

Abstract
Importance
Schizophrenia (SCZ) is a devastating psychiatric condition. Identifying the specific genetic variants and pathways that increase susceptibility to SCZ is critical to improve disease understanding and address the urgent need for new drug targets.
Objective
To identify SCZ susceptibility genes.
Design
We integrated results from a meta-analysis of 18 genome-wide association studies (GWAS) involving 1 085 772 single-nucleotide polymorphisms (SNPs) and 6 databases that showed significant informativeness for SCZ. The 9380 most promising SNPs were then specifically genotyped in an independent family-based replication study that, after quality control, consisted of 8107 SNPs.
Setting
Linkage meta-analysis, brain transcriptome meta-analysis, candidate gene database, OMIM, relevant mouse studies, and expression quantitative trait locus databases.
Patients
We included 11 185 cases and 10 768 control subjects from 6 databases and, after quality control 6298 individuals (including 3286 cases) from 1811 nuclear families.
Main Outcomes and Measures
Case-control status for SCZ.
Results
Replication results showed a highly significant enrichment of SNPs with small P values. Of the SNPs with replication values of P<.01, the proportion of SNPs that had the same direction of effects as in the GWAS meta-analysis was 89% in the combined ancestry group (sign test, P<2.20×10−16) and 93% in subjects of European ancestry only (P<2.20×10−16). Our results supported the major histocompatibility complex region showing a 3.7-fold overall enrichment of replication values of P<.01 in subjects from European ancestry. We replicated SNPs in TCF4 (P=2.53×10−10) and NOTCH4 (P=3.16×10−7) that are among the most robust SCZ findings. More novel findings included POM121L2 (P=3.51×10−7), AS3MT (P=9.01×10−7), CNNM2 (P=6.07×10−7), and NT5C2 (P=4.09×10−7). To explore the many small effects, we performed pathway analyses. The most significant pathways involved neuronal function (axonal guidance, neuronal systems, and L1 cell adhesion molecule interaction) and the immune system (antigen processing, cell adhesion molecules relevant to T cells, and translocation to immunological synapse).
Conclusions and Relevance
We replicated novel SCZ disease genes and pathogenic pathways. Better understanding the molecular and biological mechanisms involved with schizophrenia may improve disease management and may identify new drug targets.
Schizophrenia (SCZ) is a major public health problem1 that ranks ninth in the global burden of illness.2 Of a large set of prenatal and antenatal risk factors, having a first-degree relative with SCZ is one of the most important,3 and genetic factors account for most of this familial risk.4 Identifying the specific genetic variants that increase susceptibility is crucial to improve our understanding of SCZ and has the potential to address the urgent need for new drug targets.5
Although SCZ genetics have proven difficult, recent genome-wide association studies (GWASs) mega- and meta-analyses have suggested several promising loci.6,7 Our aim was to perform a comprehensive family-based replication study of promising SCZ loci. Because the results from the recent mega-analysis of the Psychiatric GWAS Consortium were not available at the time we started the present study,6 we first conducted a meta-analysis of 18 SCZ GWASs that included 21 953 subjects of European ancestry. The samples used for this meta-analysis overlapped for almost 90% with the Psychiatric GWAS Consortium study. In addition to selecting for replication the SNPs with the best P values in the GWAS meta-analysis, we selected the best SNPs after integrating existing informative SCZ data sources with the meta-analysis results. Such a convergent functional genomic approach can improve statistical power to detect biologically more meaningful and reproducible effects.8 In total, we selected 9381 SNPs for genotyping in an independent sample of 6298 subjects, including 3286 cases, from 1811 nuclear families. The use of nuclear families is a critical feature because almost all GWAS studies to date involved case-control samples, and their findings have been criticized as being possible population stratification artifacts.9 Because our family-based replication study is robust against such false-positive findings, it provides an unprecedented opportunity to shed more light on recent GWAS findings.
METHODS
GWAS META-ANALYSIS
Our SCZ GWAS meta-analysis involved 18 studies. After stringent quality control, 1 085 772 genotyped and imputed SNPs were available for 21 953 subjects of European descent (11 185 cases and 10 768 controls). To account for possible population stratification within each of the GWASs, we included the first 3 principal components from the EigenSoft package (Helix Systems)10 plus any additional components that predicted case-control status (P≤.05). Additional details about the study samples and the methods can be found in eMethods 1, eTable 1, and eFigures 1 through 6 (http://www.jamapsych.com).
SNP SELECTION FOR REPLICATION STUDIES
Single-nucleotide polymorphisms were selected for a variety of reasons (eFigure 3 and eTable 2), including having the best P values in the meta-analysis and after data integration using Mathematically based Integration of Heterogeneous Data (MIND)11 (validation studies are listed in the eMethods 2). MIND estimates the (posterior) probability that an SNP is associated with the disease after taking all data (GWAS meta-analysis and external data sets) into account. It empirically “weighs” other data according to the strength of its disease-relevant information. We used the following databases that contained significant SCZ information: SzGene12 database, summarizing the results of 1617 studies reporting on 952 SZC candidate genes (excluding findings from the GWAS used in our meta-analysis); top regions from a meta-analysis of 32 independent genome-wide linkage scans of 3255 pedigrees with 7413 SCZ cases13; a gene expression meta-analysis of 12 controlled studies across 6 different microarray platforms using postmortem brain tissue from SCZ cases and controls14; the OMIM database of disease genes; human orthologs of mouse genes associated with behavioral phenotypes relevant to neuropsychiatric outcomes15; and SNPs strongly associated with variation in transcript abundance in the cortex (http://eqtl.uchicago.edu). A total of 8107 of the 9380 selected SNPs were successfully genotyped with the Illumina iSelect assay (http://www.illumina.com/products/infinium_iselect_custom_genotyping_beadchips.ilmn), with a call rate of 99.94%.
FAMILY-BASED REPLICATION
Approximately 89% of the replication samples were families from the National Institute of Mental Health repository, and the remaining 11% were collected by one of us (L.E.D.). None of these samples was used in the prior analyses, so this replication effort was completely independent. Each family included at least 1 subject with a DSM-III-R diagnosis of SCZ. After quality control, 6298 individuals (3286 cases) from 1811 families remained.
For the analyses, we first subdivided subjects into ancestral groups based on identity-by-state sharing, as estimated using the genotyped SNPs in parents or, if they had not undergone genotyping, 1 randomly selected sibling per family. Three well-distinguished clusters, where the major self-reported ethnicity in each group was African (1262 individuals from 438 families), European (2740 individuals from 794 families), and Asian (2296 individuals from 579 families) ancestry, are shown in eFigure 7. Next, we used UNPHASED software,16 which is robust to population structure when the data are complete and has only minor loss of robustness when data are missing. To further minimize the risk of population stratification effects, we first performed the analyses within each ancestral group and then combined the 3 test statistics to obtain an overall replication P value. We limited the association testing to markers with a minor allele frequency of greater than 0.05 within each group. We preserved the direction of effects (ie, sign) so that an allele being overtransmitted in one group and undertransmitted in another would have no effect in this combined analysis (eMethods 2). Additional details are given in eTable 2 and eFigure 8.
