

Summary
Intracellular pathogens have evolved mechanisms to ensure their survival and development inside their host cells. Here, we show that glucose is a pivotal modulator of hepatic infection by the rodent malaria parasite Plasmodium berghei and that glucose uptake via the GLUT1 transporter is specifically enhanced in P. berghei‐infected cells. We further show that ATP levels of cells containing developing parasites are decreased, which is known to enhance membrane GLUT1 activity. In addition, GLUT1 molecules are translocated to the membrane of the hepatic cell, increasing glucose uptake at later stages of infection. Chemical inhibition of GLUT1 activity leads to a decrease in glucose uptake and the consequent impairment of hepatic infection, both in vitro and in vivo. Our results reveal that changes in GLUT1 conformation and cellular localization seem to be part of an adaptive host response to maintain adequate cellular nutrition and energy levels, ensuring host cell survival and supporting P. berghei hepatic development.
Introduction
Glucose is the primary source of energy and a key substrate for most cells. Glucose and other carbohydrates are transported into cells by members of a family of integral membrane glucose transporter (GLUT) molecules. To date, 14 members of this family, also called the solute carrier 2A proteins, have been identified in humans, which are divided on the basis of transport characteristics and sequence similarities into several families (Classes I to III) [reviewed in (Karim et al., 2012; Mueckler and Thorens, 2013)]. GLUT1 is a class I facilitative glucose transporter expressed in liver cells (Tal et al., 1990) and overexpressed in various tumours (Smith, 1999). GLUT1 expression is highest in the human erythrocyte membrane, and has been shown to play a critical role in cerebral glucose uptake (Koranyi et al., 1991). GLUT1 is also a receptor for the human T cell leukaemia virus (Manel et al., 2003), and GLUT1‐mediated glucose transport in T‐cells has been shown to regulate Human immunodeficiency virus infection (Loisel‐Meyer et al., 2012).
Mammalian infection by the malaria parasite is initiated when Plasmodium sporozoites, injected through the bite of an infected mosquito, cross the endothelium of the liver sinusoids and enter the liver. Sporozoites then traverse a few hepatocytes before productively invading a final one, inside which they asymptomatically differentiate into exoerythrocytic forms (EEFs) that originate thousands of red blood cell‐infective merozoites (Prudencio et al., 2006). Merozoites are eventually released to the bloodstream, initiating the blood stage of infection, and giving rise to malaria symptoms. The liver stage of a mammalian infection by Plasmodium is an obligatory uni‐directional step in the parasite's progression towards the symptomatic, erythrocytic phase of its life cycle.
Blood stages of malarial parasites are dependent on glycolysis, employed as their main energy source of Adenosine triphosphate (ATP) production (Pfaller et al., 1982; Vander Jagt et al., 1990; Kirk et al., 1996), a process that has recently been modelled for the human malaria parasite P. falciparum (Penkler et al., 2015). Glucose has been shown to be transported from human blood plasma into the erythrocyte cytosol by GLUT1 (Hellwig and Joost, 1991), and then taken up by the parasite via a parasite‐encoded facilitative hexose transporter (PfHT) (Woodrow et al., 2000; Joet et al., 2003), which limits the rate of glucose entry into the parasite's glycolytic pathways (Tjhin et al., 2013). These findings support novel chemotherapeutic interventions that target PfHT (Feistel et al., 2008; Slavic et al., 2011b). Studies in the rodent malaria model, P. berghei, further found that the orthologous hexose transporter (PbHT) is expressed not only throughout the parasite's development within the mosquito vector, but also during liver and transmission stages of P. berghei (Slavic et al., 2010; Slavic et al., 2011a). During their extensive hepatic replication, developing Plasmodium parasites require the availability of large amounts of nutrients and energy. The availability of appropriate amounts of glucose in infected hepatic cells is thus expected to play an essential role in the development of liver stage Plasmodium parasites. Inhibition of PbHT by compound 3361, a specific inhibitor of plasmodial HTs, impairs hepatic P. berghei development (Slavic et al., 2011a), supporting the essentiality of glucose uptake for Plasmodium liver stages. Accordingly, a recent study established glucose concentration in the culture medium as a key factor for liver stage parasite development (Itani et al., 2014).
In this study, we employed rodent P. berghei parasites, a well‐established model of malaria infection (Prudencio et al., 2011), to elucidate the uptake and utilisation of glucose by Plasmodium liver stages. We investigated the glucose requirements during the course of P. berghei development in hepatic cells as well as the host molecular receptors involved in glucose uptake by those cells. We demonstrate that P. berghei infection leads to intracellular ATP depletion and enhances the translocation of GLUT1 to the cell membrane of infected hepatoma cells, allowing the uptake of significantly higher amounts of glucose compared with non‐infected cells. We further show that glucose plays an essential role during the liver stage of infection by the malaria parasite, modulating its liver stage development both in vitro and in vivo.
Results
Effect of glucose on P. berghei hepatic infection
In order to evaluate glucose requirements throughout the liver stage of infection, Huh7 cells, a human hepatoma cell line, were infected with luciferase‐expressing P. berghei sporozoites in the presence of varying concentrations of glucose. Glucose concentrations ranged from 1.25 to 20 mM, which includes and expands the physiological range of glucose concentrations, 2.5 to 10 mM (Shrayyef and Gerich, 2010). Luminescence intensity, a correlate of parasite load (Ploemen et al., 2009), and cell viability were measured near the end of hepatic parasite development, at 48 h post‐infection (hpi). Our results show that an increase in glucose availability correlates with an increase of overall Plasmodium infection (Fig. 1A). In contrast, any glucose concentration below the standard medium ≈ 10 mM concentration significantly impairs infection (Fig. 1A), demonstrating that glucose is required for a successful hepatic Plasmodium infection. Cell viability was not affected by the presence of an excess of glucose in the medium, but decreased for 2.5 and 1.25 mM of glucose (Fig. 1A). Crucially, decreasing the glucose concentration from 10 to 5 mM significantly decreases parasite load but has no impact on cell viability.
Figure 1.
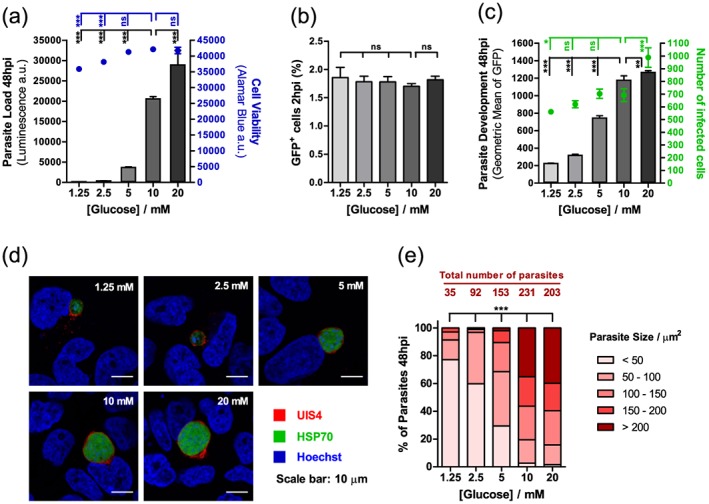
Glucose availability impacts P. berghei hepatic infection. A. Huh7 cells were infected with luciferase‐expressing P. berghei sporozoites, and the culture medium was replaced 2 hpi by medium with different concentrations of glucose. Parasite load (luminescence) and cell viability were assessed after 48 h. Representative experiment out of two independent experiments. Error bars represent standard deviation (SD). One‐way analysis of variance (ANOVA) with post‐test Dunnett. B. The culture medium of Huh7 cells was replaced by medium with different concentrations of glucose 1 h prior to infection with green fluorescent protein (GFP)‐expressing sporozoites. Cell invasion was quantified by determining the percentage of GFP+ cells 2 hpi by flow cytometry. Representative experiment out of two independent experiments. Error bars represent SD. One‐way ANOVA with post‐test Dunnett. C. Huh7 cells were infected with GFP‐expressing P. berghei sporozoites, and the culture medium was replaced 2 hpi by medium with different concentrations of glucose. Parasite development and the total number of infected cells were assessed by flow cytometry by determining the fluorescence intensity and the number of GFP+ cells at 48 hpi, respectively. Representative experiment out of five independent experiments. Error bars represent SD. One‐way ANOVA with post‐test Dunnett. D. Representative confocal images of Huh7 cells infected with sporozoites and incubated in medium with different concentrations of glucose for 48 h. Cells were immunostained with anti‐UIS4 (red), which localises to the parasitophorous vacuole membrane, anti‐heat shock protein (HSP)70 (green), which localises to the parasite's cytoplasm, and Hoechst (blue), a nuclear stain. Scale bar, 10 µm. E. For each glucose concentration, the exoerythrocytic forms in the fluorescence microscopy images acquired were counted, and their area was determined. Exoerythrocytic forms were divided into five different size classes. Pool of two independent experiments. Chi‐square test. ns, not significant; * P < 0.05, ** P < 0.01 and *** P < 0.001.
In order to dissect how glucose influences hepatic infection, we employed an established flow cytometry‐based approach that makes use of green fluorescent protein (GFP)‐expressing P. berghei parasites to determine the number of infected cells and to measure parasite development (Prudencio et al., 2008). The analysis of cells 2 h after sporozoite addition, when the invasion process is virtually completed (Prudencio et al., 2008), shows that glucose concentration does not affect the ability of parasites to traverse (data not shown) or invade (Fig. 1B) cells. Conversely, when cells were analysed 48 hpi, a glucose‐dependent increase in parasite development was observed (Fig. 1C). These results also showed that the number of infected cells is higher at 20 mM glucose concentration, and lower at glucose concentrations below the physiological range, suggesting that glucose availability influences the survival of infected cells. These results were further confirmed by immunofluorescence microscopy analysis, which demonstrated that parasite size is proportional to the amount of glucose in the medium and that the survival of liver stage parasites 48 hpi depends on the availability of glucose (Fig. 1D, E). In fact, at low glucose concentrations, not only are most parasites very small (<50 µm2) but also the number of infected cells is very low. Increasing concentrations of glucose lead to higher numbers of parasites and favour parasite growth, with approximately 40% of the parasites being larger than 200 µm2 at 10 and 20 mM glucose (Fig. 1E).
Whereas hepatoma cells are highly dependent on glucose uptake for ATP production through glycolysis (Warburg et al., 1927; Kroemer and Pouyssegur, 2008), primary hepatocytes are able to store glucose as glycogen and use oxidative phosphorylation to produce ATP. Accordingly, the requirements for glucose uptake by primary hepatic cells are significantly lower than those displayed by tumorigenic cells (O'Neil et al., 2005). In agreement with this, we showed that the decreased cell viability noted in hepatoma cells at 2.5 and 1.25 mM glucose was not observed in primary hepatocytes, even upon complete removal of glucose. Crucially, however, our data also clearly show that overall infection of mouse primary hepatocytes by P. berghei is significantly decreased in the absence of glucose in the medium (Fig. S1). Overall, these results demonstrate that glucose availability plays a crucial role in the liver stage development and survival of Plasmodium parasites.
