007 presented at the European Association for the Study of the Liver, Vienna, Austria, April 22‐26, 2015.
- 15. Afdhal N, Zeuzem S, Kwo P, Chojkier M, Gitlin N, Puoti M, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med 2014;370:1889‐1898. [DOI] [PubMed] [Google Scholar]
- 16. Afdhal N, Reddy KR, Nelson DR, Lawitz E, Gordon SC, Schiff E, et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med 2014;370:1483‐1493. [DOI] [PubMed] [Google Scholar]
- 17. Poordad F, Hezode C, Trinh R, Kowdley KV, Zeuzem S, Agarwal K, et al. ABT‐450/r‐ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med 2014;370:1973‐1982. [DOI] [PubMed] [Google Scholar]
- 18. Olysio (simeprevir) US prescribing information. Titusville, NJ: Janssen Research & Development; October 2015. http://www.olysio.com/shared/product/olysio/prescribing‐information.pdf.
- 19. European Medicines Agency . Summary of product characteristics. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/002777/WC500167867.pdf. Accessed May 27, 2015.
- 20. Jacobson IM, Dore GJ, Foster GR, Fried MW, Radu M, Rafalskiy VV, et al. Simeprevir (TMC435) with pegylated interferon alfa 2a plus ribavirin in treatment‐naive patients with chronic hepatitis C virus genotype 1 infection (QUEST‐1): a phase 3, randomised, double‐blind, placebo‐controlled trial. Lancet 2014;384:403‐413. [DOI] [PubMed] [Google Scholar]
- 21. Manns M, Marcellin P, Poordad F, de Araujo ES, Buti M, Horsmans Y, et al. Simeprevir with pegylated interferon alfa 2a or 2b plus ribavirin in treatment‐naive patients with chronic hepatitis C virus genotype 1 infection (QUEST‐2): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet 2014;384:414‐426. [DOI] [PubMed] [Google Scholar]
- 22. Kwo P, Gitlin N, Nahass R, Bernstein B, Rojter S, Schiff E, et al. A phase 3, randomised, open‐label study to evaluate the efficacy and safety of 8 and 12 weeks of simeprevir (SMV) plus sofosbuvir (SOF) in treatment‐naive and ‐experienced patients with chronic HCV genotype 1 infection without cirrhosis: OPTIMIST‐1 [Abstract]. J Hepatol 2015;62(Suppl. 2):S270. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.28422/suppinfo.
Supporting Information