PATHWAY ANALYSIS
To test whether multiple susceptibility alleles with small effects were organized into pathways, we used Consensus-PathDB, a human-centric meta-database of functional biological data compiled from 30 separate public sources of biological interactions.17–19 A list of 265 genes overlapping or flanking (±25 kilobases [kb]) the SCZ-associated SNPs with P<.05 in our analysis were included in these analysis. To account for multiple testing, we controlled the false discovery rate20 at the 0.01 level (eMethods 2 and eTable 3).
In MIND, sources of prior information used to select SNPs through data integration will only affect results to the extent that they contain disease-relevant information (ie, to the extent that genes with good P values in the sources of prior information also have good P values in the meta-analysis) (eMethods 3). Because most of the databases we used pertain to genes, bias in terms of selecting SNPs in genes as opposed to intergenic regions is expected. However, the selection was entirely based on the empirical support for SNPs in those genes. Furthermore, the pathway analyses were performed using the replication results. Therefore, our pathway findings are unlikely to reflect prior notions about SCZ-relevant pathways for which there is no empirical support (eFigure 9).
RESULTS
Figure 1 shows histograms of the P values from the association tests. Under the null hypothesis assuming no replication, the histograms would have had equal heights. The skewed histograms therefore indicated considerable enrichment of small P values. To quantify this enrichment, we divided the observed median test statistic value by the expected median under the null hypothesis. This index, which has an expected value of 1.00 if SNPs do not replicate, equaled 1.19, 1.12, and 1.07 for subjects from European, Asian, and African ancestry, respectively, and was 1.15 for the (signed) combined analysis that required the same direction of effects in all 3 groups. Next, we tested whether this enrichment for small P values was statistically significant (eMethods 3). Owing to the large number of SNPs, large sample size, and family-based association tests allowing for missing genotypes, it was computationally not feasible to perform a sufficiently large number of permutations to obtain empirical P values. Instead, we obtained the lower and upper bounds assuming no linkage disequilibrium (LD) and very high LD, respectively, among the SNPs. When we assumed no LD, the P value was so small that the statistical test in the R package returned a 0; when we assumed an extremely high LD between the SNPs, we obtained P=2.0×10−4. Thus, even in the most conservative scenario, the test indicated significant enrichment of small P values in the replication. We also performed sign tests that examined whether the direction of effects was similar in the GWAS meta-analysis and the replication study. Of the SNPs with replication values of P<.01, the proportion of SNPs that had the same direction of effect of 89% (P=2.20×10−16; 95% CI, 82%–94%) for the combined ancestry group and 93% (P=2.20 10−16; 95% CI, 88%–97%) for European subjects. Again, in both cases the statistical test in the R package returned a 0, indicating that this pattern was almost impossible to occur by change. The mean odds ratios of these SNPs were 1.2 and 1.3 in the combined and European study samples, respectively. Overall, our findings confirm the polygenetic nature of SCZ and show that we replicated a substantial number of susceptibility alleles with small effects.7,21,22
Figure 1.
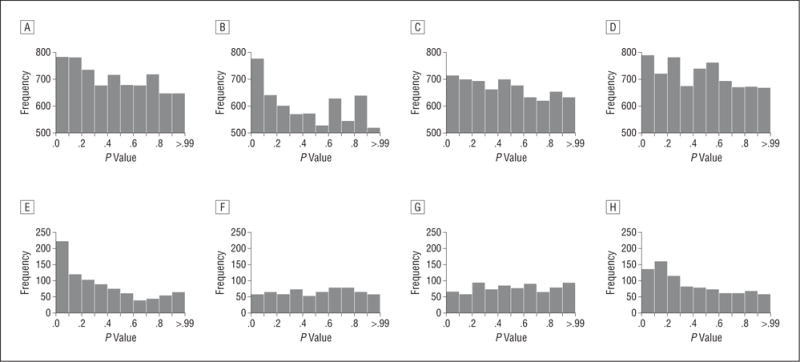
Histograms of P values from the family-based association analyses. Results are given in subjects from European (A and E), Asian (B and F), and African (C and G) ancestry. The combined analyses (D and I) present results from the test statistics across the 3 groups while taking the direction of effects into account. Plots are given for single-nucleotide polymorphisms not including (A–D) and including (F–I) the major histocompatibility complex region.
We anticipated that SNPs replicate better in the same ancestral group in which they showed their initial association signals. The GWAS meta-analysis was the main source of SNP selection and involved European subjects only, which likely explains why SNPs replicated relatively better in the European group. Although non–major histocompatibility complex (MHC) SNPs replicated in all ancestral groups, the SNPs in the MHC region did not: we observed a large 3.7-fold enrichment of values of P<.01 in European samples, but findings dissipated in non-European samples. This pattern seems consistent with the exceptionally large LD differences across ancestral groups in this region resulting from its evolutionary significance (eg, it harbors many genes affecting the immune response).
For SNPs that do not have effects, we would not expect to see any difference between markers selected with or without data integration in the replication study. To compare the 2 approaches, we therefore first selected all SNPs with P values of less than .01 to enrich for markers with effects. Figure 2 shows that SNPs selected through data integration replicated as well as SNPs selected on the basis of having the top-ranked P values in the meta-analysis without data integration. Some of the SNPs selected through data integration even had P values with ranks as high as 25 000 in the meta-analysis. The fact that such SNPs would not have been selected on the basis of their P value but do replicate demonstrates the value of considering existing data sources.
Figure 2.
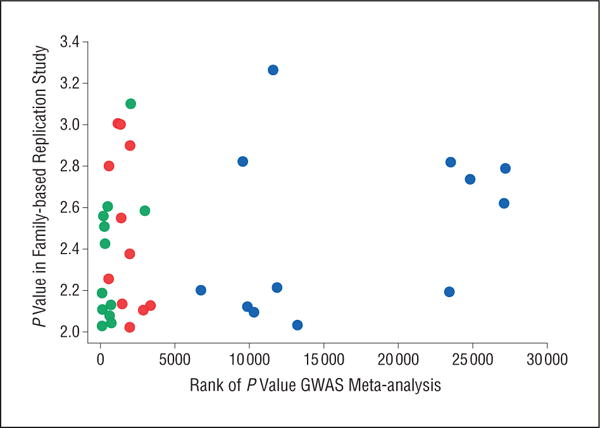
Comparison of single-nucleotide polymorphisms selected through data integration (blue), that have a top genome-wide association study (GWAS) meta-analysis P value (red), or both (green). P values in the family-based replication study are calculated on a −log10 scale.