Glucose uptake is specifically increased in hepatic cells containing replicating P. berghei parasites
Having demonstrated a requirement for glucose during liver stage P. berghei development, we then sought to determine whether glucose uptake is specifically enhanced in infected cells and whether this is linked to parasite replication within these cells. To this end, we monitored by flow cytometry the uptake of a fluorescent glucose derivative, 2‐NBDG (2‐deoxy‐2‐[(7‐nitro‐2,1,3‐benzoxadiazol‐4‐yl)amino]‐D‐glucose) (O'Neil et al., 2005; Yamada et al., 2007) by P. berghei‐infected cells. Huh7 cells were incubated with 2‐NBDG and analysed at different time points following addition of red fluorescence protein (RFP)‐expressing P. berghei sporozoites. The uptake of 2‐NBDG was measured as an increase in green fluorescence intensity, whereas parasite development was monitored as a function of red fluorescence intensity. The latter enables the population of cells containing replicating parasites to be distinguished from a smaller population of cells containing parasites that do not develop in vitro (Prudencio et al., 2008) (Fig. S2). Our results clearly show that glucose uptake is highly increased in cells containing developing parasites, from 30 hpi onwards (Fig. 2A). A smaller increase in glucose uptake is also observed at around the same time in non‐infected cells and in cells containing non‐developing parasites, present in the same well (Fig. 2A). Live fluorescence microscopy analysis of non‐infected and infected Huh7 cells 48 hpi (Fig. S3A) confirmed that 2‐NBDG fluorescence intensity is significantly higher in infected than in non‐infected cells (Fig. S3B) and further revealed the uptake of 2‐NBDG by the developing parasite (Fig. S3B).
Figure 2.
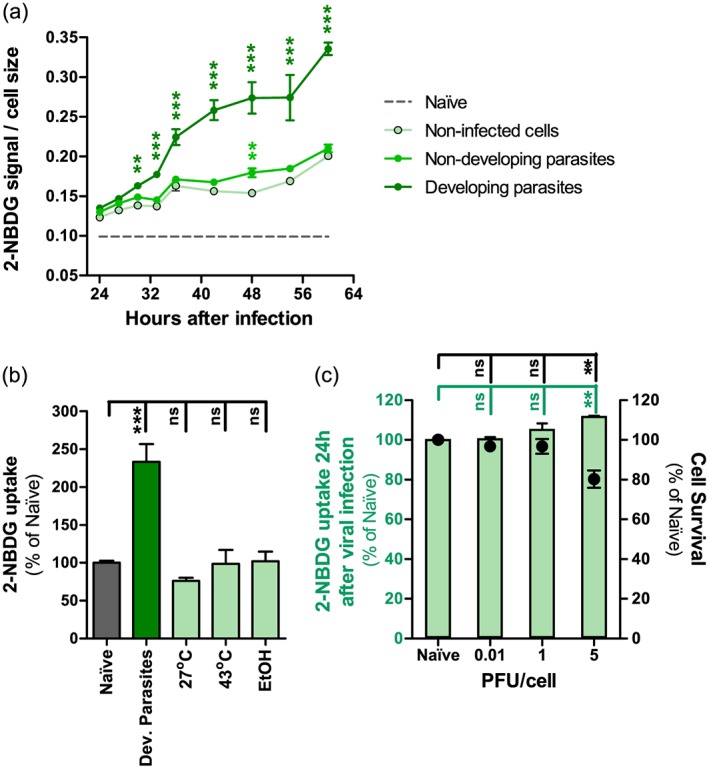
Glucose uptake is specifically increased in hepatoma cells containing developing P. berghei parasites. A. 2‐NBDG uptake by different cell populations, normalised to cell size, at different time points of infection, assessed by flow cytometry. Error bars represent standard deviation. Two‐way analysis of variance (ANOVA) with post‐test Bonferroni. B. 2‐NBDG uptake by naïve Huh7 cells, cells containing developing P. berghei parasites at 48 hpi, and cells subjected to low‐temperature, high‐temperature and ethanol‐induced oxidative stresses, assessed by flow cytometry. Pool of two independent experiments. Error bars represent standard deviation. One‐way ANOVA with post‐test Dunnet. C. 2‐NBDG uptake by naïve Huh7 cells and cells infected with MHV‐68 at different multiplicity of infection, assessed by flow cytometry. Pool of two independent experiments. Error bars represent Standard error of the mean (SEM). One‐way ANOVA with post‐test Dunnet. ns, not significant; ** P < 0.01 and *** P < 0.001. PFU, plaque‐forming unit.
Because it is known that glucose uptake may be influenced by factors such as feeding/fasting, exposure to low/high temperature, exercise, oxidative stress, several liver pathologies, such as steatosis and non‐alcoholic fatty liver disease and liver infections, for example, by hepatitis C virus (Cunningham et al., 1985; Pencek et al., 2004; Bitar et al., 2005; Bechmann et al., 2012; Moore et al., 2012; Vidyashankar et al., 2012; Yu et al., 2013), we sought to assess the specificity of the P. berghei‐induced increase in glucose uptake by infected Huh7 cells. To this end, we measured 2‐NBDG uptake by Huh7 cells subjected to conditions known to induce low‐temperature stress in various types of mammalian cells (Fujita, 1999), high‐temperature stress in a chicken hepatocellular carcinoma cell line (Sun et al., 2015) or oxidative stress in rat hepatocytes (Sergent et al., 2005; Nourissat et al., 2008) (Fig. 2B). Finally, we infected Huh7 cells with murine gammaherpesvirus 68 (MHV‐68) (Collins et al., 2009), and measured 2‐NBDG uptake at 24 hpi and at different infection loads (Fig. 2C). The efficacy of infection was confirmed by flow cytometry employing YFP‐expressing MHV‐68 in a parallel experiment (Fig. S4). Our data clearly show that there was little or no effect on 2‐NBDG uptake into the cells by any of the stress‐inducing treatments employed, or by viral infection. This indicates that the increase in glucose uptake by P. berghei‐infected cells is not the result of a non‐specific response to stress or to infection (Fig. 2B, C). Overall, our results show that P. berghei development inside Huh7 leads to a specific and marked enhancement of glucose uptake by these cells. The observed effect appears rather unique to malaria parasites, as other intracellular pathogens, such as Toxoplasma gondii do not depend on host‐derived glucose (Blume et al., 2009), and hepatitis C virus replication actually suppresses cellular glucose uptake through down‐regulation of cell surface expression of glucose transporters (Kasai et al., 2009).
RNA interference screen implicates GLUT1 in glucose uptake by P. berghei‐infected hepatic cells
The results so far suggest that infection of hepatic cells by Plasmodium modulates the uptake of glucose by these cells, possibly through the enhancement of the activity of membrane glucose transporters in the cell. To ascertain this, we decided to carry out a small‐scale RNA interference (RNAi) screen to evaluate the effect of the down‐regulation of the expression of 5 transmembrane glucose transporters upon P. berghei infection (Table S1). The screen included all class I GLUT genes (GLUT1‐4) as well as GLUT9, a major regulator of influx in HepG2 hepatoma cells (Takanaga et al., 2008).
Huh7 cells stably expressing shRNA sequences targeting the selected genes were constructed to be used throughout the RNAi screen. The screen proceeded in three consecutive steps, employing different established methods to measure hepatic infection by P. berghei. In the first step of the screen, three shRNA sequences were used to silence each of the selected genes and the resulting stable cell lines were infected with luciferase‐expressing P. berghei sporozoites. Forty‐eight h later, the parasite load in these cells was determined by measuring the bioluminescence of cell lysates. Each sequence was used in at least four independent experiments, and an shRNA targeting Scavenger receptor class B type I (SR‐BI) was employed as a positive control for infection decrease, as this receptor has been implicated in hepatic cell invasion by P. berghei parasites (Rodrigues et al., 2008). GLUT1 emerged as the gene whose knock‐down (KD) leads to the strongest decrease in infection (Fig. 3A), with GLUT4 and GLUT9 KD yielding a more moderate effect (Fig. 3A). We then carried out the second step of the RNAi screen, where only GLUT1, GLUT4 and GLUT9 were silenced prior to infection with GFP‐expressing P. berghei parasites. The extent of parasite development was assessed 48 h later by flow cytometry determination of GFP intensity (Prudencio et al., 2008). The results showed that KD of GLUT1 leads to the strongest impairment of parasite development among the three genes assessed (Fig. 3B). Therefore, GLUT1 was selected for the final confirmation screening step using the three different shRNA sequences targeting the GLUT1 gene and assessing parasite development by immunofluorescence microscopy (Fig. 3C). Interestingly, the imaging data demonstrated that parasite size correlates with the mRNA levels of GLUT1, suggesting that GLUT1‐mediated glucose uptake is required for the parasite's development (Fig. 3C and Table S1).
Figure 3.
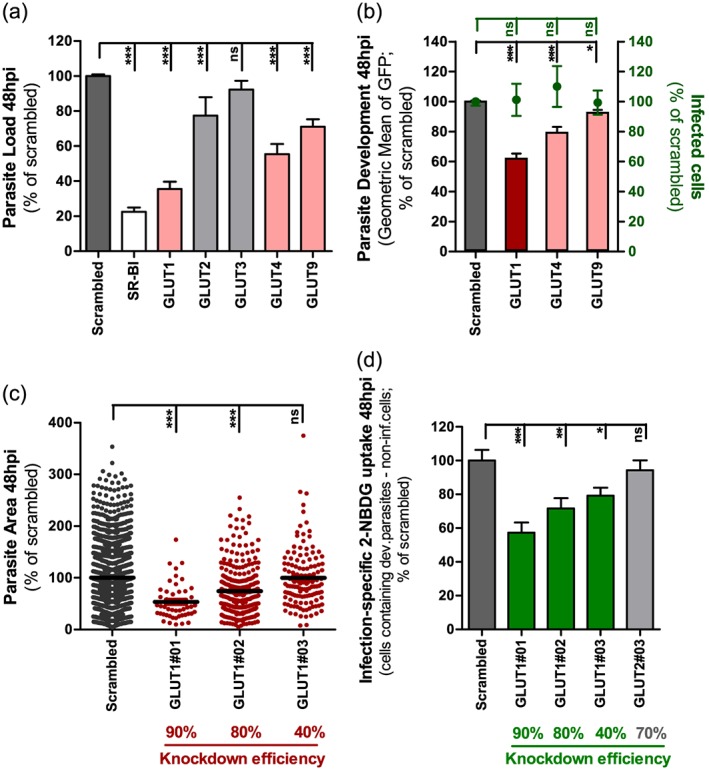
GLUT1 knockdown significantly impairs P. berghei liver stage development and glucose uptake by P. berghei‐infected cells. A. Huh7 cells with stable knockdown of one of five GLUT receptors were infected with luciferase‐expressing P. berghei sporozoites and parasite load was assessed by luminescence measurement 48 h later. A scrambled shRNA sequence was used as a negative control and a cell line with stable knockdown of SR‐BI as positive control. Pool of four independent experiments. Error bars represent SEM. B. Huh7 cells with the knockdown of GLUT1, GLUT4 or GLUT9 were infected with green fluorescent protein (GFP)‐expressing P. berghei sporozoites. The percentage of infected cells and parasite development were assessed by flow cytometry at 48 hpi. Pool of three independent experiments. Error bars represent SEM. C. Quantification of the area of the EEFs in each of the three different cell lines with GLUT1 knockdown at 48 hpi by immunofluorescence microscopy. The knockdown efficiency of each shRNA sequence is indicated. Pool of three independent experiments. D. Difference of 2‐NBDG uptake in the several cell lines with GLUT1 knockdown by developing parasites‐containing cells and non‐infected cells at 48 hpi, assessed by flow cytometry. A cell line with the knockdown of GLUT2 was used as control. Pool of four independent experiments. Error bars represent SEM. All panels: one‐way analysis of variance (ANOVA) with post‐test Dunnett. ns, not significant, * P < 0.05, ** P < 0.01 and *** P < 0.001.
To further establish a link between the uptake of glucose by GLUT1 and Plasmodium infection, we assessed the difference in 2‐NBDG uptake between infected and non‐infected Huh7 cells expressing the same three GLUT1‐targeting shRNA sequences as earlier. This difference was drastically reduced in the GLUT1‐KD stable cell lines, relative to scramble‐transduced control cells, a decrease that is proportional to the extent of the down‐modulation of GLUT1 (Fig. 3D). As an additional control, we assessed 2‐NBDG uptake following KD of GLUT2, whose expression by Huh7 cells we confirmed (2.6 ± 0.8‐fold lower than GLUT1), and can therefore contribute to glucose uptake by these cells. Nevertheless, our results showed that, contrary to GLUT1 the down‐modulation of the expression of GLUT2 does not affect 2‐NBDG uptake by cells containing developing parasites (Fig. 3D). We also confirmed that GLUT1 KD does not affect glucose uptake by non‐infected cells except for a small decrease in cells where the down‐modulation of GLUT1 expression is most pronounced (Fig. S5).