We applied a stringent definition of replication requiring the same SNP to have the same direction of effect in the replication study and meta-analysis. Table 1 (combined analysis) and Table 2 (European analysis) report the SNPs that replicated at P≤.005. The P values for all SNPs can be downloaded from http://www.people.vcu.edu/~ejvandenoord/. Compared with the meta-analysis or when combining P values from the 3 ancestral groups, sample size was ignored when calculating the overall P value across the replication study and meta-analysis. In the GWAS meta-analysis, the effect sizes will be overestimated and P values will be too optimistic owing to the winner’s curse.23–25 Furthermore, because the sample size was much larger in the meta-analysis, these P values would have dominated the combined P value. Ignoring samples sizes avoids overly optimistic P values while still providing some quantification for the combined evidence of a specific SNP across the meta- analysis and replication study. Some SNPs in Table 1 showed substantial allele frequency differences across ancestral groups. However, in addition to using nuclear families, the UNPHASED analyses were performed within each ancestral group to provide a second layer of protection against possible stratification effects.
Table 1.
SNPs With P≤.005 in Combined Replication Analysis and Direction of Effect Identical to GWAS Meta-analysis
| SNP | Chr | Coordinatea | Allele | Frequencyb
|
Oddsc
|
P Value
|
Reasong | Directionh | Genes | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| European | Asian | African | European | Asian | African | Replicationd | Meta-analysise | Overallf | |||||||
| rs2296268 | 1 | 36603165 | T | 0.29 | 0.56 | 0.68 | 1.1 | 1.2 | 1.0 | 3.30 × 10−3 | 7.02 × 10−3 | 3.21 × 10−5 | 1 | + | TRAPPC3 |
| rs589249 | 1 | 37162352 | G | 0.64 | 0.45 | 0.17 | 1.2 | 1.1 | 1.2 | 3.08 × 10−3 | 2.17 × 10−5 | 3.48 × 10−7 | 3 | + | GRIK3i |
| rs4664494 | 2 | 152499580 | T | 0.82 | 0.55 | 0.42 | 1.2 | 1.1 | 1.2 | 1.57 × 10−3 | 6.93 × 10−2 | 2.87 × 10−4 | 1 | + | NEB |
| rs10193188 | 2 | 152551210 | G | 0.18 | 0.51 | 0.57 | 0.8 | 1.0 | 0.8 | 2.19 × 10−3 | 1.40 × 10−2 | 9.46 × 10−5 | 4 | − | NEB |
| rs6739563 | 2 | 201134216 | T | 0.84 | 0.80 | 0.87 | 1.2 | 1.2 | 1.0 | 3.23 × 10−3 | 2.44 × 10−2 | 2.38 × 10−4 | 2 | + | SPATS2Li |
| rs12496073 | 3 | 25033188 | G | 0.34 | 0.51 | 0.16 | 1.2 | 1.1 | 0.9 | 4.34 × 10−3 | 3.10 × 10−3 | 3.98 × 10−5 | 2 | + | j |
| rs6898746 | 5 | 60843706 | T | 0.51 | 0.28 | 0.88 | 0.8 | 1.0 | 0.8 | 2.78 × 10−3 | 3.72 × 10−3 | 3.10 × 10−5 | 2 | − | ZSWIM6i |
| rs2857605 | 6 | 31524851 | G | 0.81 | 0.86 | 0.93 | 1.4 | 1.0 | 1.2 | 3.34 × 10−3 | 8.78 × 10−1 | 2.90 × 10−2 | 4 | + | NFKBIL1 |
| rs3130349 | 6 | 32147696 | G | 0.15 | 0.08 | 0.12 | 1.3 | 1.1 | 1.3 | 3.76 × 10−3 | 3.16 × 10−3 | 1.48 × 10−5 | 1 | + | RNF5 |
| rs3132935 | 6 | 32171075 | G | 0.82 | 0.85 | 0.83 | 0.8 | 0.9 | 1.0 | 1.51 × 10−3 | 2.17 × 10−4 | 3.16 × 10−7 | 1 | − | NOTCH4 |
| rs3132947 | 6 | 32176782 | T | 0.82 | 0.85 | 0.83 | 0.8 | 0.9 | 1.0 | 1.83 × 10−3 | 2.40 × 10−4 | 4.34 × 10−7 | 1 | − | NOTCH4 |
| rs4409766 | 10 | 104616663 | T | 0.12 | 0.27 | 0.25 | 1.2 | 1.1 | 1.5 | 1.21 × 10−3 | 1.38 × 10−2 | 5.57 × 10−5 | 4 | + | C10orf32 |
| rs3740390 | 10 | 104638480 | G | 0.11 | 0.26 | 0.07 | 1.3 | 1.1 | 1.6 | 7.90 × 10−4 | 3.30 × 10−4 | 9.01 × 10−7 | 3 | + | AS3MT |
| rs7897654 | 10 | 104662458 | T | 0.30 | 0.46 | 0.29 | 1.2 | 1.0 | 1.3 | 3.10 × 10−3 | 1.76 × 10−5 | 2.93 × 10−7 | 3 | + | AS3MTi |
| rs12221064 | 10 | 104677126 | T | 0.89 | 0.74 | 0.94 | 0.8 | 0.9 | 0.6 | 1.26 × 10−3 | 3.20 × 10−4 | 1.40 × 10−6 | 2 | − | CNNM2i |
| rs11191499 | 10 | 104764271 | T | 0.11 | 0.26 | 0.06 | 1.3 | 1.1 | 1.6 | 9.90 × 10−4 | 1.99 × 10−4 | 7.09 × 10−7 | 2 | + | CNNM2 |
| rs11191514 | 10 | 104773364 | T | 0.89 | 0.74 | 0.94 | 0.8 | 0.9 | 0.6 | 9.83 × 10−4 | 1.70 × 10−4 | 6.07 × 10−7 | 3 | − | CNNM2 |
| rs17094683 | 10 | 104851301 | T | 0.90 | 0.74 | 0.94 | 0.8 | 0.9 | 0.6 | 1.59 × 10−3 | 6.24 × 10−5 | 4.09 × 10−7 | 3 | − | NT5C2 |
| rs1564483 | 18 | 60794654 | G | 0.25 | 0.34 | 0.13 | 1.2 | 1.1 | 1.2 | 2.75 × 10−3 | 4.69 × 10−3 | 1.69 × 10−5 | 1 | + | BCL2 |
| rs12159787 | 22 | 40870699 | G | 0.91 | 0.81 | 0.78 | 0.8 | 0.9 | 0.9 | 4.07 × 10−3 | 8.45 × 10−3 | 9.89 × 10−5 | 2 | − | MKL1 |
| rs5995871 | 22 | 40922332 | G | 0.91 | 0.81 | 0.94 | 0.8 | 0.9 | 0.9 | 4.94 × 10−3 | 1.16 × 10−2 | 8.36 × 10−5 | 1 | − | MKL1 |
| rs6001980 | 22 | 41004384 | T | 0.09 | 0.19 | 0.07 | 1.3 | 1.1 | 1.2 | 4.96 × 10−3 | 3.60 × 10−3 | 5.22 × 10−5 | 2 | + | MKL1 |
| rs138880 | 22 | 50218611 | C | 0.78 | 0.89 | 0.26 | 1.1 | 1.3 | 1.2 | 2.75 × 10−3 | 9.49 × 10−6 | 1.53 × 10−7 | 3 | + | BRD1i |
Abbreviations: Chr, chromosome; GWAS, genome-wide association studies; SNP, single-nucleotide polymorphism.