Finally, we assessed infection, as well as 2‐NBDG uptake by infected cells, in the presence of WZB117, a specific inhibitor of GLUT1‐mediated glucose transport (Liu et al., 2012). Our results provide a chemical validation of the genetic approach (Fig. 3), as they show a clear dose‐dependent effect of WZB117 on overall P. berghei infection of Huh7 cells (Fig. 4A), on parasite development (Fig. 4B), and on the difference in 2‐NBDG uptake between cells infected with developing parasites and non‐infected cells (Fig. 4C). Additionally, we showed that P. berghei infection of mouse primary hepatocytes is significantly impaired by the addition of 100 μM WZB117 to the cell culture medium (Fig. S6). Most importantly, intra‐peritoneal administration of WZB117 to mouse models of Plasmodium infection significantly decreases the parasite load in the livers of P. berghei‐infected mice relative to vehicle‐treated control animals (Fig. 4D). Immunofluorescence microscopy analysis of liver sections of these mice (Fig. 4E) revealed a significant decrease in parasite areas (Fig. 4F) and numbers (Fig. 4G) in WZB117‐treated animals. These results indicate that GLUT1 inhibition by WZB117 leads to the inhibition of the hepatic parasite's development, as well as to its decreased survival in vivo.
Figure 4.
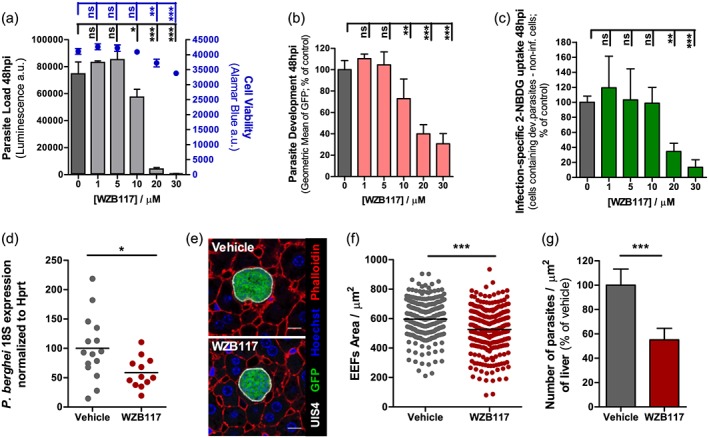
Chemical inhibition of GLUT1‐mediated glucose transport by WZB117 affects P. berghei hepatic infection in vitro (A‐C) and in vivo (D‐G). A. Huh7 cells were infected with luciferase‐expressing P. berghei sporozoites and 2 h later the culture medium was replaced by medium with increasing concentrations of WZB117. Parasite load (luminescence) and cell viability were assessed at 48 hpi. Representative experiment out of two independent experiments. Error bars represent standard deviation (SD). One‐way analysis of variance (ANOVA) with post‐test Dunnett. B. Huh7 cells were infected with red fluorescent protein‐expressing P. berghei sporozoites and 2 h later the culture medium was replaced by medium with increasing concentrations of WZB117. Parasite development was assessed by flow cytometry at 48 hpi. Pool of two independent experiments. Error bars represent SD. One‐way ANOVA with post‐test Dunnett. C. 2‐NBDG uptake by red fluorescent protein‐expressing P. berghei‐infected cells at 48 hpi after treatment with increasing concentrations of WZB117, assessed by flow cytometry. Pool of two independent experiments. One‐way ANOVA with post‐test Dunnett. D. C57BL/6 mice received three doses of 10 mg/kg of WZB117 i.p., the first immediately before infection with P. berghei sporozoites and the other two at 15 and 30 hpi. Parasite load was assessed at 44 hpi by qPCR. Pool of three independent experiments. Vehicle: n = 15 mice; WZB117‐treated: n = 12 mice. Two‐tailed Mann–Whitney test. E. Fifty µm sections of one liver lobe from one mouse of each experimental group were stained with anti‐UIS4 (white), anti‐GFP (green), phalloidin (red) and Hoechst (blue). Representative confocal images of P. berghei parasites in both experimental groups of mice. Scale bar, 10 µm. F. Area of the parasites in the control (vehicle) or the WZB117‐treated mouse, determined by immunofluorescence microscopy. Representative experiment out of two independent experiments. Two‐tailed Mann–Whitney test. G. Number of parasites per area of liver in control and WZB117‐treated mice. Representative experiment out of two independent experiments. Error bars represent SD. Two‐tailed Mann–Whitney test. ns, not significant; * P < 0.05, ** P < 0.01 and *** P < 0.001.
GLUT1 expression is not enhanced in Plasmodium‐infected cells
Having established a role for GLUT1 in the specific uptake of glucose by infected cells, we hypothesised that Plasmodium infection might lead to an increase in the expression of that transporter. To address this, we compared GLUT1 expression levels in non‐infected and infected Huh7 cells at 6, 30 and 48 hpi with GFP‐expressing P. berghei parasites. Following the separation of infected and non‐infected cells by fluorescence‐activated cell sorting (FACS) (Albuquerque et al., 2009), cells were analysed by quantitative real‐time polymerase chain reaction (qPCR), employing GLUT1‐specific primers (Table S2). The data showed no significant differences in GLUT1 expression between infected and non‐infected cells, at the selected time points (Fig. S7). As such, we concluded that the increase in GLUT1‐mediated glucose uptake by Plasmodium‐infected cells does not result from an infection‐induced enhancement of the mRNA expression of this transporter.
P. berghei development leads to cytoplasmic ATP depletion
It has been shown that GLUT1 has a cytoplasmic pocket that is postulated to allow the binding of ATP, which in turn induces conformational changes that inhibit GLUT1‐mediated glucose transport (Cloherty et al., 1996; Levine et al., 1998). This modulation of GLUT1 conformation by ATP is counteracted by the binding of Adenosine monophosphate (AMP) and Adenosine diphosphate (ADP) to the same site in GLUT1 (Blodgett et al., 2007). We thus wondered whether the observed increase in glucose uptake by Plasmodium‐infected cells would correlate to a decrease in ATP levels in these cells. To investigate this, we employed a fluorescence resonance energy transfer‐based indicator for ATP (ATeam), composed of the ε subunit of the bacterial FoF1‐ATP synthase sandwiched between the cyan‐fluorescent proteins and Venus fluorescent proteins (CFP and Venus, respectively), which enables measurement of intracellular ATP levels as a function of the Venus/CFP ratio (Imamura et al., 2009). Using RFP‐expressing P. berghei parasites and live fluorescence microscopy, we specifically monitored ATP levels in non‐infected and infected cells at 30 and 48 hpi. Interestingly, we observed that the ATP levels of infected cells were significantly lower than those of naïve cells (Fig. 5A). In agreement with the results in Fig. 2A, which show that non‐infected cells take up more 2‐NBDG than naïve cells, we also detected a smaller but significant ATP reduction in the non‐infected cells (Fig. 5A), when compared with naïve cells. These data indicate that liver stage parasite development results in a decrease in the ATP available inside the host cell, presumably leading to the well‐described ATP/ADP/AMP‐driven conformational changes in GLUT1 (Cloherty et al., 1996; Levine et al., 1998; Blodgett et al., 2007).
Figure 5.
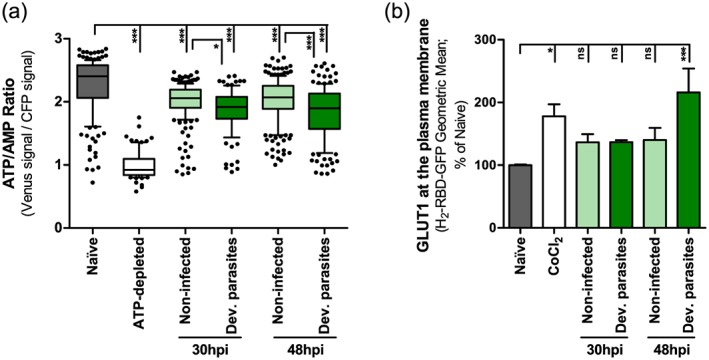
P. berghei development inside Huh7 cells results in a decreased ATP/AMP ratio and increased GLUT1 content at the plasma membrane. A. Huh7 cells were transfected with an ATP probe, AT1.03, and infected with red fluorescent protein‐expressing P. berghei sporozoites. Thirty and 48 hpi, the Venus/cyan‐flourescent proteins (CFP) emission ratio in the cytoplasm of each individual cell (non‐infected and cells containing developing parasites) was calculated from the fluorescent images acquired in both channels. Huh7 cells incubated in RPMI without glucose with 10 mM of galactose and 10 μM of Oligomycin were used as positive controls for ATP depletion. Pool of 2 and 3 independent experiments for the 30 and 48 h time points, respectively. B. Huh7 cells were infected with red fluorescent protein‐expressing sporozoites and, at 30 and 48 hpi, were incubated with the HRBD‐EGFP peptide that binds specifically to GLUT1 at the surface of the cells and were analysed by flow cytometry. Huh7 cells incubated overnight in complete RPMI with 250 μM of CoCl2 were used as positive controls. Pool of 2 and 4 independent experiments for the 30 and 48 h time points, respectively. Error bars represent SEM. Both panels: one‐way ANOVA with post‐test Tukey. ns, not significant; * P < 0.05 and *** P < 0.001.
P. berghei infection leads to GLUT1 translocation to the plasma membrane
In order to compare the amount of GLUT1 present on the surface of infected and non‐infected cells, Huh7 cells were infected with RFP‐expressing P. berghei parasites and incubated with the fusion peptide HRBD‐EGFP (consisting of the Receptor Binding Domain of the human T cell leukaemia virus fused to enhanced green fluorescent protein), which has been shown to specifically bind to GLUT1 at the extracellular surface of the cell plasma membrane (Manel et al., 2003; Kinet et al., 2007). CoCl2 was used as a positive control as it has been shown to enhance GLUT1 translocation to the plasma membrane (Koseoglu and Beigi, 1999). Flow cytometry analysis showed a similar increase in the amount of plasma membrane‐located GLUT1 in both non‐infected and infected cells at 30 hpi, relative to naïve cells (Fig. 5B). However, at 48 hpi, the amount of GLUT1 at the cell surface is specifically and significantly enhanced in infected cells (Fig. 5B), an increase whose magnitude is consistent of that observed upon stimulation of Rat2 cells by 12‐O‐tetradecanoyl‐phorbol‐13‐acetate (TPA) (Lee et al., 2015). Our results suggest that an enhancement of GLUT1 availability at the plasma membrane facilitates the uptake of glucose required for the later stages of parasite development, the time at which there is a dramatic increase in glucose uptake (Fig. 2A). Overall, our data support the notion that increased glucose uptake by P. berghei‐infected hepatic cells is mediated by GLUT1, which becomes activated by the ATP depletion and translocated into the membrane of the infected cell (Fig. 6).
Figure 6.