Indicates location of chromosome in the human genome build 19.
Indicates frequency of allele in each ancestry group. Linkage disequilibrium between adjacent markers (r2>0.8) is indicated using boldface type.
Indicates the effect in each ancestry group.
Indicates combined results from families with European, Asian, and African ancestry.
Depending on why the SNP was selected, indicates with or without data integration.
Indicates the combined P value from replication and meta-analysis.
Indicates reason for selection as only after data integration (1), only because of top P value in GWAS meta-analysis (2), both (3), or other (4).
Indicates direction of effect in the replication and meta-analysis where the allele increases (plus sign) or decreases (minus sign) the risk.
Indicates SNP is located outside of gene boundaries, and the closest gene within 100 kilobases (kb) is given.
Indicates that the closest gene is located more than 100 kb from the SNP.
Table 2.
SNPs With P≤.005 in Replication Analysis in Subjects of European Ancestry and Direction of Effect Identical to GWAS Meta-analysis
| SNPa | Chr | Coordinateb | Allele | European Ancestry
|
P Value
|
Reasonh | Directioni | Genes | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Frequencyc | Oddsd | Replicatione | Meta-analysisf | Overallg | |||||||
| rs4579747 | 1 | 154153106 | G | 0.45 | 1.2 | 2.14 × 10−3 | 2.10 × 10−2 | 7.91 × 10−5 | 1 | + | TPM3 |
| rs12140439 | 1 | 177722907 | C | 0.31 | 1.3 | 1.20 × 10−3 | 3.39 × 10−4 | 1.41 × 10−6 | 3 | + | SEC16Bj |
| rs4381823 | 2 | 48047335 | G | 0.93 | 0.7 | 1.91 × 10−3 | 2.54 × 10−3 | 5.92 × 10−6 | 1 | − | FBXO11 |
| rs11687313L | 2 | 201146399 | G | 0.13 | 0.8 | 4.04 × 10−3 | 3.92 × 10−3 | 4.64 × 10−5 | 2 | − | SPATS2Lj |
| rs12496073 | 3 | 25033188 | G | 0.34 | 1.2 | 2.01 × 10−3 | 3.10 × 10−3 | 1.91 × 10−5 | 2 | + | k |
| rs17203055 | 3 | 119084331 | G | 0.87 | 0.7 | 8.00 × 10−4 | 2.01 × 10−3 | 5.24 × 10−6 | 2 | − | ARHGAP31 |
| rs4403116L | 4 | 118281665 | G | 0.81 | 1.3 | 3.43 × 10−3 | 1.71 × 10−3 | 1.81 × 10−5 | 2 | + | k |
| rs13120709 | 4 | 169435779 | T | 0.66 | 1.3 | 8.06 × 10−4 | 9.00 × 10−2 | 3.59 × 10−4 | 4 | + | PALLD |
| rs6898746 | 5 | 60843706 | T | 0.51 | 0.8 | 3.14 × 10−3 | 3.72 × 10−3 | 3.48 × 10−5 | 2 | − | ZSWIM6 |
| rs3800316 | 6 | 27256102 | C | 0.72 | 0.8 | 4.55 × 10−3 | 2.43 × 10−5 | 6.00 × 10−7 | 3 | − | POM121L2j |
| rs16897515 | 6 | 27278020 | C | 0.16 | 1.3 | 1.71 × 10−3 | 4.76 × 10−5 | 3.51 × 10−7 | 3 | + | POM121L2 |
| rs3099840L | 6 | 31430721 | G | 0.80 | 0.8 | 2.17 × 10−3 | 1.43 × 10−3 | 9.73 × 10−6 | 2 | − | HCP5j |
| rs2857605 | 6 | 31524851 | G | 0.81 | 1.4 | 9.67 × 10−5 | 8.78 × 10−1 | 4.17 × 10−3 | 4 | + | NFKBIL1 |
| rs3130626L | 6 | 31598489 | G | 0.85 | 0.7 | 2.19 × 10−3 | 9.45 × 10−4 | 6.68 × 10−6 | 3 | − | PRRC2A |
| rs1046089 | 6 | 31602967 | G | 0.34 | 1.3 | 7.71 × 10−4 | 3.83 × 10−3 | 3.97 × 10−6 | 1 | + | PRRC2A |
| rs3117583L | 6 | 31619576 | T | 0.15 | 1.3 | 2.19 × 10−3 | 7.39 × 10−4 | 5.31 × 10−6 | 3 | + | BAG6 |
| rs2071278 | 6 | 32165444 | T | 0.12 | 1.4 | 2.70 × 10−3 | 5.39 × 10−4 | 4.91 × 10−6 | 3 | + | NOTCH4 |
| rs3132935L | 6 | 32171075 | G | 0.82 | 0.8 | 2.39 × 10−3 | 2.17 × 10−4 | 5.22 × 10−7 | 1 | − | NOTCH4 |
| rs3117137 | 6 | 32309911 | G | 0.22 | 1.3 | 3.71 × 10−3 | 2.46 × 10−3 | 2.75 × 10−5 | 2 | + | C6orf10 |
| rs2854275 | 6 | 32628428 | T | 0.89 | 0.7 | 4.99 × 10−3 | 6.87 × 10−5 | 1.59 × 10−6 | 3 | − | HLA-DQB1 |
| rs6917824L | 6 | 124342663 | G | 0.83 | 1.3 | 8.20 × 10−4 | 3.52 × 10−3 | 3.80 × 10−6 | 1 | + | NKAIN2 |
| rs10952674 | 7 | 146867339 | T | 0.61 | 0.8 | 2.34 × 10−3 | 3.31 × 10−1 | 4.52 × 10−3 | 4 | − | CNTNAP2 |
| rs7894284 | 10 | 14894901 | T | 0.18 | 0.8 | 4.78 × 10−3 | 1.08 × 10−2 | 1.46 × 10−4 | 4 | − | HSPA14 |
| rs11594111 | 10 | 14945406 | G | 0.88 | 1.4 | 3.35 × 10−3 | 4.17 × 10−3 | 1.80 × 10−5 | 1 | + | SUV39H2 |
| rs605715 | 10 | 21377866 | T | 0.61 | 1.2 | 2.18 × 10−3 | 2.61 × 10−3 | 1.74 × 10−5 | 2 | + | NEBL |
| rs478559L | 10 | 21393804 | T | 0.62 | 1.2 | 1.81 × 10−3 | 3.56 × 10−3 | 1.98 × 10−5 | 2 | + | NEBL |
| rs7907819L | 10 | 73408096 | T | 0.77 | 1.3 | 4.07 × 10−3 | 7.46 × 10−3 | 4.20 × 10−5 | 1 | + | CDH23 |
| rs2902548 | 10 | 104487382 | T | 0.83 | 0.8 | 4.73 × 10−3 | 1.46 × 10−1 | 2.48 × 10−3 | 4 | − | SFXN2 |
| rs284861 | 10 | 104572276 | T | 0.86 | 0.7 | 1.11 × 10−3 | 2.98 × 10−2 | 1.22 × 10−4 | 4 | − | C10orf26 |
| rs3750997 | 11 | 71158841 | T | 0.70 | 0.8 | 2.33 × 10−3 | 8.72 × 10−3 | 2.97 × 10−5 | 1 | − | DHCR7 |
| rs7930295 | 11 | 125313599 | G | 0.86 | 1.3 | 1.25 × 10−3 | 7.82 × 10−4 | 3.21 × 10−6 | 2 | + | FEZ1j |
| rs6590146 | 11 | 125349829 | T | 0.17 | 0.8 | 1.52 × 10−3 | 1.00 × 10−2 | 4.84 × 10−5 | 4 | − | FEZ1 |
| rs11082811 | 18 | 47763296 | T | 0.69 | 1.2 | 3.61 × 10−3 | 1.47 × 10−3 | 1.66 × 10−5 | 3 | + | CCDC11 |
| rs1261117 | 18 | 52949657 | T | 0.06 | 1.6 | 5.06 × 10−4 | 6.23 × 10−7 | 2.53 × 10−10 | 1 | + | TCF4 |
| rs17641958L | 19 | 40173197 | G | 0.30 | 0.7 | 8.10 × 10−6 | 4.29 × 10−2 | 2.48 × 10−6 | 1 | − | LGALS17A |
| rs12611334L | 19 | 40229409 | G | 0.70 | 1.4 | 1.89 × 10−5 | 2.45 × 10−2 | 1.99 × 10−6 | 1 | + | CLCj |
Abbreviations: Chr, chromosome; GWAS, genome-wide association studies; SNP, single-nucleotide polymorphism.
L Indicates that markers in strong linkage disequilibrium (r2>0.95) with the listed marker have been excluded from the Table.
Coordinate is the location of the chromosome in the human genome build 19.
Indicates frequency of allele in the ancestry group. Linkage disequilibrium between adjacent markers (r2>0.8) is indicated using boldface type.
Indicates the effect in families of European ancestry.
Indicates results from replication study including families of European ancestry.
Depending on why the SNP was selected, indicates with or without data integration.
Indicates the combined P value from replication and meta-analysis.
Indicates reason for selection as only after data integration (1), only because of top P value in GWAS meta-analysis (2), both (3), or other (4).
Indicates direction of effect in the replication and meta-analysis where the allele increases (plus sign) or decreases (minus sign) the risk.
Indicates that the SNP is located outside of gene boundaries, and the closest gene within 100 kilobases (kb) is given.
Indicates that the closest gene is located more than 100 kb from the SNP.
Table 3 shows the results from the pathway analyses. Where the same gene combination from our input list indicated multiple pathways, we show only the most significant instance to eliminate redundancy. Fourteen pathways were significant at a false discovery rate of 0.01. The 3 most significant pathways were axon guidance (P = 5.26×10−6), developmental biology (P = 1.29×10−5), and neuronal systems (P = 1.37×10−5). Larger pathways, such as these 3 top findings, which each include more than 250 genes, frequently incorporate several more specific themes, and this can be further observed in Table 3. For example, axon guidance includes the smaller L1 cell adhesion molecule (L1CAM) pathway interaction (P = 6.75×10−5), which was also significant in our analysis.
Table 3.
ConsensusPathDB Pathways That Are Significant When the False Discovery Rate Is Controlled at the .01 Level
| Pathwaya | Database | Genes Overlapping With Replication Resultsb | Sizec | Matches, No (%)d | P Value | Q Valuee |
|---|---|---|---|---|---|---|
| Axon guidance | Reactome | RPS6KA2, CACNA1C, NFASC, CACNB2, ROBO2, KCNQ3, CREB1, LY6G5B, ANK1, ANK2, ANK3, SLIT3, PAK6, CACNB4, CSNK2B | 266 | 15 (5.6) | 5.26 × 10−6 | 0.0009 |
| Developmental biology | Reactome | TCF4, RPS6KA2, ROBO2, ANK1, NFASC, CACNB2, CACNA1C, KCNQ3, CREB1, LY6G5B, PPARG, ANK2, ANK3, SLIT3, PAK6, CACNB4, CSNK2B | 360 | 17 (4.7) | 1.29 × 10−5 | 0.0009 |
| Neuronal system | Reactome | RPS6KA2, KCNK9, CACNB2, KCNQ3, CREB1, CACNB4, CHRNA5, KCNMA1, NSF, CHRNA2, CHRNA3, ALDH5A1, GABBR2, KCNQ1, SLC1A3 | 288 | 15 (5.2) | 1.37 × 10−5 | 0.0009 |
| Interaction between L1 and ankyrins | Reactome | ANK2, ANK1, NFASC, ANK3, KCNQ3 | 26 | 5 (19.2) | 2.78 × 10−5 | 0.0013 |
| L1CAM interactions | Reactome | RPS6KA2, NFASC, KCNQ3, LY6G5B, ANK1, ANK2, ANK3, CSNK2B | 97 | 8 (8.2) | 6.75 × 10−5 | 0.0021 |
| Translocation of ZAP-70 to immunological synapse | Reactome | HLA-DRA, CD247, HLA-DQB1, HLA-DQA1, HLA-DOB | 31 | 5 (16.1) | 6.78 × 10−5 | 0.0021 |
| Antigen processing and presentation | KEGG | TAP1, HLA-DRA, TAP2, CREB1, HLA-DQB1, HLA-DOB, HLA-DQA1 | 76 | 7 (9.2) | 9.68 × 10−5 | 0.0021 |
| Transmission across chemical synapses | Reactome | RPS6KA2, CACNB2, CREB1, CACNB4, CHRNA5, NSF, CHRNA2, CHRNA3, ALDH5A1, GABBR2, SLC1A3 | 195 | 11 (5.6) | 1.00 × 10−4 | 0.0021 |
| Nicotinic acetylcholine receptors | Reactome | CHRNA5, CHRNA2, CHRNA3 | 9 | 3 (33.3) | 2.21 × 10−4 | 0.0035 |
| Antigen processing and presentation | BioCarta | TAP1, HLA-DRA, TAP2 | 12 | 3 (25.0) | 5.61 × 10−4 | 0.0061 |
| Cell adhesion molecules | KEGG | HLA-DRA, NRXN2, PTPRF, NFASC, CNTNAP2, HLA-DQB1, HLA-DOB, HLA-DQA1 | 133 | 8 (6.0) | 5.97 × 10−4 | 0.0061 |
| Nuclear receptor transcription pathway | Reactome | PPARG, NR3C1, THRB, RORA, NR3C2 | 51 | 5 (9.8) | 7.47 × 10−4 | 0.0070 |
| Asthma, Homo sapiens (human) | KEGG | HLA-DRA, HLA-DQB1, HLA-DQA1, HLA-DOB | 30 | 4 (13.3) | 8.01 × 10−4 | 0.0072 |
| Hypertrophic cardiomyopathy | KEGG | ACE, SGCG, CACNB2, CACNA1C, TPM3, CACNB4 | 83 | 6 (7.2) | 1.13 × 10−3 | 0.0083 |
Abbreviations: KEGG, Kyoto Encyclopedia of Genes and Genomes; L1CAM, L1 cell adhesion molecule.