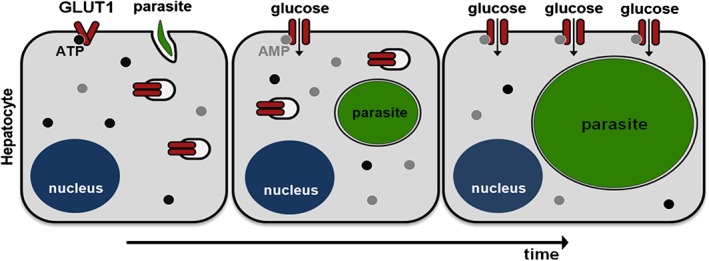
Proposed model of GLUT1‐mediated glucose uptake during hepatic infection by Plasmodium. Following the invasion of hepatic cells by Plasmodium parasites and the establishment of infection (left), parasites initiate a process of dramatic intracellular replication. Parasite development reduces the amount of host intracellular glucose, leading to depletion of the ATP pool and binding of ADP/AMP to GLUT1 transporters at the plasma membrane. This results in their activation (middle). At later stages of infection, GLUT1 transporters present in the host cell's cytoplasmic compartments are translocated to the cell membrane, leading to further enhancement of glucose uptake (right).
Discussion
Plasmodium parasites require large amounts of nutrients for their extensive replication inside liver cells. Here, we show that glucose uptake is significantly increased in infected hepatic cells, through the enhanced action and translocation of the GLUT1 membrane transporter.
Glucose is ubiquitously used as the common currency of metabolism, and the ability to transport this hexose across the plasma membrane is a feature of nearly all cells (Mueckler, 1994). Although GLUT2 is the major glucose transporter of hepatocytes, where it is involved in glucose uptake and release in the fed and fasted states, respectively (Thorens et al., 1990; Mueckler and Thorens, 2013), GLUT1 is also present in the liver (Karim et al., 2012), where it is transcribed and expressed by both periportal and perivenular hepatocytes. Of note, GLUT2 has a high capacity but a low affinity for glucose, with a K m value (the concentration of glucose at which transport is half of its maximal value) in the order of 17 mM (Uldry et al., 2002). GLUT1, on the other hand, has a higher affinity for glucose with a K m value of ~3 mM (Uldry et al., 2002), which is closer to that of PfHT (~1 mM, (Woodrow et al., 2000)). Thus, it is tempting to speculate that GLUT1 is better matched to supply glucose to the parasite, especially if local glucose concentrations decrease towards the K m of GLUT1. Interestingly, despite being expressed by all hepatocytes, membrane localization of GLUT1 under basal conditions is restricted to hepatocytes proximal to the hepatic venule (Tal et al., 1990; Thorens et al., 1990; Bilir et al., 1993; Karim et al., 2012; Mueckler and Thorens, 2013). Because the liver cell plate is perfused unidirectionally from portal to hepatic venule, the concentration of several substrates, such as oxygen and glucose, decreases as blood moves closer to the latter (Bilir et al., 1993). This is consistent with the observation that GLUT1 expression is enhanced by a decrease in circulating glucose levels (Simpson et al., 1999) as well as by hypoxia (Ebert et al., 1995). Importantly, it has recently been shown that hypoxia enhances liver stage infection by malaria parasites, an effect that is also observed following treatment with an activator of hypoxia inducer factor‐1α (HIF‐1α) or with the hypoxia mimetic CoCl2 (Ng et al., 2014). Interestingly, increased HIF‐1α levels have been shown to upregulate expression of GLUT1 (Chen et al., 2001) and CoCl2 has been shown to enhance GLUT1 translocation to the plasma membrane ((Koseoglu and Beigi, 1999) and Fig. 5B). In this context, our results suggest that GLUT1‐mediated glucose transport may provide a crucial link to explain the observed preferential infectivity of hypoxic liver cells by Plasmodium parasites.
The data presented here is consistent with our proposed model (Fig. 6), in which the extensive replication of liver stage parasites promotes the depletion of intracellular glucose and, consequently, of ATP. This effect is compensated for by an increase in glucose uptake that results from (i) the likely activation of GLUT1 transporters at the plasma membrane via AMP‐dependent conformational changes and (ii) from GLUT1 translocation to the plasma membrane, particularly towards the end of parasite development. The significant increase in glucose uptake by infected cells likely leads to a decrease in glucose availability in the vicinity of infected cells, which may result in a similar, albeit weaker, response by neighbouring non‐infected cells. In fact, these cells also display a small decrease in ATP levels, as well as some, although not statistically significant, GLUT1 translocation to the membrane (Fig. 5A,B), and a slight increase in glucose uptake from 30 hpi onwards (Fig. 2A). A question that remains open is which mechanism(s) is(are) involved in regulating GLUT1 activation and translocation into the membrane of the infected cell. It has previously been shown that enhanced glucose transport by GLUT1 transporters preexisting in the plasma membrane is associated with stimulation of AMP‐activated protein kinase (AMPK) activity (Abbud et al., 2000; Barnes et al., 2002). On the other hand, it has been shown that GLUT1 translocation to the plasma membrane may be triggered by insulin (Egert et al., 1999), in a phosphoinositide 3‐kinase(PI3K)‐dependent fashion (Egert et al., 1999; Perrini et al., 2004). Intriguingly, neither parasite development nor glucose uptake by P. berghei‐infected cells seem to be affected by the down‐regulation of either the α1 and the α2 subunits of AMPK (Fig. S8). Likewise, addition of insulin or inhibition of PI3K with Wortmannin (Nagai et al., 2011; Hsu and Yang, 2014) do not impact infection or glucose uptake by infected cells (Fig. S9). Very recently, phosphorylation of GLUT1 by protein kinase C has been shown to lead to the rapid increase in glucose uptake and enhanced cell surface localization of GLUT1 induced by TPA (Lee et al., 2015). Whether a similar mechanism may be at play during P. berghei infection of hepatic cells is currently under investigation.
Despite the fact that, under basal conditions, GLUT1 is only localised at the membrane of perivenous hepatocytes, we showed that in vivo chemical inhibition of GLUT1‐mediated glucose uptake has a substantial impact on the number of P. berghei‐infected hepatocytes. There are two possible explanations for this observation. The first is that the inhibitor only hinders GLUT1‐mediated glucose uptake on perivenous hepatocytes and, therefore, only has an effect on the parasites inside these hepatocytes. Alternatively, in accordance with our model, the replication of the parasite inside periportal hepatocytes could lead to a depletion of the cellular glucose pool and trigger the translocation of GLUT1 present in the endoplasmic reticulum (ER), Golgi and endosomal compartiments to the plasma membrane, thereby increasing the percentage of infected hepatocytes with GLUT1 at the plasma membrane and, therefore, the number of cells targeted by the inhibitor.
The (East Rutherford, NJ, USA) liver displays the capacity to remove 30–40% of the glucose presented to it following glucose ingestion and therefore must be considered a significant site of postprandial glucose removal (Pagliassotti and Cherrington, 1992). Interestingly, a recent case–control study in Ghana found that patients with type 2 diabetes mellitus had a 46% increased risk for infection with P. falciparum (Danquah et al., 2010). The authors of this study offer a number of possible explanations for this observation, including impaired immune responses, an increased number of infectious mosquito bites, and enhanced parasite growth in the blood of type 2 diabetes mellitus patients (Danquah et al., 2010). Thus, our results suggest the possibility that the elevated amounts of circulating glucose displayed by diabetic patients may also promote parasite development in the liver, thereby contributing to the increased risk of appearance of clinically relevant malaria. Further investigation of the link between diabetes and malaria is clearly warranted by the fact that sub‐Saharan Africa currently faces the world's highest increase in type 2 diabetes mellitus (Wild et al., 2004).
To the best of our knowledge, this is the first report that addresses the role of glucose during hepatic infection by Plasmodium from a molecular point of view. It identifies GLUT1 as a druggable target and a major player in glucose uptake by infected cells and shows that its function as a glucose transporter is modulated in cells containing replicating parasites. These findings contribute to an understanding of how hepatic host cells meet the energy demands imposed by the parasite's huge replication rate. Such an understanding may help identify strategies to control energy production by infected cells and thereby limit parasite development and survival.
Experimental procedures
Chemicals
Roswell Park Memorial Institute (RPMI) 1640, PBS pH 7.4, trypsin, fetal bovine serum (FBS), non‐essential amino acids, penicillin/streptomycin, glutamine, HEPES pH 7, OptiMEM and Lipofectamine RNAiMAX were purchased from Gibco‐Thermo Fisher Scientific (Waltham, MA USA). All other chemicals were obtained from Sigma‐Aldrich (St. Louis, MO, USA), unless otherwise specified.
Cells
Huh7 cells, a human hepatoma cell line, were cultured in RPMI 1640 medium supplemented with 10% v/v FBS, 0.1 mM non‐essential amino acids, 50 µg/ml penicillin/streptomycin, 2 mM glutamine and 1 mM HEPES (final concentrations), pH 7 and maintained at 37 °C with 5% CO2. Mouse primary hepatocytes were cultured in William's E medium supplemented with 4% FBS, 2 mM glutamine, 1 mM HEPES and 50 µg/ml penicillin/streptomycin, and maintained at 37 °C with 5% CO2.
Mice
C57BL/6 mice were purchased from Charles River (Lyon, France) and housed in the rodent facility of Instituto de Medicina Molecular (Lisbon, Portugal). All animal experiments were performed in strict compliance to the guidelines of our institution's animal ethics committee and the Federation of European Laboratory Animal Science Associations (FELASA).
Parasites
Green fluorescent protein (GFP)‐expressing, red fluorescent protein (RFP)‐expressing, or luciferase‐expressing P. berghei ANKA sporozoites were dissected in non‐supplemented RPMI medium from the salivary glands of infected female A. stephensi mosquitoes, bred at Instituto de Medicina Molecular, prior to being employed for in vitro and in vivo infections (Franke‐Fayard et al., 2004; Ploemen et al., 2009).
Overall in vitro infection by luminescence
Overall hepatic infection was determined by measuring the luminescence intensity in Huh7 cells or mouse primary hepatocytes infected with a firefly luciferase‐expressing P. berghei line, as previously described (Ploemen et al., 2009). Briefly, Huh7 cells or mouse primary hepatocytes (1.0 × 104 and 2.0 × 104 per well, respectively) were seeded in 96‐well plates the day before infection. Sporozoite addition was followed by centrifugation at 1800 × g for 5 min and the medium was replaced approximately 2 hpi by the appropriate medium. Parasite infection load was measured 48 hpi by a bioluminescence assay (Biotium, Hayward, CA, USA) using a multiplate reader Infinite M200 (Tecan, Männedorf, Switzerland). The effect of the different treatments on cell viability was assessed by the CellTiter‐Blue assay (Promega, Fitchburg, WI, USA) according to the manufacturer's protocol.
Quantification of P. berghei invasion and development by flow cytometry
Invasion of hepatoma cells and intracellular parasite development were assessed by determining the percentage of GFP+ cells 2 hpi with a GFP‐expressing P. berghei line and by measuring the intensity of the GFP signal of the infected cells 48 hpi, respectively, as previously described (Prudencio et al., 2008). Huh7 cells (5.0 × 104 per well) were seeded in 24‐well plates the day before infection. The medium was replaced by the appropriate medium 1 h prior or 2 hpi, for invasion and development quantification, respectively. Cells were then collected for flow cytometry analysis at 2 h or 48 hpi, respectively, and analysed on a BD Biosciences FACScalibur (BD Biosciences, Franklin Lakes, NJ, USA). Data acquisition and analysis were carried out using the cellquest (version 3.1.1 f1, BD Biosciences) and flowjo (version 6.4.7, Ashland, OR, USA) software packages, respectively.