Indicates the pathway name or description provided in the source database.
Indicates the genes from the replication results overlapping with the reference pathway.
Indicates the number of genes in the reference pathway.
Indicates the number of genes from that pathway found in our data.
Indicates the false discovery rates calculated using the P values of the individual tests as thresholds for declaring significance.
COMMENT
In our study we replicated SNPs in TCF4 (P = 2.53×10−10 in the European analysis) and NOTCH4 (P = 3.16×10−7 in the combined and P = 5.22×10−7 in the European analyses) that are among the top 10 most promising SCZ candidate genes.12 Other loci previously showing association with SCZ include GRIK326 (P = 3.48×10−7) and BRD127 (P = 1.53×10−7) in the combined analyses and FEZ128 (P = 3.21×10−6) in the European analysis. In the combined analyses, we replicated (P = 2.90×10−7) SNPs in an approximately 230-kb region on chromosome 10q24, which was recently reported to be associated in part with SCZ.6 This region encompasses an uncharacterized open reading frame (C10orf32) as well as AS3MT, CNNM2, and NT5C2. AS3MT may play a role in arsenic metabolism.29 CNNM2 is abundantly expressed in brain and functions as a divalent metal ion transporter,30 whereas NT5C2 hydrolyzes purine nucleotides and is involved in maintaining cellular nucleotide balance.31 A notable novel finding in the combined analysis (P = 1.69×10−5) is BCL2, which has been suggested as a marker for neuronal differentiation.32 Lower levels of BCL2 have been observed in the temporal cortex of patients with SCZ compared with controls.33
After TCF4, the second most significant finding (P = 3.51×10−7) in the European replication involved POM121L2, a gene with unknown function but in a location that has previously been associated with SCZ.7 In addition to NOTCH4 and POM121L2, several other genes in the MHC region replicated. The LD between the MHC SNPs reported in Table 2 (eFigure 8) showed a complex pattern and ranged from 0 to high LD (eg, SNPs in NFKBIL1, PRRC2A, and BAG6). Many of these genes are involved in immune response, although recent evidence suggests a possible role in neuronal signaling and activity-dependent changes in synaptic connectivity.34 This finding was true for some SNPs outside the MHC as well. For example, several genes with significant findings on chromosome 19 (eg, CLC) code for proteins that belong to the galectin family, which regulates immune response.
In the pathway analysis, 43 SCZ-associated genes were included in the 14 most significant pathways. Many of these are related to neuronal function and the immune system, 2 functions of potential relevance for SCZ, which are discussed in this section. The most significant pathway finding, axon guidance, includes genes involved in the process by which neurons send out axons to reach the correct targets. Growing axons sense guidance cues in the environment and respond by undergoing cytoskeletal changes that determine the direction of axon growth. Several highly conserved families of axon guidance molecules and their receptors have been identified.35 Among the SCZ-associated genes in our study that are part of this pathway are ROBO2 (roundabout, axon guidance receptor, homologue 2), which is a receptor for the axon guidance molecule SLIT2. The related gene SLIT3 is also among our findings. As mentioned already, L1CAM interactions is a subpathway of “axon guidance” that was also significantly enriched for SCZ-associated genes. The L1CAM family includes 4 structurally related proteins36 of which neurofascin (NFASC) is among our SCZ-associated genes that overlap with the L1CAM interaction reference pathway. Neurofascin has been shown to participate in neurite outgrowth and stabilization of neuronal structures, the latter particularly through interaction with ankyrins.37 This interaction was also observed in our pathway analysis (interaction between L1 and ankyrins, P = 2.78×105). Ankyrin 1, 2, and 3 (ANK1, ANK2, and ANK3) are part of our SCZ-associated genes. Ankyrins are adaptor proteins that couple membrane proteins, such as voltage-gated sodium channels, to the developing cytoskeleton.38 The observed theme of cell adhesion molecules from the KEGG (Kyoto Encyclopedia of Genes and Genomes) database (P = 5.97×10−4) replicates a previous pathway analysis of large-scale SCZ genetic studies, even to the point that we observe many of the same specific genes, including CNTNAP2 and NRXN2.39
The generic neuronal system pathway in the Reactome database consists of several subpathways, such as transmission across chemical synapses, which was also among our highly significant findings. Thus, in addition to the pathways involved in neuronal growth and projection mentioned in the previous paragraphs, we observe associations with chemical neurotransmission. Among the genes in this pathway are several nicotinic acetylcholine receptors (CHRNA5, CHRNA2, and CHRNA3) that are also significant in their own subpathway (P = 2.21×10−4); voltage-gated calcium channels, such as CACNB2 and CACNB4, γ-aminobutyric acid B2 receptor (GABBR2); and a glutamate transporter (SLC1A3). Calcium signaling in particular has previously been identified as a core theme in the etiology of SCZ through large-scale genetic studies. Additional notable genes present in the transmission across chemical synapses pathway include the cyclic adenosine monophosphate response element binding protein 1 (CREB1) and the N-ethylmaleimide–sensitive factor (NSF). CREB1 is a transcription factor involved in mediating gene regulation after signaling events,40 whereas NSF is involved in vesicle trafficking and membrane fusion.41
A final, relevant theme among the significant pathways is the immune system. Antigen processing and presentation pathways from the KEGG and Biocarta databases were significantly enriched for SCZ-associated genes. Although the composition of these pathways was somewhat different between the 2 databases, the core genes of TAP1, TAP2 and HLA-DRA were common to both. The transporter associated with antigen processing (ie, TAP) is composed of a heterodimeric complex of TAP1 and TAP2. It is a key element in the immune recognition of cells compromised by virus infection or malignant transformation. The transporter is crucial in MHC class I antigen presentation by translocating proteasomal degradation products into the lumen of the endoplasmic reticulum for loading onto MHC class I molecules.42 An additional pathway related to the immune system, translocation of ZAP-70 kinase to the immunological synapse, was also significant. The zeta chain–associated protein kinase 70-kDa (ZAP-70) is an integral part of the adaptive immune system because it initiates signaling at the immunological synapse between a T cell and an antigen-presenting cell.43 Previous large-scale genetic studies have implicated the MHC region on chromosome 6,44 and most of the genes that we observe in the present study to be involved in these immune system–related pathways, such as the TAP and HLA genes, map to the MHC region. Therefore, the evidence that ties the MHC region, and by implication the immune system, to the etiology of SCZ is increasing.