Immunofluorescence imaging of P. berghei in Huh7 cells
For immunofluorescence microscopy analyses, cells were seeded on glass coverslips in 24‐well plates and infected with sporozoites as described earlier. Forty‐eight hpi, cells were rinsed with 1x PBS and fixed with 4% v/v paraformaldehyde (Santa Cruz Biotechnology, Dallas, TX, USA) for 20 min at room temperature and stored at 4 °C in PBS 1x until being stained. Cells were incubated with the permeabilization/blocking solution (0.1% v/v Triton X‐100, 1% w/v bovine serum albumin in 1x PBS) for 30 min at room temperature. Parasites were stained with a parasite specific anti‐Heat shock protein 70 (Hsp70) antibody (2E6; dilution 1:100) and an anti‐UIS4 antibody (dilution 1:1000) for 1 h at room temperature, followed by three washes with permeabilization/blocking solution. Cells were further incubated in a 1:400 dilution of anti‐mouse Alexa‐Fluor 488 (Jackson ImmunoResearch Laboratories, West Grove, PA, USA) or anti‐goat Alexa‐Fluor 568 (Life Technologies, Carlsbad, CA, USA) secondary antibodies in the presence of a 1:1000 dilution of Hoechst 33342 (Invitrogen, Carlsbad, CA, USA) for nuclei staining. An additional three washes were carried out with permeabilization/blocking solution. Coverslips were mounted on microscope slides with Fluoromount (SouthernBiotech, Birmingham, AL, USA). Confocal images were acquired using a Zeiss LSM 710 confocal microscope (Carl Zeiss, Oberkochen, Germany). Widefield images for size determination were acquired in a Zeiss Axiovert 200 M microscope (Carl Zeiss, Oberkochen, Germany). Images were processed with imagej software (version 1.47, NIH, Bethesda, MD, USA).
Quantification of glucose uptake by flow cytometry and live microscopy
Glucose uptake into Huh7 cells was quantified by flow cytometry using a fluorescent D‐glucose derivative, 2‐[N‐(7‐nitrobenz‐2‐oxa‐1,3‐diazol‐4‐yl)amino]‐2‐deoxy‐D‐glucose (2‐NBDG; Molecular Probes, Life Technologies) as a tracer (O'Neil et al., 2005; Yamada et al., 2007). Briefly, Huh7 cells (1.0 × 104 per well) were seeded in 96‐well plates the day before infection with RFP‐expressing P. berghei sporozoites. At several time points after infection, the medium was replaced by RPMI without glucose supplemented with 1 mM of glucose and 0.1 mM of 2‐NBDG, and cells were incubated for 5 min at 37 °C to allow uptake (O'Neil et al., 2005). Cells were then collected for flow cytometry analysis and analysed on a BD LSR Fortessa flow cytometer (bd biosciences, Franklin Lakes, NJ, USA) with the diva software (version 6.2, bd biosciences, Franklin Lakes, NJ, USA). Analysis was carried out using the flowjo software (version 6.4.7, FlowJo). For live microscopy, Huh7 cells (2.0 × 105) were seeded on glass bottom microwell dishes (MatTek, Ashland, MA, USA) the day before infection with RFP‐expressing P. berghei sporozoites. At 48 hpi, the medium was replaced by RPMI without glucose supplemented with 1 mM of glucose and 0.1 mM of 2‐NBDG, and cells were incubated for 30 min at 37 °C. Cells were then visualised on a 3i Marianas SDC microscope and the acquired images were processed with imagej software (version 1.47).
Temperature shocks and ethanol‐induced oxidative stress
Huh7 cells (1.0 × 104 per well) were seeded in 96‐well plates and incubated at 37 °C for 48 h. To induce a mild cold stress response, one of the plates was incubated at 27 °C for 1 h (Fujita, 1999). To induce a heat stress response, another plate was incubated at 43 °C for 1 h (Sun et al., 2015). A control plate was maintained at 37 °C. Ethanol‐induced oxidative stress was attained by incubating Huh7 cells in complete RPMI with 50 mM of ethanol for 24 h (Sergent et al., 2005; Nourissat et al., 2008). Following temperature shocks and ethanol‐induced oxidative stress, the uptake of 2‐NBDG by the cells was determined as previously described.
Viral infection‐induced stress
Huh7 cells (5.0 × 104 per well) were seeded in 24‐well plates and, on the following day, were infected with 0.01, 1 and 5 plaque‐forming units/cell of wild type murine gammaherpesvirus 68 (MHV‐68) or YFP‐expressing MHV‐68 (MHV‐68‐YFP) (Collins et al., 2009). Twenty‐four h later, the uptake of 2‐NBDG by the MHV‐68‐infected cells was determined as previously described. The percentage of MHV‐68‐infected cells in each condition was estimated by determining the percentage of YFP+ cells after infection with the same amount of plaque‐forming units/cell of MHV‐68‐YFP.
Assessment of the impact of glucose transporters on Plasmodium infection by RNA interference (RNAi)
Down‐modulation of the genes encoding selected glucose transporters employed short hairpin RNAs (shRNAs). All shRNAs were purchased from the MISSION TRC library (Sigma) in the form of bacterial glycerol stocks which were grown to obtain the purified plasmids. Each gene was targeted by using three distinct shRNAs, used individually (Table S1). For the lentiviral production, HEK 293FT cells (2.0 × 104 per well) were seeded in 96‐well plates. On the following day, cells were transfected with the packaging vectors and each individual shRNA plasmid in a final concentration of 100 ng/well using the FuGENE 6 reagent (Promega), according to the manufacturer's instructions. The lentiviral particles were collected in the supernatant of these cells approximately 60 h after transfection and stored at −80 °C. For the transduction of Huh7 cells and subsequent generation of cell lines with stable knockdown of the genes of interest, cells (1.0 × 105 per well) were seeded in 12‐well plates. On the following day, the medium was replaced by 400 µl of supplemented RPMI with 8 µg/ml of polybrene, on top of which were added 100 µl of lentiviral particles‐containing supernantant [approximate multiplicity of infection (MOI) of 1:1] and the plates were centrifuged for 30 min at 1200 × g and 37 °C. Twenty‐four hours after transduction, the medium was replaced by supplemented RPMI with 5 µg/ml of puromycin (Calbiochem) for the selection of the transduced cells which were allowed to grow for at least a week before being used for infections with luciferase‐expressing or GFP‐expressing P. berghei sporozoites to determine overall infection and parasite development by luminescence, flow cytometry and microscopy, as described earlier. Cells transduced with lentiviral particles carrying a negative control shRNA (SHC002) not targeting any annotated gene in the human genome were used as negative control. A stable cell line with the knockdown of Scavenger receptor class B type I (SR‐BI) was used as positive control in the luminescence assays (Rodrigues et al., 2008). The knockdown efficiency of each shRNA sequence (Table S1) was assessed by quantitative PCR (qPCR) with specific primers for each gene (Table S2).
Small interfering RNA transfection
4.0 × 104 Huh7 cells were reverse‐transfected with 30 nM of target specific (human AMPK α1: ref. L‐005027‐00‐0005; human AMPK α2: ref. L‐005361‐00‐0005) or control small interfering RNA sequence pools (ON‐TARGETplus SMARTpool, Dharmacon, Lafayette, CO, USA), using Lipofectamine RNAiMAX (Gibco/Invitrogen) according to the manufacturer's instructions. Twenty‐four hours after transfection, the cells were infected with 3.0 × 104 GFP‐expressing P. berghei sporozoites. Cells were collected for flow cytometry analysis at 48 hpi and analysed on a BD LSR Fortessa flow cytometer with the diva software (version 6.2). Analysis was carried out using the flowjo software (version 6.4.7, FlowJo). The efficiency of knockdown was assessed with specific primers by qPCR (Table S2).
Fluorescence‐activated cell sorting of P. berghei‐infected and non‐infected Huh7 cells
Huh7 cells (1.0 × 105 per well) were seeded in 24‐well plates and infected 24 h later with 1.0 × 105 GFP‐expressing P. berghei sporozoites. Cells were collected at 2 hpi and FACS‐sorted on a BD FACSAria III Cell Sorter (BD Biosciences). Non‐infected and GFP‐expressing P. berghei‐infected cells were gated on the basis of their different fluorescence intensity, as previously established, and collected simultaneously (Prudencio et al., 2008; Albuquerque et al., 2009). Immediately after FACS‐sorting, both infected and non‐infected cells were washed and seeded in 24‐well plates at a density of 1.5 × 105 per well. Infected cells were diluted 1:1 with non‐infected cells to allow replicates. Cells were then incubated until collection at 30 or 48 hpi. In the case of the 6 hpi time point, P. berghei‐infected cells were FACS‐sorted at this time, diluted 1:1 with non‐infected cells, pelleted, snap‐frozen and stored until RNA extraction.
RNA extraction, complementary DNA synthesis and qPCR
RNA was extracted from cultured cells using the High Pure RNA Isolation kit (Roche, Basel, Switzerland) according to the manufacturer's instructions. The amount of RNA in each sample was assessed with a NanoDrop® ND‐1000 spectrophotometer (NanoDrop, Wilmington, DE, USA).
Complementary DNA (cDNA) was synthesised from 1 µg of RNA using the Roche cDNA synthesis kit, according to the manufacturer's instructions. The cDNA was synthesised employing the following thermocycling parameters: 25 °C for 10 min, 55 °C for 30 min, and 85 °C for 5 min. qPCR reaction was performed in a total volume of 20 µl in a ABI Prism 7500 Fast system (Applied Biosystems, Foster City, CA, USA) using the iTaqTM Universal SYBR® Green kit (BioRad, Hercules, CA, USA) as follows: 50 °C for 2 min, 95 °C for 10 min, 40 cycles at 95 °C for 15 s and 60 °C for 1 min, melting stage was done at 95 °C for 15 s, 60 °C for 1 min, and 95 °C for 30 s. Primers for hypoxanthine‐guanine phosphoribosyltransferase (Hprt), a well‐established housekeeping gene, were used for normalisation in all experiments (Table S2). The delta‐delta cycle threshold (ΔΔCT) relative quantification method was used for analysis of qPCR results.
In vivo WZB117 treatment, P. berghei sporozoite infection and quantification of parasite liver load by qPCR
Six weeks old male C57BL/6 mice were injected intraperitoneally (i.p.) with 10 mg/kg of WZB117 in PBS/DMSO 1:1 (v/v), or with vehicle alone, immediately before intravenous (i.v.) injection of 3.0 × 104 GFP‐expressing P. berghei sporozoites (Liu et al., 2012). The administration of the drug or vehicle was repeated at 15 and 30 hpi, and the livers were collected at 44 hpi and homogenised in 3 ml of denaturing solution (4 M guanidine thiocyanate; 25 mM sodium citrate pH 7, 0.5% w/v N‐lauroylsarcosine and v/v 0.7% β mercaptoethanol in DEPC‐treated water). Total RNA was extracted from the livers with the NZY Total RNA Isolation Kit (NZYTech, Lisboa, Portugal), according to the manufacturer's protocol, and converted into cDNA as described earlier. Parasite load was quantified by qPCR using primers specific to P. berghei 18S RNA (Table S2). Mouse Hprt expression was used for normalisation.
Immunohistochemical staining of liver sections
For microscopy, paraformaldehyde‐fixed liver lobes were cut in 50 µm sections and were incubated in permeabilization/blocking solution (1% w/v bovine serum albumin, 0.5% v/v Triton‐X100 in PBS) at room temperature for 1 h, followed by a 2 h incubation at room temperature with an anti‐UIS4 antibody (dilution 1:500). Slices were further incubated in a 1:300 dilution of anti‐GFP‐Alexa488 antibody (Invitrogen) and anti‐goat Alexa‐Fluor 568 (Invitrogen) in the presence of a 1:1000 dilution of Hoechst 33342 (Invitrogen) and a 1:100 dilution of Phalloidin‐660 (Invitrogen) for actin staining for 1 h. After washing, slices were mounted on microscope slides with Fluoromount (SouthernBiotech). Images were acquired and processed as described above.