CONCLUSIONS
We integrated results from a meta-analysis of 18 SCZ GWASs with existing informative SCZ databases to select SNPs for a family-based replication study. Results suggested a considerable enrichment of small P values in the replication study. Test results showed that this enrichment was statistically significant. Furthermore, of the SNPs with replication values of P<.01, the proportion of SNPs that had the same direction of effect as in the GWAS meta-analysis was about 90%, which is almost impossible to occur by chance. Finally, analyses suggested several significant pathways, which again suggested that SNPs replicated. Because the group of selected SNPs replicated as a whole, it follows that individual SNPs in the replication study replicate. A complication was that rather than a few SNPs having large effects, many SNPs appeared to have small effects. Although our pathway analyses of these SNPs implicated specific pathogenic processes, some caution is required before making strong statements about the replication status of individual SNPs. Other studies have reached a similar conclusion that for SCZ, many SNPs with small effects may be involved and these SNPs replicate as a group.21,45 Our article adds to these studies. The facts that we used a family-based replication (thereby minimizing the probability that population substructure accounts for the replication) and tested a much smaller set of SNPs pinpoints more precisely the genes that are likely involved.
Some of the SNPs selected through data integration had ranks up to 25 000 in the meta-analysis. Although these SNPs would never have been selected on the basis of their P value, they replicated as well as SNPs selected on the basis of having the top-ranked P values in the meta-analysis without data integration. This demonstrates the value of considering existing data sources. This conclusion also emerges from a study by Ayalew et al,46 who identified and prioritized genes involved in SCZ by gene-level integration of GWAS data with other genetic and gene expression studies. Some of the top findings in their study were the previously reported SCZ candidate gene TCF4 and the pathways involved in synaptic connectivity and glutamate signaling. Following the same concept but using a more statistical approach, we also identified SNPs in TCF4 and identified pathways related to cellular connectivity and signaling. Given that the number of (publicly) available databases and the tools to curate these data are increasing rapidly, future studies should be able to capitalize even more on data integration.
In addition to SNPs in two of the most promising SCZ candidate genes, we replicated several recently identified susceptibility genes for SCZ in an independent family-based replication sample. Our family-based replication also suggests that previous claims implicating the MHC region as a susceptibility region for SCZ cannot be discarded as population stratification artifacts. Pathway analyses of the many small effects reveal several biological themes involved in brain function, immune response, and biological functions of potential importance for the development of SCZ. The present investigation is, to our knowledge, the first family-based replication that confirms several of the recent mega- and meta-analyses top findings in a completely independent study sample using a different genotyping assay than what was used in the initial detection.
Supplementary Material
Acknowledgments
Funding/Support: This study was supported by grants R01HG004240, R01MH078069, and 1R01MH097283 (Drs Bukszár, Khachane, Aberg, and McClay) and grant R01MH080403 (Drs Sullivan and Liu) from the National Human Genome Research Institute.
Role of the Sponsors: The National Institute of Mental Health financially supported the design and conduct of the study but was not involved in manuscript review or approval.
Footnotes
Author Contributions: Dr van den Oord had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors contributed their critical reviews of the manuscript during its preparation and approved submission of the final manuscript. Drs Aberg and Liu contributed equally to the work.
Conflict of Interest Disclosures: None reported.
Additional Information: Dr Ophoff acted as an author on behalf of the GROUP (Genetic Risk and Outcome in Psychosis) Consortium. Members of the consortium include René S. Kahn, MD, Don H. Linszen, MD, Jim van Os, MD, PhD, Durk Wiersma, PhD, Richard Bruggeman, MD, Wiepke Cahn, MD, Lieuwe de Haan, MD, Lydia Krabbendam, PhD, and Inez Myin-Germeys, PhD.
Online-Only Material: The eMethods, eTables, and eFigures are available at http://www.jamapsych.com.
References
- 1.Collins PY, Patel V, Joestl SS, et al. Scientific Advisory Board and the Executive Committee of the Grand Challenges on Global Mental Health Grand challenges in global mental health. Nature. 2011;475(7354):27–30. doi: 10.1038/475027a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murray CJL, Lopez AD. The Global Burden of Disease. Geneva, Switzerland: World Health Organization; 1996. [Google Scholar]
- 3.Murray RM, Jones PB, Susser E, van Os J, Cannon M. The Epidemiology of Schizophrenia. Cambridge, England: Cambridge University Press; 2003. [Google Scholar]
- 4.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60(12):1187–1192. doi: 10.1001/archpsyc.60.12.1187. [DOI] [PubMed] [Google Scholar]
- 5.Miller G. Is Pharma running out of brainy ideas? Science. 2010;329(5991):502–504. doi: 10.1126/science.329.5991.502. [DOI] [PubMed] [Google Scholar]
- 6.Ripke S, Sanders AR, Kendler KS, et al. Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43(10):969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi J, Levinson DF, Duan J, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460(7256):753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niculescu AB, III, Segal DS, Kuczenski R, Barrett T, Hauger RL, Kelsoe JR. Identifying a series of candidate genes for mania and psychosis: a convergent functional genomics approach. Physiol Genomics. 2000;4(1):83–91. doi: 10.1152/physiolgenomics.2000.4.1.83. [DOI] [PubMed] [Google Scholar]
- 9.McClellan J, King MC. Genomic analysis of mental illness: a changing landscape. JAMA. 2010;303(24):2523–2524. doi: 10.1001/jama.2010.869. [DOI] [PubMed] [Google Scholar]
- 10.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 11.Bukszár J, Van den Oord E. RUTCOR Research Report: Mathematically-based Integration of Heterogeneous Data. RRR 16-2011. http://rutcor.rutgers.edu/pub/rrr/reports2011/16_2011.pdf. Accessed August 17, 2011.