Determination of ATP levels inside single cells by live microscopy
Huh7 cells (2.0 × 105 cells) were seeded on glass bottom microwell dishes (MatTek). The following day, 1 µg of ATeam plasmid carrying the ATP indicator (pRSET‐AT1.03) was transfected into the cells with FuGENE 6 (Roche) according to the manufacturer's instructions (Imamura et al., 2009). Twenty‐four hours after transfection, cells were infected with RFP‐expressing P. berghei sporozoites and, at selected time points of infection, were visualised at 37 °C on a Zeiss LSM 710 confocal microscope as described previously (Ando et al., 2012). Huh7 cells incubated for 6 h in RPMI without glucose with 10 mM of galactose and 10 μM of Oligomycin were used as positive controls for ATP depletion. Image analysis was performed using the imagej software (version 1.47). The Venus/CFP emission ratio was calculated for each cell dividing its mean intensity in the Venus channel by the mean intensity in the CFP channel.
Quantification of GLUT1 at the plasma membrane of infected cells by flow cytometry
Huh7 cells (1.0 × 104 per well) were seeded in 96‐well plates and were infected with RFP‐expressing P. berghei sporozoites on the following day. Thirty and 48 hpi, cells were detached using PBS containing 1 mM EDTA, centrifuged at 4 °C, and the cell pellet was resuspended in cold RPMI containing 10% v/v FBS. Cells were further incubated with supernatant obtained from 293 T cells transfected with a vector encoding the HRBD‐EGFP fusion protein (dilution 1:50 in PBS with 2% v/v FBS) (Manel et al., 2003; Kinet et al., 2007). Following incubation at 37 °C for 30 min, cells were harvested by centrifugation, washed one time with PBS with 2% v/v FBS, resuspended in the same buffer and analysed with a BD LSR Fortessa cytometer with the diva software (version 6.2). Huh7 cells incubated overnight in complete RPMI with 250 μM of cobalt (II) chloride (CoCl2) were used as positive controls (Koseoglu and Beigi, 1999). Analysis was carried out using the flowjo software (version 6.4.7, FlowJo).
Statistical analyses
Statistical analyses were performed using the graphpad prism 5 software (La Jolla, CA, USA). One‐way analysis of variance, Two‐way analysis of variance, Chi‐Square, Mann–Whitney U test or Student's t‐test were used for significance of differences observed, as indicated in each figure. ns, not significant; * P < 0.05, ** P < 0.01 and *** P < 0.001.
Conflict of interest
The authors declare no conflicts of interest.
Supporting information
Fig. S1. Glucose availability impacts P. berghei infection ex vivo. (A) Mouse primary hepatocytes were infected with luciferase‐expressing P. berghei sporozoites and the culture medium was replaced 2 hpi by medium with different concentrations of glucose. Parasite load (luminescence) and cell viability were assessed 48 hpi. Pool of 3 independent experiments. Error bars represent SEM. One‐way ANOVA with post‐test Dunnett. (B) Mouse primary hepatocytes were infected with GFP‐expressing P. berghei sporozoites and the culture medium was replaced by medium with different concentrations of glucose 2 hpi. Parasite development and the total number of infected cells were assessed at 48 hpi by flow cytometry by determining the fluorescence intensity and the number of GFP+ cells, respectively. One experiment. One‐way ANOVA with post‐test Dunnett. Error bars represent SD. ns ‐ not significant, * p < 0.05, ** p < 0.01 and *** p < 0.001.
Fig. S2. Gating strategy to determine 2‐NBDG uptake and parasite development by flow cytometry. Huh7 cells were infected with RFP‐expressing P. berghei sporozoites and incubated with 2‐NBDG‐containing medium at different time points of infection. Dot plot represents non‐infected and infected (RFP+) cells of one replicate from the 48 h time point. Left histogram shows the different fluorescence intensity displayed by non‐developing (RFPlow) and developing (RFPhigh) parasites. Right histogram exemplifies the 2‐NBDG uptake by the different cell populations (non‐infected, non‐developing and developing parasites), assessed as an increased fluorescence intensity in the green channel.
Fig. S3. P. berghei‐infected Huh7 cells and developing parasites uptake more 2‐NBDG than non‐infected and naïve cells. Huh7 cells were infected with RFP‐expressing P. berghei sporozoites and incubated for 30 min with 2‐NBDG‐containing medium at 48 hpi. (A) Representative images of P. berghei‐infected Huh7 cells with or without incubation with 2‐NBDG. A non‐infected cell, an infected cell and a parasite are outlined in yellow, pink and red, respectively. Scale bar, 20 μm. (B) Quantification of 2‐NBDG uptake by naïve (grey bar), non‐infected cells (yellow bar), infected cells (pink bar) and developing parasites (red bar), as assessed by live fluorescence microscopy. Representative experiment out of 2 independent experiments. One‐way ANOVA with post‐test Bonferroni and two‐tailed Mann‐Whitney test, respectively. Error bars represent SD. *** p < 0.001.
Fig. S4. Quantification of the percentage of MHV‐68‐YFP‐infected cells by flow cytometry. Percentage of YFP+ cells 24 hpi with different loads of MHV‐68‐YFP.
Fig. S5. 2‐NBDG uptake by non‐infected Huh7 cells with GLUT1 knockdown. 2‐NBDG uptake by non‐infected cells of the different cell lines with GLUT1 knockdown at 48 hpi, assessed by flow cytometry. Pool of 4 independent experiments. Error bars represent SEM. One‐way ANOVA with post‐test Dunnett. ns ‐ not significant and *** p < 0.001.
Fig. S6. Chemical inhibition of GLUT1‐mediated glucose transport by WZB117 affects P. berghei hepatic infection ex vivo. (A) Mouse primary hepatocytes were infected with luciferase‐expressing P. berghei sporozoites and 2 h later the culture medium was replaced by medium containing WZB117. Parasite load (luminescence) and cell viability were assessed at 48 hpi. Pool of 2 independent experiments. Error bars represent SEM. One‐way ANOVA with post‐test Dunnett. (B) Mouse primary hepatocytes were infected with GFP‐expressing P. berghei sporozoites and 2 h later the culture medium was replaced by medium containing WZB117. Parasite development was assessed by flow cytometry at 48 hpi. Error bars represent SD. One‐way ANOVA with post‐test Dunnett. ns ‐ not significant, * p < 0.05, ** p < 0.01 and *** p < 0.001.
Fig. S7. GLUT1 expression is not altered in P. berghei‐infected cells. qPCR quantification of GLUT1 transcriptional expression in FACS‐sorted infected and non‐infected Huh7 cells. Each time point represents a pool of 2 independent sorting experiments. Error bars represent SD. Two‐tailed Mann‐Whitney test. ns – not significant.
Fig. S8. Increased GLUT1‐dependent glucose uptake into P. berghei‐infected cells is independent of AMPK. (A) Knockdown efficiency upon transfection in Huh7 cells of the siRNAs used against the α1 and α2 subunits of AMPK. (B) Huh7 cells with the knockdown of α1 or/and α2 subunits of AMPK were infected with GFP‐expressing P. berghei sporozoites and parasite development was assessed by flow cytometry at 48 hpi. Pool of 2 independent experiments. Error bars represent SD. One‐way ANOVA with post‐test Dunnett. (C) 2‐NBDG uptake at 48 hpi by developing parasites‐containing cells with the knockdown of α1 or/and α2 subunits of AMPK, assessed by flow cytometry. Representative experiment out of 2 independent experiments. Error bars represent SD. One‐way ANOVA with post‐test Dunnett. ns ‐ not significant, *** p < 0.001.
Fig. S9. PI3K stimulation or inhibition does not impact P. berghei infection of Huh7 cells neither does it alter glucose uptake by infected cells. Huh7 cells were infected with luciferase‐expressing P. berghei sporozoites and 2 h later were treated with different concentrations of (A) insulin or (B) Wortmannin. Parasite load (luminescence) was assessed after 48 h. Representative experiment out of 2 and pool of 3 independent experiments, respectively. Error bars represent SD. To evaluate possible effects on glucose uptake, Huh7 cells were infected with RFP‐expressing P. berghei sporozoites and the treatment with different concentrations of insulin (C) and Wortmannin (D) was initiated 2 hpi. 2‐NBDG uptake by developing parasites‐containing cells was assessed at 48 hpi by flow cytometry. Pool of 2 independent experiments for both insulin and Wortmannin. Error bars represent SD. All panels: one‐way ANOVA with post‐test Dunnett. ns ‐ not significant.
Table S1. List of shRNA sequences used, with the corresponding knockdowns.
Table S2. List of primer sequences.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Acknowledgements
We are very grateful to Marc Sitbon and Julie Laval (Institut de Génétique Moléculaire de Montpellier CNRS‐UMSF, France), and Vincent Petit and Sandra Moriceau (Metafora biosystems), for kindly providing the HRBD‐EGFP peptide, to Hiromi Imamura (Graduate School of Biostudies, Kyoto University, Japan) for kindly providing the pRSET‐AT1.03 plasmid, to Maria M. Mota (Instituto de Medicina Molecular, Portugal) for critically reviewing the manuscript, to Marta Miranda for help with the viral infections, to Ana Parreira and Filipa Teixeira for producing the various lines of P. berghei‐infected mosquitoes and to the bioimaging and flow cytometry facilities of iMM Lisboa for technical support.
This work was supported by the Fundação para a Ciência e Tecnologia (www.fct.pt, FCT, Portugal) through grants PTDC/SAU/MIC/117060/2010 and PTDC/SAU‐MET/118199/2010 to MP and LMS, respectively. MP was sponsored by an Investigador FCT (2013) grant, PM by FCT fellowship SFRH/BD/71098/2010 and LMS by the European Community's Seventh Framework Programme (FP7/2007‐2013) under grant agreement N. 242095 (EVIMalaR). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Meireles P., Sales‐Dias J., Andrade C. M., Mello‐Vieira J., Mancio‐Silva L., Pedro Simas J., Staines H. M., and Prudêncio M. (2017) GLUT1‐mediated glucose uptake plays a crucial role during Plasmodium hepatic infection, Cellular Microbiology, 19: e12646. doi: 10.1111/cmi.12646.
References
- Abbud, W. , Habinowski, S. , Zhang, J.Z. , Kendrew, J. , Elkairi, F.S. , Kemp, B.E. , et al. (2000) Stimulation of AMP‐activated protein kinase (AMPK) is associated with enhancement of Glut1‐mediated glucose transport. Arch Biochem Biophys 380: 347–352. [DOI] [PubMed] [Google Scholar]
- Albuquerque, S.S. , Carret, C. , Grosso, A.R. , Tarun, A.S. , Peng, X. , Kappe, S.H. , et al. (2009) Host cell transcriptional profiling during malaria liver stage infection reveals a coordinated and sequential set of biological events. BMC Genomics 10: 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando, T. , Imamura, H. , Suzuki, R. , Aizaki, H. , Watanabe, T. , Wakita, T. , and Suzuki, T. (2012) Visualization and measurement of ATP levels in living cells replicating hepatitis C virus genome RNA. PLoS Pathog 8: e1002561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes, K. , Ingram, J.C. , Porras, O.H. , Barros, L.F. , Hudson, E.R. , Fryer, L.G. , et al. (2002) Activation of GLUT1 by metabolic and osmotic stress: potential involvement of AMP‐activated protein kinase (AMPK). J Cell Sci 115: 2433–2442. [DOI] [PubMed] [Google Scholar]
- Bechmann, L.P. , Hannivoort, R.A. , Gerken, G. , Hotamisligil, G.S. , Trauner, M. , and Canbay, A. (2012) The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol 56: 952–964. [DOI] [PubMed] [Google Scholar]
- Bilir, B.M. , Gong, T.W. , Kwasiborski, V. , Shen, C.S. , Fillmore, C.S. , Berkowitz, C.M. , and Gumucio, J.J. (1993) Novel control of the position‐dependent expression of genes in hepatocytes. The GLUT‐1 transporter. J Biol Chem 268: 19776–19784. [PubMed] [Google Scholar]
- Bitar, M.S. , Al‐Saleh, E. , and Al‐Mulla, F. (2005) Oxidative stress‐‐mediated alterations in glucose dynamics in a genetic animal model of type II diabetes. Life Sci 77: 2552–2573. [DOI] [PubMed] [Google Scholar]
- Blodgett, D.M. , De Zutter, J.K. , Levine, K.B. , Karim, P. , and Carruthers, A. (2007) Structural basis of GLUT1 inhibition by cytoplasmic ATP. J Gen Physiol 130: 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume, M. , Rodriguez‐Contreras, D. , Landfear, S. , Fleige, T. , Soldati‐Favre, D. , Lucius, R. , and Gupta, N. (2009) Host‐derived glucose and its transporter in the obligate intracellular pathogen Toxoplasma gondii are dispensable by glutaminolysis. Proc Natl Acad Sci U S A 106: 12998–13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Pore, N. , Behrooz, A. , Ismail‐Beigi, F. , and Maity, A. (2001) Regulation of glut1 mRNA by hypoxia‐inducible factor‐1. Interaction between H‐ras and hypoxia. J Biol Chem 276: 9519–9525. [DOI] [PubMed] [Google Scholar]
- Cloherty, E.K. , Diamond, D.L. , Heard, K.S. , and Carruthers, A. (1996) Regulation of GLUT1‐mediated sugar transport by an antiport/uniport switch mechanism. Biochemistry 35: 13231–13239. [DOI] [PubMed] [Google Scholar]
- Collins, C.M. , Boss, J.M. , and Speck, S.H. (2009) Identification of infected B‐cell populations by using a recombinant murine gammaherpesvirus 68 expressing a fluorescent protein. J Virol 83: 6484–6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham, J.J. , Gulino, M.A. , Meara, P.A. , and Bode, H.H. (1985) Enhanced hepatic insulin sensitivity and peripheral glucose uptake in cold acclimating rats. Endocrinology 117: 1585–1589. [DOI] [PubMed] [Google Scholar]
- Danquah, I. , Bedu‐Addo, G. , and Mockenhaupt, F.P. (2010) Type 2 diabetes mellitus and increased risk for malaria infection. Emerg Infect Dis 16: 1601–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert, B.L. , Firth, J.D. , and Ratcliffe, P.J. (1995) Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter‐1 via distinct Cis‐acting sequences. J Biol Chem 270: 29083–29089. [DOI] [PubMed] [Google Scholar]
- Egert, S. , Nguyen, N. , and Schwaiger, M. (1999) Myocardial glucose transporter GLUT1: translocation induced by insulin and ischemia. J Mol Cell Cardiol 31: 1337–1344. [DOI] [PubMed] [Google Scholar]
- Feistel, T. , Hodson, C.A. , Peyton, D.H. , and Landfear, S.M. (2008) An expression system to screen for inhibitors of parasite glucose transporters. Mol Biochem Parasitol 162: 71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke‐Fayard, B. , Trueman, H. , Ramesar, J. , Mendoza, J. , van der Keur, M. , van der Linden, R. , et al. (2004) A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol 137: 23–33. [DOI] [PubMed] [Google Scholar]
- Fujita, J. (1999) Cold shock response in mammalian cells. J Mol Microbiol Biotechnol 1: 243–255. [PubMed] [Google Scholar]
- Hellwig, B. , and Joost, H.G. (1991) Differentiation of erythrocyte‐(GLUT1), liver‐(GLUT2), and adipocyte‐type (GLUT4) glucose transporters by binding of the inhibitory ligands cytochalasin B, forskolin, dipyridamole, and isobutylmethylxanthine. Mol Pharmacol 40: 383–389. [PubMed] [Google Scholar]
- Hsu, P.Y. , and Yang, Y.W. (2014) Gene delivery via the hybrid vector of recombinant adeno‐associated virus and polyethylenimine. Eur J Pharm Sci 52: 62–68. [DOI] [PubMed] [Google Scholar]
- Imamura, H. , Nhat, K.P. , Togawa, H. , Saito, K. , Iino, R. , Kato‐Yamada, Y. , et al. (2009) Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer‐based genetically encoded indicators. Proc Natl Acad Sci U S A 106: 15651–15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itani, S. , Torii, M. , and Ishino, T. (2014) d‐Glucose concentration is the key factor facilitating liver stage maturation of Plasmodium. Parasitol Int 63: 584–590. [DOI] [PubMed] [Google Scholar]
- Joet, T. , Eckstein‐Ludwig, U. , Morin, C. , and Krishna, S. (2003) Validation of the hexose transporter of Plasmodium falciparum as a novel drug target. Proc Natl Acad Sci U S A 100: 7476–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim, S. , Adams, D.H. , and Lalor, P.F. (2012) Hepatic expression and cellular distribution of the glucose transporter family. World J Gastroenterol 18: 6771–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai, D. , Adachi, T. , Deng, L. , Nagano‐Fujii, M. , Sada, K. , Ikeda, M. , et al. (2009) HCV replication suppresses cellular glucose uptake through down‐regulation of cell surface expression of glucose transporters. J Hepatol 50: 883–894. [DOI] [PubMed] [Google Scholar]
- Kinet, S. , Swainson, L. , Lavanya, M. , Mongellaz, C. , Montel‐Hagen, A. , Craveiro, M. , et al. (2007) Isolated receptor binding domains of HTLV‐1 and HTLV‐2 envelopes bind Glut‐1 on activated CD4+ and CD8+ T cells. Retrovirology 4: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk, K. , Horner, H.A. , and Kirk, J. (1996) Glucose uptake in Plasmodium falciparum‐infected erythrocytes is an equilibrative not an active process. Mol Biochem Parasitol 82: 195–205. [DOI] [PubMed] [Google Scholar]
- Koranyi, L. , Bourey, R.E. , James, D. , Mueckler, M. , Fiedorek, F.T., Jr. , and Permutt, M.A. (1991) Glucose transporter gene expression in rat brain: Pretranslational changes associated with chronic insulin‐induced hypoglycemia, fasting, and diabetes. Mol Cell Neurosci 2: 244–252. [DOI] [PubMed] [Google Scholar]
- Koseoglu, M.H. , and Beigi, F.I. (1999) Mechanism of stimulation of glucose transport in response to inhibition of oxidative phosphorylation: analysis with myc‐tagged Glut1. Mol Cell Biochem 194: 109–116. [DOI] [PubMed] [Google Scholar]
- Kroemer, G. , and Pouyssegur, J. (2008) Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell 13: 472–482. [DOI] [PubMed] [Google Scholar]
- Lee, E.E. , Ma, J. , Sacharidou, A. , Mi, W. , Salato, V.K. , Nguyen, N. , et al. (2015) A protein kinase C Phosphorylation motif in GLUT1 affects glucose transport and is mutated in GLUT1 deficiency syndrome. Mol Cell 58: 845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, K.B. , Cloherty, E.K. , Fidyk, N.J. , and Carruthers, A. (1998) Structural and physiologic determinants of human erythrocyte sugar transport regulation by adenosine triphosphate. Biochemistry 37: 12221–12232. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Cao, Y. , Zhang, W. , Bergmeier, S. , Qian, Y. , Akbar, H. , et al. (2012) A small‐molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell‐cycle arrest, and inhibits cancer cell growth in vitro and in vivo . Mol Cancer Ther 11: 1672–1682. [DOI] [PubMed] [Google Scholar]
- Loisel‐Meyer, S. , Swainson, L. , Craveiro, M. , Oburoglu, L. , Mongellaz, C. , Costa, C. , et al. (2012) Glut1‐mediated glucose transport regulates HIV infection. Proc Natl Acad Sci U S A 109: 2549–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manel, N. , Kim, F.J. , Kinet, S. , Taylor, N. , Sitbon, M. , and Battini, J.L. (2003) The ubiquitous glucose transporter GLUT‐1 is a receptor for HTLV. Cell 115: 449–459. [DOI] [PubMed] [Google Scholar]
- Moore, M.C. , Coate, K.C. , Winnick, J.J. , An, Z. , and Cherrington, A.D. (2012) Regulation of hepatic glucose uptake and storage in vivo . Adv Nutr 3: 286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueckler, M. (1994) Facilitative glucose transporters. Eur J Biochem 219: 713–725. [DOI] [PubMed] [Google Scholar]
- Mueckler, M. , and Thorens, B. (2013) The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med 34: 121–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai, T. , Arao, T. , Furuta, K. , Sakai, K. , Kudo, K. , Kaneda, H. , et al. (2011) Sorafenib inhibits the hepatocyte growth factor‐mediated epithelial mesenchymal transition in hepatocellular carcinoma. Mol Cancer Ther 10: 169–177. [DOI] [PubMed] [Google Scholar]
- Ng, S. , March, S. , Galstian, A. , Hanson, K. , Carvalho, T. , Mota, M.M. , and Bhatia, S.N. (2014) Hypoxia promotes liver‐stage malaria infection in primary human hepatocytes in vitro . Dis Model Mech 7: 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourissat, P. , Travert, M. , Chevanne, M. , Tekpli, X. , Rebillard, A. , Le Moigne‐Muller, G. , et al. (2008) Ethanol induces oxidative stress in primary rat hepatocytes through the early involvement of lipid raft clustering. Hepatology 47: 59–70. [DOI] [PubMed] [Google Scholar]
- O'Neil, R.G. , Wu, L. , and Mullani, N. (2005) Uptake of a fluorescent deoxyglucose analog (2‐NBDG) in tumor cells. Mol Imaging Biol 7: 388–392. [DOI] [PubMed] [Google Scholar]
- Pagliassotti, M.J. , and Cherrington, A.D. (1992) Regulation of net hepatic glucose uptake in vivo . Annu Rev Physiol 54: 847–860. [DOI] [PubMed] [Google Scholar]
- Pencek, R.R. , James, F.D. , Lacy, D.B. , Jabbour, K. , Williams, P.E. , Fueger, P.T. , and Wasserman, D.H. (2004) Exercise‐induced changes in insulin and glucagon are not required for enhanced hepatic glucose uptake after exercise but influence the fate of glucose within the liver. Diabetes 53: 3041–3047. [DOI] [PubMed] [Google Scholar]
- Penkler, G. , du Toit, F. , Adams, W. , Rautenbach, M. , Palm, D.C. , van Niekerk, D.D. , and Snoep, J.L. (2015) Construction and validation of a detailed kinetic model of glycolysis in Plasmodium falciparum. FEBS J 282: 1481–1511. [DOI] [PubMed] [Google Scholar]
- Perrini, S. , Natalicchio, A. , Laviola, L. , Belsanti, G. , Montrone, C. , Cignarelli, A. , et al. (2004) Dehydroepiandrosterone stimulates glucose uptake in human and murine adipocytes by inducing GLUT1 and GLUT4 translocation to the plasma membrane. Diabetes 53: 41–52. [DOI] [PubMed] [Google Scholar]
- Pfaller, M.A. , Krogstad, D.J. , Parquette, A.R. , and Nguyen‐Dinh, P. (1982) Plasmodium falciparum: stage‐specific lactate production in synchronized cultures. Exp Parasitol 54: 391–396. [DOI] [PubMed] [Google Scholar]
- Ploemen, I.H. , Prudencio, M. , Douradinha, B.G. , Ramesar, J. , Fonager, J. , van Gemert, G.J. , et al. (2009) Visualisation and quantitative analysis of the rodent malaria liver stage by real time imaging. PLoS One 4: e7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio, M. , Mota, M.M. , and Mendes, A.M. (2011) A toolbox to study liver stage malaria. Trends Parasitol 27: 565–574. [DOI] [PubMed] [Google Scholar]