- 12.Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40(7):827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- 13.Ng MY, Levinson DF, Faraone SV, et al. Meta-analysis of 32 genome-wide linkage studies of schizophrenia. Mol Psychiatry. 2009;14(8):774–785. doi: 10.1038/mp.2008.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Higgs BW, Elashoff M, Richman S, Barci B. An online database for brain disease research. BMC Genomics. 2006;7:70. doi: 10.1186/1471-2164-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konneker T, Barnes T, Furberg H, Losh M, Bulik CM, Sullivan PF. A searchable database of genetic evidence for psychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2008;147B(6):671–675. doi: 10.1002/ajmg.b.30802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudbridge F. Likelihood-based association analysis for nuclear families and unrelated subjects with missing genotype data. Hum Hered. 2008;66(2):87–98. doi: 10.1159/000119108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39:D712–D717. doi: 10.1093/nar/gkq1156. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pentchev K, Ono K, Herwig R, Ideker T, Kamburov A. Evidence mining and novelty assessment of protein-protein interactions with the ConsensusPathDB plugin for Cytoscape. Bioinformatics. 2010;26(21):2796–2797. doi: 10.1093/bioinformatics/btq522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: a database for integrating human functional interaction networks. Nucleic Acids Res. 2009;37:D623–D628. doi: 10.1093/nar/gkn698. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57(1):289–300. [Google Scholar]
- 21.International Schizophrenia Consortium. Purcell SM, Wray NR, Stone JL, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stefansson H, Ophoff RA, Steinberg S, et al. Genetic Risk and Outcome in Psychosis (GROUP) Common variants conferring risk of schizophrenia. Nature. 2009;460(7256):744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kraft P. Curses—winner’s and otherwise—in genetic epidemiology. Epidemiology. 2008;19(5):649–651. 657–658. doi: 10.1097/EDE.0b013e318181b865. [DOI] [PubMed] [Google Scholar]
- 24.Zhong H, Prentice RL. Bias-reduced estimators and confidence intervals for odds ratios in genome-wide association studies. Biostatistics. 2008;9(4):621–634. doi: 10.1093/biostatistics/kxn001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zollner S, Pritchard JK. Overcoming the winner’s curse: estimating penetrance parameters from case-control data. Am J Hum Genet. 2007;80(4):605–615. doi: 10.1086/512821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cherlyn SY, Woon PS, Liu JJ, Ong WY, Tsai GC, Sim K. Genetic association studies of glutamate, GABA and related genes in schizophrenia and bipolar disorder: a decade of advance. Neurosci Biobehav Rev. 2010;34(6):958–977. doi: 10.1016/j.neubiorev.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 27.Severinsen JE, Bjarkam CR, Kiaer-Larsen S, et al. Evidence implicating BRD1 with brain development and susceptibility to both schizophrenia and bipolar affective disorder. Mol Psychiatry. 2006;11(12):1126–1138. doi: 10.1038/sj.mp.4001885. [DOI] [PubMed] [Google Scholar]
- 28.Sakae N, Yamasaki N, Kitaichi K, et al. Mice lacking the schizophrenia-associated protein FEZ1 manifest hyperactivity and enhanced responsiveness to psychostimulants. Hum Mol Genet. 2008;17(20):3191–3203. doi: 10.1093/hmg/ddn215. [DOI] [PubMed] [Google Scholar]
- 29.Lin S, Shi Q, Nix FB, et al. A novel S-adenosyl-L-methionine:arsenic(III) methyltransferase from rat liver cytosol. J Biol Chem. 2002;277(13):10795–10803. doi: 10.1074/jbc.M110246200. [DOI] [PubMed] [Google Scholar]
- 30.Goytain A, Quamme GA. Functional characterization of ACDP2 (ancient conserved domain protein), a divalent metal transporter. Physiol Genomics. 2005;22(3):382–389. doi: 10.1152/physiolgenomics.00058.2005. [DOI] [PubMed] [Google Scholar]
- 31.Careddu MG, Allegrini S, Pesi R, Camici M, Garcia-Gil M, Tozzi MG. Knockdown of cytosolic 5′-nucleotidase II (cN-II) reveals that its activity is essential for survival in astrocytoma cells. Biochim Biophys Acta. 2008;1783(8):1529–1535. doi: 10.1016/j.bbamcr.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 32.Beveridge NJ, Tooney PA, Carroll AP, Tran N, Cairns MJ. Down-regulation of miR-17 family expression in response to retinoic acid induced neuronal differentiation. Cell Signal. 2009;21(12):1837–1845. doi: 10.1016/j.cellsig.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 33.Ohi K, Hashimoto R, Yasuda Y, et al. No association between the Bcl2-interacting killer (BIK) gene and schizophrenia. Neurosci Lett. 2009;463(1):60–63. doi: 10.1016/j.neulet.2009.07.063. [DOI] [PubMed] [Google Scholar]
- 34.Shatz CJ. MHC class I: an unexpected role in neuronal plasticity. Neuron. 2009;64(1):40–45. doi: 10.1016/j.neuron.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vitriol EA, Zheng JQ. Growth cone travel in space and time: the cellular ensemble of cytoskeleton, adhesion, and membrane. Neuron. 2012;73(6):1068–1081. doi: 10.1016/j.neuron.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maness PF, Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat Neurosci. 2007;10(1):19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- 37.Kriebel M, Wuchter J, Trinks S, Volkmer H. Neurofascin: a switch between neuronal plasticity and stability. Int J Biochem Cell Biol. 2012;44(5):694–697. doi: 10.1016/j.biocel.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 38.Bennett V, Chen L. Ankyrins and cellular targeting of diverse membrane proteins to physiological sites. Curr Opin Cell Biol. 2001;13(1):61–67. doi: 10.1016/s0955-0674(00)00175-7. [DOI] [PubMed] [Google Scholar]
- 39.O’Dushlaine C, Kenny E, Heron E, Donohoe G, Gill M, Morris D, International Schizophrenia Consortium Molecular pathways involved in neuronal cell adhesion and membrane scaffolding contribute to schizophrenia and bipolar disorder susceptibility. Mol Psychiatry. 2011;16(3):286–292. doi: 10.1038/mp.2010.7. [DOI] [PubMed] [Google Scholar]
- 40.Sakamoto K, Karelina K, Obrietan K. CREB: a multifaceted regulator of neuronal plasticity and protection. J Neurochem. 2011;116(1):1–9. doi: 10.1111/j.1471-4159.2010.07080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morgan A, Burgoyne RD. Membrane traffic: controlling membrane fusion by modifying NSF. Curr Biol. 2004;14(22):R968–R970. doi: 10.1016/j.cub.2004.10.045. [DOI] [PubMed] [Google Scholar]
- 42.Oancea G, O’Mara ML, Bennett WF, Tieleman DP, Abele R, Tampé R. Structural arrangement of the transmission interface in the antigen ABC transport complex TAP. Proc Natl Acad Sci U S A. 2009;106(14):5551–5556. doi: 10.1073/pnas.0811260106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Billadeau DD. T cell activation at the immunological synapse: vesicles emerge for LATer signaling. Sci Signal. 2010;3(121):e16. doi: 10.1126/scisignal.3121pe16. [DOI] [PubMed] [Google Scholar]
- 44.Sullivan PF, Daly MJ, O’Donovan M. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 2012;13(8):537–551. doi: 10.1038/nrg3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee SH, DeCandia TR, Ripke S, et al. Schizophrenia Psychiatric Genome-Wide Association Study Consortium (PGC-SCZ); International Schizophrenia Consortium (ISC); Molecular Genetics of Schizophrenia Collaboration (MGS) Estimating the proportion of variation in susceptibility to schizophrenia captured by common SNPs. Nat Genet. 2012;44(3):247–250. doi: 10.1038/ng.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ayalew M, Le-Niculescu H, Levey DF, et al. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction. Mol Psychiatry. 2012;17(9):887–905. doi: 10.1038/mp.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.