- Prudencio, M. , Rodrigues, C.D. , Ataide, R. , and Mota, M.M. (2008) Dissecting in vitro host cell infection by Plasmodium sporozoites using flow cytometry. Cell Microbiol 10: 218–224. [DOI] [PubMed] [Google Scholar]
- Prudencio, M. , Rodriguez, A. , and Mota, M.M. (2006) The silent path to thousands of merozoites: the Plasmodium liver stage. Nat Rev Microbiol 4: 849–856. [DOI] [PubMed] [Google Scholar]
- Rodrigues, C.D. , Hannus, M. , Prudencio, M. , Martin, C. , Goncalves, L.A. , Portugal, S. , et al. (2008) Host scavenger receptor SR‐BI plays a dual role in the establishment of malaria parasite liver infection. Cell Host Microbe 4: 271–282. [DOI] [PubMed] [Google Scholar]
- Sergent, O. , Pereira, M. , Belhomme, C. , Chevanne, M. , Huc, L. , and Lagadic‐Gossmann, D. (2005) Role for membrane fluidity in ethanol‐induced oxidative stress of primary rat hepatocytes. J Pharmacol Exp Ther 313: 104–111. [DOI] [PubMed] [Google Scholar]
- Shrayyef, M.Z. , and Gerich, J.E. (2010) Normal glucose homeostasis In Principles of Diabetes Mellitus. Poretsky L. (ed). Springer, Berlin, Germany. [Google Scholar]
- Simpson, I.A. , Appel, N.M. , Hokari, M. , Oki, J. , Holman, G.D. , Maher, F. , et al. (1999) Blood‐brain barrier glucose transporter: effects of hypo‐ and hyperglycemia revisited. J Neurochem 72: 238–247. [DOI] [PubMed] [Google Scholar]
- Slavic, K. , Delves, M.J. , Prudencio, M. , Talman, A.M. , Straschil, U. , Derbyshire, E.T. , et al. (2011a) Use of a selective inhibitor to define the chemotherapeutic potential of the plasmodial hexose transporter in different stages of the parasite's life cycle. Antimicrob Agents Chemother 55: 2824–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavic, K. , Krishna, S. , Derbyshire, E.T. , and Staines, H.M. (2011b) Plasmodial sugar transporters as anti‐malarial drug targets and comparisons with other protozoa. Malar J 10: 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavic, K. , Straschil, U. , Reininger, L. , Doerig, C. , Morin, C. , Tewari, R. , and Krishna, S. (2010) Life cycle studies of the hexose transporter of Plasmodium species and genetic validation of their essentiality. Mol Microbiol 75: 1402–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, T.A. (1999) Facilitative glucose transporter expression in human cancer tissue. Br J Biomed Sci 56: 285–292. [PubMed] [Google Scholar]
- Sun, L. , Lamont, S.J. , Cooksey, A.M. , McCarthy, F. , Tudor, C.O. , Vijay‐Shanker, K. , et al. (2015) Transcriptome response to heat stress in a chicken hepatocellular carcinoma cell line. Cell Stress Chaperones 20: 939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takanaga, H. , Chaudhuri, B. , and Frommer, W.B. (2008) GLUT1 and GLUT9 as major contributors to glucose influx in HepG2 cells identified by a high sensitivity intramolecular FRET glucose sensor. Biochim Biophys Acta 1778: 1091–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal, M. , Schneider, D.L. , Thorens, B. , and Lodish, H.F. (1990) Restricted expression of the erythroid/brain glucose transporter isoform to perivenous hepatocytes in rats. Modulation by glucose. J Clin Invest 86: 986–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorens, B. , Cheng, Z.Q. , Brown, D. , and Lodish, H.F. (1990) Liver glucose transporter: a basolateral protein in hepatocytes and intestine and kidney cells. Am J Physiol 259: C279–285. [DOI] [PubMed] [Google Scholar]
- Tjhin, E.T. , Staines, H.M. , van Schalkwyk, D.A. , Krishna, S. , and Saliba, K.J. (2013) Studies with the Plasmodium falciparum hexokinase reveal that PfHT limits the rate of glucose entry into glycolysis. FEBS Lett 587: 3182–3187. [DOI] [PubMed] [Google Scholar]
- Uldry, M. , Ibberson, M. , Hosokawa, M. , and Thorens, B. (2002) GLUT2 is a high affinity glucosamine transporter. FEBS Lett 524: 199–203. [DOI] [PubMed] [Google Scholar]
- Vander Jagt, D.L. , Hunsaker, L.A. , Campos, N.M. , and Baack, B.R. (1990) D‐lactate production in erythrocytes infected with Plasmodium falciparum. Mol Biochem Parasitol 42: 277–284. [DOI] [PubMed] [Google Scholar]
- Vidyashankar, S. , Sharath Kumar, L.M. , Barooah, V. , Sandeep Varma, R. , Nandakumar, K.S. , and Patki, P.S. (2012) Liv.52 up‐regulates cellular antioxidants and increase glucose uptake to circumvent oleic acid induced hepatic steatosis in HepG2 cells. Phytomedicine 19: 1156–1165. [DOI] [PubMed] [Google Scholar]
- Warburg, O. , Wind, F. , and Negelein, E. (1927) The Metabolism of Tumors in the Body. J Gen Physiol 8: 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild, S. , Roglic, G. , Green, A. , Sicree, R. , and King, H. (2004) Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27: 1047–1053. [DOI] [PubMed] [Google Scholar]
- Woodrow, C.J. , Burchmore, R.J. , and Krishna, S. (2000) Hexose permeation pathways in Plasmodium falciparum‐infected erythrocytes. Proc Natl Acad Sci U S A 97: 9931–9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada, K. , Saito, M. , Matsuoka, H. , and Inagaki, N. (2007) A real‐time method of imaging glucose uptake in single, living mammalian cells. Nat Protoc 2: 753–762. [DOI] [PubMed] [Google Scholar]
- Yu, J.W. , Sun, L.J. , Liu, W. , Zhao, Y.H. , Kang, P. , and Yan, B.Z. (2013) Hepatitis C virus core protein induces hepatic metabolism disorders through down‐regulation of the SIRT1‐AMPK signaling pathway. Int J Infect Dis 17: e539–545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Glucose availability impacts P. berghei infection ex vivo. (A) Mouse primary hepatocytes were infected with luciferase‐expressing P. berghei sporozoites and the culture medium was replaced 2 hpi by medium with different concentrations of glucose. Parasite load (luminescence) and cell viability were assessed 48 hpi. Pool of 3 independent experiments. Error bars represent SEM. One‐way ANOVA with post‐test Dunnett. (B) Mouse primary hepatocytes were infected with GFP‐expressing P. berghei sporozoites and the culture medium was replaced by medium with different concentrations of glucose 2 hpi. Parasite development and the total number of infected cells were assessed at 48 hpi by flow cytometry by determining the fluorescence intensity and the number of GFP+ cells, respectively. One experiment. One‐way ANOVA with post‐test Dunnett. Error bars represent SD. ns ‐ not significant, * p < 0.05, ** p < 0.01 and *** p < 0.001.
Fig. S2. Gating strategy to determine 2‐NBDG uptake and parasite development by flow cytometry. Huh7 cells were infected with RFP‐expressing P. berghei sporozoites and incubated with 2‐NBDG‐containing medium at different time points of infection. Dot plot represents non‐infected and infected (RFP+) cells of one replicate from the 48 h time point. Left histogram shows the different fluorescence intensity displayed by non‐developing (RFPlow) and developing (RFPhigh) parasites. Right histogram exemplifies the 2‐NBDG uptake by the different cell populations (non‐infected, non‐developing and developing parasites), assessed as an increased fluorescence intensity in the green channel.
Fig. S3. P. berghei‐infected Huh7 cells and developing parasites uptake more 2‐NBDG than non‐infected and naïve cells. Huh7 cells were infected with RFP‐expressing P. berghei sporozoites and incubated for 30 min with 2‐NBDG‐containing medium at 48 hpi. (A) Representative images of P. berghei‐infected Huh7 cells with or without incubation with 2‐NBDG. A non‐infected cell, an infected cell and a parasite are outlined in yellow, pink and red, respectively. Scale bar, 20 μm. (B) Quantification of 2‐NBDG uptake by naïve (grey bar), non‐infected cells (yellow bar), infected cells (pink bar) and developing parasites (red bar), as assessed by live fluorescence microscopy. Representative experiment out of 2 independent experiments. One‐way ANOVA with post‐test Bonferroni and two‐tailed Mann‐Whitney test, respectively. Error bars represent SD. *** p < 0.001.
Fig. S4. Quantification of the percentage of MHV‐68‐YFP‐infected cells by flow cytometry. Percentage of YFP+ cells 24 hpi with different loads of MHV‐68‐YFP.
Fig. S5. 2‐NBDG uptake by non‐infected Huh7 cells with GLUT1 knockdown. 2‐NBDG uptake by non‐infected cells of the different cell lines with GLUT1 knockdown at 48 hpi, assessed by flow cytometry. Pool of 4 independent experiments. Error bars represent SEM. One‐way ANOVA with post‐test Dunnett. ns ‐ not significant and *** p < 0.001.
Fig. S6. Chemical inhibition of GLUT1‐mediated glucose transport by WZB117 affects P. berghei hepatic infection ex vivo. (A) Mouse primary hepatocytes were infected with luciferase‐expressing P. berghei sporozoites and 2 h later the culture medium was replaced by medium containing WZB117. Parasite load (luminescence) and cell viability were assessed at 48 hpi. Pool of 2 independent experiments. Error bars represent SEM. One‐way ANOVA with post‐test Dunnett. (B) Mouse primary hepatocytes were infected with GFP‐expressing P. berghei sporozoites and 2 h later the culture medium was replaced by medium containing WZB117. Parasite development was assessed by flow cytometry at 48 hpi. Error bars represent SD. One‐way ANOVA with post‐test Dunnett. ns ‐ not significant, * p < 0.05, ** p < 0.01 and *** p < 0.001.
Fig. S7. GLUT1 expression is not altered in P. berghei‐infected cells. qPCR quantification of GLUT1 transcriptional expression in FACS‐sorted infected and non‐infected Huh7 cells. Each time point represents a pool of 2 independent sorting experiments. Error bars represent SD. Two‐tailed Mann‐Whitney test. ns – not significant.
Fig. S8. Increased GLUT1‐dependent glucose uptake into P. berghei‐infected cells is independent of AMPK. (A) Knockdown efficiency upon transfection in Huh7 cells of the siRNAs used against the α1 and α2 subunits of AMPK. (B) Huh7 cells with the knockdown of α1 or/and α2 subunits of AMPK were infected with GFP‐expressing P. berghei sporozoites and parasite development was assessed by flow cytometry at 48 hpi. Pool of 2 independent experiments. Error bars represent SD. One‐way ANOVA with post‐test Dunnett. (C) 2‐NBDG uptake at 48 hpi by developing parasites‐containing cells with the knockdown of α1 or/and α2 subunits of AMPK, assessed by flow cytometry. Representative experiment out of 2 independent experiments. Error bars represent SD. One‐way ANOVA with post‐test Dunnett. ns ‐ not significant, *** p < 0.001.
Fig. S9. PI3K stimulation or inhibition does not impact P. berghei infection of Huh7 cells neither does it alter glucose uptake by infected cells. Huh7 cells were infected with luciferase‐expressing P. berghei sporozoites and 2 h later were treated with different concentrations of (A) insulin or (B) Wortmannin. Parasite load (luminescence) was assessed after 48 h. Representative experiment out of 2 and pool of 3 independent experiments, respectively. Error bars represent SD. To evaluate possible effects on glucose uptake, Huh7 cells were infected with RFP‐expressing P. berghei sporozoites and the treatment with different concentrations of insulin (C) and Wortmannin (D) was initiated 2 hpi. 2‐NBDG uptake by developing parasites‐containing cells was assessed at 48 hpi by flow cytometry. Pool of 2 independent experiments for both insulin and Wortmannin. Error bars represent SD. All panels: one‐way ANOVA with post‐test Dunnett. ns ‐ not significant.
Table S1. List of shRNA sequences used, with the corresponding knockdowns.
Table S2. List of primer sequences.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item